

Abstract
Antineutrophil cytoplasm antibody (ANCA)-associated vasculitides are small-vessel vasculitides that include granulomatosis with polyangiitis (formerly Wegener’s granulomatosis), microscopic polyangiitis, and eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome). Renal-limited ANCA-associated vasculitides can be considered the fourth entity. Despite their rarity and still unknown cause(s), research pertaining to ANCA-associated vasculitides has been very active over the past decades. The pathogenic role of antimyeloperoxidase ANCA (MPO-ANCA) has been supported using several animal models, but that of antiproteinase 3 ANCA (PR3-ANCA) has not been as strongly demonstrated. Moreover, some MPO-ANCA subsets, which are directed against a few specific MPO epitopes, have recently been found to be better associated with disease activity, but a different method than the one presently used in routine detection is required to detect them. B cells possibly play a major role in the pathogenesis because they produce ANCAs, as well as neutrophil abnormalities and imbalances in different T-cell subtypes [T helper (Th)1, Th2, Th17, regulatory cluster of differentiation (CD)4+ CD25+ forkhead box P3 (FoxP3)+ T cells] and/or cytokine–chemokine networks. The alternative complement pathway is also involved, and its blockade has been shown to prevent renal disease in an MPO-ANCA murine model. Other recent studies suggested strongest genetic associations by ANCA type rather than by clinical diagnosis. The induction treatment for severe granulomatosis with polyangiitis and microscopic polyangiitis is relatively well codified but does not (yet) really differ by precise diagnosis or ANCA type. It comprises glucocorticoids combined with another immunosuppressant, cyclophosphamide or rituximab. The choice between the two immunosuppressants must consider the comorbidities, past exposure to cyclophosphamide for relapsers, plans for pregnancy, and also the cost of rituximab. Once remission is achieved, maintenance strategy following cyclophosphamide-based induction relies on less toxic agents such as azathioprine or methotrexate. The optimal maintenance strategy following rituximab-based induction therapy remains to be determined. Preliminary results on rituximab for maintenance therapy appear promising. Efforts are still under way to determine the optimal duration of maintenance therapy, ideally tailored according to the characteristics of each patient and the previous treatment received.
Keywords: Antineutrophil cytoplasmic antibody-associated vasculitis, granulomatosis with polyangiitis (Wegener’s granulomatosis), microscopic polyangiitis, Churg-Strauss syndrome (eosinophilic granulomatosis with polyangiitis), cyclophosphamide, rituximab
Introduction
Antineutrophil cytoplasm antibody (ANCA)-associated vasculitides are small-vessel vasculitides that include granulomatosis with polyangiitis (GPA; formerly Wegener’s granulomatosis), microscopic polyangiitis (MPA), and eosinophilic granulomatosis with polyangiitis (EGPA; also known as Churg–Strauss syndrome) (Table 1) (1–4). Renal-limited ANCA-associated vasculitis can be considered the fourth entity in this group, although it eventually corresponds to a kidney-limited form of MPA or GPA in practice. Despite the rarity and still unknown cause(s) of ANCA-associated vasculitides, research pertaining to these diseases has been very active and steadily increasing over the past three decades. The results of several clinical and more basic fundamental studies demonstrated that each of these diseases has some different pathogenic mechanisms and genetic associations (5–8). From a therapeutic point of view, treatment strategies have been gradually better defined, and several targeted biologic agents, initially developed for other diseases, have been studied and/or are still under investigation (9–13). One of them is the monoclonal antiCD20 antibody rituximab, which was demonstrated in two randomized controlled trials to be a possible alternative to the conventional cytotoxic cyclophosphamide to induce remission in adults with severe GPA or MPA combined with glucocorticoids (14–16). Investigations have already been conducted and some others are ongoing to evaluate rituximab for maintenance therapy (17). Several other trials are under way to further optimize the treatment strategies for patients with more specific forms of ANCA-associated vasculitides or at different times of their disease course (18, 19). This article summarizes the results of some of the main studies recently published on ANCA-associated vasculitides that may impact practice.
Table 1.
Classification criteria and definitions of the ANCA-associated vasculitides according to the American College of Rheumatology (ACR, 1990; microscopic polyangiitis was not yet individualized as a specific entity at that time) and the 2012 Chapel Hill nomenclature (1–4)
| 1990 ACR classification criteria for Wegener’s granulomatosis |
For purposes of classification, a patient shall be said to have Wegener’s granulomatosis if at least 2 of these 4 criteria are present. The presence of any 2 or more criteria yields a sensitivity of 88.2% and a specificity of 92.0%.
|
| 1990 ACR classification criteria for Churg–Strauss syndrome |
For purposes of classification, a patient shall be said to have Churg–Strauss syndrome if at least 4 of these 6 criteria are present. The presence of any 4 or more criteria yields a sensitivity of 85% and a specificity of 99.7%.
|
| Definition of ANCA-associated vasculitides in the nomenclature of systemic vasculitis adopted in 2012 by the Chapel Hill consensus conference |
| Large-vessel vasculitis: Giant-cell arteritis; Takayasu arteritis |
| Medium-sized-vessel vasculitis: Polyarteritis nodosa; Kawasaki disease |
| Small-vessel vasculitis*: |
| ANCA-associated vasculitides† |
| Granulomatosis with polyangitiis (Wegener’s granulomatosis) |
| Necrotizing granulomatous inflammation usually involving the upper and lower respiratory tract, and necrotizing vasculitis affecting predominantly small to medium vessels (e.g., capillaries, venules, arterioles, arteries, and veins). Necrotizing glomerulonephritis is common. |
| Eosinophilic granulomatosis with polyangiitis (Chrug-Strauss syndrome) |
| Eosinophil-rich and necrotizing granulomatous inflammation often involving the respiratory tract. Necrotizing vasculitis predominantly affecting small to medium vessels and associated with asthma and eosinophilia. ANCA is more frequent when glomerulonephritis is present. |
| Microscopic polyangiitis |
| Necrotizing vasculitis with few or no immune deposits predominantly affecting small vessels (i.e., capillaries, venules, or arterioles). |
| Necrotizing arteritis involving small and medium arteries may be present. Necrotizing glomerulonephritis is very common. Pulmonary capillaritis often occurs. Granulomatous inflammation is absent. |
| Immune-complex small-vessel vasculitides |
| IgA vasculitis (Henoch–Schönlein purpura) |
| Cryoglobulinemic vasculitis |
| Hypocomplementemic urticarial vasculitis (anti-C1q vasculitis) |
| Variable vessel vasculitis: Behcet’s disease; Cogan’s syndrome |
| Single-organ vasculitis: cutaneous leukocytoclastic angiitis; cutaneous arteritis; primary central nervous system vasculitis; isolated aortitis; others |
| Vasculitis associated with systemic disease: lupus vasculitis; rheumatoid vasculitis; sarcoid vasculitis; others |
| Vasculitis associated with probable etiology: hepatitis C virus–associated cryoglobulinemic vasculitis; hepatitis B virus-associated vasculitis; syphilis-associated aortitis; drug-associated immune complex vasculitis; drug-associated ANCA-associated vasculitis; cancer-associated vasculitis; others |
Large vessels are the aorta and its major branches and the analogous veins. Medium vessels are the main visceral arteries and veins and their initial branches. Small vessels are intraparenchymal arteries, arterioles, capillaries, venules, and veins.
Necrotizing vasculitis with few or no immune deposits predominantly affecting small vessels (i.e., capillaries, venules, arterioles, and small arteries) associated with myeloperoxidase (MPO) ANCA or proteinase 3 (PR3) ANCA. Not all patients have ANCA. Add a prefix indicating ANCA reactivity, e.g., MPO-ANCA, PR3-ANCA, ANCA-negative.
ACR: American College of Rheumatology; ANCA: antineutrophil cytoplasm antibody
Epidemiology
ANCA-associated vasculitides affect both genders equally. The average age at diagnosis is in the fifth decade, but young children and older adults can be affected too. Most patients (93%–98%) are white (Caucasian and Hispanics). However, recent studies of MPA, the most common ANCA-associated vasculitis in Asians, showed that its annual incidence in Japan is similar to that in the United Kingdom (20). The estimated annual incidences of GPA or MPA are close and vary from 2 to 12 cases per million population, with prevalences from 23 to 160 cases per million population (21). EGPA is much rarer than GPA or MPA, with an incidence of 1–4 cases per million population and a prevalence of approximately 10–20 cases per million population (8, 22, 23).
Intriguingly, the incidence of GPA, and perhaps that of the two other ANCA-associated vasculitides, seems to have increased over the past decades, according to several European studies. These changes could be explained in part by the better awareness of the disease and thus more frequent and earlier diagnosis (24). However, a recent British study suggested peaks of incidence every 8 to 10 years for GPA (17.4 vs 4.53 cases/million/year during vs not during peaks), with no such periodicity for MPA (25). Many studies suggested some seasonal variations in the incidence of GPA but not reproducibly during the same periods of the year (21, 26).
Genetics
ANCA-associated vasculitides are not inherited or genetic diseases. Familial forms are extremely rare. Several international teams have conducted studies on genome-wide associations. The most reproducible genetic associations reported are for molecules of the major histocompatibility complex HLA-DPB1*0401 for patients with ANCA (odds ratio 3.38) and allele deficiency of alpha-1 antitrypsin for GPA (serpin A1; PI*Z alleles in 5%–27% of GPA patients, PI*S alleles in 11.58%, homozygosity for deficiency ZZ, SS, or SZ associated with more severe forms) (27, 28). The other main and most recent finding is that the strongest genetic associations are with ANCA antigenic specificity rather than with clinical syndrome (MPA vs GPA): antiproteinase 3 ANCA (PR3-ANCA)-associated vasculitis is associated with HLA-DP, the genes encoding alpha 1-antitrypsin (SERPINA1) and PR3 (PRTN3), whereas antimyeloperoxidase ANCA (MPO-ANCA)-associated vasculitis is more often associated with HLA-DQ. As detailed later in this article, this finding underscores important differences between PR3- and MPO-ANCA-associated vasculitides. One may eventually categorize patients with ANCA-associated vasculitides according to their ANCA status rather than as having GPA or MPA (23). Genes associated with EGPA have been more challenging to study, mainly because of the small number of patients analyzed so far and may include HLA-DRB1*04 and *07 alleles (27, 29).
