

ABSTRACT
Pulmonary alveolar proteinosis is associated with impaired alveolar macrophage differentiation due to genetic defects in the granulocyte macrophage colony-stimulating factor (GM-CSF) axis or autoantibody blockade of GM-CSF. The anti-GM-CSFRα antibody mavrilimumab has shown clinical benefit in patients with rheumatoid arthritis, but with no accompanying pulmonary pathology observed to date. We aimed to model systemic versus pulmonary pharmacodynamics of an anti-GM-CSFRα antibody to understand the pharmacology that contributes to this therapeutic margin. Mice were dosed intraperitoneal with anti-GM-CSFRα antibody, and pharmacodynamics bioassays for GM-CSFRα inhibition performed on blood and bronchoalveolar lavage (BAL) cells to quantify coverage in the circulation and lung, respectively. A single dose of 3 mg/kg of the anti-GM-CSFRα antibody saturated the systemic cellular pool, but dosing up to 10 times higher had no effect on the responsiveness of BAL cells to GM-CSF. Continued administration of this dose of anti-GM-CSFRα antibody for 7 consecutive days also had no inhibitory effect on these cells. Partial inhibition of GM-CSFRα function on cells from the BAL was only observed after dosing for 5 or 7 consecutive days at 30 mg/kg, 10-fold higher than the proposed therapeutic dose. In conclusion, dosing with anti-GM-CSFRα antibody using regimes that saturate circulating cells, and have been shown to be efficacious in inflammatory arthritis models, did not lead to complete blockade of the alveolar macrophages response to GM-CSF. This suggests a significant therapeutic window is possible with GM-CSF axis inhibition.
KEYWORDS: Antibody exposure, GM-CSF, GM-CSF Receptor, lung partitioning, pharmacodynamics, pulmonary alveolar proteinosis, rheumatoid arthritis
Abbreviations
- BAL
bronchoalveolar lavage
- GM-CSF
granulocyte macrophage colony-stimulating factor
- GM-CSFRα
granulocyte macrophage colony-stimulating factor receptor α subunit
- i.n
intranasal
- LLOQ
Lower Limit of Quantitation
- ND
not detectable
- PAP
pulmonary alveolar proteinosis
- PD
pharmacodynamics
- PK
pharmacokinetics
- RA
Rheumatoid arthritis
Introduction
Rheumatoid arthritis (RA), a chronic systemic autoimmune disease characterized by inflammation of synovial joints, affects approximately 1% of the population. The debilitating, painful joint swelling and damage can be refractory to, or incompletely modified by, current therapies, including both small molecule and biologic disease modifying anti-rheumatic drugs.1 Consequently, there is still need for novel treatments that are more efficacious.
Antibody blockade of granulocyte macrophage colony-stimulating factor (GM-CSF) and knock-out mice have revealed that the cytokine has a pivotal role in inflammation,2 and especially in models of autoimmune inflammation such as collagen-induced arthritis3,4 and experimental autoimmune encephalomyelitis.5 GM-CSF mediates its effects by specifically binding the GM-CSF receptor α subunit (GM-CSFRα), and then recruiting the signaling common β-chain. Antibody blockade of GM-CSFRα has a similarly anti-inflammatory effect on arthritis models.6 GM-CSFRα is widely expressed on myeloid cells and granulocytes, which are strongly associated with the inflammation in RA.7 GM-CSF is raised in synovial fluid from patients with RA.8,9 These data make the GM-CSF/GM-CSFRα axis an attractive target for therapeutic intervention in RA and other autoimmune diseases.
Mavrilimumab, an antagonistic antibody targeting GM-CSFRα, recently completed a Phase 2b trial in patients with RA who have had an inadequate response to methotrexate.10 Highly significant improvements in the signs and symptoms of arthritis were observed in this study, which replicated the results of an earlier Phase 2 study.11
In addition to its role in inflammation, studies in knock out mice have demonstrated that GM-CSF has a limited role in hematopoiesis, with steady-state effects being limited to some tissue-resident dendritic cell populations.12-15 This was surprising because GM-CSF was originally identified as a molecule that can expand myeloid progenitors.16 Further analysis did demonstrate a requirement for GM-CSF in alveolar macrophage function.12,13 The alveolar macrophages from mice deficient in GM-CSF or the common β chain are less able to catabolise surfactant lipids, which leads to the formation of foamy alveolar macrophage and the accumulation of lipoproteinaceous material in the lung.17,18 The lung phenotype of these mice and its similarity to a very rare lung condition, known then as idiopathic pulmonary alveolar proteinosis (PAP), led to the discovery that defects in this pathway underlie this disease.19,20 Subsequently PAP has been shown to be strongly associated with anti-GM-CSF autoantibodies,21 or with mutations in either of the GM-CSF receptor subunits.22-25 Additionally, transfer of auto-antibodies from patients with anti-GM-CSF-associated PAP to non-human primates led to the formation of foamy macrophages.26 These results indicate that GM-CSF plays an important role in the terminal differentiation of alveolar macrophages in mice and humans.
Dosing of very high levels (≥ 30 mg/kg/week for 11 weeks) of mavrilimumab in preclinical non-human primate studies was associated with the formation of a very small number of foamy macrophages within the lungs.27 However, there were no demonstrable changes in lung function or lung safety in either of the mavrilimumab trials. Notably the highest and most efficacious dose tested in patients, 150 mg every 2 weeks,10 was considerably lower than that which gave the subtle lung pathology preclinically.
Therefore, the aim of this study was to investigate the pharmacokinetic and pharmacodynamic characteristics of a systemically dosed anti-GM-CSFRα antibody in the periphery and in the lung. Antibodies are known to have lower penetrance into the lung, but we wanted to specifically explore the coverage on alveolar macrophages compared to circulating monocytes. Thereby, we could establish whether it was possible to dose an anti-GM-CSFRα antibody to get maximal blockade in the periphery, but minimize the potential lung toxicity by sparing alveolar macrophage function.
