

ABSTRACT
Antibodies are an important class of biotherapeutics that offer specificity to their antigen, long half-life, effector function interaction and good manufacturability. The immunogenicity of non-human-derived antibodies, which can be a major limitation to development, has been partially overcome by humanization through complementarity-determining region (CDR) grafting onto human acceptor frameworks. The retention of foreign content in the CDR regions, however, is still a potential immunogenic liability. Here, we describe the humanization of an anti-myostatin antibody utilizing a 2-step process of traditional CDR-grafting onto a human acceptor framework, followed by a structure-guided approach to further reduce the murine content of CDR-grafted antibodies. To accomplish this, we solved the co-crystal structures of myostatin with the chimeric (Protein Databank (PDB) id 5F3B) and CDR-grafted anti-myostatin antibody (PDB id 5F3H), allowing us to computationally predict the structurally important CDR residues as well as those making significant contacts with the antigen. Structure-based rational design enabled further germlining of the CDR-grafted antibody, reducing the murine content of the antibody without affecting antigen binding. The overall “humanness” was increased for both the light and heavy chain variable regions.
KEYWORDS: Antibody, antibody engineering, complementarity-determining region, humanization, in silico design, immunogenicity, myostatin, X-ray crystallography
Abbreviations
- ActRIIB
activin receptor IIB
- ADA
Anti-Drug Antibody
- CDR
complementarity-determining region
- EDL
Extensor digitorum longus
- GDF8
growth differentiation factor 8
- GDF11
growth differentiation factor 11
- HAHA
human anti-human antibody
- HAMA
human-anti-mouse-antibody
- HSC
Human String Content
- KD
dissociation constant
- PDB
Protein Databank
- RGA
reporter gene assay
- RMSD
root mean squared deviation
- SDR
specificity-determining residues
- SPR
surface plasmon resonance
- TGF-β
transforming growth factor β
Introduction
Biologics, particularly antibodies, represent an increasing trend in therapeutics development, and over 44 antibody biotherapeutics are currently approved in the United States and/or Europe.1-3 Antibodies offer specificity to antigens, as well as long serum half-life and effector functions, which are both mediated by specific sites on the constant domain.4 Despite their obvious benefits, one major liability has been the tendency for many non-human-derived antibodies, e.g., from mouse or rat, to show a significant immunogenic response. Generally, a strong immune response (i.e., formation of anti-drug antibodies (ADA)) is mounted against the foreign (non-self) sequences of murine antibodies, producing a marked human-anti-mouse-antibody (HAMA) response.5,6 Moreover, it is believed that the higher the identity to natural human B cell antibodies, the lower the risk for HAMA response in the clinic.2,3
Given the apparent link between HAMA and non-human sequence content, one popular method to address this immunogenicity risk is to reduce the murine content by humanizing the mouse antibody through complementarity-determining region (CDR) grafting onto a human acceptor germline framework. This method leaves only the CDRs with xenogeneic residues.7-10 Despite this, the human anti-human antibody (HAHA) responses can still be problematic when treating with humanized antibodies.2,10-14 There is still murine content in the CDR and the HAHA response is usually associated with epitopes that at least partially contain CDR residues.15 Therefore, to further reduce the potential risk of immunogenicity, the removal of the murine content in the CDRs is also desirable. This is potentially difficult as the CDR encodes the binding region for antibody:antigen interactions. Care must be taken to maintain these interacting and loop supporting residues. Despite this, some additional humanization is achievable because not all of the residues in the CDR region are required for binding. It has been postulated that only 20–33% of the residues within the CDRs are essential for the antibody to bind to the antigen.16
There are currently several different approaches to minimize the foreign CDR content, including methods that select for reduced content in the humanization process and those that remove the content after CDR grafting. The former approach includes use of only the murine residues from the specificity-determining residues (SDRs), i.e., the residues that are essential for the binding of the antibody to the ligand, along with multiple human germline sequences as templates.9,17,18 “Super-humanization” uses a CDR homology based approach to antibody humanization and effectively selects a germline with CDRs that most closely match the murine CDRs.19,20 An additional method includes screening for germline segments while only grafting the CDR3 of the heavy and light chain.21 This method can also be combined with affinity optimization using somatic hypermutation.22 Finally, immunologically relevant antibody humanness, or human string content, is also a method that has been used to select humanized antibodies.23 Here, framework segments are selected from multiple germlines to make the overall variable region more humanlike.
There are alternative approaches to reduce the potential immunogenicity risk associated with the remaining non-human content after CDR grafting. One method reduces CDR-containing immunogenic epitopes through mutations of residues informed by alanine scanning and guided selection.15 Similarly, the method of CDR repair utilizes soft-randomization of CDR position with care taken to focus the library on germline amino acids.24 Finally, a method of generating a binary substitution library between mouse and germline CDR allows for germlining any non-essential CDR residue.25
Beyond the potential risk of immunogenicity associated with non-human sequence content, the additional constraint of manufacturability must be considered when selecting germlines. Properties such as stability and expression vary among particular families of V-genes. V-genes from the VH1, VH3 and VH5 families are found to be better biotherapeutic candidates than those from other families.26 However, CDR grafting onto a subset of frameworks does not always result in stable antibodies because there may be some incompatibility between the CDR and the framework residues. This is often compensated for by mutations in the framework region to recover the native mouse structure pair.27,28
In this study, we examined a structure guided approach to reduce the potential risk of immunogenicity of a rodent derived antibody. The antibody selected for humanization was the anti-myostatin murine antibody RK35. Myostatin (growth differentiation factor 8/GDF8) is a negative regulator of skeletal muscle growth and a member of the TGF-β (transforming growth factor β) superfamily. The biological function of myostatin is well conserved, where there is 100% amino acid sequence identity between mouse and human homologs. Myostatin signaling deficiency through mutation or knock-out results in the increase of the skeletal muscles mass where the muscles are 100–200% larger than normal through hyperplasia and hypertrophy, as seen in mammals including humans.29-36 In dogs with only a single functional myostatin allele, improved muscle function was observed, although not as an overt increase in muscle mass as seen with homozygotes.34 Pharmacological inhibition of myostatin activity in rodents from either myostatin neutralizing antibodies, mutant myostatin propeptides or even decoy myostatin receptor-fusion proteins, results in increased muscle mass and improved muscle function.37-39 As previously reported, a murine antibody termed RK35 was obtained through hybridoma technology. RK35 neutralizes mature myostatin signaling through activin receptor IIB (ActRIIB), and showed promising in vivo results, including slowing of muscle atrophy and grip strength loss in rodent models of ALS.38,39
In this work, we initially demonstrate the successful humanization of the anti-myostatin antibody with a traditional CDR grafting approach onto clinically validated germline frameworks.40 Following the traditional CDR grafting, we explored the further reduction of murine CDR content guided by co-crystal structures of the antibody:antigen complex. The co-crystal structures allowed us to understand the epitope and paratope of this antibody, as well as model the presentation and stability of the CDRs. Reducing the murine content of antibodies by rational design is a way to mitigate the possible associated risk of immunogenicity, while opening further optimization/engineering for therapeutic use, such as improvements in stability and other biophysical characteristics.
Results
Humanization by CDR grafting and characterization of RK35
RK35 is a mouse monoclonal antibody against mature myostatin. It was generated from myostatin knockout mice immunized with human myostatin. This antibody neutralized myostatin's interaction with the high affinity receptor ActRIIB, as shown with a competition ELISA and a reporter gene cell-based activity assay (RGA), which monitors the activation of the SMAD-regulated signaling pathway (Table 1). Additionally, this antibody increased muscle mass in vivo (Fig. 1). RK35 was also compared to a reference clinical human myostatin inhibitor, MYO-029,41 showing an increased binding affinity in surface plasmon resonance (SPR) analysis when comparing the chimeric RK35 (mouse variable region with human IgG1 constant region) to MYO-029 (Table 1). Importantly, RK35 significantly increased the maximum tetanic force, cross-sectional area and mass of the Extensor digitorum longus (EDL) muscle, whereas mice treated with MYO-029 did not show an increase in any of these parameters (Fig. 1).
Table 1.
In vitro activity comparison of RK35.
| Construct | Biacore KD (pM) | ELISA IC50 (nM) | RGA IC50 (ng/mL) |
|---|---|---|---|
| murine RK35 | 21.8 # | 0.17+/−0.02 (4) | 33.5 +/ −21.2 (2) |
| chimera RK35 | 3.0 | 0.17+/−0.02 (4) | 24.3 +/− 2.5 (2) |
| RK35 1.0/1.0 | 2.6 +/− 1.8 (3) | 0.14 | 27.3 +/− 8.5 (3) |
| MYO-029 |
11.4 |
|
|
| Construct VH/VL |
Biacore KD (pM) |
ELISA vs 1.0/1.0 |
RGA vs 1.0/1.0 |
| RK35 1.0/1.2 | 1.5 | 0.91X | 1.0X |
| RK35 1.0/1.3 | 3.2 | 0.96X | 0.9X |
| RK35 1.0/1.4 | 2.5 | 0.61X | 1.5X |
| RK35 1.0/1.5 | 57 | 5.94X | Not active |
| RK35 1.2/1.0 | N/A | Not active | N/A |
| RK35 1.3/1.0 | N/A | Not active | N/A |
| RK35 1.4/1.0 | 284 | 1.71X | 7.5X |
| RK35 1.5/1.0 | 9.2 | 0.88X | 1.3X |
| RK35 1.5/1.4 | 5.8 +/− 0.5 (4) | 0.92X | 1.8X |
Results from Biacore, Elisa and RGA assays are shown on top for the murine, chimeric and CDR-grafted RK35 (RK35 1.0/1.0) along with reference antibody MYO-029. Results for the same assays are shown on the bottom for mutant variants of the humanized RK35 1.0/1.0. These results are shown as a ratio of mutant to wild-type affinity or activity. All measurements were done with a single replicate except where noted by the standard deviation and number of replicates in parenthesis. The Biacore measurement for murine RK35 (#) was done using a different capture antibody (anti-murine IgG) so the results are not directly comparable to the other measurements. “Not Active” means that no activity detected in the assay. “N/A” means that this antibody was not measured because no activity was seen in the Elisa.
Figure 1.
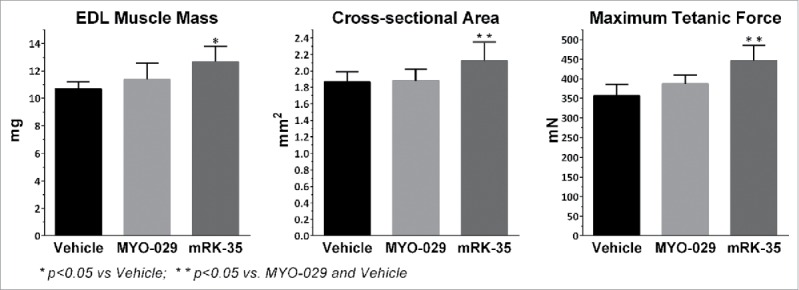
In vivo comparison of RK35 to benchmark MYO-29. Mouse EDL muscle phenotypic and contractile properties following treatment with anti-myostatin antibodies. Mice treated for 2 weeks with 10 mg/kg/wk of murine RK-35 (mRK-35) showed significant increase in muscle weight compared to vehicle treated animals. 10mg/kg/wk MYO-029 treatment did not show a significant muscle weight change. The calculated cross-sectional area and measured maximum tetanic force significantly increased in mice treated with mRK-35 when compared to vehicle or MYO-029, while treatment with MYO-029 did not show a significant change in either measurement. *p < 0.05 vs Vehicle; * * p < 0.05 vs Vehicle and MYO-029. Statistical analysis was done using a one-way ANOVA with Tukey's Post Test performed using GraphPad Prism.
Due to its superior properties compared to MYO-029 in the in vitro and in vivo models, the antibody RK35 was selected as a candidate for optimization. Although no experimental in vivo study was performed, it is well known that there is strong risk of an HAMA response for mouse-derived monoclonal antibodies.5,6 This risk is highlighted using the Epivax in silico ADA prediction tool which indicates that murine RK35 would be at risk for potential immunogenicity based upon comparison to 23 other antibodies with known immune response (Fig. S1).42,43 The RK35 antibody was selected for humanization to reduce this potential risk. The framework template selection was based upon sequence similarity to close human germline sequences, as well as homology to clinically validated germline sequences.40 The CDRs of the heavy and light chains were grafted onto VH3 and VK1 germlines respectively. This CDR-grafted heavy chain (VH 1.0) and CDR-grafted light chain (VL 1.0) retained full activity as assayed by competition ELISA, RGA and SPR (Table 1). Knowing the high homology of myostatin to GDF11 (growth differentiation factor 11), SPR analysis was utilized to confirm that the dissociation constant (KD) of RK35 1.0/1.0 (VH1.0 heavy chain and VL1.0 light chain) to recombinant mature GDF11 was not statistically different from the KD to recombinant mature myostatin (KD of binding to GDF11 is 4.7 pM).
Co-crystal structure of RK35 Fab and mature myostatin dimer
To aid in analysis and further humanization of the RK35 antibody, X-ray crystal structures of the RK35 chimeric antibody as well as the humanized version (RK35 1.0/1.0) in complex with myostatin were obtained. To do this, both the chimeric and humanized antibodies were processed with papain to obtain Fabs. The purified Fabs were complexed with recombinant mature myostatin dimer and screened for crystal formation. Crystals were obtained for the Fab:myostatin complex for both the humanized and chimeric RK35. The humanized complex was resolved to 2.70 Å (Protein Databank (PDB) id 5F3H) and the chimeric murine complex was resolved to 1.76 Å (PDB id 5F3B) using x-ray crystallography (See Methods and Table 2).