Clinical and biological findings, diagnosis
The main characteristics and differences of GPA, MPA, and EGPA are summarized in Table 2, and Figures 1–7 show some of the most typical manifestations. ANCA-associated vasculitides are all potentially life-threatening, but less severe forms exist. For example, GPA can remain localized to the upper airway where it is often persistent, multi-relapsing, and/or refractory to treatments.
Table 2.
Main characteristics of the 3 ANCA-associated vasculitides
| Microscopic polyangiitis | Eosinophilic granulomatosis with polyangiitis (Churg–Strauss syndrome) | Granulomatosis with polyangiitis (Wegener’s granulomatosis) | |
|---|---|---|---|
| Clinical manifestations | |||
| Constitutional symptoms (fever, arthralgia, myalgia) | 55–80% | 30–50% | 70–100% |
| Skin | Purpura (35–60%) | Purpura, pseudourticarial rash (50–70%) | Purpura (10–50%) |
| ENT manifestations | Few patients (2–30%), not specific, not destructive, and not granulomatous | Frequent (20–80%): allergic rhinitis, sinus polyposis (not destructive) | Frequent (50–95%): crusting rhinitis, destructive sinusitis, saddle-nose deformity, nasal septum deformity, otitis media |
| Lung involvement | Frequent (60–80%): alveolar hemorrhage | Frequent (50%): transient patchy infiltrates, eosinophil pleural effusion, rarely nodules | Frequent (60–80%): lung plain and/or excavated nodules, alveolar hemorrhage, bronchial and/or subglottic stenosis |
| Asthma | No | Yes (approcimately 100%) | No |
| Kidney involvement | Very frequent: glomerulonephritis (necrotizing extra-capillary), 80% | Not frequent: glomerulonephritis (necrotizing extra-capillary), 20% | Frequent: glomerulonephritis (necrotizing extra-capillary), 60–80% |
| Peripheral neuropathy (mononeuritis multiplex) | Possible (35%) | Very frequent (65–75%) | Possible (25%) |
| Other “classical” manifestations | Venous thrombosis (7–8%) | Cardiac manifestations (10–50%; cardiomyopathy), venous thrombosis (7–8%) | Eye manifestations (scleritis, orbital tumor), pachymeningitis, venous thrombosis (7–8%) |
| Biology | |||
| Standard | Non-specific inflammatory syndrome; check creatinine and urine analysis (red blood cell casts?) | Eosinophilia, often >3,000/mm3, Non-specific inflammatory syndrome | Non-specific inflammatory syndrome; check creatinine and urine analysis (red blood cell casts?) |
| ANCA | Yes (60–80%): mainly MPO-ANCA (perinuclear labeling pattern in immunofluorescence) | Yes (30–40%): mainly MPO-ANCA (perinuclear labeling pattern in immunofluorescence) | Yes (90% if systemic disease): mainly PR3-ANCA (cytoplasmic labeling pattern in immunofluorescence) |
| Radiology | |||
| Check chest X-ray and/or CT scan for alveolar hemorrhage (ground-glass opacities); other imaging studies according to clinical presentation | Check chest X-ray and/or CT scan for labile and transient lung infiltrates (rarely alveolar hemorrhage or nodules); sinus X-ray and/or CT scan for non-erosive sinusitis, polyps; other imaging studies according to clinical presentation | Check chest X-ray and/or CT scan for alveolar hemorrhage (ground-glass opacities), lung nodules, excavated or not, and subglottic and/or bronchial stenosis; sinus X-ray and/or CT scan for erosive sinusitis, pseudotumor; other imaging according to clinical presentation | |
| Histology | |||
| Necrotizing vasculitis of small-sized vessels; no granuloma (rare cases) | Granuloma, including eosinophils (frequent); necrotizing vasculitis of small-sized vessels | Granuloma (frequent but not always); necrotizing vasculitis of small-sized vessels | |
ANCA: antineutrophil cytoplasm antibody; CT scan: computerized tomography; ENT: ear, nose, throat; MPO: myeloperoxidase: PR3: proteinase 3
Figure 1.
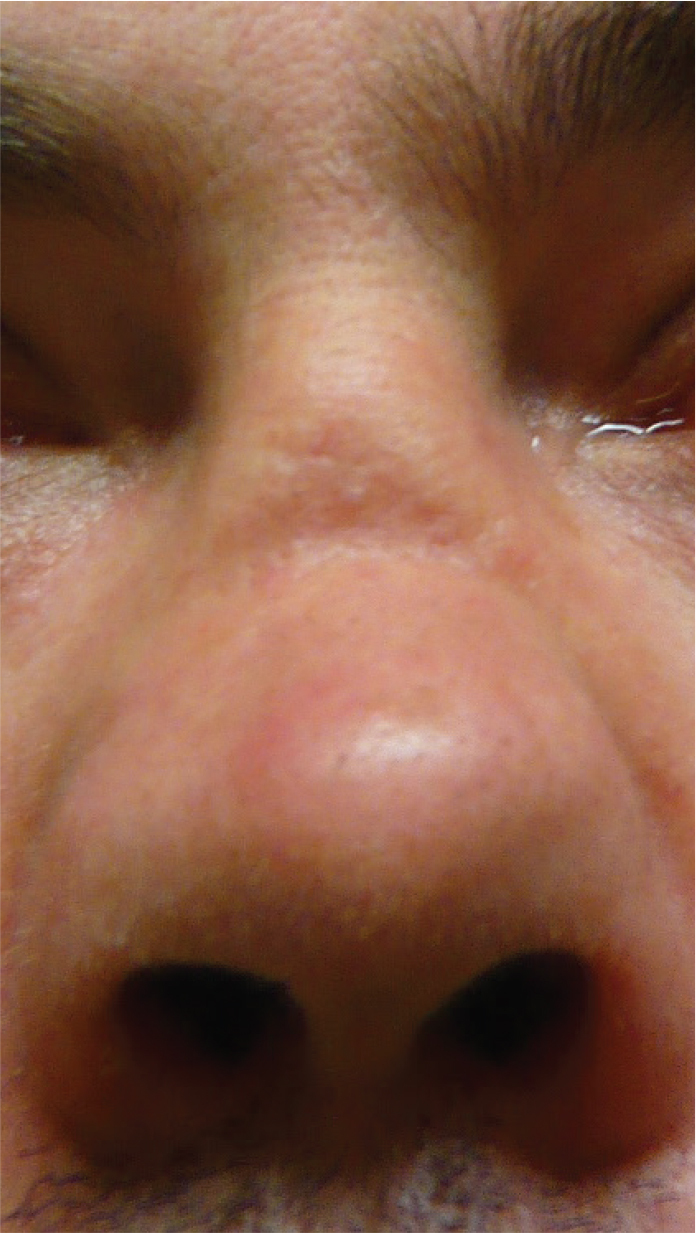
Saddle-nose deformity in a patient with granulomatosis with polyangiitis (GPA)
Figure 2.

Nasal septum perforation in a patient with polyangiitis (GPA)
(Note the light going from one nostril to the other through the perforated nasal septum and the crusty bleedy posterior wall of the sino-nasal cavity)
Figure 3.

Purpuro-ecchymotic skin lesions on the legs of a patient with eosinophilic granulomatosis with polyangiitis (EGPA)
(Legs are the most commonly involved areas for skin lesions in antineutrophil cytoplasm antibody (ANCA)-associated vasculitides)
Figure 4.

Nodular cutaneous lesions in a patient with granulomatosis with polyangiitis (GPA)
Elbows are a common location for such skin lesions in granulomatosis with polyangiitis (GPA) and eosinophilic granulomatosis with polyangiitis (EGPA).
Figure 5.

Computerized tomography (CT) scan of sinuses in a patient with granulomatosis with polyangiitis (GPA)
(Note the perforated nasal septum and bilateral fulfillment of maxillary sinuses-sinusitis)
Figure 6.

Chest computerized tomography (CT) in a patient with granulomatosis with polyangiitis (GPA)
(Note the multiple plain lung nodules surrounded by ground glass opacities suggestive of associated peri-nodular alveolar hemorrhage)
Figure 7.

Chest computerized tomography (CT) in a patient with microscopic polyangiitis (MPA)
(Note the diffuse ground-glass opacities, which are suggestive of alveolar hemorrhage, with some pseudonodular consolidation appearance in the left lung)
The diagnosis of ANCA-associated vasculitis relies on the combination of clinical findings and results of imaging studies and basic and nonspecific biology tests (inflammatory markers such as erythrocyte sedimentation rate and C-reactive protein level, complete blood count, renal parameters, and urine sediment analysis). In addition, more specific methods such as ANCA testing and, when feasible, biopsy of the affected organ can also be performed (Figure 8). Ruling out the differentials or complications of therapy, including infections or malignancy, is mandatory during the entire disease course. Some drugs can induce (propylthiouracil is the most famous one) and/or mimic (levamisole–cocaine) ANCA-associated vasculitides (30–33). Most routinely used methods for ANCA testing include indirect immunofluorescence (to detect c-ANCA with a cytoplasmic labeling pattern, p-ANCA with a perinuclear pattern, and sometimes x-ANCA for an atypical pattern) and enzyme-linked immunosorbent assay (ELISA; to detect PR3- and/or MPO-ANCA; non-routine ELISA tests can also detect ANCA with other specificities such as anti-elastase or anti-cathepsin G). Newer detection techniques include multiplex (“bead”) technology, automated image analysis of immunofluorescence patterns, and “third-generation PR3-ANCA and MPO-ANCA ELISA.” It should be kept in mind that a positive ANCA test result can be observed in other conditions, such as auto-immune hepatitis, ulcerative colitis, infection with hepatitis C virus or HIV, or infectious endocarditis, without associated vasculitis. In addition, with the presently available and routine ANCA detection and measurement tests, ANCA status and titer monitoring should not be used to adjust therapy, and up to 20% of GPA and MPA patients and more than 60% of those with EGPA are ANCA-negative. These ANCA-negative GPA patients most often have limited disease, although it may progress later on to a more severe and diffused form and sometimes become ANCA-positive. The ANCA-negative EGPA patients may more frequently have cardiac manifestations but less renal or peripheral nerve involvement (34–38).