Results
Developing systemic and lung pharmacodynamic assays for anti-GM-CSFRα
To model the lung partitioning of anti-GM-CSFRα in the mouse, we developed systemic and lung pharmacodynamic bioassays. First, we established a bioassay for GM-CSF added exogenously to blood. GM-CSF is a poor direct activator of blood leucocytes, but does potently prime these cells for lipopolysaccharide (LPS) stimulation.28 In this assay, the addition of GM-CSF alone to blood did not induce interleukin (IL)-6 production, and LPS alone induced a low amount. Priming with GM-CSF for 4 hrs followed by LPS stimulation induced a high level of IL-6 in the range of 518 +/− 254 pg/ml (Fig. 1A). To demonstrate that this would act as a pharmacodynamic bioassay for the mavrilimumab surrogate anti-mouse GM-CSFRα antagonistic antibody, CAM-3003, we titrated the antibody in the blood assay prior to GM-CSF addition. CAM-3003 dose dependently reduced the level of IL-6 induced by GM-CSF and LPS to that induced by LPS alone. To represent the inhibitory effect of CAM-3003 in a blood sample, the amount of IL-6 due to GM-CSF potentiation (the level induced by LPS deducted from that induced by LPS and GM-CSF) was expressed as a percentage of the average GM-CSF potentiation in the no-antibody controls (Fig. 1B).
Figure 1.

GM-CSF potentiation of LPS induced IL-6 as a readout of GM-CSFRα inhibition. (A) Mouse whole blood, diluted in media, was primed with 10 pg/ml GM-CSF or not for 4 hrs followed by overnight stimulation with or without 10 ng/ml LPS. IL-6 levels in the supernatant were determined. GM-CSF potentiation was calculated by subtracting LPS stimulated IL-6 levels from that induced by GM-CSF primed blood. (B) Anti-GM-CSFRα, CAM-3003, or an irrelevant isotype control were titrated in prior to GM-CSF priming, and inhibition of the GM-CSF potentiated IL-6 release presented as a percentage of blood without antibody treatment. (C) Mouse bronchoalveolar lavage (BAL) cells were primed and stimulated as in A. The GM-CSF contribution calculated as above. (D) IL-6 levels in C were normalized for BAL cell number by dividing the IL-6 concentration by Cell-Titer Glo quantification of cell number after a freeze/thaw. (E) Ten nM anti-GM-CSFRα, CAM-3003, or an irrelevant isotype control were added prior to GM-CSF priming, and inhibition of the GM-CSF potentiated IL-6 release presented as a percentage of blood without antibody treatment. All measurements were duplicates and data shown are mean +/− SEM of 4 animals. * P < 0.05 and ** P < 0.01.
We set up a similar pharmacodynamic bioassay using bronchoalveolar lavage (BAL) cells because GM-CSF similarly enhances LPS induction of IL-6 from macrophages. Alveolar macrophages typically constituted greater than 95% of BAL cells as determined by F4/80 staining (data not shown). As shown previously, GM-CSF did not induce IL-6 production. LPS stimulated a small release (149 +/− 56 pg/ml); however, in combination high levels were detected (694 +/− 168 pg/ml, Fig. 1C), similar to that observed for blood when taking into consideration absolute cell numbers. CAM-3003, but not isotype control antibody, was able to inhibit the GM-CSF potentiated release of IL-6 (Fig. 1E). Because BAL cell recovery varied between mice, subsequent experiments with BAL cells accounted for this by normalizing each well to total cell counts as determined by Cell-titer Glo measurements as in Fig. 1D. Thus both bioassays acted as pharmacodynamic assays for CAM-3003, anti-GM-CSFRα.
Validating systemic and lung pharmacodynamic assays for anti-GM-CSFRα
To understand the utility of the systemic pharmacodynamics bioassay, mice were intraperitoneally (i.p.) administered various doses of CAM-3003. Pharmacokinetic analysis of CAM-3003 showed that the antibody was only detectable in plasma at the 2 highest doses, 1 mg/kg and 3 mg/kg, and these resulted in partial and complete blockade of the systemic bioassay, respectively (Fig. 2A and B).
Figure 2.

Ex vivo bioassays of GM-CSF potentiation of LPS induced IL-6 function as a pharmacodynamic readout of GM-CSFRα inhibition. (A) Mice were dosed i.p. with anti-GM-CSFRα, CAM-3003, and antibody levels in plasma measured 48 hr later. LLOQ = 13 ng/ml (B) GM-CSFRα inhibition on the blood cells was quantified by stimulation with 10 ng/ml LPS overnight with and without priming with 10 pg/ml GM-CSF. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. (C) Mice were dosed i.n. with anti-GM-CSFRα, CAM-3003, and antibody levels in the bronchoalveolar lavage (BAL) were measured 2 hr later. Four, 40 or 400 μg CAM-3003 equate to approximately 0.22, 2.2 and 22 mg/kg. LLOQ = 31 ng/ml (D) GM-CSFRα inhibition on the BAL cells was quantified by stimulation with 10 ng/ml LPS overnight with and without priming with 10 pg/ml GM-CSF. IL-6 levels in the supernatant were determined and normalized to the number of cells as determined by Cell-Titer Glo. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. All measurements were duplicates and data shown are mean +/− SEM of 4–6 animals. * P < 0.05 and ** P < 0.01.
To validate the lung pharmacodynamics assay, we wanted to achieve maximal coverage in the lung, so mice were administered anti-GM-CSFRα antibody via the intranasal (i.n.) route. Two hours after administration of 4, 40 or 400 μg CAM-3003 (equating to ∼0.22, 2.2 and 22 mg/kg), the antibody could be detected at increasing concentrations in the BAL at all dose levels (Fig. 2C). Dose-dependent inhibition could be seen in the BAL cell bioassay in these mice, with full inhibition being seen at the 40 and 400 μg dose (Fig. 2D). These demonstrate that the BAL cells bioassay functions as a pharmacodynamic assay for the lung compartment, and complete coverage could be achieved.
Lung and systemic pharmacodynamics following a single dose of anti-GM-CSFRα
To understand the pharmacodynamics of anti-GM-CSFRα in the lung following systemic administration, we dosed mice i.p. with 3, 10 and 30 mg/kg CAM-3003. As expected, even at the lowest dose, 3 mg/kg, CAM-3003 was detectable in plasma 48 hrs after administration, and completely inhibited the GM-CSF potentiation of the blood bioassay. The two higher doses showed increased systemic exposure that equated to dose, and similarly saturated pharmacodynamics (Fig. 3A and B), but no functional effect on BAL cells was observed (Fig. 3C). This shows there was no pharmacodynamic effect of systemic anti-GM-CSFRα in the lung, even when the antibody was dosed 10 times higher than that required to fully inhibit in the periphery.