Table 2.
Data collection and refinement statistics.
| |
Chimera (5F3B) |
Humanized (5F3H) |
|---|---|---|
| Space group | P1 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 66.09, 67.62, 78.56 | 75.14, 80.92, 101.40 |
| α, β, γ (°) | 78.98, 88.77, 67.44 | 88.33, 103.78, 92.40 |
| Resolution (Å) | 49.17 – 1.76 | 19.99 – 2.70 |
| No. reflections | 104791 | 40287 |
| Completeness for Range | 85.9% | 63.5% |
| I / σI | 13.4 (1.42) | 6.0 (1.5) |
| Rwork / Rfree* | 0.1794 / 0.2057 | 0.2182 / 0.2699 |
| No. atoms | ||
| Protein | 8385 | 15925 |
| Ligand/ion | 30 | 0 |
| Water | 1047 | 0 |
| B-factors | ||
| Protein | 27.4 | 39.1 |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | 0.008 |
| Bond angles (°) | 1.08 | 1.18 |
*Rwork = Σ||Fobs| - |Fcalc|/ Σ|Fobs|. Rfree is equivalent to Rwork, except that it is calculated for a randomly chosen 5% of reflections omitted from the refinement process. I / σI = Σ|I/σI| over 20 resolution shell bins and the number in parenthesis is the |I/σI| in the lowest resolution shell bin (1.831 Å to 1.760 Å for the Chimera and 2.838 Å to 2.701 Å for the Humanized).
Both crystal structures of the mature myostatin in complex with either the chimeric Fab or the humanized 1.0/1.0 Fab show 2 Fabs interacting with each myostatin dimer (Fig. 2). There is one copy of this 2:2 complex in the unit cell of the chimera and 2 copies in the humanized. Each Fab is interacting on the end of one of the myostatin monomers and only makes contact with one of the 2 myostatin chains in the dimer. It is important to note that the structure of the myostatin dimer is noticeably different than those previously reported, and does not show the typical butterfly structure of other myostatin structures44 or closely related TGF-β family members.45-47 There are 2 crystal structures in the PDB with 100% sequence identity in the myostatin region. These are 3hh2, which contains a myostatin dimer in complex with follistatin 288,44 and 3sek which contains a myostatin monomer in complex with follistatin-like 3 complex.48 Looking at an alignment of the monomers of myostatin from 3hh2, 3sek, the humanized structure, and chimeric structure (Fig. 3a), we see that most of the β-strand “Fingers” region of myostatin aligns well, but there is a large shift in the regions identified as the “Pre-helix” region and the “Wrist-helix” region. This causes a large change in the dimer formation seen when comparing the dimer in 3hh2 with the chimeric structure (Fig. 3b-d). When one monomer is used for the alignment, the other monomer appears to bind with a vastly different conformation due to the repositioning of the wrist-helix, although we see that the wrist-helix from chain 2 for each dimer docks in approximately the same location onto chain one. This repositioning of the structure alters the conformation from the butterfly like dimer reported previously for myostatin44 and other TGF-β family proteins 49 (Fig. 3d) to a more linear dimer (Fig. 3c). Analysis of these myostatin structures and several additional TGF-β family proteins suggests that the flexibility of the pre-helix/wrist helix region could enable this change in conformation associated with different binding partners and crystal environments (see Supplementary Results, Figs. S2 and S3 and Table S1).
Figure 2.

Co-crystal structure of (A) Chimeric RK35 Fab and (B) Humanized RK35 Fab in complex with dimeric myostatin. In panel A, 2 chimeric RK35 Fabs (cyan/green and tan/gray) interact with one dimer of myostatin (magenta/yellow). In panel B, there are 2 copies of this interaction. One has the 2 humanized RK35 Fabs (cyan/green and magenta/yellow) interacting with a myostatin dimer (dark green/light green). The second copy contains the 2 Fabs (orange/blue and tan/gray) interacting with a second myostatin dimer (pink/gold).
Figure 3.
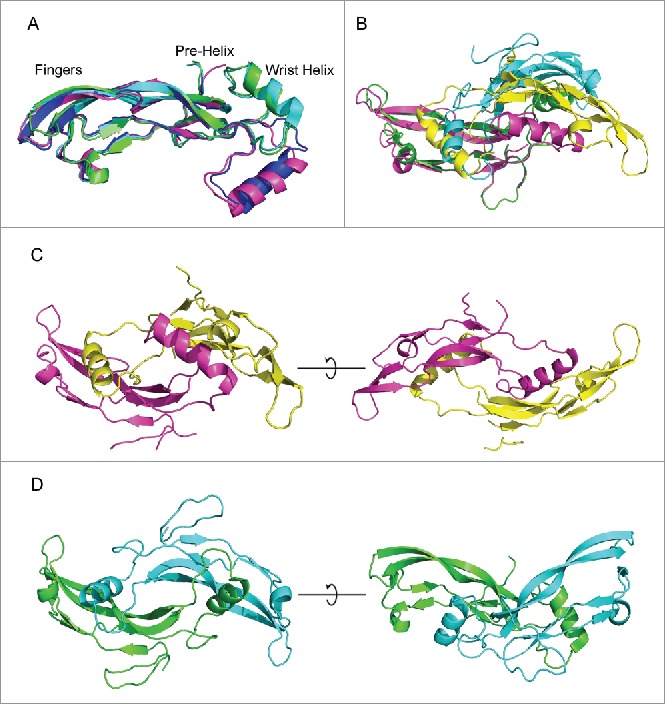
Comparison of myostatin with PDB crystal structures. (A) myostatin from the humanized (blue) and chimeric (magenta) co-crystal structures are aligned with myostatin from PDBID:3hh2 (green) and PDBID3sek (cyan). The pre-helix and wrist helix are labeled for myostatin from the chimeric and humanized structures. These are shifted downwards for the 3hh2 and 3sek structures. (B) Alignment of dimeric myostatin from chimeric structure (magenta and yellow) with 3hh2 (green and cyan). (C) Linear structure of dimeric myostatin from the chimeric crystal structure. (D) Butterfly conformation of the dimeric myostatin from the 3hh2 crystal structure.
The chimeric structure was used to determine the residues in the CDRs that were making contact with the binding epitope on myostatin (<5 .0 Å), and cross referenced to the RK35 1.0/1.0 co-crystal structure (Fig. 4a–b). A list of these is found in Supplementary Table 2. In addition, the structure gave us more information behind the ease of humanization for the straight CDR grafting method. This was done by looking at the structural similarity of the human versus murine variable domain, particularly in the CDR region (Fig. 4c). The Cα-root mean squared deviation (RMSD) of several components were compared, including the entire Fab, the variable regions, the variable frameworks, and each individual CDR. The overall similarity was quite high, with only a 0.88 Å deviation between the Fabs, 0.54 Å for the variable domain, 0.40 Å for the variable heavy, 0.56 Å for the variable light, 0.56 Å for the frameworks, and 0.32–0.76 Å for the CDRs. This is not much different than what one would expect when comparing multiple crystal structures of the same protein, which have an average variation of about 0.3 Å.50
Figure 4.
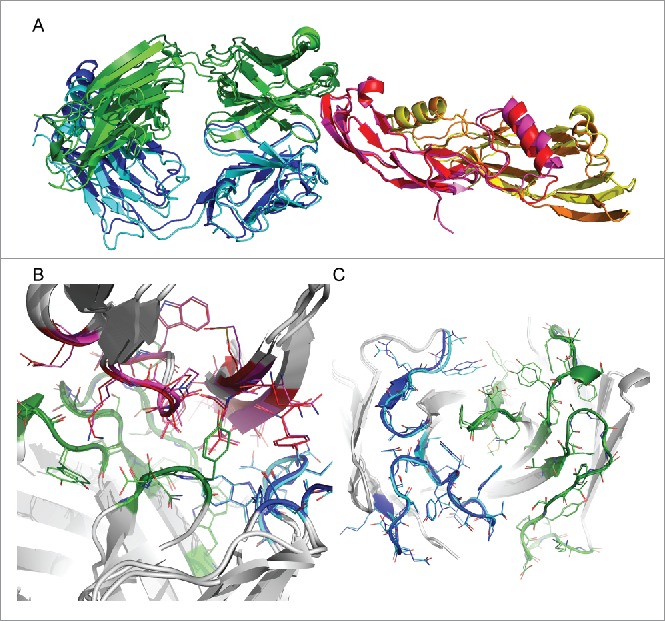
Ribbon diagram showing the superposition of the chimeric and humanized co-crystal structures with myostatin. Here for the Chimeric Fab, the heavy chain is shown in green, the light chain is shown in cyan and the myostatin dimer is shown in magenta and yellow. For the humanized Fab, the heavy chain is shown in dark green, the light chain is shown in blue, and the myostatin dimer is shown in red and yellow. (A) The structures are aligned using the interface residues. (B) A close-up of the interface from panel A with only the interface residues in color and sidechains shown as lines. (C) The Fab structures is aligned using the CDR residues. Here only the CDR residues are shown in color and the sidechains are shown as lines.
Epitope overlap
In vivo, myostatin binds to the ActRIIB and co-receptors, leading to the activation of SMAD pathway.51 From the RGA assay, we can see that RK35 is able to block myostatin from activating this pathway (Table 1). To further investigate the biological function of RK35, we wanted to determine whether it would be able to directly block the epitope of ActRIIB on the surface of myostatin. For this, we aimed to generate a homology model of the ActRIIB:myostatin complex. Besides myostatin, there are a number of other TGF-β family members that signal through the ActRIIB receptor, including activin A, BMP-2, BMP-6, BMP-7, BMP-9, GDF-5, GDF-11, inhibin A, inhibin B and nodal.52,53 Of these, there are several available complex crystal structures showing a conserved binding mode of ActRIIB binding to the fingers region of activin A,54,55 BMP-2,56 and BMP-9.57 Analysis of available structural data showing the conserved binding modes to ActRIIB, the sequence homology of the predicted binding interface of these homologs to myostatin, similar binding affinities, and correlated changes of affinity associated with ActRIIB mutations predicted to be in the binding interface, suggests that myostatin should bind to ActRIIB with the same binding mode as these other TGF-β family members (See Supplementary Results and Figs. S3 and S4). Therefore, available complex structures should provide reasonable templates to generate a homology model.
The template structure used to generate the homology model complex was the ActRIIB:Activin A complex (1nyu).55 To generate a homology model, myostatin from the human RK35:myostatin complex was structurally aligned to the activin A domain of the crystal structure 1nyu. Using this ActRIIB:myostatin model and the human RK35:myostatin crystal structure, binding epitopes were determined as residues on myostatin that were within 5.0 Å of their respective binding partners. The epitope of RK35 contained 17 residues and ActRIIB contained 19 (see Supplementary Table 3). Of these residues on ActRIIB, 11 of them (58%) overlapped with the binding epitope on RK35 (see Fig. 5). Such a large overlap of the epitope suggests that the function of the RK35 antibody would be to directly compete with ActRIIB on the same binding patch on myostatin and inhibit the activation of the SMAD pathway. Additionally, since the core of the epitopes overlap, it would be impossible for ActRIIB to bind myostatin without first removing RK35.
Figure 5.
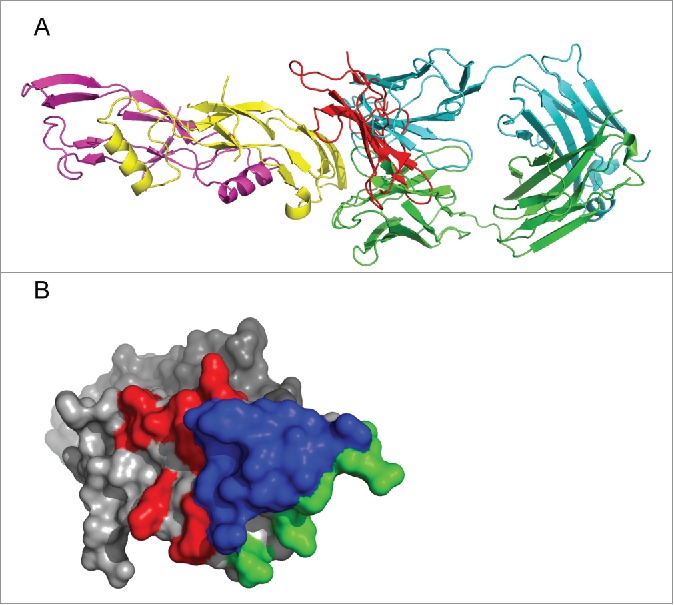
Analysis of RK35 epitope versus ActRIIB binding site on mature myostatin (A) Alignment of the human RK35:myostatin crystal structure with the ActRIIB:myostatin homology model using the myostatin domain. Here myostatin is shown in yellow and magenta, the RK35 Fab in Cyan and Green and the ActRIIB ECD is shown in red. When the myostatin domains are aligned there is significant overlap between the ActRIIB ECD and the RK35 Fab. (B) The binding epitope of ActRIIB and RK35 are shown on the surface of myostatin. The epitopes were determined as residues on myostatin within 5 Å of the 2 binding partners. These were calculated from the human crystal structure for RK35 and a homology model from the crystal structure of Activin A bound to the ECD of ActRIIB (PDBID: 1nyu) for ActRIIB. Residues that are contained in both epitopes are shown in blue (11 residues), residues that are only in the ActRIIB epitope are in red (8 residues) and those only in the RK35 epitope are in green (6 residues).