Figure 8.

Histology of muscle–nerve biopsy in a patient with eosinophilic granulomatosis with polyangiitis (EGPA)
(Note the massive vessel wall and perivascular infiltrate by inflammatory cells, mainly eosinophils, the vessel wall necrosis, and subsequent vessel lumen occlusion)
Biopsies of skin purpuric lesions are easy to perform and will usually reveal vasculitis, although most medium- or small-sized vessel vasculitis can cause the same type of histological lesions. Vascular and extravascular eosinophilic granulomas are more suggestive of EGPA. Nasal or sinus biopsies have low sensitivity (<40%), even when performed by trained surgeons on ulcerated areas and with deep mucosa samples. Open lung biopsy, with some lung involvement on imaging, yields high sensitivity along with renal biopsy but is an invasive procedure (39, 40). Renal biopsy is easier to perform and can show the classical pauci-immune crescentic and necrotizing glomerulonephritis in patients with renal involvement. The histological pattern can also help determine the renal prognosis because the presence of sclerosis at diagnosis already reflects intense damage that may not be reversible (41). Focal glomerular lesions (>50% of normal glomeruli) and crescentic categories (crescents in >50% of the glomeruli) have better prognosis than sclerotic (>50% of glomeruli) or mixed categories. In practice, the need for an invasive biopsy in an ANCA-positive patient with typical clinical manifestations of GPA or MPA and no evidence of any infection, cancer, or drug-induced disease remains a case-based decision.
Pathogenesis
The precise cause(s) of ANCA-associated vasculitides remain(s) unknown. The hypothesis of an infectious agent, such as Staphylococcus aureus for GPA, (over)activating the immune system has been repeatedly suggested; however, even if this is implicated, it can barely be sufficient to cause or explain the full-blown disease and its multiple facets by itself (42–44). B cells possibly play a major role in the pathogenesis because they produce ANCAs. Imbalances in different T-cell subtypes (Th1, Th2, Th17, regulatory CD4+ CD25+ FoxP3+ T cells, etc.) and/or cytokine–chemokine networks can also lead to or at least participate in the rupture of tolerance, triggering auto-immunity and/or oxidative burst aggressive toward endothelial cells. An excessive and abnormally sustained antigenic presentation of PR3 and/or MPO has also been implicated in patients with GPA or MPA through their overexpression on neutrophils, genetically determined, as well as on endothelial cell membranes and the formation of neutrophil extracellular traps (NETs) and microparticles by neutrophils, which include the PR3 and/or MPO molecules (43, 45–47).
The pathogenic role of MPO-ANCAs has been supported by animal models by the active transfer of MPO-ANCAs and a single case of MPA in the newborn of a mother with anti-MPO-ANCAs (passive transplacental transfer) (48, 49). Additional experiments demonstrated the major importance of neutrophils, neutrophil activation pathway, and alternative complement pathway (mainly through C5a) in the induction of the MPO-ANCA-induced animal model (50, 51). Recent studies showed that the blockade of the C5a receptor (C5aR, CD88) with oral CCX168 could not only prevent but also limit renal disease in this murine model (18). Based on the promising results of an exploratory open-label study on GPA or MPA with renal disease, a randomized placebo-controlled trial is now ongoing to further investigate this C5aR-blocking agent in patients with GPA or MPA (Clinicaltrials.gov Identifier: NCT02222155).
Conversely, we lack convincing evidence to support the pathogenic role of PR3-ANCAs or to explain why some patients with biopsy-confirmed GPA or MPA have typical clinical manifestations despite being ANCA-negative. There may be several explanations. First, homology between human and murine PR3 is less than that for MPO. Hence, animal models are more difficult to generate, and complex alterations in mice are required before achieving some vasculitic changes close to those seen in GPA (52–54). Second, only a fraction of ANCAs are pathogenic in an individual, i.e., only those ANCAs directed toward one or a few specific epitope(s) are pathogenic. A recent study of MPO-ANCA-positive patients demonstrated that only a subset of MPO-ANCAs associated more strongly and specifically with disease activity than others that were directed toward different MPO epitopes (55). Moreover, these specific ANCAs, which were directed toward the linear amino-acid sequence 447–459 of the MPO molecule, were detected in many ANCA-negative patients with MPA and correlated well with disease activity. When they were injected in mice, they triggered the development of (proliferative, but not necrotizing) glomerulonephritis. Routine ANCA tests do not properly detect these specific MPO-epitopes-ANCA447–459 because such tests are based on total serum. Serum immunoglobulins (Ig) had to be first purified to allow for the detection of these specific MPO-epitopes-ANCAs447–459 in this study. The authors showed that a serum factor (likely a fraction of ceruloplasmin) indeed bound to these specific MPO-epitopes-ANCAs447–459, thereby preventing the detection of the most clinically relevant ANCAs with routine tests, particularly in ANCA-negative patients. More recently, a Japanese group reported that some MPO-ANCAs reacted with moesin, a heparin-binding protein in the plasma membrane of the cellular renal cortex, to activate glomerular endothelial cells in SCG/Kj mice, which spontaneously develop MPO-ANCA-associated renal disease. Such ANCAs with anti-moesin specificity can be detected in patients with MPO-ANCA-associated vasculitis (56). Specific alterations and “maturation” of ANCAs may also be necessary for them to become pathogenic, including the selection of higher-affinity PR3-ANCAs in nasal mucosa granulomas or modulation of their sialylation levels (57, 58).
Apart from this hypothesis of a few more-pathogenic subsets of ANCAs, other auto-antibodies may be involved. Kain et al. (59, 60) found antilysosome-associated membrane protein 2 antibodies in more than 90% of patients with PR3- as well as MPO-ANCA-related glomerulonephritis. However, these results were not replicated by another group (61).
Antiendothelial cell antibodies can be detected in many GPA and MPA patients, but whether they can cause vessel lesions or simply occur as a consequence of vessel damage remains debated (62–64). The precise molecular targets of these anti-endothelial cell auto-antibodies remain to be better identified.
Finally, in EGPA, only 30%–40% of the patients are ANCA-positive, mainly with perinuclear MPO-ANCAs (8). We lack an animal model of EGPA, and MPO-ANCA induced murine models do not show any of the main features of EGPA (i.e., blood eosinophilia, tissue eosinophilic granulomas, or obstructive lung disease). Eosinophils possibly play a central and/or additional role in the development of EGPA directly or through their granule degradation products. Several cytokine network imbalances may be at stake (65, 66). Overall, the pathogenic mechanisms of EGPA are much less known than those that are implicated in GPA or MPA and are probably quite different, at least for ANCA-negative EGPA. For this reason, and some important clinical and genetic differences, whether EGPA should be distinguished more clearly from GPA and MPA remains debated, thereby removing the concept of ANCA-associated vasculitides as a group of three diseases.
Treatment
Treatment of severe ANCA-associated vasculitides comprises two phases: remission induction therapy based on the combination of glucocorticoids and another immunosuppressive agent, and once remission is achieved, maintenance therapy (to maintain remission). Besides potent therapies used for more than 50 years to induce remission such as cyclophosphamide, others (biologic agents) have been more recently found to be promising or even (for rituximab) as effective as and possibly less toxic than cyclophosphamide, at least for the risks of infertility and late cancers. However, a few patients still experience refractory disease and/or unrelenting relapses, which underscores the continuing need for newer and more effective therapies (67).
Over the past decades, research into therapies for ANCA-associated vasculitis has evolved from good monocentric studies, although small sample-sized or not always generalizable, to multicentric national, then continental, and now world-wide controlled trials, which can thus aim at enrolling more than 250 patients, thereby capable of answering more questions (13, 68).
Glucocorticoids remain the cornerstone of therapy for ANCA-associated vasculitides, but efforts are still ongoing to identify potent glucocorticoid-sparing agents. The initial dosage of glucocorticoids for patients with active and severe disease is usually 1 mg/kg/day prednisone-equivalent, sometimes preceded by 1 to 3 boluses of methylprednisolone (7.5 to 15 mg/kg/day) (69). After the first 2–4 weeks of treatment, the dose of glucocorticoids is reduced by approximately 10% every 1 to 2 weeks to achieve a half-dose (0.5 mg/kg/d) at approximately the third month of treatment. In the United States, many centers aim to stop glucocorticoids at 6 months (70, 71). The optimal schedules and pace for the initial dose taper and the duration of low-dose treatment with glucocorticoids remain controversial (72, 73). The PEXIVAS study (13) may help to answer the first question about tapering, whereas another international study (TAPIR; Clinicaltrials.gov Identifier: NCT01940094) may provide some evidence for the effectiveness or not of prolonged low-dose glucocorticoid use.
Glucocorticoids can be used alone as first-line therapy to effectively induce remission in patients with non-severe MPA or non-severe EGPA (with a five-factor score of 0), on the basis of few cohort studies (74, 75). However, more than half of these patients eventually require the addition of another immunosuppressant because of progressive, refractory, or relapsing disease. An ongoing controlled study is evaluating the effectiveness of a combination of glucocorticoids and azathioprine (vs glucocorticoids and a placebo of azathioprine) as first-line treatment for these types of patients (CHUSPAN2; Clinicaltrials.gov Identifier: NCT00647166). Importantly, all GPA patients with limited as well as severe forms and those with severe MPA or EGPA [with life-threatening manifestation(s) or any major organ involvement] must receive a combination of glucocorticoids and another immunosuppressant (19, 69, 76).
Cyclophosphamide and rituximab, combined with glucocorticoids, are now two possible agents for remission induction in patients with severe GPA or MPA. They can induce a response or remission in >80% of patients. For patients with limited GPA, methotrexate (0.3 mg/kg/week, orally or subcutaneously) can be considered instead of cyclophosphamide (77) because methotrexate is as effective as cyclophosphamide in achieving remission in such patients but is associated with a higher subsequent rate of relapse (78). For patients with severe EGPA, data are too limited at this time to consider rituximab, a potential alternative to cyclophosphamide for first-line induction therapy, but it could be used for refractory cases (79).