Figure 3.
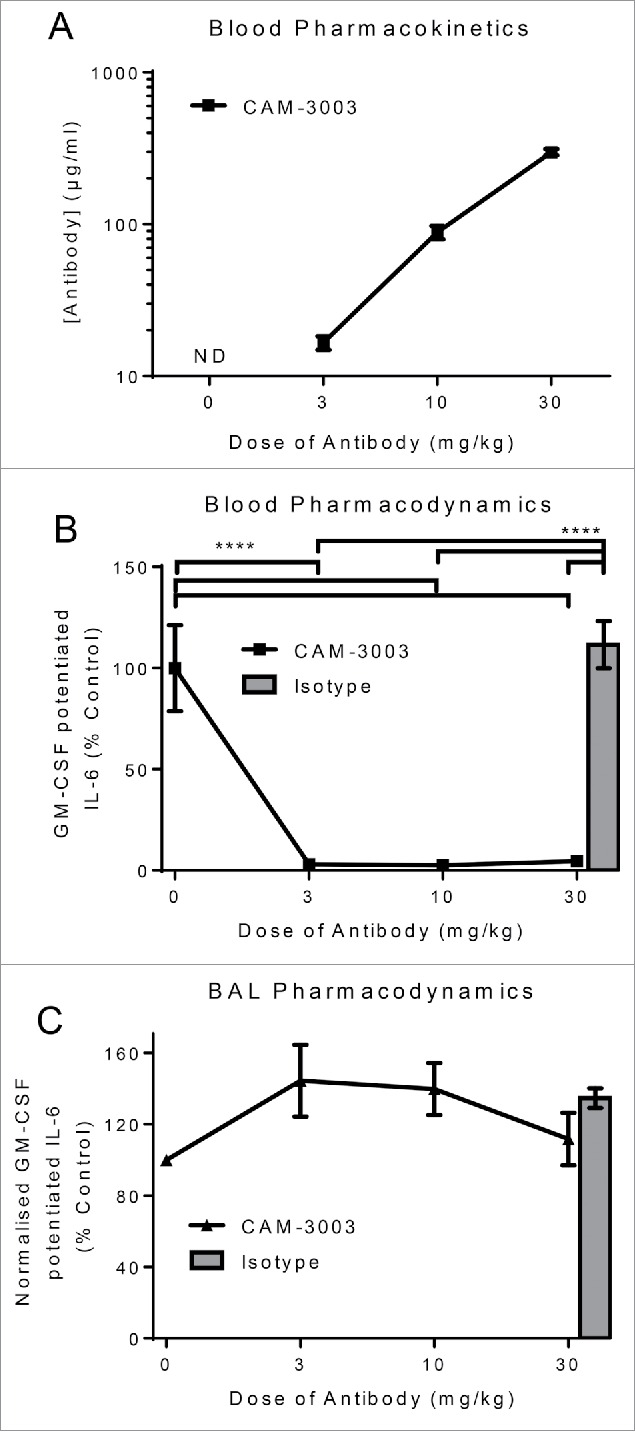
Single dose of anti-GM-CSFRα inhibits blood but not BAL cells. Mice were dosed i.p. with anti-GM-CSFRα, CAM-3003, or isotype control and anti-GM-CSFRα antibody levels in plasma measured 48 hr later. LLOQ = 97 ng/ml (A). (B) GM-CSFRα inhibition on the blood cells was quantified by stimulation with 10 ng/ml LPS overnight with and without priming with 10 pg/ml GM-CSF. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. (C) GM-CSFRα inhibition on the bronchoalveolar lavage (BAL) cells was quantified by stimulation as in B. IL-6 levels in the supernatant were determined and normalized to the number of cells as determined by Cell-Titer Glo. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. All measurements were duplicates and data shown are mean +/− SEM of 4–6 animals. **** P < 0.0001 and ** P < 0.01.
Lung and systemic pharmacodynamics following multiple doses of CAM-3003
Next we tested whether repeat doses of 30 mg/kg CAM-3003 daily for 1, 3, 5 or 7 d affected the lung compartment. The levels of the antibody continued to increase in the plasma as expected. Likewise, the antibody was detectable in the BAL even following the single administration, and the levels increased with the number of doses (Fig. 4A). Repeated administration of 30 mg/kg completely inhibited GM-CSF activity in the systemic bioassay (Fig. 4B). Interestingly, significant pharmacodynamics effects of the antibody on the lung cells could be observed after 5 daily doses of 30 mg/kg CAM-3003 (53% ± 23% inhibition). However, even after 7 d of antibody treatment only a partial blockade of the GM-CSF potentiation of IL-6 from BAL cells was observed (72% ± 11% inhibition, Fig. 4C).
Figure 4.
