

ABSTRACT
Metastatic melanoma is an aggressive cancer with a poor prognostic, and the design of new targeted drugs to treat melanoma is a therapeutic challenge. A promising approach is to produce monoclonal antibodies (mAbs) against the endothelin B receptor (ETB), which is known to be overexpressed in melanoma and to contribute to proliferation, migration and vasculogenic mimicry associated with invasiveness of this cancer.
We previously described rendomab-B1, a mAb produced by DNA immunization. It is endowed with remarkable characteristics in term of affinity, specificity and antagonist properties against human ETB expressed by the endothelial cells, but, surprisingly, had poor affinity for ETB expressed by melanoma cells. This characteristic strongly suggested the existence of a tumor-specific ETB form. In the study reported here, we identified a new mAb, rendomab-B4, which, in contrast to rendomab-B1, binds ETB expressed on UACC-257, WM-266-4 and SLM8 melanoma cells. Moreover, after binding to UACC-257 cells, rendomab-B4 is internalized and colocalizes with the endosomal protein EEA-1. Interestingly, rendomab-B4, despite its inability to compete with endothelin binding, is able to inhibit phospholipase C pathway and migration induced by endothelin. By contrast, rendomab-B4 fails to decrease ERK1/2 phosphorylation induced by endothelin, suggesting a biased effect on ETB.
These particular properties make rendomab-B4 an interesting tool to analyze ETB-structure/function and a promising starting point for the development of new immunological tools in the field of melanoma therapeutics.
KEYWORDS: Cancer, endothelin, endothelin B receptor, melanoma, monoclonal antibody, migration, phospholipase C, MAPK
Introduction
Endothelins (ETs) constitute a family of 3 21-amino acid peptides, ET-1, ET-2 and ET-3, which bind to 2 distinct 7 transmembrane domain receptors ETA and ETB belonging to the G protein-coupled receptor (GPCR) family. The endothelin axis (endothelins and their receptors) is strongly involved in physiological and pathological processes. ET-1 plays a crucial role in the regulation of physiological smooth muscle motility,1-3 but ET-1 is also implicated in a large variety of pathologies, including hypertension, heart failure, kidney disorders and infectious diseases.4-6 In addition, the ET axis is overexpressed in cancer of different organs contributing to tumor growth by acting on cell proliferation, survival, migration, differentiation, angiogenesis and inflammatory cell recruitment.7,8 ETA are upregulated in prostate,9 ovary10 and breast cancers while ETB is overexpressed in melanoma.11-13
Melanoma is an aggressive cancer that presents an increased incidence rate.14 This cancer is characterized by its capacity to metastasize promptly, leading to an increase in mortality rates in many countries.15 Somatic mutations have been found in BRAF and N-RAS genes in about 50% and 20% of melanomas, respectively, resulting in constitutive activation of ERK1/2 MAPK pathway.16 Moreover, gene expression profiling and targeted approaches have demonstrated that ETB expression is upregulated in melanoma.12,17 The upregulation of ETB is involved in proliferation, migration and angiogenesis associated with tumor growth and invasiveness. In melanoma, ET-1 via ETB expressed on cancer cells modulates migration and formation of vasculogenic mimicry via the upregulation of HIF/VEGF/VEGFR pathway.18 These data implicate ETB as a potential driver of melanoma progression and an important marker of aggressive phenotype.7,12
An ETB-specific peptidic antagonist (BQ788) has been used in basic research to reduce the proliferation of cancer cells.19,20 Preclinical trial confirmed the efficacy of BQ788 on melanoma growth.21 However, the dual ETB-specific antagonist bosentan used as a monotherapy has a low effect on melanoma progression, and no additional effect when combined with a chemotherapeutic agent (dacarbazine).22,23 Therefore, the development of new therapeutic molecules targeting ETB is needed to block the upregulated signaling pathways that occur in melanoma.
The use of therapeutic monoclonal antibodies (mAbs) is now established as a very attractive alternative to conventional cancer treatment. Compared to small pharmacological molecules, mAbs can detect fine antigenic differences between normal and pathologic cells, inhibiting different functions involved in cell growth, migration, angiogenesis or metastasis. Moreover, mAbs display various cytotoxic actions through the immune system, and they can be coupled to several imaging tracers and markers or cytotoxic molecules. Trastuzumab exemplifies the successful application of mAbs to cancer. Directed against the human epidermal growth factor receptor HER-2 often overexpressed in breast cancer, trastuzumab has been shown to significantly improve the overall survival of HER2-positive cancer patients.24 Like HER-2 in breast cancer, ETB overexpressed in melanoma, can be targeted by mAbs. Based on rapid ligand-mediated internalization, anti-ETB antibodies that would be co-internalized represent a useful tool to carry cytotoxic drugs, and induce cancer cell death.