Further reduction of murine content by germlining of CDRs
In order to further humanize the antibody, we first wanted to determine which residues could be mutated to the human germline. This was done using the crystal structure contacts, as well as computational prediction of changes in binding affinity and stability upon mutation. For the heavy chain, the CDR-H1 was identical to the germline, and so only the CDR-H2 and the end of CDR-H3 were examined. For the light chain, all 3 CDRs were examined for potential mutations to the germline.
The CDR-H2 region for RK35-VH1.0 was defined as Kabat position H50 to H65, and the alignment with human germline is shown in Fig. 6. From this segment, 10 residues are conserved and 7 residues are not. We looked at the effect of mutation on each of the 7 non-conserved residues from the RK35-VH1.0 sequence to the human germline sequence. First, the change in mutation stability was determined using the stability prediction algorithm in Discovery Studio. The results are shown in Table 3. In this method, changes of >0 .5 kcal/mol are classified as significant changes. Sites 50, 52a, 55, and 60 all showed a loss of stability upon mutations, sites 53 and 56 showed no significant change in stability, and site 58 showed an increase in stability upon mutation. Next, we looked at the change in binding affinity upon mutation using 3 different methods (Discovery Studio, MacroModel and Prime) described in the methods section. For the Discovery Studio method, a change of >0 .5 kcal/mol were classified as a significant change. The other 2 methods required larger changes of >1 .0 kcal/mol to be classified as significant. The results of these calculations are shown in Table 3. For site 50, the Thr to Ala mutation showed a loss of binding affinity associated with some loss in the packing interaction, as suggested by only the Prime method. For site 52a, the Ser to Gly mutation was predicted to be neutral by all methods, showing some loss in the packing interaction, but made up for by a decrease in electrostatic energy. Both sites 53 (Gly to Ser) and 60 (Pro to Ala) are predicted to be neutral by all methods and do not show a strong interaction at all. Site 55 is the only one that was predicted to have a significant increase in binding affinity as predicted by the Prime method. This method suggests that the mutation has a decrease in solvation energy without a loss in packing. Finally, mutations sites 56 (Tyr to Ser) and 58 (Ser to Tyr) were both predicted by all 3 methods to reduce affinity. For site 56, this mutation loses a strong packing interaction that is not compensated for by its decrease in unfavorable electrostatics energy, whereas, the mutation of site 58 Ser to Tyr creates a large steric clash that cannot be overcome by minimization.
Figure 6.
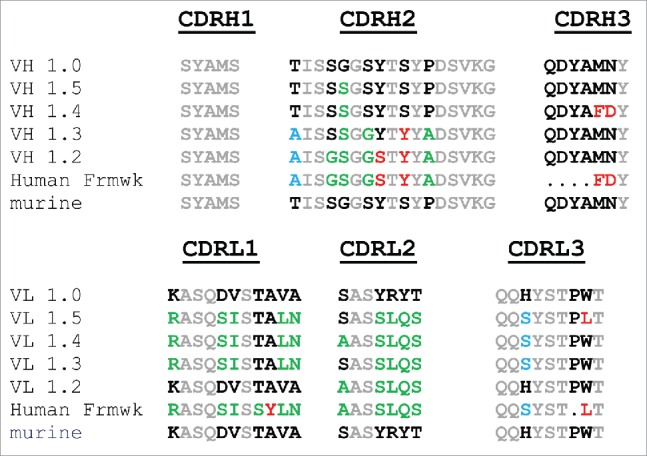
Alignment of further humanized RK35 constructs CDR regions to the murine and germline sequences. Gray lettering is used for residues that are the same in mouse and human sequence, black lettering is used where the murine amino acid is different from the human germline, green are residues predicted to be acceptable to germline to human, red are residues predicted to be not acceptable to germline and blue residues are likely acceptable, but not all prediction methods agree.
Table 3.
Predicted changes in stability and binding affinity upon mutations to germline.
| Site | DP-47 | RK35 VH 1.0 | DS3 Stability | DS3 Binding | MM Binding | Prime Binding | Stability Class | Affinity Class | Description |
|---|---|---|---|---|---|---|---|---|---|
| H50 | A | T | 1.19 | 0.21 | 0.90 | 4.26 | - | - | Small loss in packing |
| H52A | G | S | 1.73 | −0.01 | 0.56 | 0.01 | - | 0 | Small loss in packing offset by small gain in electrostatics |
| H53 | S | G | −0.13 | −0.15 | −0.34 | 0.58 | 0 | 0 | Not a strong interaction |
| H55 | G | S | 1.17 | 0.41 | 0.08 | −3.96 | - | + | Small gain in solvation |
| H56 | S | Y | 0.04 | 2.19 | 2.37 | 1.26 | 0 | −− | Small desolvation gain with large loss of packing interactions |
| H58 | Y | S | −1.64 | 8.49 | 181.3 | 88.64 | + | −− | Creates a large clash not accommodated by rearrangement |
| H60 | A | P | 2.28 | −0.04 | 0.35 | −0.12 | - | 0 | Not a strong interaction |
| H99 | F | M | −0.67 | −0.04 | 0.92 | 0.03 | + | 0 | Not a strong interaction |
| H101 | D | N | −0.15 | −0.22 | 1.16 | 2.29 | 0 | −− | Mutation to Asp could destabilize salt-bridge formed by H96-Asp |
| Site | DPK9 | RK35 VL 1.0 | DS3 Stability | DS3 Binding | MM Binding | Prime Binding | Stability Class | Affinity Class | Description |
|---|---|---|---|---|---|---|---|---|---|
| L24 | R | K | −1.03 | −0.01 | −0.23 | −0.09 | + | 0 | Not a strong interaction |
| L28 | S | D | −0.91 | 0.22 | −0.05 | 0.58 | + | 0 | Not a strong interaction |
| L29 | I | V | −3.12 | −0.11 | 0.11 | −0.67 | + | 0 | Not a strong interaction |
| L31 | S | T | 1.48 | −0.23 | −0.11 | 1.03 | - | 0 | Hbond maintained |
| L32 | Y | A | 0.79 | 2.18 | 544.0 | 1547.9 | - | −− | Creates a large clash not accommodated by rearrangement |
| L33 | L | V | −0.65 | 0.09 | −0.12 | −0.12 | + | 0 | Not a strong interaction, minor change in internal packing |
| L34 | N | A | 1.74 | −0.07 | 1.03 | −0.50 | - | 0 | Not a strong interaction, minor change in VH/VL packing |
| L50 | A | S | −0.08 | 0.13 | 0.95 | 0.76 | 0 | 0 | Loss of Hbond offset by decreased desolvation. |
| L53 | S | Y | −0.62 | 0.23 | −2.20 | −0.40 | + | 0 | Not a strong interaction |
| L54 | L | R | 0.98 | −0.16 | −1.65 | −0.28 | - | 0 | Not a strong interaction |
| L55 | Q | Y | 2.01 | −0.04 | 1.43 | −0.08 | −− | 0 | Not a strong interaction, minor change in VH/VL packing |
| L56 | S | T | 0.12 | 0.01 | −0.06 | 0.33 | 0 | 0 | Not a strong interaction |
| L91 | S | H | 4.26 | 0.66 | 0.95 | 3.07 | −− | - | Small loss in packing |
| L96 | L | W | 2.62 | 1.59 | 0.01 | 5.41 | −− | − | Large loss of hydrophobic packing and packing at the VH/VL interface |
All energy values are in approximate kcal/mol. DS3: are calculation performed using Discovery Studio 3.0. Prime: are calculations using the Schrodinger Prime program, and MM: are calculations using the Schrodinger MacroModel program. Stability and Binding affinity classes were defined by the following categories: (+) = slightly favorable to humanize, (0) = neutral, (−) = slightly unfavorable to humanize. (−−) = unfavorable to humanize, and (−−−) = very unfavorable to humanize
Of the 7 possible mutations, only the G53S mutation was predicted to not decrease the stability or the binding affinity, suggesting that this would be safest mutation to make. Three mutations (S52aG, S55G and P60A) were seen to have a small decrease in stability with either no change or a slight increase in binding affinity, with a fourth (T50A) having a slight decrease in stability and binding affinity. The prediction suggests that these could be mutated without a large effect on binding or stability. The final 2 mutations (Y56S and S58Y) are both predicted to greatly decrease the binding affinity and should be avoided.
There is not a direct alignment of the CDR-H3 to the germline, but possible mutations of the Kabat positions H99 and H101 were examined to enable mutation to more common human residues. The mutations modeled were M99F and N101D. Here, the M99F was predicted to be slightly stabilizing, but does not show a large change in binding affinity by any of the 3 methods. For N100D, this mutation was predicted to decrease the affinity by 2 of the methods, as it will interfere with the Asp at H96 that is making a salt bridge with myostatin.
There are 14 possible mutations in the CDRs of the VL that are different between the germline and VL1.0. We performed the same analysis as for the H2 and H3 loops to determine which germline mutations would affect the binding and which could be tolerated. The results are shown in Table 3. From this, we predict that 11 of these sites could be mutated to the germline without any effect on binding (L24, L28, L29, L31, L33, L34, L50, L53, L54, L55 and L56). Of these, the L50 mutation of Ser to Ala is predicted to be neutral, but does alter the type of binding interaction. Here it replaces a hydrogen bond to myostatin with hydrophobic packing. Only one of the mutations was predicted have a small effect on binding (L91) and 2 are predicted to reduce binding greatly (L32 and L96).
Experimental validation of predicted CDR mutations
RK35 VH1.0/VL1.0, which was both well expressed and fully functional after CDR grafting (Table 1), was used as the starting point in an effort to further humanize this antibody by reducing the murine content of the CDRs in the variable light and variable heavy chains. Initially, we attempted to introduce fully human germline residues into CDRH2 and CDRL2. These constructs were designated RK35 VH 1.2 and RK35 VL 1.2 respectively. Amino acid sequence alignment is shown in Fig. 6. For all variants made, quantitated conditioned medium (described in the Methods section) was used to assess binding activity in the competition ELISA. If the conditioned medium did not show any activity in the ELISA, then the construct was not moved forward into purification and further analysis. RK35 VH 1.0/VL 1.2 was purified and the purified material was used to confirm that indeed germlining VL2 did not result in a loss of activity as measured by competition ELISA, Biacore KD, or the RGA (Table 1). This correlates well with the prediction that none of the germline mutations should decrease the binding affinity. The heavy chain CDR-H2 germline construct RK35 VH1.2/VL 1.0 did not show any activity in the conditioned media, which also corresponds to the prediction that there are several mutations that could significantly lower the binding affinity.
For the light chain, we continued to further germline the construct since the initial CDR-L2 germlining was successful. Guided by the computational predictions and the RK35 CDR contacts with mature myostatin seen in the crystal structure, the constructs RK35 VL1.3 and VL1.4 were designed. Here both constructs further germlined the CDR-L1 and CDR-L3 residues that were not predicted to decrease the affinity. This includes 5 CDR-L1 mutations and one CDR-L3 mutation. The only difference between the 2 was that the L50 position was the murine residue for VL1.3 and the germline for VL1.4. This was done because this residue makes contact in the crystal structure and could alter the type of binding interaction. Both constructs showed little change in the binding affinity (Table 1). The VL1.4 construct retained all but 3 of the murine residues. One of these did not align to the germline and 2 were predicted to decrease the binding affinity. Finally, RK35 VL 1.5 was designed to attempt to further germline the construct. This was identical to VL 1.3 but mutated W96L. However, as predicted, RK35 VH 1.0/VL 1.5 showed reduced binding activity by ELISA and Biacore, and complete loss of neutralization activity in the RGA (Table 1). This Trp makes a large hydrophobic contact at the interface, and mutating to Leu removed a large amount of predicted van der Waals interaction.
The initial VH1.2 construct fully germlined the CDR-H2 and did not retain any activity. The new construct, VH 1.3 only retained murine amino acids if the co-crystal structure indicated that the residue was making direct contact with mature myostatin. The remaining residues in CDR-H2 not shown to make direct contact in the CDRs were germlined (Fig. 6). Again, this construct did not retain any activity. This is most likely due in part to the H56 mutation. Though the murine serine is not making a direct contact, the mutation to Tyr is predicted to clash.
The next set of constructs was more conservative for the CDR-H2. RK35 VH 1.4 contained substitutions at positions M99F and N101D, and RK35 VH 1.5 only has the substitution G53S in CDR-H2. Previous attempts to change CDR-H2 resulted in severe loss of activity, since CDR-H2 makes a number of important contacts with myostatin. RK35 VH 1.4/VL 1.0 purified showed 1.71X reduction in activity in the competition ELISA and 7.5X in the RGA assay compared to RK35 1.0/1.0. This correlated with the KD reduction seen by Biacore. RK35 VH 1.5/VL 1.0 showed retained activity by ELISA, Biacore and RGA (Table 1) and correlated with the prediction based on structural modeling (Table 3).