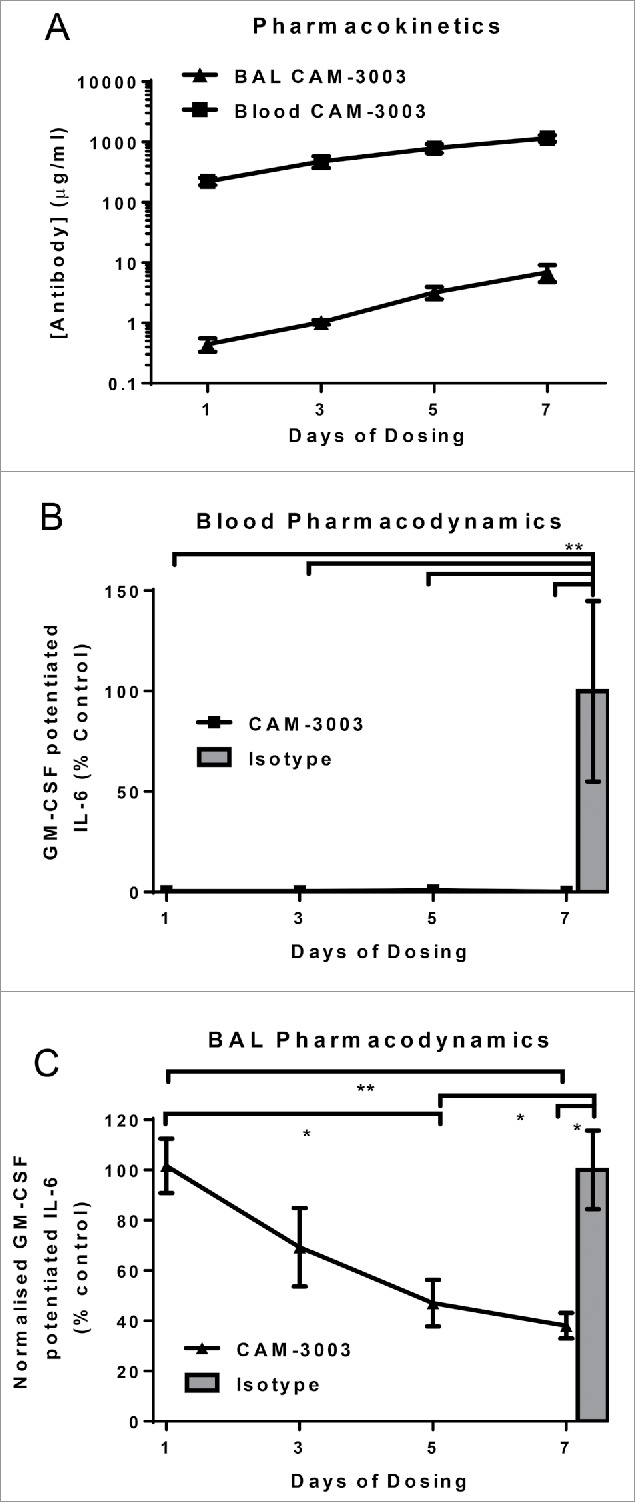
Multiple doses of anti-GM-CSFRα completely inhibits blood but not BAL cells. Mice were dosed i.p. with 30 mg/kg anti-GM-CSFRα, CAM-3003, or isotype control every day for 1, 3, 5 or 7 consecutive days. (A) Anti-GM-CSFRα antibody levels in plasma and bronchoalveolar lavage (BAL) were measured 48 hr later. Plasma LLOQ = 19.5 μg/ml and BAL LLOQ = 97 ng/ml. (B) GM-CSFRα inhibition on the blood cells was quantified by stimulation with 10 ng/ml LPS overnight with and without priming with 10 pg/ml GM-CSF. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. (C) GM-CSFRα inhibition on the BAL cells was quantified by stimulation as in B. IL-6 levels in the supernatant were determined and normalized to the number of cells as determined by Cell-Titer Glo. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. All measurements were duplicates and data shown are mean +/− SEM of 4–6 animals. * P < 0.05 and ** P < 0.01.
The lung pharmcodynamic effects seen after multiple doses of 30 mg/kg CAM-3003 could be a function of the length of exposure rather than the amount of antibody administered. To test this, we compared the systemic and lung pharmacokinetics and pharmacodynamics following 7 daily doses of 3 or 30 mg/kg CAM-3003. As would be predicted, dosing at 30 mg/kg resulted in higher antibody levels in both plasma and BAL compared to the lower dose (Fig. 5A), and, consistent with previous data, both resulted in blockade of the GM-CSF potentiation of IL-6 in blood (Fig. 5B). In contrast, daily administration of 3 mg/kg did not affect the BAL cell bioassay, with the 30 mg/kg group again showing partial coverage (34% ± 13% inhibition, Fig. 5C). This indicates repeated, very high dose systemic administration far beyond that necessary to completely block the axis in the periphery is required for the anti-GM-CSFRα antibody to have an inhibitory effect on alveolar macrophages.
Figure 5.
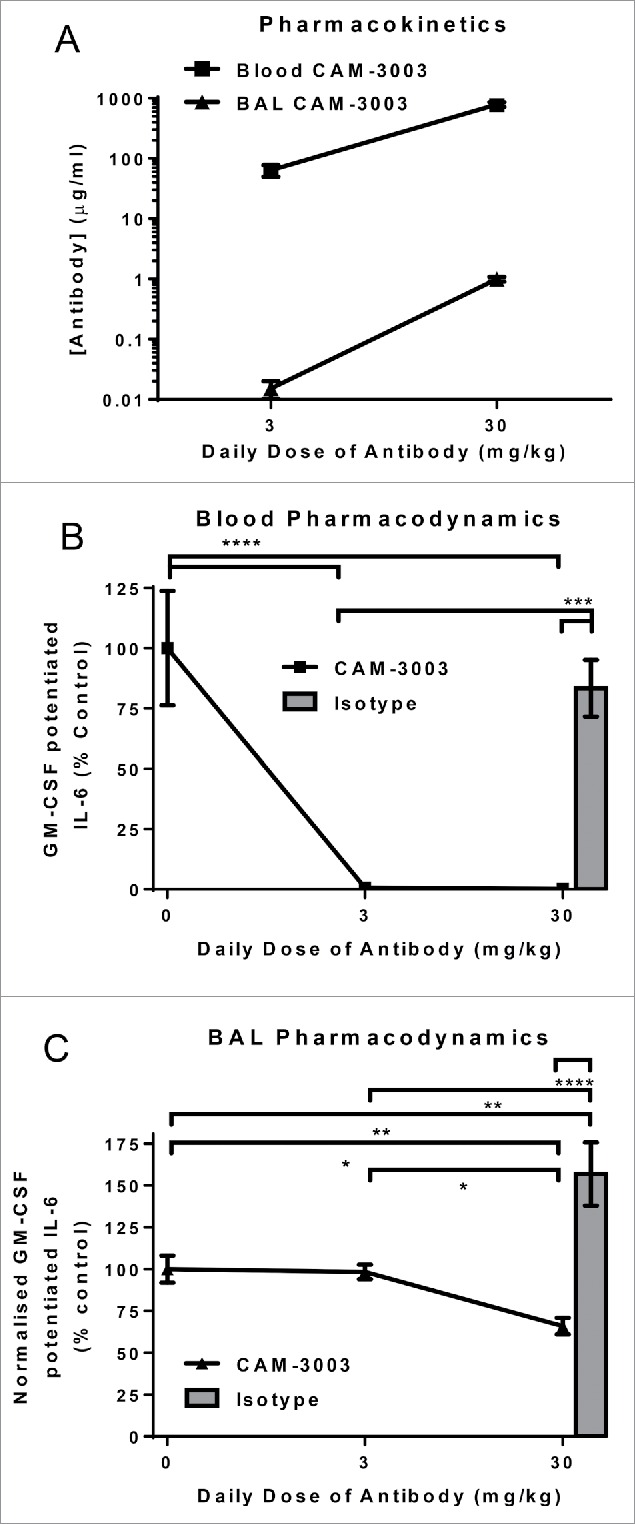
Multiple doses of anti-GM-CSFRα completely inhibits blood but not BAL cells. Mice were dosed i.p. with 3 or 30 mg/kg anti-GM-CSFRα, CAM-3003, or isotype control every day for 7 consecutive days. (A) Anti-GM-CSFRα antibody levels in plasma and bronchoalveolar lavage (BAL) were measured 48 hr later. Plasma LLOQ = 1.9 μg/ml and BAL LLOQ = 4.9 ng/ml. (B) GM-CSFRα inhibition on the blood cells was quantified by stimulation with 10 ng/ml LPS overnight with and without priming with 10 pg/ml GM-CSF. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. (C) GM-CSFRα inhibition on the BAL cells was quantified by stimulation as in B. IL-6 levels in the supernatant were determined and normalized to the number of cells as determined by Cell-Titer Glo. GM-CSF potentiated IL-6 release was the increased levels due to GM-CSF priming, and expressed as a percentage of untreated mice. All measurements were duplicates and data shown are mean +/− SEM of 6–8 animals. * P < 0.05; ** P < 0.01; *** P < 0.001 and **** P < 0.0001.