Our group25 and others17 have recently developed mAbs directed against ETB. However, the higher affinity of the antibody that we describe here, associated with a fast internalization of ETB, might make it a good candidate for antibody-drug conjugate (ADC) development to target ETB in melanoma.17 Previously, our group described rendomab-B1,25 a mAb that specifically recognizes human ETB. This antibody is a strong antagonist and inhibits ETB functions in endothelial cells. However, rendomab-B1 does not bind to melanoma ETB, suggesting that specific melanoma conformation of ETB might exist.
Using genetic immunization, an original approach to produce antibodies against the tridimensional conformations of membrane receptors, we obtained new mAbs directed against the ETB expressed on normal or cancer cells. One of them, rendomab-B4, is particularly interesting because this mAb is able to recognize UACC-257 melanoma ETB with high affinity, internalizes with ETB and inhibits ETB-mediated migration in melanoma cells.
Results
Characterization of anti-human ETB specific monoclonal antibodies
As described previously, rendomab-B1 was initially selected from 24 antibodies obtained through genetic immunization based on its unique property to behave as a remarkably potent antagonist of human ETB.25 However this mAb displayed a very low affinity for ETB expressed on tumor cells, which prompted us to look for the potential presence, among the 23 other anti-ETB mAbs that we produced, of antibodies that could recognize the tumor-associated form of human ETB. Toward that end, the binding properties of all the mAbs on Chinese hamster ovary (CHO) cells stably transfected with human ETB (CHO-ETB) and different melanoma cell lines were investigated. As expected, all mAbs were able to bind CHO-ETB, although with different intensities, since this cell line was used for the screening of anti-ETB antibodies-expressing hybridomas. This is illustrated in Fig. 1A for 9 mAbs selected here for their high Bmax value on CHO- ETB (rendomab B4 and 8 other rendomabs, BS1 to BS8); 3 mAbs belonged to the IgG2B subclass, 3 to the IgG1 and 3 to the IgG3 subclass. The affinities of these 9 mAbs for CHO- ETB are shown in Fig. 1B, and, as can be observed, no correlation was found between the isotypic class of the mAbs, Bmax, and affinity.
Figure 1.
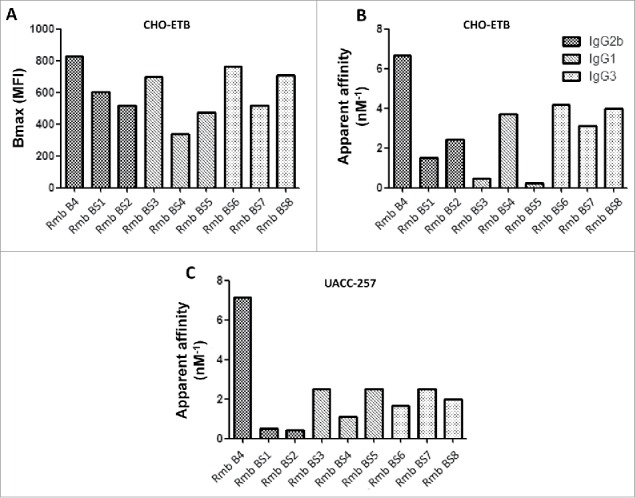
Binding properties of anti-human ETB specific monoclonal antibodies. To evaluate maximal binding (A) and apparent affinities (B, C), CHO and UACC-257 cells expressing hETB were incubated for 24 h at 4°C with increasing concentrations of ETB directed antibodies. Binding of anti-ETB antibodies were revealed using R-PE-labeled anti-mouse antibody and quantified by flow cytometry. Each antibody's maximal binding (Bmax) and apparent affinity (1/apparent Kd) were determined using GraphPad Prism software.