Cyclophosphamide can be administered as intravenous “pulses” (boluses at regular intervals; 15 mg/kg, with a maximum of 1200 mg/pulse, every 14 days for 1 month then every 3 weeks) or continuous oral tablets (2 mg/kg/day, with a maximum of 200 mg/day) with doses adjusted to patient age and glomerular filtration rate. Both routes are equally effective in achieving remission, but oral daily cyclophosphamide is associated with increased frequency of neutropenia (80) and in some (but not all) studies, infections (81). Patients receiving oral cyclophosphamide indeed receive a higher cumulative dose, which is about 16 g compared with 8 g for those receiving the intravenous “pulse” regimen for the same 3-month duration. This difference in cumulative dose may also explain why, with a longer follow-up (median, 4.3 years), a study comparing pulsed intravenous and continuous oral routes for induction (followed by azathioprine maintenance) showed that oral continuous cyclophosphamide use could be associated with lower subsequent relapse rate (20% instead of 40%) (82). Conversely, the risk of infertility and/or late complications (i.e., cancers, mainly of the bladder, but also lymphomas or skin cancers) is also directly linked with the cumulative dose of cyclophosphamide. We lack a consensual threshold for the maximum “safe” cumulative dose of cyclophosphamide. However, most cases of cancers in GPA patients exposed to cyclophosphamide occurred in those who had received more than 36 g (83).
Rituximab is a chimeric monoclonal antiCD20 (B-cell) antibody that has been evaluated in two randomized trials (RAVE and RITUXVAS) and subsequently approved in April 2011 by the United States Food and Drug Administration, as an alternative to cyclophosphamide to treat severe forms of GPA and MPA in adults with a history of ANCA positivity and combined with glucocorticoids (14, 16). In the two studies, rituximab was found not inferior to cyclophosphamide in inducing remission at 6 months, with (disappointingly) comparable rates of side effects including infections (mainly community-acquired upper and lower respiratory-tract infections). The doses of rituximab used in these studies were 4 infusions of 375 mg/m2 each given at 1-week intervals. When choosing between rituximab and cyclophosphamide for induction, one should consider the patient’s plans for pregnancy, particularly for females of childbearing age, and comorbidities, apart from the high cost of rituximab. Rituximab is clearly indicated for patients with contraindications to cyclophosphamide and those who have already received large cumulative doses of cyclophosphamide (>20 or 30 g, but consensus is lacking on the threshold dose). According to the RAVE trial results, the response to rituximab, compared to cyclophsophamide, may be superior in relapsers who are naive of rituximab and possibly those PR3-ACAN positive patients (as compared to MPO-ANCA positive ones) (14, 15). However, no other MPA or GPA patient subsets would overly benefit more from one induction agent than the other. Studies yielded some conflicting results on whether rituximab action would be less or slower to achieve an effect for granulomatous manifestations in GPA compared with its effect for vasculitic manifestations (84).
Plasma exchange (7 sessions over 2 weeks) combined with induction treatment can be considered for patients with severe ANCA-associated vasculitis with active glomerulonephritis and/or alveolar hemorrhage, particularly as a rescue treatment for patients who did not respond well and/or rapidly to the induction therapy (85). However, the benefit of plasma exchange in such patients has not been formally studied or demonstrated. A randomized international trial (PEXIVAS) aims at determining more clearly whether or not plasma exchange is beneficial in terms of overall survival and renal recovery at 3 years (13, 86).
Following induction with cyclophosphamide, patients who achieve clinical remission can be switched to a less toxic immunosuppressant for maintenance, usually after 3 months to a maximum of 6 months of cyclophosphamide (11). The maintenance treatment should last at least 18–24 months (11). The most commonly used maintenance agents are azathioprine (2 mg/kg/day, orally) and methotrexate (0.3 mg/kg/week, orally, intramuscularly, or subcutaneously), both equally effective and safe (10, 11). In patients with renal insufficiency, methotrexate may accumulate faster and therefore may be more hazardous to use. Leflunomide (20 mg/day, orally) is an option for patients with intolerance to azathioprine or methotrexate. Mycophenolate mofetil was found to be associated with higher relapse rate than azathioprine in the European IMPROVE trial (at 4 years, 55% vs 38% relapses) and thus should be used only when no other option is possible (12). As for optimal duration of low-dose prednisone, the optimal duration of maintenance with these other agents remains unknown (87). Past studies with maintenance for 1–3 years showed that irrespective of the induction and maintenance regimen, the relapse rate in GPA could still reach 51%–64% at 7 years (10, 11, 88–90). The preliminary results of the European REMAIN trial, which compared 2 vs 4 years of maintenance, suggest that the continuation of maintenance for 4 years may be associated with fewer relapses, in particular for patients with persistent ANCA positivity at remission (results announced at the 2015 ANCA Workshop, London, UK–May 2015).
The maintenance strategy following the rituximab-based induction treatment currently lacks consensus. In the RAVE trial, no maintenance therapy was given after the fourth rituximab infusion. The relapse rates were comparable at 18 months in the rituximab and cyclophosphamide–azathioprine arms, but they remained unsatisfactorily high, at approximately 30% (15). Several options are therefore possible following rituximab-based induction therapy. Some groups suggested that re-treatment with rituximab should be considered according to B-cell and/or CD19+ CD20+ lymphocyte count monitoring (with a repeat full course of four rituximab infusions with B-lymphocyte reconstitution and/or ANCA reappearance or significant titer increase). In one single-center study, such an approach was efficient (91). However, the reliability of CD19+ B-cell and ANCA monitoring to predict a flare has been challenged in several other studies, including the RAVE and RITUXVAS trials. Several other groups reported their promising experience with systematic maintenance infusions at regular intervals every 6–12 months independent of ANCA status or B-cell count and using different dosages (92–96). The French Vasculitis Study Group MAINRITSAN trial, a prospective randomized open-label study to compare azathioprine and rituximab (500 mg every 6 months) for maintenance in GPA or MPA patients following a glucocorticoid-cyclophosphamide-based induction, found a lower rate of major relapses with rituximab at 28 months (5.3% vs 29.3%)(97). A somewhat similar international study (RITAZAREM; Clinicaltrials.gov Identifier: NCT 01697267) is evaluating rituximab, 1000 mg every 4 months (vs azathioprine), for maintenance in relapsing ANCA-positive patients with GPA or MPA following a glucocorticoid–rituximab-based induction. Because none of these rituximab-based maintenance options have been validated and/or officially approved yet, the remaining choices following rituximab-based induction are, in practice, to retreat (with rituximab) only for clinical relapse or to give a conventional maintenance immunosuppressive agent such as azathioprine or methotrexate (92, 98).
Other or additional (“on-top-of”) agents might further improve the rate of sustained remission and limit the risk of subsequent relapses. Cotrimoxazole (trimethoprim–sulfamethoxazole) cannot substitute for immunosuppressive treatment (99), but given at a high dose (320 mg/day trimethoprim, 1600 mg/day sulfamethoxazole) and combined with the usual treatments of GPA, it could further reduce the rate of localized ENT relapse by 40% at 1 year, regardless of the presence or absence of S. aureus on nasal swabs (100). Importantly, cotrimoxazole must also be prescribed but at a lower dose (160 mg trimethoprim and 800 mg sulfamethoxazole, 3 days/week) for prophylaxis against Pneumocystis jiroveci pneumonia in patients who are receiving induction therapy with cyclophosphamide or rituximab and for several months after their discontinuation (69). Patients allergic to cotrimoxazole should be given alternative prophylaxis with oral dapsone (100 mg/day, in the absence of glucose-6-phosphate dehydrogenase deficit) or atovaquone (1500 mg/day) (101). Etanercept in addition to conventional treatment has been investigated in GPA, but its use was found to be associated with increased occurrence of malignancies (102). Since then, antitumor necrosis factor agents have rarely been used to treat ANCA-associated vasculitis, except for some refractory cases and as a last choice. Belimumab, a monoclonal antibody directed against B-lymphocyte stimulator/B-cell activating factor (BLyS/BAFF) combined with conventional maintenance agents (azathioprine or methotrexate; BREVAS, Clinicaltrials.gov Identifier: NCT01663623) is under investigation. As mentioned above, a placebo-controlled trial is ongoing to evaluate the addition of CCX168, a new oral C5aR-blocking agent, to standard induction treatments (glucocorticoids and rituximab or cyclophosphamide) for remission induction in patients with systemic GPA or MPA (Clinicaltrials.gov Identifier: NCT02222155).
Treatment of multi-relapsing or refractory disease, with or without some complex manifestations, such as subglottic and tracheobronchial stenoses, orbital tumor, or pachymeningitis, goes beyond the scope of this review article. Such diseases should ideally be managed in reference centers with expertise in vasculitis. Studies are ongoing for these patient populations, including those with rituximab or abatacept (for limited relapsing GPA; ABROGATE, Clinicaltrials.gov Identifier: NCT02108860).
Treatment for EGPA is at present not very different from that for MPA. Non-severe forms are often easily controlled with glucocorticoids first, but more than half the patients will become glucocorticoid-dependent and will relapse and/or will need to continue glucocorticoids because of asthma or sinusitis. Patients with severe EGPA, particularly those with cardiomyopathy, need more aggressive treatment with a combination of glucocorticoids and cyclophosphamide. However, more than half the patients will need to continue glucocorticoids because of persistent asthma or sinusitis. Data are limited for rituximab, but its use can be considered in refractory and/or relapsing cases (79, 103). A randomized-controlled study from the French vasculitis study group should start soon to further determine its place in the therapeutic armamentarium for EGPA. Anti-interleukin 5 (IL-5) monoclonal antibodies (mepolizumab) are also under investigation for relapsing and/or glucocorticoid-dependent EGPA patients (Clinicaltrials.gov Identifier: NCT02020889) after two small open-label studies showed some promising results (104).