Discussion
We developed similar pharmacodynamics bioassays to understand functional coverage of a mouse anti-GM-CSRα monoclonal antibody systemically and on the alveolar macrophages of the lung. The data from the systemic pharmacodynamic bioassay demonstrated that administration of 1 mg/kg CAM-3003 i.p. was only sufficient to partially block GMCSRα in blood, and that doses of 3 mg/kg or higher gave complete coverage. This is consistent with our previous in vivo observations. Subcutaneous injection of recombinant GM-CSF induces margination of both monocytes and neutrophils. One mg/kg CAM-3003 dosed i.p. partially blocked this, and 10 mg/kg completely reversed it.6 Daily administration of 10 mg/kg CAM-3003, and to a lesser extent 1 mg/kg, over a 14-day period can reduce the clinical score in a collagen-induced arthritis model.6 Therefore, we believe that this assay accurately reflects systemic efficacy in vivo. Interestingly, despite the difference in potency between the anti-mouse GM-CSFRα antibody and mavrilimumab, the 1 mg/kg dose is not dissimilar to the highest and most efficacious dose of mavrilimumab tested, 150 mg.10
In contrast, total coverage in the lung pharmacodynamics bioassay was only achieved by i.n. delivery of CAM-3003. Partial blockade was observed when 30 mg/kg CAM-3003 was administered i.p. daily for 5 days, a dose tenfold higher than that necessary to get complete coverage systemically. Indeed, dosing at 3 mg/kg, which gives complete systemic coverage, consecutively for 7 d did not affect the alveolar macrophage response to GM-CSF. The lung pharmacodynamics correlated well with the pharmacokinetics in the BAL. The BAL pharmacokinetics, however, could not be compared directly to the blood because of the dilution effect of the lavage fluid.
This indicates a true disconnect between systemic and lung effects of an anti-GM-CSFRα antibody that would indicate a lung partitioning of considerably lower than 10% of the serum concentration. Importantly, in this study, lung refers to the luminal space where the alveolar macrophages reside because these are the cells responsible for the PAP pathology. This would explain the apparent disconnect with previous determinations of the lung partitioning of monoclonal antibodies by physiology-based pharmacokinetics using labeled antibodies in both mice and humans, as these measured interstitial lung tissue measurements.29-33 This comparison indicates the lung epithelium further limits therapeutic antibody exposure. Conversely, pharmacokinetic studies that uses BAL measurements to quantify partitioning into the lung lumen under-estimate the partitioning due to dilution with the lavage fluid.34,35 In contrast synovial concentration of a therapeutic antibody in RA patients has been estimated to be within 3-6-fold of the plasma concentration.36 This further indicates a large therapeutic window.
Transcytosis of IgGs across the lung epithelium is predominately mediated by the neonatal Fc Receptor, FcRn,37,38 but FcRn expression is predominantly apical,37 and transport weighted in the apical to basolateral direction.38 Despite this, absorptive rates across the lung epithelium are low,39 and presumably even lower from basolateral to apical. Interestingly, modulating FcRn binding did increase BAL levels of a human antibody in a non-human primate study, but not to a greater extent than the increase in serum concentration.35 Modulating FcRn binding, therefore, may not enhance lung penetrance of a therapeutic antibody when this is considered desirable, e.g., respiratory indications.
RA can be associated with extra-articular manifestations, including interstitial lung disease. Indeed, in some cases pulmonary pathology precedes the joint involvement, which might indicate the disease can initiate in the lung.40 The more common lung complications, such as interstitial lung disease and pleural disease, do not present in the airspaces, so the observations here may not be directly applicable. However, it is possible that mavrilimumab will be less effective at treating lung manifestations. Patients with symptomatic or uncontrolled lung disease were excluded from the mavrilimumab studies, so we do not know how this drug affects these co-morbidities. Even with established biologics like the anti-tumor necrosis factor products and rituximab, it is unclear whether these improve or worsen lung co-morbidities;40 based on our observations, it is likely that these drugs probably also have poor penetrance into the lung.
If GM-CSF neutralization in the alveolar space is so challenging, how do the anti-GM-CSF autoantibodies drive the PAP pathology? Possible explanations are either local production in the lung or longevity of exposure. A recent report suggests the polyclonal nature of GM-CSF autoantibodies in PAP patients leads to immune complex formation, which has the 2 effects of very potent neutralization and rapid clearance. Indeed, this led to a greater overall potency than a high affinity therapeutic monoclonal antibody.41
In summary we demonstrated that the lung partitioning of a mouse anti-GM-CSFRα is so low that we were only able to partly neutralize the receptor on alveolar macrophages, even when dosing mice on 7 consecutive days with a dose 10 times higher than that need to saturate blood leukocytes. This difference in exposure could in part contribute to the lack of any pulmonary changes observed to date following treatment with efficacious doses of mavrilimumab, in ongoing clinic trials.
Materials and methods
Antibodies
The antibodies used were CAM-3003, a mouse IgG1 anti-mouse GM-CSFRa, and CAT-004, a mouse IgG1 raised against an irrelevant antigen. These antibodies were made in house at MedIm-mune.