Most interestingly, all 9 selected mAbs appeared to be able to bind ETB expressed at the surface of the 3 melanoma cell lines tested: UACC-257, WM-266-4 and SLM8. Fig. 1C shows their apparent affinities for UACC-257, and a detailed analysis of rendomab-B4 binding on the 3 different melanoma cells is shown below. In addition, among the 23 new antibodies that we tested, some other mAbs were totally unable to bind ETB expressed on any tumoral cell line, as was observed for the rendomab B1 that we already described,25 whereas others recognized ETB expressed only on a given tumoral cell line (results not shown). Since, as illustrated in Fig. 1B and C, rendomab B4 (Rmb B4) exhibited the highest apparent affinity values not only for CHO-ETB, but above all for the tumoral UACC-257 cell line, it was selected for further characterization of its functional properties.
Rendomab-B4 specifically recognizes with high affinity human ETB overexpressed in CHO cell line
It is well established that the 3 ETs bind ETB with comparable affinities. First, we checked the binding properties of ETs on CHO-ETB by using fluorescent ET-1 and ET-3 (ET-1-FAM and ET-3-FAM, respectively). Flow cytometry experiments showed that the 2 labeled ETs bound to CHO-ETB with the same Kd value, ∼1 nM, and the maximal binding was obtained at 100 nM for the 2 ligands (Fig. 2A and B). Data presented in Fig. 2C show that rendomab-B4 binding was dose-dependent, saturable, and reached a plateau for a 50 nM concentration. Half maximal binding (apparent Kd : Kd*) was about 0.15 ± 0.03 nM (mean ± s.d), reflecting the high affinity of rendomab-B4 for hETB. Additional experiments show that rendomab-B4 bound neither to untransfected CHO cells that do not express ETB, nor to a rat leiomyoma cell line (ELT-3) that endogenously expresses ETB at high level,26 and did not cross-react with ETA expressed in CHO cells (data not shown). Altogether, these data indicated that rendomab-B4 was specific to human ETB. This specific binding of rendomab-B4 to ETB was visualized by immunofluorescence experiments showing that the mAb displayed a marked membrane labeling in CHO-ETB cells, but not on control untransfected cells (Fig. 2D).
Figure 2.
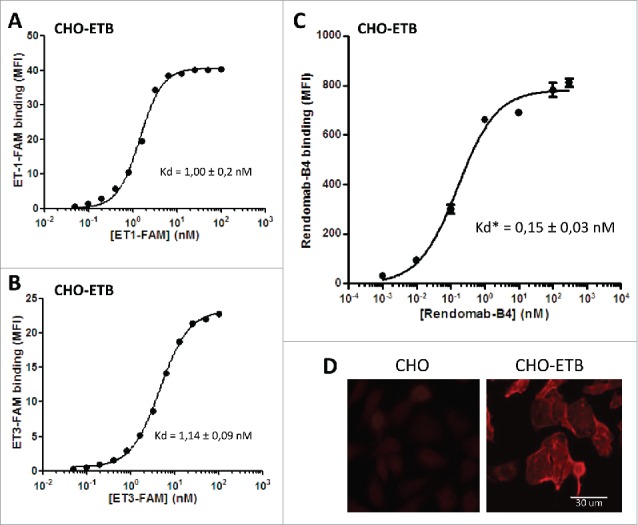
Rendomab-B4 specifically recognizes with high affinity human ETB overexpressed in CHO cell line. To check human ETB expression on CHO-ETB surface, cells were incubated with increasing concentrations of ET-1-FAM (A) and ET-3-FAM (B) for 24 h at 4°C in the dark. To determine rendomab-B4 affinity, increasing concentrations of the monoclonal antibody were incubated with CHO-ETB cells for 24 h at 4°C (C). Rendomab-B4 binding was revealed using R-PE-labeled anti-mouse antibody. The binding of labeled-peptides and monoclonal antibody were measured on a GUAVA flow cytometer and resulting curves (A, B and C), which correspond to mean fluorescence intensity (MFI) as a function of ligand concentrations, were plotted and fitted using GraphPad Prism software. Kd or apparent Kd (Kd*) corresponding to half maximal effective concentration values correspond to the mean ± s.d. of three (A and B) or 5 (C) independent experiments. (D) CHO and CHO-ETB cells were fixed and incubated with 100 nM of rendomab-B4, followed by Cy3-conjugated anti-mouse IgG antibody incubation.