Conclusion
Although the real cause(s) of ANCA-associated vasculitides remain(s) unknown and we still lack a definitive cure, major advances have been achieved over the past decade in the understanding and treatment of these diseases. The pathogenic mechanisms are multiple but are better understood, and new potential targets for therapy, such as the C5a receptor antagonist for GPA and MPA, are continually being identified. The detection of ANCA subsets directed against specific MPO epitopes that correlate better with disease activity and can be found in many patients who were previously considered ANCA-negative, through methods different from those used in routine practice, may also change our practice. A good biological marker to assess disease activity and predict flares might finally be found, at least for MPO-ANCA-positive patients. Evidence for the pathogenic role of PR3-ANCA-associated disease remains weaker than for MPO-ANCA-associated disease, and whether similar epitope-specific and possibly more pathogenic PR3-ANCA subsets can be detected will be interesting. Obviously, many differences exist between each of the three main ANCA-associated vasculitides, first clinically, then biologically (ANCA status) and as recently confirmed, genetically (stronger genetic association by ANCA type than clinical diagnosis). However, at present, treatment for GPA and MPA (or PR3- and MPO-ANCA-associated vasculitides) remains globally the same with the same drugs. Whether all these differences between ANCA-associated vasculitides will eventually affect how we treat them first need to be investigated. The optimal duration of glucocorticoid and maintenance treatments could perhaps also be tailored to each patient’s disease characteristics.
Remaining questions that researchers in the vasculitis field are currently seeking to answer include how to treat patients with very severe disease (early mortality rate remains around 10% for patients with severe GPA or MPA), with difficult-to-treat and/or refractory manifestations (e.g., subglottic stenosis, orbital tumors) and/or continually relapsing disease. Other questions of importance are how to further decrease the damage associated with the disease (end-stage renal disease, peripheral nerve damage, saddle-nose deformity in GPA) or its treatments and how to best treat associated non-vasculitic manifestations such as asthma in EGPA.
Footnotes
Peer-review: Externally peer-reviewed.
Conflict of Interest: Dr. Pagnoux reports having received fees for serving on advisory boards from Roche, Genzyme, Sanofi, ChemoCentryx and GlaxoSmithKline, lecture fees from Roche, Bristol-Myers Squibb, and EuroImmune, and grant support from Roche, Euroimmune and Terumo BCT.
Financial Disclosure: The author declared that this work has received no financial support.
References
- 1.Jennette JC, Falk RJ, Andrassy K, Bacon PA, Churg J, Gross WL, et al. Nomenclature of systemic vasculitides. Proposal of an international consensus conference. Arthritis Rheum. 1994;37:187–92. doi: 10.1002/art.1780370206. http://dx.doi.org/10.1002/art.1780370206. [DOI] [PubMed] [Google Scholar]
- 2.Jennette JC, Falk RJ, Bacon PA, Basu N, Cid MC, Ferrario F, et al. 2012 revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Arthritis Rheum. 2013;65:1–11. doi: 10.1002/art.37715. http://dx.doi.org/10.1002/art.37715. [DOI] [PubMed] [Google Scholar]
- 3.Masi AT, Hunder GG, Lie JT, Michel BA, Bloch DA, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Churg-Strauss syndrome (allergic granulomatosis and angiitis) Arthritis Rheum. 1990;33:1094–100. doi: 10.1002/art.1780330806. http://dx.doi.org/10.1002/art.1780330806. [DOI] [PubMed] [Google Scholar]
- 4.Leavitt RY, Fauci AS, Bloch DA, Michel BA, Hunder GG, Arend WP, et al. The American College of Rheumatology 1990 criteria for the classification of Wegener’s granulomatosis. Arthritis Rheum. 1990;33:1101–7. doi: 10.1002/art.1780330807. http://dx.doi.org/10.1002/art.1780330807. [DOI] [PubMed] [Google Scholar]
- 5.Craven A, Robson J, Ponte C, Grayson PC, Suppiah R, Judge A, et al. ACR/EULAR-endorsed study to develop Diagnostic and Classification Criteria for Vasculitis (DCVAS) Clin Exp Nephrol. 2013;17:619–21. doi: 10.1007/s10157-013-0854-0. http://dx.doi.org/10.1007/s10157-013-0854-0. [DOI] [PubMed] [Google Scholar]
- 6.Kallenberg CG. Pathogenesis of ANCA-associated vasculitides. Ann Rheum Dis. 2011;70(Suppl 1):i59–63. doi: 10.1136/ard.2010.138024. http://dx.doi.org/10.1136/ard.2010.138024. [DOI] [PubMed] [Google Scholar]
- 7.Kallenberg CG, Zhao MH. Evolving concepts in pathogenesis and treatment of ANCA-associated systemic vasculitides. Nephrology (Carlton) 2009;14:1–2. doi: 10.1111/j.1440-1797.2009.01093.x. http://dx.doi.org/10.1111/j.1440-1797.2009.01093.x. [DOI] [PubMed] [Google Scholar]
- 8.Pagnoux C. Churg-Strauss syndrome: evolving concepts. Discov Med. 2010;9:243–52. [PubMed] [Google Scholar]
- 9.Pagnoux C, Quéméneur T, Ninet J, Diot E, Kyndt X, de Wazières B, et al. Treatment of systemic necrotizing vasculitides in patients aged sixty-five years or older: results of a multicenter, open-label, randomized controlled trial of corticosteroid and cyclophosphamide-based induction therapy. Arthritis Rheumatol. 2015;67:1117–27. doi: 10.1002/art.39011. http://dx.doi.org/10.1002/art.39011. [DOI] [PubMed] [Google Scholar]
- 10.Pagnoux C, Mahr A, Hamidou MA, Boffa JJ, Ruivard M, Ducroix JP, et al. Azathioprine or methotrexate maintenance for ANCA-associated vasculitis. N Engl J Med. 2008;359:2790–803. doi: 10.1056/NEJMoa0802311. http://dx.doi.org/10.1056/NEJMoa080231. [DOI] [PubMed] [Google Scholar]
- 11.Jayne D, Rasmussen N, Andrassy K, Bacon P, Tervaert JW, Dadoniene J, et al. A randomized trial of maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. N Engl J Med. 2003;349:36–44. doi: 10.1056/NEJMoa020286. http://dx.doi.org/10.1056/NEJMoa020286. [DOI] [PubMed] [Google Scholar]
- 12.Hiemstra TF, Walsh M, Mahr A, Savage CO, de Groot K, Harper L, et al. Mycophenolate mofetil vs azathioprine for remission maintenance in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized controlled trial. JAMA. 2010;304:2381–8. doi: 10.1001/jama.2010.1658. http://dx.doi.org/10.1001/jama.2010.1658. [DOI] [PubMed] [Google Scholar]
- 13.Walsh M, Merkel PA, Peh CA, Szpirt W, Guillevin L, Pusey CD, et al. Plasma exchange and glucocorticoid dosing in the treatment of anti-neutrophil cytoplasm antibody associated vasculitis (PEXIVAS): protocol for a randomized controlled trial. Trials. 2013;14:73. doi: 10.1186/1745-6215-14-73. http://dx.doi.org/10.1186/1745-6215-14-73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Stone JH, Merkel PA, Spiera R, Seo P, Langford CA, Hoffman GS, et al. Rituximab versus cyclophosphamide for ANCA-associated vasculitis. N Engl J Med. 2010;363:221–32. doi: 10.1056/NEJMoa0909905. http://dx.doi.org/10.1056/NEJMoa0909905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Specks U, Merkel PA, Seo P, Spiera R, Langford CA, Hoffman GS, et al. Efficacy of remission-induction regimens for ANCA-associated vasculitis. N Engl J Med. 2013;369:417–27. doi: 10.1056/NEJMoa1213277. http://dx.doi.org/10.1056/NEJMoa1213277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones RB, Tervaert JW, Hauser T, Luqmani R, Morgan MD, Peh CA, et al. Rituximab versus cyclophosphamide in ANCA-associated renal vasculitis. N Engl J Med. 2010;363:211–20. doi: 10.1056/NEJMoa0909169. http://dx.doi.org/10.1056/NEJMoa0909169. [DOI] [PubMed] [Google Scholar]
- 17.Guillevin L, Pagnoux C, Karras A, Khoutra C, Aumaitre O, Cohen P, et al. Rituximab Versus Azathioprine for Maintenance in Antineutrophil Cytoplasmic Antibodies (ANCA)-Associated Vasculitis. A prospective study in 117 patients [abstract] Presse Med. 2013;42:679. http://dx.doi.org/10.1016/j.lpm.2013.02.068. [Google Scholar]
- 18.Xiao H, Dairaghi DJ, Powers JP, Ertl LS, Baumgart T, Wang Y, et al. C5a Receptor (CD88) Blockade Protects against MPO-ANCA GN. J Am Soc Nephrol. 2014;25:225–31. doi: 10.1681/ASN.2013020143. http://dx.doi.org/10.1681/ASN.2013020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schonermarck U, Gross WL, de Groot K. Treatment of ANCA-associated vasculitis. Nature reviews. 2013;10:25–36. doi: 10.1038/nrneph.2013.225. http://dx.doi.org/10.1038/nrneph.2013.225. [DOI] [PubMed] [Google Scholar]
- 20.Fujimoto S, Watts RA, Kobayashi S, Suzuki K, Jayne DR, Scott DG, et al. Comparison of the epidemiology of anti-neutrophil cytoplasmic antibody-associated vasculitis between Japan and the U.K. Rheumatology (Oxford, England) 2011;50:1916–20. doi: 10.1093/rheumatology/ker205. http://dx.doi.org/10.1093/rheumatology/ker205. [DOI] [PubMed] [Google Scholar]