Dosing
Female BALB/c mice (6–8 weeks) were purchased from Harlan and housed at the Argenta Animal facility. All work was carried out to UK Home Office ethical and husbandry standards under the authority of an appropriate project license, which was reviewed and approved by animal welfare and ethical review board.
Antibodies were diluted in phosphate-buffered saline (PBS) to a concentration designed to give the defined dose and instilled either i.p. (10 ml/kg) or i.n. (in 50 μl). Forty-eight hours following i.p. or 2 hrs after i.n. dosing, animals were sacrificed by anesthetic overdose (pentobarbitone Na). A blood sample was taken from each animal in turn via the sub-clavian vein, and placed in a lithium-heparin coated tube. A BAL was then performed using 3 separate 0.4 ml volumes of PBS, which were combined.
Blood pharmacodynamic bioassay
One hundred μl of whole blood from each animal was added to 400 μl of RPMI media supplemented with penicillin and streptomycin (All Invitrogen). The blood/medium mixture from each animal was plated out at 50 μl/well into 96-well tissue culture plates. Relevant wells were stimulated with 10 pg/ml recombinant mouse GM-CSF (R&D Systems) or nothing for 4 hr, followed by 10 ng/ml LPS (Salmonella minnesota R595, Calbiochem) overnight at 37°C. Supernatants were removed following centrifugation (1500 × g for 10 mins) and IL-6 concentration was determined (Mesoscale Discovery).
GM-CSF potentiated IL-6 was calculated by subtracting the concentration of IL-6 induced by LPS alone from GM-CSF primed LPS stimulation. The effect of antibody installation was expressed as a percentage of vehicle (PBS) treated mice.
Lung pharmacodynamic bioassay
BAL fluid cells were spun down and resuspended in 450 μl RPMI media supplemented with 10% fetal calf serum, penicillin and streptomycin (Invitrogen). After plating out at 50 μl/well, cells were stimulated and supernatants assayed as above. After a freeze/thaw cycle, 100 μl of Cell Titer-Glo substrate (Promega) was added to each well containing BALF cells and read as per manufacturer's instructions.
Variation in BAL cell numbers harvested was accounted for by normalization of GM-CSF potentiated IL-6 to cell number by dividing IL-6 concentration by Cell Titer-Glo measurement. When Cell Titer-Glo value dropped below the normal range (<1000 ), data was deemed unreliable and excluded. Normalized GM-CSF potentiated IL-6 was calculated by subtracting the cell-normalized concentration of IL-6 induced by LPS alone from GM-CSF primed LPS stimulation. The effect of antibody installation was expressed as a percentage of vehicle (PBS) treated mice.
Pharmacokinetic assay
Maxisorb plates (Nunc) were coated overnight with 1 μg/ml recombinant murine GM-CSFRα. After washing with PBS containing 0.5% Tween, plates were blocked with 1% non-fat milk for 1 hr. The samples, BAL fluid or plasma, or standard were serially diluted (1 in 2) in the blocking buffer, and incubated in the wells for 2 hrs. After washing, goat anti-mouse HRP (The Binding Site) was added for 30 mins. The plates were washed extensively and developed with tetramethylbenzidine (Sigma). The reaction was stopped with 1 M sulfuric acid and the plates were read absorption at 450 nm. CAM-3003 was used as the standard starting at 500 ng/ml diluted to 0.48 or 0.24 ng/ml.
Statistics
Results are expressed as mean+/− SEM analyzed by one-way ANOVA followed by Bonferroni post-test comparing all condi-tions.
Disclosure of potential conflicts of interest.
All contributors were employed by MedImmune Ltd during these studies.
Acknowledgments
We would like to sincerely thank all the staff at Argenta, especially Vince Russell for conducting the in life phase of these studies. Dominic Corkill was of considerable assistance for all the in vivo studies.
Funding
This work was funded by MedImmune Ltd, a subsidiary of AstraZeneca.
References
- 1.McInnes IB, Schett G. The pathogenesis of rheumatoid arthritis. N Eng J Med 2011; 365:2205-19; PMID:22150039; http://dx.doi.org/ 10.1056/NEJMra1004965 [DOI] [PubMed] [Google Scholar]
- 2.Hamilton JA. Colony-stimulating factors in inflammation and autoimmunity. Nat Rev Immunol 2008; 8:533-44; PMID:18551128; http://dx.doi.org/ 10.1038/nri2356 [DOI] [PubMed] [Google Scholar]
- 3.Cook AD, Braine EL, Campbell IK, Rich MJ, Hamilton JA. Blockade of collagen-induced arthritis post-onset by antibody to granulocyte-macrophage colony-stimulating factor (GM-CSF): requirement for GM-CSF in the effector phase of disease. Arthritis Res 2001; 3:293-8; PMID:11549370; http://dx.doi.org/ 10.1186/ar318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Campbell IK, Rich MJ, Bischof RJ, Dunn AR, Grail D, Hamilton JA. Protection from collagen-induced arthritis in granulocyte-macrophage colony-stimulating factor-deficient mice. J Immunol 1998; 161:3639-44; PMID:975988711581310 [PubMed] [Google Scholar]