Finally, we wanted to test if these heavy and light chain mutants could be used in combination. RK35 VH1.5/VL1.0 and RK35 VH1.0/VL1.4 were the fully active heavy and light chain mutants with the most germline mutations. To determine if the activity of these 2 mutants would be retained when combined, we generated the combination clone RK35 VH1.5/VL1.4. This combination shows retained activity by ELISA, Biacore and RGA (Table 1).
Biophysical characterization of RK35 constructs
In addition to function, it is important to monitor the biophysical characterization of antibodies because those properties can be indicative of manufacturability, thus influencing the potential of a drug as a therapeutic. The heavy and light chain mutants described above had their thermal stability and pH stability measured (Supplementary Table 4). The onset of unfolding, T1%, for all the proteins was above the threshold of 50°C, although it can be seen that RK35 VH1.0/VL1.0 is more stable than the other humanized variants. Variations in the pH stability assay were relatively small, although again version RK35 VH1.0/VL1.0 was among the most stable.
Increased humanness
In this work, we introduced germline mutations to reduce the amount of non-human sequence content in the CDRs based upon the known correlation of non-human sequence content with an immune response.2,3,15 Comparing the CDR grafted RK35 VH1.0/VL1.0 with the clones that had the least amount of murine content with retained activity by in vitro analysis (RK35 VL 1.4 and RK35 VH 1.5), we were able to remove 11 of 28 non-germline residues. To further demonstrate the potential decrease in immunogenicity risk, we used the Lazar et al. method, which quantifies the level of potential MHC/T-cell epitopes in an antibody sequence, to determine the “humanness” of the antibody sequence.23 Here, the humanness was defined as the proportion of peptide strands in an antibody that are found within the set of human germline sequences. We calculated 2 different metrics of increased humanness from this method: the number of Perfect 9mers (9-mers that can be found in a human germline) and the Human String Content (HSC) score. To validate whether similarity to germline sequences correlated to reduced immunogenicity we analyzed a dataset of 43 clinical antibodies with reported immunogenicity.6,15,58,59 From this we can see that the number of 9-mers and the HSC score do in fact show significant correlation with a decrease in immunogenicity. (Table S5 and Fig. S5). We then evaluated the HSC and 9-mers scores along with the percent sequence identity and percent sequence similarity of our designed sequences (Table 4). As can be seen, the RK35 VH1.0 and RK35 VL1.0 clones greatly increased their HSC score and the number of perfect 9mers over the chimera. Surprisingly though, the heavy chain shows an increased humanness over the light chain, which is the opposite of what is typically observed for humanized antibodies.60 However, in this case the antibody has a relatively short 7 amino acid long CDR-H3, (80% of murine CDRs are longer than 7 amino acids)61 and has a CDR-H1 that already matches the human germline. The further optimized clones, RK35 VL 1.4 and RK35 VH 1.5, have HSC scores of 96 and 91, respectively. These are increased from the heavy and light chain CDR grafted scores (90 and 89), as well as the original murine construct (87 and 79). Moreover, going from the murine to the CDR-grafted to further optimized constructs greatly reduces the amount of non-human sequence content present on the surface of these constructs (Fig. 7).
Table 4.
Humanness of RK35 Constructs.
| Variable Domain | Perfect 9mers | HSC | SeqID | SeqSim | T20 |
|---|---|---|---|---|---|
| RK35_VH_murine | 51 | 87 | 78 | 89 | 80.43 |
| RK35-VH_1.0 | 74 | 90 | 89 | 91 | 89.18 |
| RK35-VH_1.2 | 94 | 95 | 95 | 97 | 95.35 |
| RK35-VH_1.3 | 81 | 93 | 93 | 95 | 93.66 |
| RK35-VH_1.4 | 75 | 92 | 91 | 91 | 90.69 |
| RK35-VH_1.5 | 74 | 91 | 90 | 92 | 89.66 |
| RK35_VL_murine | 10 | 79 | 70 | 86 | 73.79 |
| RK35-VL_1.0 | 54 | 89 | 86 | 92 | 89.44 |
| RK35-VL_1.2 | 67 | 93 | 91 | 94 | 90.89 |
| RK35-VL_1.3 | 71 | 96 | 95 | 97 | 95.28 |
| RK35-VL_1.4 | 79 | 96 | 96 | 97 | 96.31 |
| RK35-VL_1.5 | 72 | 96 | 96 | 98 | 95.93 |
Perfect 9mers are the number of 9-mers in the sequence that can be found in a human germline.23 HSC is the Human String Content score.23 SeqID and SeqSim are the sequence identity or sequence similarity to the DP47 or DPK9 germlines. T20 is the humanness of monoclonal antibody variable region sequences as compared to the human antibody repitoire.59
Figure 7.
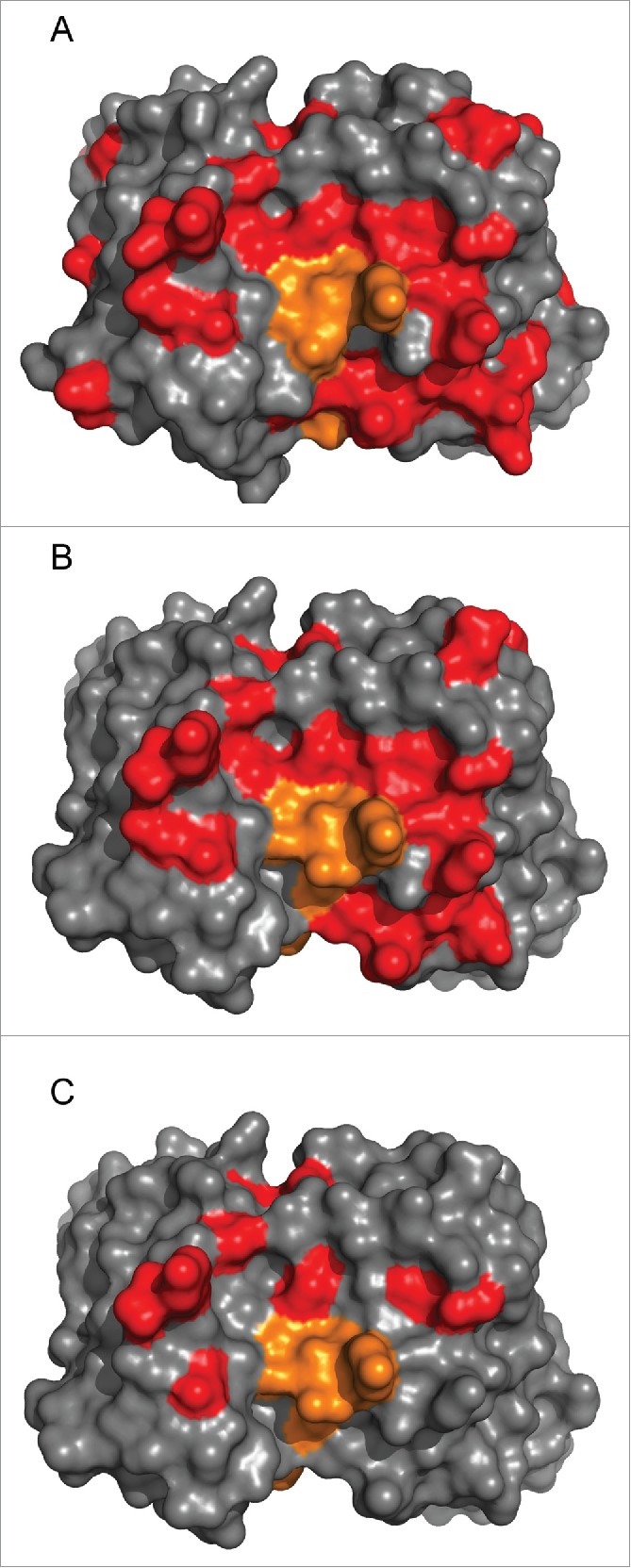
Reduction in non-human sequence content. The non-human sequence content, defined by alignments to DP47 and DPK9, is shown for the variable regions of (a) murine RK35 (b) RK35 1.0/1.0 and (c) RK35 1.5/1.4. Here red is the non-human sequence content and orange is the CDRH3 residues which do not align to germline.
The definition of humanness can be defined more broadly to compare the similarity of the sequence to antibodies in the entire human repertoire.59,62 We evaluated the humanness of these sequences using this definition with the T20 humanness calculator described in Gao et al., which has been shown to correlate with the immunogenicity of a similar set of clinical antibodies.59 Here we found that the ranking of the T20 results were consistent with the 9-mer and HSC score germline definition of humanness (Table 4). Therefore, by either definition these mutants have increased humanness.
We examined the CDR regions more in-depth specifically because it has been shown that they are a significant cause of immune response in humanized antibodies.15 Here we calculated the HSC and Perfect 9mers for sequence regions around each CDR that contained one or more CDR residues (Table 5). The majority of the increase in humanness is seen in the light chain CDRs, where, in clone VL1.4, mutations in L2 remove all the non-human sequence content and L1 and L3 humanness are greatly increased. For CDR-L1, an additional 8 of 19 potential 9-mer epitopes match human germlines, while in the CDR-L3 4 additional 9-mers are germlined. For the heavy chain, CDR-H1 is completely germlined in the CDR grafted version and only a small amount of non-human content was removed. This was accounted for by a small overall increase in the HSC score of CDR-H2.
Table 5.
Humanness of CDR regions.
| CDR-H1 | CDR-H2 | CDR-H3 | ||||
|---|---|---|---|---|---|---|
| Variable Domain | Perfect 9mers | HSC | Perfect 9mers | HSC | Perfect 9mers | HSC |
| RK35_VH_murine | 12 | 95 | 5 | 79 | 1 | 63 |
| RK35-VH_1.0 | 18 | 100 | 5 | 83 | 1 | 63 |
| RK35-VH_1.2 | 18 | 100 | 25 | 100 | 1 | 63 |
| RK35-VH_1.3 | 18 | 100 | 12 | 92 | 1 | 63 |
| RK35-VH_1.4 | 18 | 100 | 5 | 83 | 2 | 70 |
| RK35-VH_1.5 |
18 |
100 |
5 |
85 |
1 |
63 |
| CDR-L1 | CDR-L2 | CDR-L3 | ||||
| Variable Domain |
Perfect 9mers |
HSC |
Perfect 9mers |
HSC |
Perfect 9mers |
HSC |
| RK35_VL_murine | 1 | 73 | 0 | 70 | 1 | 83 |
| RK35-VL_1.0 | 1 | 76 | 1 | 76 | 3 | 85 |
| RK35-VL_1.2 | 1 | 76 | 14 | 100 | 3 | 85 |
| RK35-VL_1.3 | 9 | 89 | 6 | 94 | 7 | 91 |
| RK35-VL_1.4 | 9 | 89 | 14 | 100 | 7 | 91 |
| RK35-VL_1.5 | 9 | 89 | 6 | 94 | 8 | 94 |
Perfect 9mers are the number of 9-mers in the sequence that can be found in a human germline.23 HSC is the Human String Content score.23
Evaluation of prediction methods
As described above we utilized a computational method to successfully identify tolerated CDR mutations to enhance humanness of RK35, while maintaining stability and affinity. Based upon this, we wanted to evaluate the expected predictive power of this method on a broader set of data and get a better understanding of its general utility beyond this single example. The design goal of this method was to differentiate mutations that would destabilize the antibody from those that would be neutral or stabilizing, and differentiate those that would reduce affinity from those that would maintain or increase affinity. To understand how well these methods performed at this classification, we evaluated all 3 affinity prediction methods and the single stability method on larger data sets of experimentally determined ΔΔG of mutations with corresponding X-ray crystal structures. For affinity measurements we utilized AbBind,63 a large dataset that contains >1000 antibody:antigen interactions. To determine the predictive power of each method, we calculated their performance at correctly identifying affinity reducing mutations (ΔΔG >=1.0 kcal/mol). This is similar to the hot-spot predictions to determine key interface residues.64-66 We generated ΔΔG predictions for the Prime and MacroModel methods on this data set and utilized predictions using the Discovery Studio method previously determined.63 The results of these ΔΔG predictions are shown in Supplementary Table 6. We analyzed these predictions for how well each method could identify an affinity reducing mutation, and whether the combination of methods added to the predictive performance. The results of this analysis are shown in Table 6. We used the same prediction cutoff as described above. For Discovery Studio, >0.5kcal was predicted to be affinity reducing and for Prime and MacroModel >1.0 kcal/mol was used. Those below this cutoff were defined as tolerated. For the individual predictions, Discovery Studio was able to correctly classify 62% of the affinity reducing mutations and 78% of the tolerated mutations, Prime was able to correctly classify 49% of the affinity reducing mutations, and 79% of the tolerated mutations, and MacroModel was able to correctly classify 50% of the affinity reducing mutations, and 86% of the tolerated mutations. In addition, we looked at the prediction accuracy of the 3 methods as measured by the percentage of predicted affinity reducing mutations that were also experimentally affinity reducing. For Discovery Studio, Prime and MacroModel these were 64%, 59% and 69%, respectively. The same prediction accuracy for tolerated mutations was 77%, 71% and 73%, respectively.
Table 6.