- 21.Mohammad AJ, Jacobsson LT, Westman KW, Sturfelt G, Segelmark M. Incidence and survival rates in Wegener’s granulomatosis, microscopic polyangiitis, Churg-Strauss syndrome and polyarteritis nodosa. Rheumatology (Oxford, England) 2009;48:1560–5. doi: 10.1093/rheumatology/kep304. http://dx.doi.org/10.1093/rheumatology/kep304. [DOI] [PubMed] [Google Scholar]
- 22.Sinico RA, Bottero P. Churg-Strauss angiitis. Best practice & research. Clinical rheumatology. 2009;23:355–66. doi: 10.1016/j.berh.2009.02.004. http://dx.doi.org/10.1016/j.berh.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 23.Mahr A, Moosig F, Neumann T, Szczeklik W, Taille C, Vaglio A, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): evolutions in classification, etiopathogenesis, assessment and management. Current opinion in rheumatology. 2014;26:16–23. doi: 10.1097/BOR.0000000000000015. http://dx.doi.org/10.1097/BOR.0000000000000015. [DOI] [PubMed] [Google Scholar]
- 24.van der Woude FJ. Anticytoplasmic antibodies in Wegener’s granulomatosis. Lancet. 1985;2:48. doi: 10.1016/s0140-6736(85)90105-9. http://dx.doi.org/10.1016/S0140-6736(85)90105-9. [DOI] [PubMed] [Google Scholar]
- 25.Watts RA, Mooney J, Skinner J, Scott DG, Macgregor AJ. The contrasting epidemiology of granulomatosis with polyangiitis (Wegener’s) and microscopic polyangiitis. Rheumatology (Oxford, England) 2012;51:926–31. doi: 10.1093/rheumatology/ker454. http://dx.doi.org/10.1093/rheumatology/ker454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mohammad AJ, Jacobsson LT, Mahr AD, Sturfelt G, Segelmark M. Prevalence of Wegener’s granulomatosis, microscopic polyangiitis, polyarteritis nodosa and Churg-Strauss syndrome within a defined population in southern Sweden. Rheumatology (Oxford, England) 2007;46:1329–37. doi: 10.1093/rheumatology/kem107. http://dx.doi.org/10.1093/rheumatology/kem107. [DOI] [PubMed] [Google Scholar]
- 27.Wieczorek S, Holle JU, Epplen JT. Recent progress in the genetics of Wegener’s granulomatosis and Churg-Strauss syndrome. Current opinion in rheumatology. 2010;22:8–14. doi: 10.1097/BOR.0b013e3283331151. http://dx.doi.org/10.1097/BOR.0b013e3283331151. [DOI] [PubMed] [Google Scholar]
- 28.Xie G, Roshandel D, Sherva R, Monach PA, Lu EY, Kung T, et al. Association of granulomatosis with polyangiitis (Wegener’s) with HLA-DPB1*04 and SEMA6A gene variants: evidence from genome-wide analysis. Arthritis and rheumatism. 2013;65:2457–68. doi: 10.1002/art.38036. http://dx.doi.org/10.1002/art.38036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vaglio A, Casazza I, Grasselli C, Corradi D, Sinico RA, Buzio C. Churg-Strauss syndrome. Kidney international. 2009;76:1006–11. doi: 10.1038/ki.2009.210. http://dx.doi.org/10.1038/ki.2009.210. [DOI] [PubMed] [Google Scholar]
- 30.Pearson T, Bremmer M, Cohen J, Driscoll M. Vasculopathy related to cocaine adulterated with levamisole: A review of the literature. Dermatol Online J. 2012;18:1. [PubMed] [Google Scholar]
- 31.Khan TA, Cuchacovich R, Espinoza LR, Lata S, Patel NJ, Garcia-Valladares I, et al. Vasculopathy, hematological, and immune abnormalities associated with levamisole-contaminated cocaine use. Seminars in arthritis and rheumatism. 2011;41:445–54. doi: 10.1016/j.semarthrit.2011.04.010. http://dx.doi.org/10.1016/j.semarthrit.2011.04.010. [DOI] [PubMed] [Google Scholar]
- 32.Bateman H, Rehman A, Valeriano-Marcet J. Vasculitis-like Syndromes. Current rheumatology reports. 2009;11:422–9. doi: 10.1007/s11926-009-0062-9. http://dx.doi.org/10.1007/s11926-009-0062-9. [DOI] [PubMed] [Google Scholar]
- 33.Molloy ES, Langford CA. Vasculitis mimics. Current opinion in rheumatology. 2008;20:29–34. doi: 10.1097/BOR.0b013e3282f1dcf2. http://dx.doi.org/10.1097/BOR.0b013e3282f1dcf2. [DOI] [PubMed] [Google Scholar]
- 34.Comarmond C, Pagnoux C, Khellaf M, Cordier JF, Hamidou M, Viallard JF, et al. Eosinophilic granulomatosis with polyangiitis (Churg-Strauss): clinical characteristics and long-term followup of the 383 patients enrolled in the French Vasculitis Study Group cohort. Arthritis and rheumatism. 2013;65:270–81. doi: 10.1002/art.37721. http://dx.doi.org/10.1002/art.37721. [DOI] [PubMed] [Google Scholar]
- 35.Pagnoux C, Guillevin L. Churg-Strauss syndrome: evidence for disease subtypes? Current opinion in rheumatology. 2010;22:21–8. doi: 10.1097/BOR.0b013e328333390b. http://dx.doi.org/10.1097/BOR.0b013e328333390b. [DOI] [PubMed] [Google Scholar]
- 36.Sinico RA, Di Toma L, Maggiore U, Bottero P, Radice A, Tosoni C, et al. Prevalence and clinical significance of antineutrophil cytoplasmic antibodies in Churg-Strauss syndrome. Arthritis and rheumatism. 2005;52:2926–35. doi: 10.1002/art.21250. http://dx.doi.org/10.1002/art.21250. [DOI] [PubMed] [Google Scholar]
- 37.Baldini C, Della Rossa A, Grossi S, Catarsi E, Talarico R, d’Ascanio A, et al. [Churg-Strauss syndrome: outcome and long-term follow-up of 38 patients from a single Italian centre]. Reumatismo. 2009;61:118–24. doi: 10.4081/reumatismo.2009.118. [DOI] [PubMed] [Google Scholar]
- 38.Stone JH. Limited versus severe Wegener’s granulomatosis: baseline data on patients in the Wegener’s granulomatosis etanercept trial. Arthritis and rheumatism. 2003;48:2299–309. doi: 10.1002/art.11075. http://dx.doi.org/10.1002/art.11075. [DOI] [PubMed] [Google Scholar]
- 39.Hoffman GS, Kerr GS, Leavitt RY, Hallahan CW, Lebovics RS, Travis WD, et al. Wegener granulomatosis: an analysis of 158 patients. Annals of internal medicine. 1992;116:488–98. doi: 10.7326/0003-4819-116-6-488. http://dx.doi.org/10.7326/0003-4819-116-6-488. [DOI] [PubMed] [Google Scholar]
- 40.Duna GF, Calabrese LH. Limitations of invasive modalities in the diagnosis of primary angiitis of the central nervous system. The Journal of rheumatology. 1995;22:662–7. [PubMed] [Google Scholar]
- 41.Bajema IM. Pathological classification of anti-neutrophil cytoplasmic antibody (ANCA)-associated glomerulonephritis. Clinical and experimental immunology. 2011;164(Suppl 1):14–6. doi: 10.1111/j.1365-2249.2011.04359.x. http://dx.doi.org/10.1111/j.1365-2249.2011.04359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kallenberg CG. Pathogenesis of ANCA-associated vasculitis, an update. Clinical reviews in allergy & immunology. 2011;41:224–31. doi: 10.1007/s12016-011-8258-y. http://dx.doi.org/10.1007/s12016-011-8258-y. [DOI] [PubMed] [Google Scholar]
- 43.Jennette JC, Falk RJ, Gasim AH. Pathogenesis of antineutrophil cytoplasmic autoantibody vasculitis. Current opinion in nephrology and hypertension. 2011;20:263–70. doi: 10.1097/MNH.0b013e3283456731. http://dx.doi.org/10.1097/MNH.0b013e3283456731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Willcocks LC, Lyons PA, Rees AJ, Smith KG. The contribution of genetic variation and infection to the pathogenesis of ANCA-associated systemic vasculitis. Arthritis research & therapy. 2010;12:202. doi: 10.1186/ar2928. http://dx.doi.org/10.1186/ar2928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kessenbrock K, Krumbholz M, Schonermarck U, Back W, Gross WL, Werb Z, et al. Netting neutrophils in autoimmune small-vessel vasculitis. Nature medicine. 2009;15:623–5. doi: 10.1038/nm.1959. http://dx.doi.org/10.1038/nm.1959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Brogan PA, Dillon MJ. Endothelial microparticles and the diagnosis of the vasculitides. Internal medicine. 2004;43:1115–9. doi: 10.2169/internalmedicine.43.1115. http://dx.doi.org/10.2169/internalmedicine.43.1115. [DOI] [PubMed] [Google Scholar]
- 47.Hong Y, Eleftheriou D, Hussain AA, Price-Kuehne FE, Savage CO, Jayne D, et al. Anti-neutrophil cytoplasmic antibodies stimulate release of neutrophil microparticles. J Am Soc Nephrol. 2012;23:49–62. doi: 10.1681/ASN.2011030298. http://dx.doi.org/10.1681/ASN.2011030298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bansal PJ, Tobin MC. Neonatal microscopic polyangiitis secondary to transfer of maternal myeloperoxidase-antineutrophil cytoplasmic antibody resulting in neonatal pulmonary hemorrhage and renal involvement. Annals of allergy, asthma & immunology : official publication of the American College of Allergy, Asthma, & Immunology. 2004;93:398–401. doi: 10.1016/S1081-1206(10)61400-7. http://dx.doi.org/10.1016/S1081-1206(10)61400-7. [DOI] [PubMed] [Google Scholar]