Rendomab-B4 does not compete with ET-1 and ET-3 for binding on CHO-ETB
Competition experiments were performed to characterize the interaction of rendomab-B4 with ETB expressed in CHO in the presence of ET-1-FAM or ET-3-FAM. Data depicted in Fig. 3 show that ET-1 and ET-3 inhibited ET-1-FAM (Fig. 3A) and ET-3-FAM (Fig. 3B), respectively, with IC50 estimated at 0.17 ± 0.07 and 0.31 ± 0.01 nM, respectively. The specific ETA antagonist BQ123 failed to reduce ET-1-FAM and ET-3-FAM binding in CHO-ETB. In contrast, BQ788, the specific antagonist for ETB, inhibited ET-1-FAM and ET-3-FAM binding in a dose-dependent manner, with IC50 values around 25.2 ± 2.8 and 8.9 ± 4.0 nM, respectively. Rendomab-B4, and its corresponding isotype control mAb, failed to impair the binding of ET-1-FAM (Fig. 3A) and ET-3-FAM (Fig. 3B). Conversely, rendomab-B4 binding was not inhibited by ET-1, ET-3 or random control peptide (Fig. 3C). These data demonstrated that rendomab-B4 and ETs are not competing for receptor binding, suggesting that their binding sites on ETB are distinct.
Figure 3.

Rendomab-B4 does not compete with ET-1 and ET-3 for binding CHO-ETB. CHO-ETB cells were incubated with 10 nM of ET-1-FAM (A) or ET-3-FAM (B) and simultaneously with increasing concentrations of isotype control antibody, rendomab-B4, selective receptor antagonists (BQ123 or BQ788) and the corresponding unlabelled endothelin (ET-1 or ET-3).(C) Cells were incubated simultaneously with 100 nM of rendomab-B4 and with increasing concentrations of endothelin (ET-1 or ET-3) or control peptide. Rendomab-B4 binding on cell surface was detected with a R-PE-labeled anti-mouse antibody. The fluorescence (A, B and C) was quantified by flow cytometry. Inhibition binding curves, corresponding to MFI relative to competitor concentration, were plotted and fitted with GraphPad Prism software. Two independent experiments were performed.
Rendomab-B4 recognizes a discontinuous epitope on hETB N-terminal domain
To further characterize the binding site of rendomab-B4 on hETB, epitope mapping experiments were performed. Pepscan membranes spotted with overlapping dodecapeptides, frameshifted by one residue, and corresponding to the N-terminal part and extracellular loops (E1, E2, E3) of the hETB were used. The immunization protocol that was carried out was expected to lead to the production of antibodies directed against the extracellular parts of the ETB.
As shown in Fig. 4A, the membrane incubated with rendomab-B4 displayed 2 immunoreactive regions, one from A21 to B8 and another from C17 to C24 spots, corresponding to ETB residues 28 to 38 and 70 to 77, respectively. The two series of immunoreactive peptides are part of the N-terminal domain, as represented in Fig. 4B and 4C. The deduced minimal sequences were ERGFPPDRATP and EVPKGDRT. The first sequence corresponds to the most N-terminal region of the mature receptor, once signal peptide is cleaved. The absence of any similarity between the 2 sequences suggested that rendomab-B4 recognized a conformational epitope formed by the juxtaposition of 2 regions of the receptor. These two sequences are unique to human ETB and are not found in any other human proteins. Interestingly, the 2 sequences forming the epitope are not conserved between human and rodents, and each peptide differs by 3 amino acids (residues noted with a * in Fig. 4C). This may explain why rendomab-B4 is unable to bind ETB-expressing rat cells, and suggest that these residues are crucial for rendomab-B4 binding.
Figure 4.