Analysis of Binding Affinity Prediction Methods.
| Number of Methods Predicted as Affinity Reducing | ||||||
|---|---|---|---|---|---|---|
| Prediction | All 3 | Only 2 | Only 1 | Majority | Minority | None |
| Affinity Reducing | 108 | 102 | 97 | 210 | 179 | 82 |
| Tolerated | 27 | 63 | 147 | 90 | 528 | 381 |
| Fraction Correct | 0.80 | 0.62 | 0.40 | 0.70 | 0.75 | 0.82 |
| Fraction Affinity Reducing | 0.28 | 0.26 | 0.25 | 0.54 | 0.46 | 0.21 |
| Fraction Tolerated |
0.04 |
0.10 |
0.24 |
0.15 |
0.85 |
0.62 |
| Discovery Studio | Prime | MacroModel | ||||
| Prediction |
AR |
Tol |
AR |
Tol |
AR |
Tol |
| Affinity Reducing | 243 | 146 | 189 | 200 | 193 | 196 |
| Tolerated | 135 | 483 | 131 | 487 | 88 | 530 |
| Fraction Correct | 0.64 | 0.77 | 0.59 | 0.71 | 0.69 | 0.73 |
| Fraction Affinity Reducing | 0.62 | 0.38 | 0.49 | 0.51 | 0.50 | 0.50 |
| Fraction Tolerated | 0.22 | 0.78 | 0.21 | 0.79 | 0.14 | 0.86 |
The total number of affinity reducing and tolerated mutants found in each prediction group is shown for: (top) number of methods predicting a mutant to be affinity reducing, and (bottom) individual methods predictions of affinity reducing (AR) and tolerated (Tol) mutations. The Fraction correct is the true positive rate for “All 3” AR (n=3), “Only 2” AR (n=2), “Only 1” AR (n=1) and “Majority” AR (n>=2) predictions and “AR” predictions for individual methods, and the true negative rate for “Minority” AR (n<=1) and “None” AR n=0 predictions and “Tol” predictions for individual methods. The fraction affinity reducing and fraction tolerated are number of experimentally determined affinity reducing and tolerated mutants in that group divided by the total number of affinity reducing or tolerated mutants, respectively.
In addition to examining the methods individually, we also investigated whether a combined effect had more predictive power. This was indeed the case, as when 2 or more methods agreed, they accurately predicted the affinity reducing mutations 70% of the time and tolerated mutations 75% of the time. Moreover, when all 3 methods agreed, this accuracy increased to 80% and 82%, respectively. This enables us to better understand why the prediction accuracy of RK35 mutants was quite high, as 16 of 23 mutations fall into the combined slightly favorable/neutral groups (all predicted as tolerated) and 2 of 23 in the very unfavorable group (all predicted as affinity reducing) (See Supplementary Results for further analysis of affinity prediction of individual mutations).
For stability measurements, we could not identify a database similar to AbBind that contains antibody specific mutations, so we evaluated a large dataset of >1000 mutations from 35 different protein types with available experimental stability data and crystal structures.67 We predicted the change of stability for this data set using the Discovery Studio method (Supplementary Table 7). As before, we defined destabilizing mutations as those with an experimental ΔΔG >= 1.0 kcal/mol. We used the same type of analysis here as for the binding affinity dataset, but with only the single Discovery Studio method (Table 7). Here the prediction cutoff was 0.5 kcal/mol to separate destabilizing from tolerated mutations. For stability mutations, Discovery Studio was able to correctly classify 85% of destabilizing mutations, and 48% of tolerated mutations. The accuracy of predicting destabilizing mutations was 67% and for predicting tolerated mutations was 72%. The classification and accuracy of the stability measurements was similar to that of affinity, but with only a single method on which to base the categorization, the expected performance on the designed mutants would be somewhat reduced (See Supplementary Results for further analysis of stability prediction of individual mutations).
Table 7.
Analysis of Stability Prediction Method.
| Discovery Studio | ||
|---|---|---|
| Prediction | Des | Tol |
| Destabilizing | 482 | 83 |
| Tolerated | 239 | 217 |
| Fraction Correct | 0.67 | 0.72 |
| Fraction Destabilizing | 0.85 | 0.15 |
| Fraction Tolerated | 0.52 | 0.48 |
The table shows total number of destabilizing (Des) and tolerated (Tol) mutants found in each prediction group for the Discovery Studio stability prediction method. The Fraction correct is the true positive rate for destabilizing predictions and true negative rate for Tolerated predictions. The fraction destabilizing and fraction tolerated are the number of experimentally determined destabilizing and tolerated mutants in that group divided by the total number of destabilizing or tolerated mutants, respectively.
Discussion
In this work we described a method of antibody humanization utilized to reduce the non-human sequence content in the frameworks and CDR regions. The initial step of humanization by CDR grafting onto a human germline frequently requires one or more back-mutations to restore activity.16,68-70 However, in the case of humanizing RK35, no back-mutations were required. The ease of humanization of this antibody was not overly surprising given the similarity between the chimeric and CDR-grafted variants. Several methods of humanization of murine antibodies have been described previously, demonstrating a list of sites that have been frequently identified to be important to maintain the CDR loop conformation.16,68-70 Of the sites that differ between the humanized and murine RK35 variable domains, McCafferty et al.70 included only the H94 and L48 positions as residues being important for canonical conformation. Both the R_H94_K and I_L48_L mutation are very conservative. In addition, 2 residues, H49 and L48, are part of the Vernier zone.69 These sites are described as being residues directly underneath the CDRs. L48 has a conservative mutation as described above and H49 is also a small change with a S_H49_A mutation. All the other mutations are not identified as critical for humanization by these methods. It is somewhat unique in this case to have both the chimeric and humanized crystal structures available to understand the humanization process. Given the similarity of the 2 structures, it becomes more apparent why the humanization was so straight-forward. The CDR positions are seen to be nearly identical between the 2 constructs, particularly the residues making contact with myostatin. Thus the conservative differences in the H94, L48 and H49 positions do not alter the structure significantly, and were therefore not required to be back-mutated.
The ability to obtain co-crystal structures to determine the amino acids in the CDRs that form the antibody paratope allowed for the rational design approach to further humanize and optimize the RK35 antibody. For the most part, we accurately predicted which mutations would be tolerated and which would be unfavorable. The confidence in the affinity predictions was increased when different prediction methods agreed and had less confidence when they differed. The difference in the predicted ΔΔG is associated with the construction of each individual method in terms of force fields, solvation models, sidechain placement and minimization. As has been reported before, individual score functions perform differently on different types of predictions.63,71 Despite the different prediction methods, these methods appear to be somewhat complementary as there is an increased predictive power when all 3 make the same categorization. This enhancement based upon the agreement of the methods is probably due to a combination of factors, including the identification of residue positions that are easier to classify and those that are not associated with inaccurate biases in any particular method.
The reduction of xenogenic content in antibodies beyond CDR grafting increases the confidence of reduced immunogenicity risk before entering the clinic. Experimental conformation of this risk during preclinical assessment of immunogenicity could be performed in several ways,72 including the use of T cell proliferation assays. These assays use healthy donor peripheral blood mononuclear cells to investigate if the drug of interest, or specific peptide from the drug, is presented to T cells and results in the proliferation of those T cells. These methods are most appropriately used as indicative of the risk of inducing an immune response, rather than definitive measures of which peptides will induce the immune response. However, at present, these assays are neither technically nor clinically validated, and therefore are not as of yet reliable predictors of the expected immune response in patients.72 Without such validated pre-clinical models, utilization of humanness as a metric, which has been demonstrated here and in the literature to correlate with decreased immunogenicity,59 gives us further confidence in reducing this potential immunogenicity risk. Ultimate assessment, however, would require clinical validation of these antibodies.
The current work on further germlining the CDR-grafted RK35 antibody is another demonstration that not all residues in the CDRs are necessary for retained antigen binding, making additional optimization of the potential immunogenicity risk associated with non-human sequence content, stability and other biophysical properties possible. This approach of removing murine content through mutations of tolerated germline residues is possible without the knowledge of the antibody:antigen complex crystal structure. This can be accomplished through experimental screening and selection methods like those described in Townson et al.57 There they described a method of phage display using germline substitutions in a binary selection library. From their CDR germlining of 3 different antibodies they were able to remove 32 to 53 % of the non-human residues (11 to 16 total residues) from the CDRs H1, H2, L1, L2 and L3 left after CDR grafting. These results are similar to the content removed in the methods described here, where we were able to remove 50 % of the non-human sequence content (11 residues). The precise amount of murine content removed will undoubtedly vary by antibody and the nature of the interaction with the antigen, but each method has similar success in terms of reduction of potential immunogenic residues.
In the method described here, the x-ray structures were successfully solved, but in general this can sometimes be a labor intensive and time-consuming process, and some antigens or antibody:antigen complexes may not be suitable for crystallization. Despite the up-front resources, the overall design process, as compared to experimental screening, was quick and straight-forward, requiring only the prediction of 23 different mutations, and the generation of 10 different antibody constructs. This resulted in the removal of a similar amount of sequence content as in the phage display experiment. Therefore, when an X-ray structure is available, this method would be an efficient way to reduce the potential immunogenic risk associated with non-human sequence content. Moreover, the utility of a solved crystal structure is not limited to optimization of the germline sequence. In silico optimization can be used for many other properties of antibodies required for successful development. Properties such as viscosity, aggregation propensity, chemical instability, phase separation and reduced clearance rates benefit from a co-crystal structure for direct prediction,73-76 as well as to prevent the introduction of destabilizing and affinity reducing mutations. Additionally, as described previously, determining the epitope of interaction can be useful in understanding the biological activity and function of the antibody. Given all of these benefits, determining the crystal structure of antibody:antigen complex is frequently an activity performed independent of the germline optimization of the CDR residues. Therefore, methods that can take advantage of this effort with minimal additional resources are well warranted.
Given the diversity of doses, diseases and allotype tolerance of patients in the clinic, predicting immunogenicity continues to be a complex goal that the field is striving toward. While in silico and other prediction methods for immunogenicity continue to be explored, it is the general belief that the reduction of xenogenic content of antibodies will improve the chances that low immunogenicity will be seen. In this work, we demonstrated a straightforward approach to significantly reduce the non-human sequence content of an antibody with a minimal number of constructs screened. This should allow for the mitigation of some potential immunogenicity risk for the development of antibodies as biotherapeutics.
Material and methods
Cell culture expression and purification
Transient expression of antibodies was performed in COS-1 M6 cells77 by co-transfection of RK35 heavy and light chain V-regions cloned into mammalian expression vectors. To each 100 mm tissue culture dish (Corning 430176), 40µl of Trans-IT (Mirus MIR2306) was added to 2 mls of room temperature Optimem (Invitrogen-Gibco 11058-021) + glutamine 2 mM final concentration. This mixture of Optimem and Trans-IT was vortexed and incubated at room temperature for 15 minutes. Maxiprep DNA was added (8 µg of heavy chain and 8 µg of light chain) to the mixture and incubated at room temperature for 15 minutes. This solution was then added to the p100 containing ∼8 ml of growth media (DMEM + HI FBS + Penn + Strep + Glutamine). After 24 hours at 37°C, 10% CO2, the cells were rinsed with R1CD1 (serum free growth media) and, 10 ml of R1CD1 + PSG was added to each p100. Conditioned medium was harvested after 48 hours at 37°C, 10% CO2, spun down to pellet cells and the supernatant was removed to a new tube.
The conditioned media was then filtered through a 0.22 μm filter and loaded onto a Protein A FF Sepharose column equilibrated with phosphate-buffered saline-calcium-magnesium free (PBS-CMF) buffer (137mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 2.7 mM KH2PO4, pH 7.2). The column was washed with 5 CVs of PBS-CMF pH 7.2 before the antibody was eluted over a gradient (0–100%) using a Protein A Eluting Buffer (20 mM citric acid, 150 mM NaCl, pH 2.5) and peak fractions were filter pooled. Peak fractions from the Protein A capture were loaded onto a 4.5 L size exclusion column packed with Superdex 200. The purified antibody was then loaded onto a 1.1 L G-25 buffer exchange column equilibrated with 10 mM histidine, 5% sucrose, pH 6.0. The antibody was then concentrated using an Amicon stir cell equipped with a 30,000MWCO filter.
Myostatin was expressed from a Chinese hamster ovary (CHO) cell line and purified similar to the method described in Thies et al.78
Crystallization of the RK35 and myostatin complex
Chimeric RK35 was cleaved with immobilized papain for about 16 hours. Following cleavage, papain was removed using a disposable column. The digested antibody fragments were dialyzed to a low salt buffer and the Fab and Fc components were separated using a HiTrap SP ion exchange column. The Fab was subsequently exchanged to a standard myostatin binding buffer containing 2% CHAPS and myostatin was added to a final ratio of 1:2.25 Fab. The complex was subsequently loaded to a Superdex 200 sizing column. Chimeric RK35 Fab and myostatin were purified and the protein complex was concentrated to 10.75 mg/ml in a buffer of 50 mM tris hydrochloride pH 7.5 and 100 mM sodium chloride. Crystals were obtained using the hanging drop method with equilibration at 18°C against a solution containing 20% PEG MME 5000 and 100 mM bis-tris pH 6.5.
Crystals containing the humanized RK35 Fab and myostatin were obtained in a similar manner, except the protein solution was equilibrated against an unbuffered solution containing 20% PEG 3350 and 200 mM sodium chloride.