- 49.Xiao H, Heeringa P, Hu P, Liu Z, Zhao M, Aratani Y, et al. Antineutrophil cytoplasmic autoantibodies specific for myeloperoxidase cause glomerulonephritis and vasculitis in mice. The Journal of clinical investigation. 2002;110:955–63. doi: 10.1172/JCI15918. http://dx.doi.org/10.1172/JCI0215918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xiao H, Schreiber A, Heeringa P, Falk RJ, Jennette JC. Alternative complement pathway in the pathogenesis of disease mediated by anti-neutrophil cytoplasmic autoantibodies. The American journal of pathology. 2007;170:52–64. doi: 10.2353/ajpath.2007.060573. http://dx.doi.org/10.2353/ajpath.2007.060573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Xiao H, Heeringa P, Liu Z, Huugen D, Hu P, Maeda N, et al. The role of neutrophils in the induction of glomerulonephritis by anti-myeloperoxidase antibodies. The American journal of pathology. 2005;167:39–45. doi: 10.1016/S0002-9440(10)62951-3. http://dx.doi.org/10.1016/S0002-9440(10)62951-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Primo VC, Marusic S, Franklin CC, Goldmann WH, Achaval CG, Smith RN, et al. Anti-PR3 immune responses induce segmental and necrotizing glomerulonephritis. Clinical and experimental immunology. 2010;159:327–37. doi: 10.1111/j.1365-2249.2009.04072.x. http://dx.doi.org/10.1111/j.1365-2249.2009.04072.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Little MA, Al-Ani B, Ren S, Al-Nuaimi H, Leite M, Jr, Alpers CE, et al. Anti-proteinase 3 anti-neutrophil cytoplasm autoantibodies recapitulate systemic vasculitis in mice with a humanized immune system. PloS one. 2012;7:e28626. doi: 10.1371/journal.pone.0028626. http://dx.doi.org/10.1371/journal.pone.0028626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.van Timmeren MM, Heeringa P. Pathogenesis of ANCA-associated vasculitis: recent insights from animal models. Current opinion in rheumatology. 2012;24:8–14. doi: 10.1097/BOR.0b013e32834bde57. http://dx.doi.org/10.1097/BOR.0b013e32834bde57. [DOI] [PubMed] [Google Scholar]
- 55.Roth AJ, Ooi JD, Hess JJ, van Timmeren MM, Berg EA, Poulton CE, et al. Epitope specificity determines pathogenicity and detectability in ANCA-associated vasculitis. The Journal of clinical investigation. 2013;123:1773–83. doi: 10.1172/JCI65292. http://dx.doi.org/10.1172/JCI65292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Suzuki K, Suzuki K, Nagao T, Nakayama T. Proposal of anti-moesin as a novel biomarker for ANCA-associated vasculitis. Clinical and experimental nephrology. 2013;17:638–41. doi: 10.1007/s10157-013-0861-1. http://dx.doi.org/10.1007/s10157-013-0861-1. [DOI] [PubMed] [Google Scholar]
- 57.Voswinkel J, Muller A, Lamprecht P. Is PR3-ANCA formation initiated in Wegener’s granulomatosis lesions? Granulomas as potential lymphoid tissue maintaining autoantibody production. Ann N Y Acad Sci. 2005;1051:12–9. doi: 10.1196/annals.1361.042. http://dx.doi.org/10.1196/annals.1361.042. [DOI] [PubMed] [Google Scholar]
- 58.Espy C, Morelle W, Kavian N, Grange P, Goulvestre C, Viallon V, et al. Sialylation levels of anti-proteinase 3 antibodies are associated with the activity of granulomatosis with polyangiitis (Wegener’s) Arthritis and rheumatism. 2011;63:2105–15. doi: 10.1002/art.30362. http://dx.doi.org/10.1002/art.30362. [DOI] [PubMed] [Google Scholar]
- 59.Kain R, Exner M, Brandes R, Ziebermayr R, Cunningham D, Alderson CA, et al. Molecular mimicry in pauci-immune focal necrotizing glomerulonephritis. Nature medicine. 2008;14:1088–96. doi: 10.1038/nm.1874. http://dx.doi.org/10.1038/nm.1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kain R, Tadema H, McKinney EF, Benharkou A, Brandes R, Peschel A, et al. High prevalence of autoantibodies to hLAMP-2 in anti-neutrophil cytoplasmic antibody-associated vasculitis. J Am Soc Nephrol. 2012;23:556–66. doi: 10.1681/ASN.2011090920. http://dx.doi.org/10.1681/ASN.2011090920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Roth AJ, Brown MC, Smith RN, Badhwar AK, Parente O, Chung H, et al. Anti-LAMP-2 antibodies are not prevalent in patients with antineutrophil cytoplasmic autoantibody glomerulonephritis. J Am Soc Nephrol. 2012;23:545–55. doi: 10.1681/ASN.2011030273. http://dx.doi.org/10.1681/ASN.2011030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Del Papa N, Guidali L, Sironi M, Shoenfeld Y, Mantovani A, Tincani A, et al. Anti-endothelial cell IgG antibodies from patients with Wegener’s granulomatosis bind to human endothelial cells in vitro and induce adhesion molecule expression and cytokine secretion. Arthritis and rheumatism. 1996;39:758–66. doi: 10.1002/art.1780390507. http://dx.doi.org/10.1002/art.1780390507. [DOI] [PubMed] [Google Scholar]
- 63.Guilpain P, Mouthon L. Antiendothelial cells autoantibodies in vasculitis-associated systemic diseases. Clinical reviews in allergy & immunology. 2008;35:59–65. doi: 10.1007/s12016-007-8069-3. http://dx.doi.org/10.1007/s12016-007-8069-3. [DOI] [PubMed] [Google Scholar]
- 64.Sebastian JK, Mahr AD, Ahmed SS, Stone JH, Romay-Penabad Z, Davis JC, et al. Antiendothelial cell antibodies in patients with Wegener’s granulomatosis: prevalence and correlation with disease activity and manifestations. The Journal of rheumatology. 2007;34:1027–31. [PubMed] [Google Scholar]
- 65.Hellmich B, Csernok E, Gross WL. Proinflammatory cytokines and autoimmunity in Churg-Strauss syndrome. Ann N Y Acad Sci. 2005;1051:121–31. doi: 10.1196/annals.1361.053. http://dx.doi.org/10.1196/annals.1361.053. [DOI] [PubMed] [Google Scholar]
- 66.Guilpain P, Guillevin L, Mouthon L. [Eosinophil granule cationic proteins: eosinophil activation markers]. La Revue de medecine interne / fondee par la Societe nationale francaise de medecine interne. 2006;27:406–8. doi: 10.1016/j.revmed.2006.01.008. http://dx.doi.org/10.1016/j.revmed.2006.01.008. [DOI] [PubMed] [Google Scholar]
- 67.Smith RM, Jones RB, Jayne DR. Progress in treatment of ANCA-associated vasculitis. Arthritis research & therapy. 2012;14:210. doi: 10.1186/ar3797. http://dx.doi.org/10.1186/ar3797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Merkel PA. L50. The future of international clinical trials in vasculitis. Presse Med. 2013;42:637–41. doi: 10.1016/j.lpm.2013.02.001. http://dx.doi.org/10.1016/j.lpm.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 69.Mukhtyar C, Guillevin L, Cid MC, Dasgupta B, de Groot K, Gross W, et al. EULAR recommendations for the management of primary small and medium vessel vasculitis. Annals of the rheumatic diseases. 2009;68:310–7. doi: 10.1136/ard.2008.088096. http://dx.doi.org/10.1136/ard.2008.088351. [DOI] [PubMed] [Google Scholar]
- 70.McGregor JAC, Hogan SL, Hu Y, Jennette CE, Falk RJ, Nachman PH. GC use beyond 6 months does not prevent relapses but increases risk of infection. Clinical and experimental immunology. 2011;164:60–1. [Google Scholar]
- 71.WGET. Wegener’s Granulomatosis Etanercept Trial Research Group. Etanercept plus standard therapy for Wegener’s granulomatosis. N Engl J Med. 2005;352:351–61. doi: 10.1056/NEJMoa041884. http://dx.doi.org/10.1056/NEJMoa041884. [DOI] [PubMed] [Google Scholar]
- 72.Walsh M, Merkel PA, Mahr A, Jayne D. Effects of duration of glucocorticoid therapy on relapse rate in antineutrophil cytoplasmic antibody-associated vasculitis: A meta-analysis. Arthritis care & research. 2010;62:1166–73. doi: 10.1002/acr.20176. http://dx.doi.org/10.1002/acr.20176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGregor JG, Hogan SL, Hu Y, Jennette CE, Falk RJ, Nachman PH. Glucocorticoids and relapse and infection rates in anti-neutrophil cytoplasmic antibody disease. Clinical journal of the American Society of Nephrology : CJASN. 2012;7:240–7. doi: 10.2215/CJN.05610611. http://dx.doi.org/10.2215/CJN.05610611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Samson M, Puechal X, Devilliers H, Ribi C, Cohen P, Bienvenu B, et al. Long-term follow-up of a randomized trial on 118 patients with polyarteritis nodosa or microscopic polyangiitis without poor-prognosis factors. Autoimmunity reviews. 2014;13:197–205. doi: 10.1016/j.autrev.2013.10.001. http://dx.doi.org/10.1016/j.autrev.2013.10.001. [DOI] [PubMed] [Google Scholar]
- 75.Samson M, Puechal X, Devilliers H, Ribi C, Cohen P, Stern M, et al. Long-term outcomes of 118 patients with eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) enrolled in two prospective trials. Journal of autoimmunity. 2013;43:60–9. doi: 10.1016/j.jaut.2013.03.003. http://dx.doi.org/10.1016/j.jaut.2013.03.003. [DOI] [PubMed] [Google Scholar]
- 76.Jayne D. Treatment of ANCA-associated systemic small-vessel vasculitis. APMIS Suppl. 2009:3–9. doi: 10.1111/j.1600-0463.2009.02470.x. [DOI] [PubMed] [Google Scholar]
- 77.de Groot K, Rasmussen N, Bacon PA, Tervaert JW, Feighery C, Gregorini G, et al. Randomized trial of cyclophosphamide versus methotrexate for induction of remission in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and rheumatism. 2005;52:2461–9. doi: 10.1002/art.21142. http://dx.doi.org/10.1002/art.21142. [DOI] [PubMed] [Google Scholar]