- 5.McQualter JL, Darwiche R, Ewing C, Onuki M, Kay TW, Hamilton JA, Reid HH, Bernard CC. Granulocyte macrophage colony-stimulating factor: a new putative therapeutic target in multiple sclerosis. J Exp Med 2001; 194:873-82; PMID:11581310; http://dx.doi.org/ 10.1084/jem.194.7.873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Greven DE, Cohen ES, Gerlag DM, Campbell J, Woods J, Davis N, Van Nieuwenhuijze A, Lewis A, Heasmen S, McCourt M, et al.. Preclinical characterisation of the GM-CSF receptor as a therapeutic target in rheumatoid arthritis. Annals Rheumatic Dis 2015; 74:1924-30; PMID:24936585; http://dx.doi.org/ 10.1136/annrheumdis-2014-205-234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haringman JJ, Gerlag DM, Zwinderman AH, Smeets TJ, Kraan MC, Baeten D, McInnes IB, Bresnihan B, Tak PP. Synovial tissue macrophages: a sensitive biomarker for response to treatment in patients with rheumatoid arthritis. Annals Rheumatic Dis 2005; 64:834-8; PMID:15576415; http://dx.doi.org/ 10.1136/ard.2004.029751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bell AL, Magill MK, McKane WR, Kirk F, Irvine AE. Measurement of colony-stimulating factors in synovial fluid: potential clinical value. Rheumatol Int 1995; 14:177-82; PMID:7536953; http://dx.doi.org/ 10.1007/BF00262295 [DOI] [PubMed] [Google Scholar]
- 9.Wright HL, Bucknall RC, Moots RJ, Edwards SW. Analysis of SF and plasma cytokines provides insights into the mechanisms of inflammatory arthritis and may predict response to therapy. Rheumatol 2012; 51:451-9; PMID:22179732; http://dx.doi.org/ 10.1093/rheumatology/ker338 [DOI] [PubMed] [Google Scholar]
- 10.Burmester GR, McInnes IB, Kremer JM, Miranda P, Korkosz M, Vencovsky J, et al.. Efficacy and safety/tolerability of mavrilimumab, a Human GM-CSFRa, monoclonal antibody in patients with rheumatoid arthritis. Arthritis Rheumatol 2014; 66:S1231-2; http://dx.doi.org/ 10.1002/art.38914 [DOI] [Google Scholar]
- 11.Burmester GR, Weinblatt ME, McInnes IB, Porter D, Barbarash O, Vatutin M, Szombati I, Esfandiari E, Sleeman MA, Kane CD, et al.. Efficacy and safety of mavrilimumab in subjects with rheumatoid arthritis. Annals Rheumatic Dis 2013; 72:1445-52; PMID:23234647; http://dx.doi.org/ 10.1136/annrheumdis-2012-202450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stanley E, Lieschke GJ, Grail D, Metcalf D, Hodgson G, Gall JA, Maher DW, Cebon J, Sinickas V, Dunn AR. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoiesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci U S A 1994; 91:5592-6; PMID:8202532; http://dx.doi.org/ 10.1073/pnas.91.12.5592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dranoff G, Crawford AD, Sadelain M, Ream B, Rashid A, Bronson RT, Dickersin GR, Bachurski CJ, Mark EL, Whitsett JA, et al.. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994; 264:713-6; PMID:8171324; http://dx.doi.org/ 10.1126/science.8171324 [DOI] [PubMed] [Google Scholar]
- 14.King IL, Kroenke MA, Segal BM. GM-CSF-dependent, CD103+ dermal dendritic cells play a critical role in Th effector cell differentiation after subcutaneous immunization. J Exp Med 2010; 207:953-61; PMID:20421390; http://dx.doi.org/ 10.1084/jem.20091844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bogunovic M, Ginhoux F, Helft J, Shang L, Hashimoto D, Greter M, Liu K, Jakubzick C, Ingersoll MA, Leboeuf M, et al.. Origin of the lamina propria dendritic cell network. Immunity 2009; 31:513-25; PMID:19733489; http://dx.doi.org/ 10.1016/j.immuni.2009.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgess AW, Metcalf D. The nature and action of granulocyte-macrophage colony stimulating factors. Blood 1980; 56:947-58; PMID:7002232 [PubMed] [Google Scholar]
- 17.Ikegami M, Ueda T, Hull W, Whitsett JA, Mulligan RC, Dranoff G, Jobe AH. Surfactant metabolism in transgenic mice after granulocyte macrophage-colony stimulating factor ablation. Am J Physiol 1996; 270:L650-8; PMID:8928826 [DOI] [PubMed] [Google Scholar]
- 18.Robb L, Drinkwater CC, Metcalf D, Li R, Kontgen F, Nicola NA, Begley CG. Hematopoietic and lung abnormalities in mice with a null mutation of the common β subunit of the receptors for granulocyte-macrophage colony-stimulating factor and interleukins 3 and 5. Proc Natl Acad Sci U S A 1995; 92:9565-9; PMID:7568173; http://dx.doi.org/ 10.1073/pnas.92.21.9565 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carey B, Trapnell BC. The molecular basis of pulmonary alveolar proteinosis. Clin Immunol 2010; 135:223-35; PMID:20338813; http://dx.doi.org/ 10.1016/j.clim.2010.02.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Trapnell BC, Whitsett JA, Nakata K. Pulmonary alveolar proteinosis. N Eng J Med 2003; 349:2527-39; PMID:14695413; http://dx.doi.org/ 10.1056/NEJMra023226 [DOI] [PubMed] [Google Scholar]
- 21.Kitamura T, Tanaka N, Watanabe J, Uchida , Kanegasaki S, Yamada Y, Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med 1999; 190:875-80; PMID:10499925; http://dx.doi.org/ 10.1084/jem.190.6.875 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tanaka T, Motoi N, Tsuchihashi Y, Tazawa R, Kaneko C, Nei T, Yamamoto T, Hayashi T, Tagawa T, Nagayasu T, et al.. Adult-onset hereditary pulmonary alveolar proteinosis caused by a single-base deletion in CSF2RB. J Med Genetics 2011; 48:205-9; PMID:21075760; http://dx.doi.org/ 10.1136/jmg.2010.082586 [DOI] [PubMed] [Google Scholar]