Data collection and refinement
Single-wavelength (1.0 Å) data for each crystal was collected at beamline 22-ID (Southeast Regional Collaborative Access Team, SER-CAT). A single crystal, cooled to −180°C, was used for each data set. Data processing was carried out with the HK2000 program.79
The structure of the chimeric RK35 in complex with myostatin was solved by molecular replacement using the program AMORE.80 The probe used in the molecular replacement search was PDB entry 1HZH. Prior to refinement, 5% of the data were randomly selected and designated as an Rfree test set to monitor the progress of the refinement. The protein model was rebuilt using iterative cycles of Coot 81 with subsequent refinement with Buster.82 Once RK35 had been rebuilt, the coordinates for myostatin as found in PDB entry 3HH2 were placed manually into difference density. Additional cycles of rebuilding models with Coot followed by refinement with Buster resulted in a structure with good geometry and statistics (Table 2).
The structure of the humanized RK35 in complex with myostatin was solved by molecular replacement using the structure of the chimeric RK35 and myostatin complex as the probe. The humanized RK35 molecules were rebuilt using iterative cycles of Coot with refinement using Buster yielding a structure with good geometry and statistics (Table 2).
Structure preparation for molecular modeling
The x-ray crystal structure used for this set of calculations was the chimera RK35 plus myostatin, which had a resolution of 1.76 Å. This structure contained 6 protein chains per unit cell corresponding to 2 light chains (B and F), 2 heavy chains (A and E) and 2 chains of myostatin (C and D). For input structures, only chains A and B were used for stability calculations, and chains A, B, C and D were used for binding affinity calculations. These represent a single Fab dimer and the Fab dimer/myostatin dimer complex, respectively. To prepare the structure for different calculation methods, 2 different preparation methods were used. First the dsv formatted structure was constructed by applying the “Prepare Protein” protocol of Discovery Studio 3.0 (Accelrys Inc.) to the stability and binding affinity input structures with default parameters and the CHARMPLR forcefield. Next the mae formatted structure was prepared from the binding affinity structure using the “prepwizard” script of the Schrödinger 2010 software suite (Schrödinger LLC) with default parameters and the OPLS2005 forcefield.
Calculation of change in stability upon mutation
The change in stability upon mutation was calculated by applying the “Calculate Mutation Energy (Stability)” protocol from Discovery Studio 3.0 to the dsv formatted Fab dimer structure. The default parameters were used with the exception of the Non-polar Surface Coefficient, which was set to 0.007.
Calculation of change in binding affinity upon mutation
Three different methods were used to calculate the change in binding affinity upon mutation using the Fab dimer/myostatin dimer complex.
Discovery studio method
The change in binding affinity upon mutation was calculated by applying the “Calculate Mutation Energy (Binding)” protocol from Discovery Studio 3.0 to the dsv formatted structure. The default parameters were used with the exception of the Non-polar Surface Coefficient which was set to 0.007. The ligand was defined as the C and D chains.
MacroModel method
The change in binding affinity upon mutation was calculated by using a script that ran different components of the MacroModel package from Schrödinger 2010. The method of the script was to mutate the residue of interest and then minimize the side-chain conformation using a combination of a Monte Carlo search algorithm of the selected side chain and a local continuous structure energy minimization that included the selected side chain and the local structural environment. This method used the OPLS2005 forcefield. The binding affinity was calculated by applying this method to the mae structure with chains A, B, C and D present (bound state) followed by 2 states that only contained chains A+B and C+D (unbound states). The binding energy was the difference between the bound state and sum of the unbound states. The change in binding affinity upon mutation was calculated by taking the difference in binding energies.
Prime method
The change in binding affinity upon mutation was calculated using a script that ran different components of the Prime package from Schrödingerr 2010. The method of the script was to mutate the residue of interest and then minimize the side-chain conformation using a discrete side-chain search algorithm that included the target side chain and other local side chains. This method used the OPLS2005 forcefield. The binding affinity was defined by applying this method to the mae structure with chains A, B, C and D present (bound state) followed by 2 states that only contained chains A+B and C+D (unbound states). The binding energy was the difference between the bound state and the unbound state. The change in binding affinity upon mutation was calculated by taking the difference in binding energies.
Total human IgG ELISA
Conditioned medium from transient transfection was quantitated by total human IgG-Fc- specific ELISA. Briefly, a flat-bottom ELISA plate (Costar 3590) was coated overnight at room temperature with 100µl of 1µg/ml goat anti-human IgG in PBS (Pierce 31125) in each well. Plates were blocked with 100 µl/well of a 0.02% Casein Solution in PBS for a minimum of 3 hours or up to 24 hours at room temperature. If the plates were not used immediately, they were stored for up to a month at 4°C in storage buffer (0.02% NaN3 in PBS). Standard and samples were run in serial dilution series in assay buffer (0.5% BSA + 0.02%Tween-20 in PBS) with 100 µl added to the washed wells of the ELISA plate and incubated for 3 to 24 hours at room temperature. After the plate was washed, 100 µl of goat anti-human IgG (Pierce 31413) diluted 1:5000 in assay buffer was added to well and allowed to incubate for 15 minutes at room temperature. The plate was washed and developed in 100 µl per well BioFX TMB (TMBW-0100-01). The reaction was then stopped in 100 µl per well 0.18 N H2S04 and the plate was read at 450 nm on Molecular Devices vMax plate reader. The unknowns were calculated from the linear range of the curve from the dilution series of the standard.
Competition ELISA and RGA cell based activity assay
Standard Competition ELISA techniques, as well as a pGL3-(CAGA)12 reporter assay (described below) were used to determine the IC50 for inhibition of binding of myostatin to its receptor, ActRIIB, and inhibition of downstream signaling. A recombinant ActRIIBFc chimera (generated by fusing the extracellular domain of the human ActRIIB receptor with human IgG1 Fc region) was coated on 96well flat-bottom assay plates (Costar, Cat. No. 3590) at 1 μg/ml in 0.2 M sodium carbonate buffer overnight at 4°C. Plates were then blocked with 1 mg/ml BSA in PBS 0.1% tween, 200 µl/well, for 1 hour at room temperature or overnight at 4°C and washed following a standard ELISA protocol. To analyze the inhibitory activity, RK35 was tested at various concentrations by pre-incubation with 10 ng/ml biotinylated mature myostatin. After incubation for 45 minutes at room temperature, 100 μl of a RK35 and biotinylated myostatin mixture was added to the plate and incubated for 1 hour at room temperature. After a wash step, streptavidin-horseradish peroxidase (SA-HRP, Southern Biotech 7100-05) diluted at 1:5000 was added and incubated for 30 minutes, followed by the addition of TMB (BioFX TMB TMBW-0100-01). Colorimetric measurements were taken at 450 nm in a Molecular Devices microplate reader.
To demonstrate the activity of myostatin, a reporter gene assay (RGA) was developed using a reporter vector pGL3(CAGA)12 expressing luciferase under control of a TGFβ induced promoter. The CAGA is a TGFβ-responsive sequence within the promoter of the TGFβ-induced gene PAI1. A reporter vector containing 12 CAGA boxes was made using the basic luciferase reporter plasmid pGL3 (Promega). The TATA box and transcription initiation site from the adenovirus major later promoter (35/+10) was inserted between the BglII and HindIII sites. Oligonucleotides containing 12 repeats of the CAGA boxes, i.e., AGCCAGACA, were annealed and cloned into the XhoI restriction site. The human rhabdomyosarcoma cell line A204 (ATCC HTB82) was transiently transfected with pGL3(CAGA)12 using FuGENE 6 transfection reagent (Boehringer). Following transfection, cells were cultured in 96well plates in McCoy's 5A medium supplemented with 2 mM glutamine, 100 U/ml streptomycin, 100 µg/ml penicillin and 10% fetal calf serum for 16 hours. Cells were then treated for 6 hours at 37°C with or without 10 ng/ml myostatin in McCoy's 5A medium containing glutamine, streptomycin, penicillin, and 1 mg/ml bovine serum albumin. Luciferase was quantified in the treated cells using the Luciferase Assay System (Promega). To test the inhibitory activity of RK35, myostatin was pre-incubated with the antibody for 1 hour at room temperature. This mixture was then added to the transfected cells and incubated for 6 hours at 37°C. Luciferase was quantified using the Luciferase Assay System (Promega).
SPR analysis
SPR analysis was performed at 25°C using a BIACORE 3000 (GE Healthcare). Protein A was immobilized on all 4 flow cells of a CM5 sensor-chip using amine coupling chemistry. The surface was activated by injecting a solution of 0.2 M N-ethyl-N-dimethylaminopropylcarbodiimide (EDC) and 50 mM N-hydroxysuccinimide (NHS) for 7 minutes. Protein A was diluted to 50 μg/ml in 10 mM sodium acetate buffer pH 5.0 and injected for 3 minutes at a flow rate of 10 μl per minute. The surface was then blocked using 1 M ethanolamine (ETH) for 7 minutes. Final immobilization levels of protein A were between 1000–1200 Response Unit (RU). The immobilization procedure was followed by several washes with running buffer to equilibrate the surface (0.01 M HEPES pH 7.4, 0.15 M NaCl, 3 mM EDTA, 0.005% P20). The RK 35 antibodies were diluted to 0.25 μg/ml in HBS-EP buffer. 5 μl of each antibody was injected over Protein A coated flow cells 2, 3, or 4 at a rate of 10 μl/minute, yielding approximately 200 RU of captured antibody. A titration series of mature myostatin was prepared in 0.01 M sodium acetate pH 5.0, 0.15 M NaCl, 3 mM EDTA, 0.005% P20, which was also used as the assay running buffer. The concentrations of the 2-fold dilutions of myostatin ranged from 4.0 nM to 0.125 nM and were injected over the captured antibody for 2 minutes at a flow rate of 50 μl/minute and allowed to dissociate for 30 minutes. After each cycle of antibody capture and myostatin injection the sensor chip surface was regenerated with 30 μl of 10 mM NaPO4, 0.5 M NaCl pH 2.5 at a flow rate of 50 μl/minute. BIAevaluation software version 4.1.1 (GE Healthcare) was used for data analysis. Data were double referenced,83 subtracting the signal contributed by the buffer and the reference surface. The Langmuir 1:1 model was used to globally fit the sensogram data and calculate KD values. An example sensogram is shown in Fig. S6.
Sypro Orange experiment for thermostability
This assay has been described previously by King et al.84 Briefly, samples were diluted to 0.1 mg/ml in PBS and Sypro Orange dye (5000x stock solution in dimethyl sulfoxide, Invitrogen) was added to a final concentration of 2.5x. Samples were placed in a 96 well plate in quadruplicate and heated from 20°C to 95°C at 1°C/min on an ABI Prism 7000 sequence detection system (Applied Biosystems). Fluorescence was detected using the SYBR detector. The raw data, fluorescence vs. temperature, was background-subtracted and fitted to 3 Gaussians in Excel. Based on the fitting, at each temperature the un-foldedness of the protein could be calculated. The temperature at which the protein is 1% unfolded, T1%, was used as the onset of unfolding.84
pH stability solutions
As described before,84 a protein A loading buffer (650 mM sodium sulfate, 20 mM sodium citrate, 20 mM boric acid and 20 mM sodium phosphate, pH 9) and protein A elution buffer (20 mM citric acid and 150 mM sodium chloride, pH 1.5), were titrated resulting in 24 solutions from pH 9 to pH 1.5. For fluorescence experiments, 98 μl of each of the pH buffers was placed in black, clear-bottom 96-well plates (Corning Inc.). Antibody solutions were concentrated to 5 mg/ml where necessary, using MicroCon 30 kDa cut-off filters (Millipore) and 2 μl aliquots were added to the 96-well plate, for a final protein concentration of 0.1 mg/ml (∼0.67 µM for an antibody). Following sealing and storage at 4°C for 24 hours, the plate with different pH solutions and protein was equilibrated at room temperature for 30 minutes. Aliquots of 5 µl 8-anilinonaphthalene-1-sulfonic acid ammonium salt (ANS; Sigma-Aldrich) in RODI water were added to a final ANS concentration of 67 µM, 100-fold molar excess over antibody. Fluorescence intensity was read immediately on an Infinite M1000 plate reader (Tecan Systems Inc.) exciting at 360 nm and reading the emission at 500 nm (20 nm bandwidth). The fluorescence signal of ANS in the various buffers was negligible and therefore not subtracted from the protein samples. Plotting fluorescence intensity against pH, a Boltzman sigmoidal curve was fitted to the data using Origin 7 SR2 (OriginLab). The pH50 value corresponded to the pH value where the fluorescence was at half maximum.
In vivo experiment
Male C57Bl/6 mice purchased from Charles River Laboratories were housed in a facility with 12 hour light-dark cycle and fed standard mouse diet (Lab Diet 5053) and water ad libitum. All procedures performed on animals in this study were in accordance with established guideline and regulations, and were reviewed and approved by Pfizer Institutional Animal Care and Use Committee. Pfizer care facilities that supported this work are fully accredited by AAALAC International.
At 8 weeks of age, mice were given weekly IP injections of either 10 mg/kg mRK-35, 10 mg/kg MYO-029, or Vehicle (PBS). After two weeks of treatment, mice were anesthetized with ketamine/xylazine and the EDL muscle was removed and placed in an oxygenated Ringer's bath. A silk suture was tied to the proximal and distal ends of the EDL just beyond the myotendinous junction, and the muscle was suspended between the lever arm of a force transducer (Aurora Scientific) and an immobile post. The muscle was set to resting length (L0) by a series of twitches. To determine isometric tetanic force, an electrical pulse at 120 Hz for one half second was applied to the muscle 3 times with a 5 minute rest period between each pulse and the maximum force at each pulse was recorded. Cross-sectional area was calculated by dividing the muscle mass by the mammalian muscle density (1.06 mg/mm3) multiplied by L0 times the ratio of muscle length to fiber length (0.45).85 Statistical analysis was done using a one-way ANOVA with Tukey's Post Test performed using GraphPad Prism version 6.03 for Windows, GraphPad Software, La Jolla California USA (www.graphpad.com).