- 78.Faurschou M, Westman K, Rasmussen N, de Groot K, Flossmann O, Hoglund P, et al. Brief Report: long-term outcome of a randomized clinical trial comparing methotrexate to cyclophosphamide for remission induction in early systemic antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and rheumatism. 2012;64:3472–7. doi: 10.1002/art.34547. http://dx.doi.org/10.1002/art.34547. [DOI] [PubMed] [Google Scholar]
- 79.Thiel J, Hassler F, Salzer U, Voll RE, Venhoff N. Rituximab in the treatment of refractory or relapsing eosinophilic granulomatosis with polyangiitis (Churg-Strauss syndrome) Arthritis research & therapy. 2013;15:R133. doi: 10.1186/ar4313. http://dx.doi.org/10.1186/ar4313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.de Groot K, Harper L, Jayne DR, Flores Suarez LF, Gregorini G, Gross WL, et al. Pulse versus daily oral cyclophosphamide for induction of remission in antineutrophil cytoplasmic antibody-associated vasculitis: a randomized trial. Annals of internal medicine. 2009;150:670–80. doi: 10.7326/0003-4819-150-10-200905190-00004. http://dx.doi.org/10.7326/0003-4819-150-10-200905190-00004. [DOI] [PubMed] [Google Scholar]
- 81.Guillevin L, Cordier JF, Lhote F, Cohen P, Jarrousse B, Royer I, et al. A prospective, multicenter, randomized trial comparing steroids and pulse cyclophosphamide versus steroids and oral cyclophosphamide in the treatment of generalized Wegener’s granulomatosis. Arthritis and rheumatism. 1997;40:2187–98. doi: 10.1002/art.1780401213. http://dx.doi.org/10.1002/art.1780401213. [DOI] [PubMed] [Google Scholar]
- 82.Harper L, Morgan MD, Walsh M, Hoglund P, Westman K, Flossmann O, et al. Pulse versus daily oral cyclophosphamide for induction of remission in ANCA-associated vasculitis: long-term follow-up. Annals of the rheumatic diseases. 2012;71:955–60. doi: 10.1136/annrheumdis-2011-200477. http://dx.doi.org/10.1136/annrheumdis-2011-200477. [DOI] [PubMed] [Google Scholar]
- 83.Faurschou M, Sorensen IJ, Mellemkjaer L, Loft AG, Thomsen BS, Tvede N, et al. Malignancies in Wegener’s granulomatosis: incidence and relation to cyclophosphamide therapy in a cohort of 293 patients. The Journal of rheumatology. 2008;35:100–5. [PubMed] [Google Scholar]
- 84.Holle JU, Dubrau C, Herlyn K, Heller M, Ambrosch P, Noelle B, et al. Rituximab for refractory granulomatosis with polyangiitis (Wegener’s granulomatosis): comparison of efficacy in granulomatous versus vasculitic manifestations. Annals of the rheumatic diseases. 2011 doi: 10.1136/ard.2011.153601. [DOI] [PubMed] [Google Scholar]
- 85.de Joode AA, Sanders JS, Smid MW, Stegeman CA. Plasmapheresis rescue therapy in progressive systemic ANCA-associated vasculitis: Single-center results of stepwise escalation of immunosuppression. Journal of clinical apheresis. 2014 doi: 10.1002/jca.21318. http://dx.doi.org/10.1002/jca.21318. [DOI] [PubMed] [Google Scholar]
- 86.Walsh M, Catapano F, Szpirt W, Thorlund K, Bruchfeld A, Guillevin L, et al. Plasma exchange for renal vasculitis and idiopathic rapidly progressive glomerulonephritis: a meta-analysis. American journal of kidney diseases : the official journal of the National Kidney Foundation. 2011;57:566–74. doi: 10.1053/j.ajkd.2010.10.049. http://dx.doi.org/10.1053/j.ajkd.2010.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Springer J, Nutter B, Langford CA, Hoffman GS, Villa-Forte A. Outcomes in patients with granulomatosis with polyangiitis (Wegener’s) treated with short vs. long-term maintenace therapy [abstract] Arthritis and rheumatism. 2012;64(Suppl):S706. [Google Scholar]
- 88.Holle JU, Gross WL, Latza U, Nolle B, Ambrosch P, Heller M, et al. Improved outcome in 445 patients with Wegener’s granulomatosis in a German vasculitis center over four decades. Arthritis and rheumatism. 2011;63:257–66. doi: 10.1002/art.27763. http://dx.doi.org/10.1002/art.27763. [DOI] [PubMed] [Google Scholar]
- 89.Sanders JS, Slot MC, Stegeman CA. Maintenance therapy for vasculitis associated with antineutrophil cytoplasmic autoantibodies. The New England journal of medicine. 2003;349:2072–3. doi: 10.1056/NEJM200311203492116. author reply -3. [DOI] [PubMed] [Google Scholar]
- 90.Springer J, Nutter B, Langford CA, Hoffman GS, Villa-Forte A. Granulomatosis with polyangiitis (Wegener’s): impact of maintenance therapy duration. Medicine. 2014;93:82–90. doi: 10.1097/MD.0000000000000020. http://dx.doi.org/10.1097/MD.0000000000000020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cartin-Ceba R, Golbin JM, Keogh KA, Peikert T, Sanchez-Menendez M, Ytterberg SR, et al. Rituximab for remission induction and maintenance in refractory granulomatosis with polyangiitis (Wegener’s): ten-year experience at a single center. Arthritis and rheumatism. 2012;64:3770–8. doi: 10.1002/art.34584. http://dx.doi.org/10.1002/art.34584. [DOI] [PubMed] [Google Scholar]
- 92.Jones RB, Ferraro AJ, Chaudhry AN, Brogan P, Salama AD, Smith KG, et al. A multicenter survey of rituximab therapy for refractory antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and rheumatism. 2009;60:2156–68. doi: 10.1002/art.24637. http://dx.doi.org/10.1002/art.24637. [DOI] [PubMed] [Google Scholar]
- 93.Smith RM, Jones RB, Guerry MJ, Laurino S, Catapano F, Chaudhry A, et al. Rituximab for remission maintenance in relapsing antineutrophil cytoplasmic antibody-associated vasculitis. Arthritis and rheumatism. 2012;64:3760–9. doi: 10.1002/art.34583. http://dx.doi.org/10.1002/art.34583. [DOI] [PubMed] [Google Scholar]
- 94.Charles P, Neel A, Tieulie N, Hot A, Pugnet G, Decaux O, et al. Rituximab for induction and maintenance treatment of ANCA-associated vasculitides: a multicentre retrospective study on 80 patients. Rheumatology (Oxford, England) 2014;53:532–9. doi: 10.1093/rheumatology/ket381. http://dx.doi.org/10.1093/rheumatology/ket381. [DOI] [PubMed] [Google Scholar]
- 95.Besada E, Koldingsnes W, Nossent JC. Long-term efficacy and safety of pre-emptive maintenance therapy with rituximab in granulomatosis with polyangiitis: results from a single centre. Rheumatology (Oxford, England) 2013;52:2041–7. doi: 10.1093/rheumatology/ket257. http://dx.doi.org/10.1093/rheumatology/ket257. [DOI] [PubMed] [Google Scholar]
- 96.Roubaud-Baudron C, Pagnoux C, Meaux-Ruault N, Grasland A, Zoulim A, JLG, et al. Rituximab maintenance therapy for granulomatosis with polyangiitis and microscopic polyangiitis. The Journal of rheumatology. 2012;39:125–30. doi: 10.3899/jrheum.110143. http://dx.doi.org/10.3899/jrheum.110143. [DOI] [PubMed] [Google Scholar]
- 97.Guillevin L, Pagnoux C, Karras A, Khouatra C, Aumaitre O, Cohen P, et al. Rituximab versus Azathioprine for Maintenance in ANCA-Associated Vasculitis [Abstract] Arthritis and rheumatism. 2012;64:S706. doi: 10.1056/NEJMoa1404231. [DOI] [PubMed] [Google Scholar]
- 98.Azar L, Springer J, Langford CA, Hoffman GS. Rituximab with or without a conventional maintenance agent in the treatment of relapsing granulomatosis with polyangiitis (Wegener’s): a retrospective single-center study. Arthritis & rheumatology. 2014;66:2862–70. doi: 10.1002/art.38744. http://dx.doi.org/10.1002/art.38744. [DOI] [PubMed] [Google Scholar]
- 99.de Groot K, Reinhold-Keller E, Tatsis E, Paulsen J, Heller M, Nolle B, et al. Therapy for the maintenance of remission in sixty-five patients with generalized Wegener’s granulomatosis. Methotrexate versus trimethoprim/sulfamethoxazole. Arthritis and rheumatism. 1996;39:2052–61. doi: 10.1002/art.1780391215. http://dx.doi.org/10.1002/art.1780391215. [DOI] [PubMed] [Google Scholar]
- 100.Stegeman CA, Tervaert JW, de Jong PE, Kallenberg CG. Trimethoprim-sulfamethoxazole (co-trimoxazole) for the prevention of relapses of Wegener’s granulomatosis. Dutch Co-Trimoxazole Wegener Study Group. The New England journal of medicine. 1996;335:16–20. doi: 10.1056/NEJM199607043350103. http://dx.doi.org/10.1056/NEJM199607043350103. [DOI] [PubMed] [Google Scholar]
- 101.Pagnoux C, Guillevin L. How can patient care be improved beyond medical treatment? Best practice & research Clinical rheumatology. 2005;19:337–44. doi: 10.1016/j.berh.2004.11.005. http://dx.doi.org/10.1016/j.berh.2004.11.005. [DOI] [PubMed] [Google Scholar]
- 102.WGET. Etanercept plus standard therapy for Wegener’s granulomatosis. The New England journal of medicine. 2005;352:351–61. doi: 10.1056/NEJMoa041884. http://dx.doi.org/10.1056/NEJMoa041884. [DOI] [PubMed] [Google Scholar]
- 103.Mohammad AJ, Hot A, Arndt F, Moosig F, Guerry MJ, Amudala N, et al. Rituximab for the treatment of eosinophilic granulomatosis with polyangiitis (Churg-Strauss) Annals of the rheumatic diseases. 2014 doi: 10.1136/annrheumdis-2014-206095. [DOI] [PubMed] [Google Scholar]
- 104.Herrmann K, Gross WL, Moosig F. Extended follow-up after stopping mepolizumab in relapsing/refractory Churg-Strauss syndrome. Clinical and experimental rheumatology. 2012;30:S62–5. [PubMed] [Google Scholar]