- 23.Suzuki T, Maranda B, Sakagami T, Catellier P, Couture CY, Carey BC, Chalk C, Trapnell BC. Hereditary pulmonary alveolar proteinosis caused by recessive CSF2RB mutations. Eur Respir J 2011; 37:201-4; PMID:21205713; http://dx.doi.org/ 10.1183/09031936.00090610 [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Moczygemba M, Doan ML, Elidemir O, Fan LL, Cheung SW, Lei JT, Moore JP, Tavana G, Lewis LR, Zhu Y, et al.. Pulmonary alveolar proteinosis caused by deletion of the GM-CSFRalpha gene in the X chromosome pseudoautosomal region 1. J Exp Med 2008; 205:2711-6; PMID:18955567; http://dx.doi.org/ 10.1084/jem.20080759 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Suzuki T, Sakagami T, Rubin BK, Nogee LM, Wood RE, Zimmerman SL, Smolarek T, Dishop MK, Wert SE, Whitsett JA, et al.. Familial pulmonary alveolar proteinosis caused by mutations in CSF2RA. J Exp Med 2008; 205:2703-10; PMID:18955570; http://dx.doi.org/ 10.1084/jem.20080990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sakagami T, Beck D, Uchida K, Suzuki T, Carey BC, Nakata K, Keller G, Wood RE, Wert SE, Ikegami M, et al.. Patient-derived granulocyte/macrophage colony-stimulating factor autoantibodies reproduce pulmonary alveolar proteinosis in nonhuman primates. Am J Respir Critical Care Med 2010; 182:49-61; PMID:20224064; http://dx.doi.org/ 10.1164/rccm.201001-0008OC [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ryan PC, Sleeman MA, Rebelatto M, Wang B, Lu H, Chen X, Wu CY, Hinrichs MJ, Roskos L, Towers H, et al.. Nonclinical safety of mavrilimumab, an anti-GMCSF receptor α monoclonal antibody, in cynomolgus monkeys: relevance for human safety. Toxicol Appl Pharma-col 2014; 279:230-9; PMID:24937321; http://dx.doi.org/ 10.1016/j.taap.2014.06.002 [DOI] [PubMed] [Google Scholar]
- 28.Tiegs G, Barsig J, Matiba B, Uhlig S, Wendel A. Potentiation by granulocyte macrophage colony-stimulating factor of lipopolysaccharide toxicity in mice. J Clin Invest 1994; 93:2616-22; PMID:8201000; http://dx.doi.org/ 10.1172/JCI117274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Heiskanen T, Heiskanen T, Kairemo K. Development of a PBPK model for monoclonal antibodies and simulation of human and mice PBPK of a radiolabelled monoclonal antibody. Curr Pharmaceutical Design 2009; 15:988-1007; PMID:19275663; http://dx.doi.org/ 10.2174/138161209787581968 [DOI] [PubMed] [Google Scholar]
- 30.Shah DK, Betts AM. Antibody biodistribution coefficients: inferring tissue concentrations of monoclonal antibodies based on the plasma concentrations in several preclinical species and human. Mabs 2013; 5:297-305; PMID:23406896; http://dx.doi.org/ 10.4161/mabs.23684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garg A, Balthasar JP. Physiologically-based pharmacokinetic (PBPK) model to predict IgG tissue kinetics in wild-type and FcRn-knockout mice. J Pharmacokinetics Pharmacodynamics 2007; 34:687-709; PMID:17636457; http://dx.doi.org/ 10.1007/s10928-007-9065-1 [DOI] [PubMed] [Google Scholar]
- 32.Davda JP, Jain M, Batra SK, Gwilt PR, Robinson DH. A physiologically based pharmacokinetic (PBPK) model to characterize and predict the disposition of monoclonal antibody CC49 and its single chain Fv constructs. Int Immunopharmacol 2008; 8:401-13; PMID:182-79794; http://dx.doi.org/ 10.1016/j.intimp.2007.10.023 [DOI] [PubMed] [Google Scholar]
- 33.Abuqayyas L, Balthasar JP. Application of knockout mouse models to investigate the influence of FcgammaR on the tissue distribution and elimination of 8C2, a murine IgG1 monoclonal antibody. Int J Pharmaceutics 2012; 439:8-16; PMID:23018115; http://dx.doi.org/ 10.1016/j.ijpharm.2012.09.042 [DOI] [PubMed] [Google Scholar]
- 34.Hart TK, Cook RM, Zia-Amirhosseini P, Minthorn E, Sellers TS, Maleeff BE, Eustis S, Schwartz LW, Tsui P, Appelbaum ER, et al.. Preclinical efficacy and safety of mepolizumab (SB-240563), a humanized monoclonal antibody to IL-5, in cynomolgus monkeys. J Aller Clin Immunol 2001; 108:250-7; PMID:11496242; http://dx.doi.org/ 10.1067/mai.2001.116576 [DOI] [PubMed] [Google Scholar]
- 35.Dall'Acqua WF, Kiener PA, Wu H. Properties of human IgG1s engineered for enhanced binding to the neonatal Fc receptor (FcRn). J Biol Chem 2006; 281:23514-24; PMID:16793771; http://dx.doi.org/ 10.1074/jbc.M604292200 [DOI] [PubMed] [Google Scholar]
- 36.Choy EH, Connolly DJ, Rapson N, Jeal S, Brown JC, Kingsley GH, Panayi GS, Johnston JM. Pharmacokinetic, pharmacodynamic and clinical effects of a humanized IgG1 anti-CD4 monoclonal antibody in the peripheral blood and synovial fluid of rheumatoid arthritis patients. Rheumatol 2000; 39:1139-46; PMID:11035136; http://dx.doi.org/ 10.1093/rheumatology/39.10.1139 [DOI] [PubMed] [Google Scholar]
- 37.Spiekermann GM, Finn PW, Ward ES, Dumont J, Dickinson BL, Blumberg RS, Lencer WI. Receptor-mediated immunoglobulin G transport across mucosal barriers in adult life: functional expression of FcRn in the mammalian lung. J Exp Med 2002; 196:303-10; PMID:12163559; http://dx.doi.org/ 10.1084/jem.20020400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kim KJ, Fandy TE, Lee VH, Ann DK, Borok Z, Crandall ED. Net absorption of IgG via FcRn-mediated transcytosis across rat alveolar epithelial cell monolayers. Am J Physiol Lung Cell Mol Physiol 2004; 287:L616-22; PMID:15169676; http://dx.doi.org/ 10.1152/ajplung.00121.2004 [DOI] [PubMed] [Google Scholar]
- 39.Sakagami M, Omidi Y, Campbell L, Kandalaft LE, Morris CJ, Barar J, Gumbleton M. Expression and transport functionality of FcRn within rat alveolar epithelium: a study in primary cell culture and in the isolated perfused lung. Pharm Res 2006; 23:270-9; PMID:16382279; http://dx.doi.org/ 10.1007/s11095-005-9226-0 [DOI] [PubMed] [Google Scholar]
- 40.Shaw M, Collins BF, Ho LA, Raghu G. Rheumatoid arthritis-associated lung disease. Eur Respir Rev 2015; 24:1-16; PMID:25726549; http://dx.doi.org/ 10.1183/09059180.00008014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Piccoli L, Campo I, Fregni CS, Rodriguez BM, Minola A, Sallusto F, Luisetti M, Corti D, Lanzavecchia A. Neutralization and clearance of GM-CSF by autoantibodies in pulmonary alveolar proteinosis. Nat Commun 2015; 6:7375; PMID:26077231; http://dx.doi.org/ 10.1038/ncomms8375 [DOI] [PMC free article] [PubMed] [Google Scholar]