Accession numbers
The structures generated in this study were deposited in the Protein Data Bank (PDB) under accession numbers 5F3B and 5F3H, as described in Table 2.
Supplementary Material
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
X-ray data were collected at Southeast Regional Collaborative Access Team (SER-CT) 22-ID (or 22-BM) beamline at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
References
- 1.Ecker DM, Jones SD, Levine HL. The therapeutic monoclonal antibody market. MAbs 2015; 7:9-14; PMID:25529996; http://dx.doi.org/ 10.4161/19420862.2015.989042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lu ZJ, Deng SJ, Huang DG, He Y, Lei M, Zhou L, Jin P. Frontier of therapeutic antibody discovery: The challenges and how to face them. World J Biol Chem 2012; 3:187-96; PMID:23275803; http://dx.doi.org/ 10.4331/wjbc.v3.i12.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weiner LM. Fully human therapeutic monoclonal antibodies. J Immunother 2006; 29:1-9; PMID:16365595; http://dx.doi.org/ 10.1097/01.cji.0000192105.24583.83 [DOI] [PubMed] [Google Scholar]
- 4.Borrebaeck CA. Antibody engineering. New York: Oxford University Press, 1995 [Google Scholar]
- 5.Carter P. Improving the efficacy of antibody-based cancer therapies. Nat Rev Cancer 2001; 1:118-29; PMID:11905803; http://dx.doi.org/ 10.1038/35101072 [DOI] [PubMed] [Google Scholar]
- 6.Hwang WY, Foote J. Immunogenicity of engineered antibodies. Methods 2005; 36:3-10; PMID:15848070; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.001 [DOI] [PubMed] [Google Scholar]
- 7.Queen C, Schneider WP, Selick HE, Payne PW, Landolfi NF, Duncan JF, Avdalovic NM, Levitt M, Junghans RP, Waldmann TA. A humanized antibody that binds to the interleukin 2 receptor. Proc Natl Acad Sci U S A 1989; 86:10029-33; PMID:2513570; http://dx.doi.org/ 10.1073/pnas.86.24.10029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Riechmann L, Clark M, Waldmann H, Winter G. Reshaping human antibodies for therapy. Nature 1988; 332:323-7; PMID:3127726; http://dx.doi.org/ 10.1038/332323a0 [DOI] [PubMed] [Google Scholar]
- 9.Kashmiri SV, De Pascalis R, Gonzales NR, Schlom J. Sdr grafting–a new approach to antibody humanization. Methods 2005; 36:25-34; PMID:15848072; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.003 [DOI] [PubMed] [Google Scholar]
- 10.Ritter G, Cohen LS, Williams C Jr, Richards EC, Old LJ, Welt S. Serological analysis of human anti-human antibody responses in colon cancer patients treated with repeated doses of humanized monoclonal antibody a33. Cancer Res 2001; 61:6851-9; PMID:11559561 [PubMed] [Google Scholar]
- 11.Stephens S, Emtage S, Vetterlein O, Chaplin L, Bebbington C, Nesbitt A, Sopwith M, Athwal D, Novak C, Bodmer M. Comprehensive pharmacokinetics of a humanized antibody and analysis of residual anti-idiotypic responses. Immunology 1995; 85:668-74; PMID:7558164 [PMC free article] [PubMed] [Google Scholar]
- 12.Richards J, Auger J, Peace D, Gale D, Michel J, Koons A, Haverty T, Zivin R, Jolliffe L, Bluestone JA. Phase i evaluation of humanized okt3: Toxicity and immunomodulatory effects of hokt3gamma4. Cancer Res 1999; 59:2096-101; PMID:10232594 [PubMed] [Google Scholar]
- 13.Schneider WP, Glaser SM, Kondas JA, Hakimi J. The anti-idiotypic response by cynomolgus monkeys to humanized anti-tac is primarily directed to complementarity-determining regions h1, h2, and l3. J Immunol 1993; 150:3086-90; PMID:8454876 [PubMed] [Google Scholar]
- 14.Lonberg N. Fully human antibodies from transgenic mouse and phage display platforms. Curr Opin Immunol 2008; 20:450-9; PMID:18606226; http://dx.doi.org/ 10.1016/j.coi.2008.06.004 [DOI] [PubMed] [Google Scholar]
- 15.Harding FA, Stickler MM, Razo J, DuBridge RB. The immunogenicity of humanized and fully human antibodies: Residual immunogenicity resides in the cdr regions. MAbs 2010; 2:256-65; PMID:20400861; http://dx.doi.org/ 10.4161/mabs.2.3.11641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Padlan EA. Anatomy of the antibody molecule. Mol Immunol 1994; 31:169-217; PMID:8114766; http://dx.doi.org/ 10.1016/0161-5890(94)90001-9 [DOI] [PubMed] [Google Scholar]
- 17.Gonzales NR, Padlan EA, De Pascalis R, Schuck P, Schlom J, Kashmiri SV. Sdr grafting of a murine antibody using multiple human germline templates to minimize its immunogenicity. Mol Immunol 2004; 41:863-72; PMID:15261458; http://dx.doi.org/ 10.1016/j.molimm.2004.03.041 [DOI] [PubMed] [Google Scholar]
- 18.Tamura M, Milenic DE, Iwahashi M, Padlan E, Schlom J, Kashmiri SV. Structural correlates of an anticarcinoma antibody: Identification of specificity-determining residues (sdrs) and development of a minimally immunogenic antibody variant by retention of sdrs only. J Immunol 2000; 164:1432-41; PMID:10640759; http://dx.doi.org/ 10.4049/jimmunol.164.3.1432 [DOI] [PubMed] [Google Scholar]
- 19.Hwang WY, Almagro JC, Buss TN, Tan P, Foote J. Use of human germline genes in a cdr homology-based approach to antibody humanization. Methods 2005; 36:35-42; PMID:15848073; http://dx.doi.org/ 10.1016/j.ymeth.2005.01.004 [DOI] [PubMed] [Google Scholar]
- 20.Tan P, Mitchell DA, Buss TN, Holmes MA, Anasetti C, Foote J. “Superhumanized” antibodies: Reduction of immunogenic potential by complementarity-determining region grafting with human germline sequences: Application to an anti-cd28. J Immunol 2002; 169:1119-25; PMID:12097421; http://dx.doi.org/ 10.4049/jimmunol.169.2.1119 [DOI] [PubMed] [Google Scholar]
- 21.Baer M, Sawa T, Flynn P, Luehrsen K, Martinez D, Wiener-Kronish JP, Yarranton G, Bebbington C. An engineered human antibody fab fragment specific for pseudomonas aeruginosa pcrv antigen has potent antibacterial activity. Infect Immun 2009; 77:1083-90; PMID:19103766; http://dx.doi.org/ 10.1128/IAI.00815-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bowers PM, Neben TY, Tomlinson GL, Dalton JL, Altobell L, Zhang X, Macomber JL, Wu BF, Toobian RM, McConnell AD, et al.. Humanization of antibodies using heavy chain complementarity-determining region 3 grafting coupled with in vitro somatic hypermutation. J Biol Chem 2013; 288:7688-96; PMID:23355464; http://dx.doi.org/ 10.1074/jbc.M112.445502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lazar GA, Desjarlais JR, Jacinto J, Karki S, Hammond PW. A molecular immunology approach to antibody humanization and functional optimization. Mol Immunol 2007; 44:1986-98; PMID:17079018; http://dx.doi.org/ 10.1016/j.molimm.2006.09.029 [DOI] [PubMed] [Google Scholar]
- 24.Dennis MS. Cdr repair: A novel approach to antibody humanization In: Shire SJ GW, Bechtold-Peters K, Andya J, ed. Current trends in monoclonal antibody development and manufacturing. New York: Springer, 2010:9-28; http://dx.doi.org/ 10.1007/978-0-387-76643-0_2 [DOI] [Google Scholar]
- 25.Townsend S, Fennell BJ, Apgar JR, Lambert M, McDonnell B, Grant J, Wade J, Franklin E, Foy N, Ni Shuilleabhain D, et al.. Augmented binary substitution: Single-pass cdr germ-lining and stabilization of therapeutic antibodies. Proc Natl Acad Sci U S A 2015; PMID:26621728; http://dx.doi.org/12498801 10.1073/pnas.1510944112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ewert S, Huber T, Honegger A, Pluckthun A. Biophysical properties of human antibody variable domains. J Mol Biol 2003; 325:531-53; PMID:12498801; http://dx.doi.org/ 10.1016/S0022-2836(02)01237-8 [DOI] [PubMed] [Google Scholar]
- 27.Honegger A, Malebranche AD, Rothlisberger D, Pluckthun A. The influence of the framework core residues on the biophysical properties of immunoglobulin heavy chain variable domains. Protein Eng Des Sel 2009; 22:121-34; PMID:19136675; http://dx.doi.org/ 10.1093/protein/gzn077 [DOI] [PubMed] [Google Scholar]
- 28.Kugler M, Stein C, Schwenkert M, Saul D, Vockentanz L, Huber T, Wetzel SK, Scholz O, Pluckthun A, Honegger A, et al.. Stabilization and humanization of a single-chain fv antibody fragment specific for human lymphocyte antigen cd19 by designed point mutations and cdr-grafting onto a human framework. Protein Eng Des Sel 2009; 22:135-47; PMID:19188138; http://dx.doi.org/ 10.1093/protein/gzn079 [DOI] [PubMed] [Google Scholar]
- 29.McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new tgf-β superfamily member. Nature 1997; 387:83-90; PMID:9139826; http://dx.doi.org/ 10.1038/387083a0 [DOI] [PubMed] [Google Scholar]
- 30.McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci U S A 1997; 94:12457-61; PMID:9356471; http://dx.doi.org/ 10.1073/pnas.94.23.12457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mateescu RG, Thonney ML. Gene expression in sexually dimorphic muscles in sheep. J Anim Sci 2002; 80:1879-87; PMID:12162655; http://dx.doi.org/ 10.2527/2002.8071879x [DOI] [PubMed] [Google Scholar]
- 32.Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (gdf8) in double-muscled belgian blue and piedmontese cattle. Genome Res 1997; 7:910-6; PMID:9314496 [DOI] [PubMed] [Google Scholar]
- 33.Xu C, Wu G, Zohar Y, Du SJ. Analysis of myostatin gene structure, expression and function in zebrafish. J Exp Biol 2003; 206:4067-79; PMID:14555747; http://dx.doi.org/ 10.1242/jeb.00635 [DOI] [PubMed] [Google Scholar]
- 34.Mosher DS, Quignon P, Bustamante CD, Sutter NB, Mellersh CS, Parker HG, Ostrander EA. A mutation in the myostatin gene increases muscle mass and enhances racing performance in heterozygote dogs. PLoS Genet 2007; 3:e79; PMID:17530926; http://dx.doi.org/ 10.1371/journal.pgen.0030079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shelton GD, Engvall E. Gross muscle hypertrophy in whippet dogs is caused by a mutation in the myostatin gene. Neuromuscul Disord 2007; 17:721-2; PMID:17651971; http://dx.doi.org/ 10.1016/j.nmd.2007.06.008 [DOI] [PubMed] [Google Scholar]
- 36.Schuelke M, Wagner KR, Stolz LE, Hubner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med 2004; 350:2682-8; PMID:15215484; http://dx.doi.org/ 10.1056/NEJMoa040933 [DOI] [PubMed] [Google Scholar]
- 37.Bradley L, Yaworsky PJ, Walsh FS. Myostatin as a therapeutic target for musculoskeletal disease. Cell Mol Life Sci 2008; 65:2119-24; PMID:18425412; http://dx.doi.org/ 10.1007/s00018-008-8077-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Holzbaur EL, Howland DS, Weber N, Wallace K, She Y, Kwak S, Tchistiakova LA, Murphy E, Hinson J, Karim R, et al.. Myostatin inhibition slows muscle atrophy in rodent models of amyotrophic lateral sclerosis. Neurobiol Dis 2006; 23:697-707; PMID:16837207; http://dx.doi.org/ 10.1016/j.nbd.2006.05.009 [DOI] [PubMed] [Google Scholar]
- 39.Walsh FS, Rutkowski JL. Myostatin as a therapeutic target in amyotrophic lateral sclerosis. Neurochem Int 2012; 61:931-5; PMID:22841860; http://dx.doi.org/ 10.1016/j.neuint.2012.07.016 [DOI] [PubMed] [Google Scholar]
- 40.Tabs therapeutic antibody database. Http://tabs.Craic.Com, 2015 [Google Scholar]
- 41.Wagner KR, Fleckenstein JL, Amato AA, Barohn RJ, Bushby K, Escolar DM, Flanigan KM, Pestronk A, Tawil R, Wolfe GI, et al.. A phase i/iitrial of myo-029 in adult subjects with muscular dystrophy. Ann Neurol 2008; 63:561-71; PMID:18335515; http://dx.doi.org/ 10.1002/ana.21338 [DOI] [PubMed] [Google Scholar]
- 42.De Groot AS, Martin W. Reducing risk, improving outcomes: Bioengineering less immunogenic protein therapeutics. Clin Immunol 2009; 131:189-201; PMID:19269256; http://dx.doi.org/ 10.1016/j.clim.2009.01.009 [DOI] [PubMed] [Google Scholar]
- 43.Koren E, De Groot AS, Jawa V, Beck KD, Boone T, Rivera D, Li L, Mytych D, Koscec M, Weeraratne D, et al.. Clinical validation of the “in silico” prediction of immunogenicity of a human recombinant therapeutic protein. Clin Immunol 2007; 124:26-32; PMID:17490912; http://dx.doi.org/ 10.1016/j.clim.2007.03.544 [DOI] [PubMed] [Google Scholar]
- 44.Cash JN, Rejon CA, McPherron AC, Bernard DJ, Thompson TB. The structure of myostatin:Follistatin 288: Insights into receptor utilization and heparin binding. EMBO J 2009; 28:2662-76; PMID:19644449; http://dx.doi.org/ 10.1038/emboj.2009.205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Allendorph GP, Isaacs MJ, Kawakami Y, Izpisua Belmonte JC, Choe S. Bmp-3 and bmp-6 structures illuminate the nature of binding specificity with receptors. Biochemistry 2007; 46:12238-47; PMID:17924656; http://dx.doi.org/ 10.1021/bi700907k [DOI] [PubMed] [Google Scholar]
- 46.Weiskirchen R, Meurer SK, Gressner OA, Herrmann J, Borkham-Kamphorst E, Gressner AM. Bmp-7 as antagonist of organ fibrosis. Front Biosci (Landmark Ed) 2009; 14:4992-5012; PMID:19482601; http://dx.doi.org/ 10.2741/3583 [DOI] [PubMed] [Google Scholar]
- 47.Kotzsch A, Nickel J, Seher A, Sebald W, Muller TD. Crystal structure analysis reveals a spring-loaded latch as molecular mechanism for gdf-5-type i receptor specificity. EMBO J 2009; 28:937-47; PMID:19229295; http://dx.doi.org/ 10.1038/emboj.2009.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cash JN, Angerman EB, Kattamuri C, Nolan K, Zhao H, Sidis Y, Keutmann HT, Thompson TB. Structure of myostatin Follistatin-like 3: N-terminal domains of follistatin-type molecules exhibit alternate modes of binding. J Biol Chem 2012; 287:1043-53; PMID:22052913; http://dx.doi.org/ 10.1074/jbc.M111.270801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Brown MA, Zhao Q, Baker KA, Naik C, Chen C, Pukac L, Singh M, Tsareva T, Parice Y, Mahoney A, et al.. Crystal structure of bmp-9 and functional interactions with pro-region and receptors. J Biol Chem 2005; 280:25111-8; PMID:15851468; http://dx.doi.org/ 10.1074/jbc.M503328200 [DOI] [PubMed] [Google Scholar]
- 50.Zhao S, Goodsell DS, Olson AJ. Analysis of a data set of paired uncomplexed protein structures: New metrics for side-chain flexibility and model evaluation. Proteins 2001; 43:271-9; PMID:11288177; http://dx.doi.org/ 10.1002/prot.1038 [DOI] [PubMed] [Google Scholar]
- 51.Sako D, Grinberg AV, Liu J, Davies MV, Castonguay R, Maniatis S, Andreucci AJ, Pobre EG, Tomkinson KN, Monnell TE, et al.. Characterization of the ligand binding functionality of the extracellular domain of activin receptor type iib. J Biol Chem 2010; 285:21037-48; PMID:20385559; http://dx.doi.org/ 10.1074/jbc.M110.114959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gordon KJ, Blobe GC. Role of transforming growth factor-β superfamily signaling pathways in human disease. Biochim Biophys Acta 2008; 1782:197-228; PMID:18313409; http://dx.doi.org/ 10.1016/j.bbadis.2008.01.006 [DOI] [PubMed] [Google Scholar]
- 53.Upton PD, Davies RJ, Trembath RC, Morrell NW. Bone morphogenetic protein (bmp) and activin type ii receptors balance bmp9 signals mediated by activin receptor-like kinase-1 in human pulmonary artery endothelial cells. J Biol Chem 2009; 284:15794-804; PMID:19366699; http://dx.doi.org/ 10.1074/jbc.M109.002881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Greenwald J, Vega ME, Allendorph GP, Fischer WH, Vale W, Choe S. A flexible activin explains the membrane-dependent cooperative assembly of tgf-β family receptors. Mol Cell 2004; 15:485-9; PMID:15304227; http://dx.doi.org/ 10.1016/j.molcel.2004.07.011 [DOI] [PubMed] [Google Scholar]
- 55.Thompson TB, Woodruff TK, Jardetzky TS. Structures of an actriib:Activin a complex reveal a novel binding mode for tgf-β ligand:Receptor interactions. EMBO J 2003; 22:1555-66; PMID:12660162; http://dx.doi.org/ 10.1093/emboj/cdg156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Weber D, Kotzsch A, Nickel J, Harth S, Seher A, Mueller U, Sebald W, Mueller TD. A silent h-bond can be mutationally activated for high-affinity interaction of bmp-2 and activin type iib receptor. BMC Struct Biol 2007; 7:6; PMID:17295905; http://dx.doi.org/ 10.1186/1472-6807-7-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Townson SA, Martinez-Hackert E, Greppi C, Lowden P, Sako D, Liu J, Ucran JA, Liharska K, Underwood KW, Seehra J, et al.. Specificity and structure of a high affinity activin receptor-like kinase 1 (alk1) signaling complex. J Biol Chem 2012; 287:27313-25; PMID:22718755; http://dx.doi.org/ 10.1074/jbc.M112.377960 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baker MP, Reynolds HM, Lumicisi B, Bryson CJ. Immunogenicity of protein therapeutics: The key causes, consequences and challenges. Self Nonself 2010; 1:314-22; PMID:21487506; http://dx.doi.org/ 10.4161/self.1.4.13904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Gao SH, Huang K, Tu H, Adler AS. Monoclonal antibody humanness score and its applications. BMC Biotechnol 2013; 13:55; PMID:23826749; http://dx.doi.org/ 10.1186/1472-6750-13-55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Choi Y, Hua C, Sentman CL, Ackerman ME, Bailey-Kellogg C. Antibody humanization by structure-based computational protein design. MAbs 2015; 7:1045-57; PMID:26252731; http://dx.doi.org/ 10.1080/19420862.2015.1076600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Abysis version 2.3.3: A fully integrated antibody discovery system Http://www.Bioinf.Org.Uk/abysis/. 2015 [Google Scholar]
- 62.Abhinandan KR, Martin AC. Analyzing the “degree of humanness” of antibody sequences. J Mol Biol 2007; 369:852-62; PMID:17442342; http://dx.doi.org/ 10.1016/j.jmb.2007.02.100 [DOI] [PubMed] [Google Scholar]
- 63.Sirin S, Apgar JR, Bennett EM, Keating AE. Ab-bind: Antibody binding mutational database for computational affinity predictions. Protein Sci 2016; 25:393-409; PMID:26473627; http://dx.doi.org/ 10.1002/pro.2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kortemme T, Kim DE, Baker D. Computational alanine scanning of protein-protein interfaces. Sci STKE 2004; 2004:pl2; PMID:14872095 [DOI] [PubMed] [Google Scholar]
- 65.Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci U S A 2002; 99:14116-21; PMID:12381794; http://dx.doi.org/ 10.1073/pnas.202485799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Morrow JK, Zhang S. Computational prediction of protein hot spot residues. Curr Pharm Des 2012; 18:1255-65; PMID:22316154; http://dx.doi.org/ 10.2174/138161212799436412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Zhou H, Zhou Y. Stability scale and atomic solvation parameters extracted from 1023 mutation experiments. Proteins 2002; 49:483-92; PMID:12402358; http://dx.doi.org/ 10.1002/prot.10241 [DOI] [PubMed] [Google Scholar]
- 68.Chothia C, Novotny J, Bruccoleri R, Karplus M. Domain association in immunoglobulin molecules. The packing of variable domains. J Mol Biol 1985; 186:651-63; PMID:4093982; http://dx.doi.org/ 10.1016/0022-2836(85)90137-8 [DOI] [PubMed] [Google Scholar]
- 69.Foote J, Winter G. Antibody framework residues affecting the conformation of the hypervariable loops. J Mol Biol 1992; 224:487-99; PMID:1560463; http://dx.doi.org/ 10.1016/0022-2836(92)91010-M [DOI] [PubMed] [Google Scholar]
- 70.McCafferty J, Hoogenboom H, Chiswell D. Antibody engineering : A practical approach. Oxford: New York: IRL Press at Oxford University Press, 1996 [Google Scholar]
- 71.Ó Conchúir S, Barlow KA, Pache RA, Ollikainen N, Kundert K, O'Meara MJ, Smith CA, Kortemme T. A web resource for standardized benchmark data sets, metrics, and rosetta protocols for macromolecular modeling and design. PLoS One 2015; 10:e0130433; PMID:26335248; http://dx.doi.org/ 10.1371/journal.pone.0130433 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jawa V, Cousens LP, Awwad M, Wakshull E, Kropshofer H, De Groot AS. T-cell dependent immunogenicity of protein therapeutics: Preclinical assessment and mitigation. Clin Immunol 2013; 149:534-55; PMID:24263283; http://dx.doi.org/ 10.1016/j.clim.2013.09.006 [DOI] [PubMed] [Google Scholar]
- 73.Chow CK, Allan BW, Chai Q, Atwell S, Lu J. Therapeutic antibody engineering to improve viscosity and phase separation guided by crystal structure. Mol Pharm 2016; 13:915-23; PMID:26849155; http://dx.doi.org/ 10.1021/acs.molpharmaceut.5b00817 [DOI] [PubMed] [Google Scholar]
- 74.Sharma VK, Patapoff TW, Kabakoff B, Pai S, Hilario E, Zhang B, Li C, Borisov O, Kelley RF, Chorny I, et al.. In silico selection of therapeutic antibodies for development: Viscosity, clearance, and chemical stability. Proc Natl Acad Sci U S A 2014; 111:18601-6; PMID:25512516; http://dx.doi.org/ 10.1073/pnas.1421779112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voynov V, Chennamsetty N, Kayser V, Helk B, Trout BL. Predictive tools for stabilization of therapeutic proteins. MAbs 2009; 1:580-2; PMID:20068399; http://dx.doi.org/ 10.4161/mabs.1.6.9773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sydow JF, Lipsmeier F, Larraillet V, Hilger M, Mautz B, Molhoj M, Kuentzer J, Klostermann S, Schoch J, Voelger HR, et al.. Structure-based prediction of asparagine and aspartate degradation sites in antibody variable regions. PLoS One 2014; 9:e100736; PMID:24959685; http://dx.doi.org/ 10.1371/journal.pone.0100736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Horowitz M, Cepko CL, Sharp PA. Expression of chimeric genes in the early region of sv40. J Mol Appl Genet 1983; 2:147-59; PMID:6192195 [PubMed] [Google Scholar]
- 78.Thies RS, Chen T, Davies MV, Tomkinson KN, Pearson AA, Shakey QA, Wolfman NM. Gdf-8 propeptide binds to gdf-8 and antagonizes biological activity by inhibiting gdf-8 receptor binding. Growth Factors 2001; 18:251-9; PMID:11519824; http://dx.doi.org/ 10.3109/08977190109029114 [DOI] [PubMed] [Google Scholar]
- 79.Otwinowski Z, Minor W. Processing of x-ray diffraction data collected in oscillation mode In: Carter CW Jr, Sweet RM, ed. Methods in enzymology. New York: Academic Press; 1997:p307-26; http://dx.doi.org/ 10.1016/S0076-6879(97)76066-X [DOI] [PubMed] [Google Scholar]
- 80.Navaza J. Implementation of molecular replacement in amore. Acta Crystallogr D Biol Crystallogr 2001; 57:1367-72; PMID:11567147; http://dx.doi.org/ 10.1107/S0907444901012422 [DOI] [PubMed] [Google Scholar]
- 81.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of coot. Acta Crystallogr D Biol Crystallogr 2010; 66:486-501; PMID:20383002; http://dx.doi.org/ 10.1107/S0907444910007493 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bricogne G, Blance E, Brandl M, Flensburg C, Keller P, Paciorek W, Roversi P, Sharff A, Smart OS, Vonrhein C, et al.. Buster version 2.9.4 Cambridge, United Kingdom: Global phasing ltd; 2011 [Google Scholar]
- 83.Myszka DG. Improving biosensor analysis. J Mol Recognit 1999; 12:279-84; PMID:10556875; http://dx.doi.org/ 10.1002/(SICI)1099-1352(199909/10)12:5%3c279::AID-JMR473%3e3.0.CO;2-3 [DOI] [PubMed] [Google Scholar]
- 84.King AC, Woods M, Liu W, Lu Z, Gill D, Krebs MR. High-throughput measurement, correlation analysis, and machine-learning predictions for ph and thermal stabilities of pfizer-generated antibodies. Protein Sci 2011; 20:1546-57; PMID:21710487; http://dx.doi.org/ 10.1002/pro.680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Brooks SV, Faulkner JA. Contractile properties of skeletal muscles from young, adult and aged mice. J Physiol 1988; 404:71-82; PMID:3253447; http://dx.doi.org/ 10.1113/jphysiol.1988.sp017279 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.