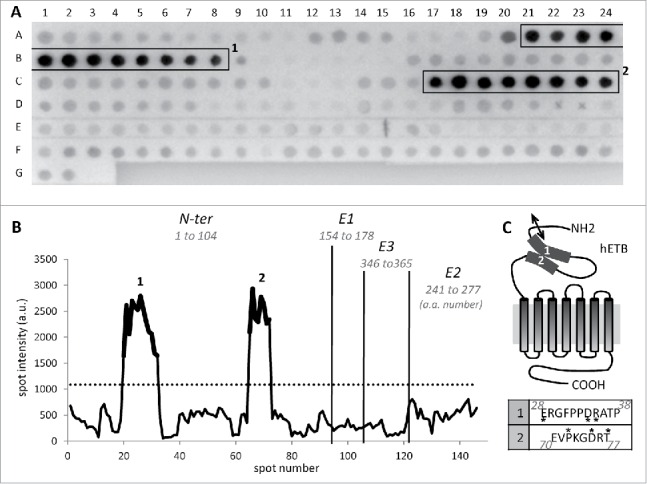
Rendomab-B4 recognizes a discontinuous epitope on hETB N-terminal tail. (A) To determine rendomab-B4 epitope, a Pepscan membrane spotted with dodecapeptides corresponding to extracellular domain and loops of the ETB receptor, was used. Hybridized monoclonal antibody was revealed using an alkaline phosphatase conjugated secondary antibody. The highest consecutive reactive spots are in boxes 1 and 2. Each spot signal obtained on the membrane was quantified. (B) The profile of hybridization of rendomab-B4, corresponding to the spot intensity as a function of the spot number on the membrane, was drawn. Vertical lines symbolize the separation between N-terminal (N-ter) domain and extracellular loops (E1, E2 and E3) of the receptor. The numbers of the amino acids at the extremities of each domain are indicated in italics. The dotted line corresponds to 3 times the background calculated as the mean intensity of all the spots except those in boxes. Parts of the curve in bold print (1 and 2) correspond to spots in boxes drawn in A. (C) Schematic representation of the human ETB and putative localization of the rendomab-B4 regions of recognition (1 and 2). The double arrow symbolizes the cleavage site of ETB signal peptide. Corresponding amino acid sequences of regions 1 and 2 are indicated in the table below. Amino acid noted * are not conserved between human and rodent (rat and mouse) ETB. This experiment was done twice with 2 independent membranes. Isotype control antibody generated no staining (data not shown).
To confirm this, human and rat ETB were expressed in CHO cells and tested for their ability to bind rendomab-B4 in an in vitro immunoprecipitation assay. Because no anti-ETB antibody that works well in western blot was available, human and rat ETB were expressed as GFP fusion proteins and detected with an anti-GFP antibody. The results in Fig. 5 (lower panel) show that human (hETB) and rat (rETB) GFP-fused ETB, immunoprecipitated with an anti-GFP nanobody followed by protein gel blot analysis with another anti GFP antibody, appear as several immunoreactive bands. For each receptor, the band of higher apparent molecular mass is likely to represent the full-length GFP-tagged receptor as its mass is consistent with the calculated theoretical mass of the protein (about 75 kDa). The other bands of lower molecular mass may represent N-terminally truncated forms of the receptor since the receptors were immunoprecipitated through their C-terminal GFP fusion.
Figure 5.
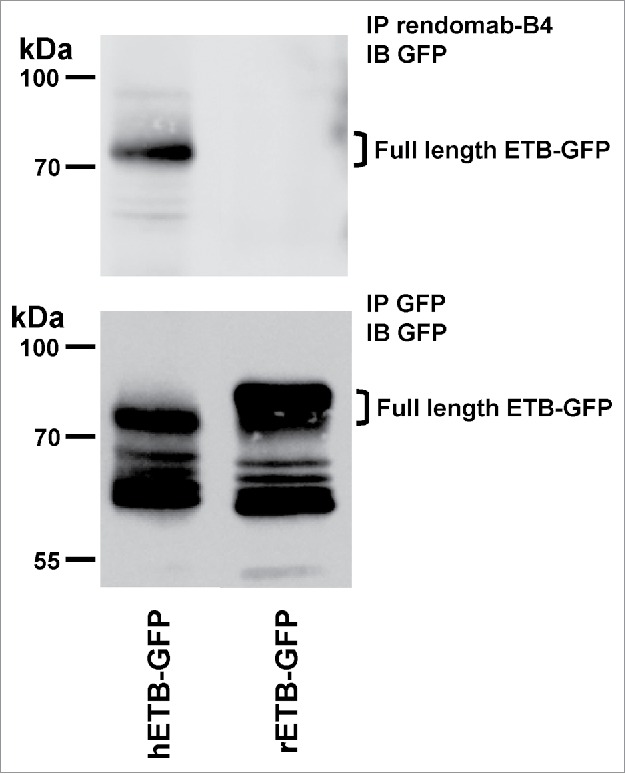
Rendomab-B4 is able to immunoprecipitate hETB. CHO transfected cells expressing either the human (hETB-GFP) or rat GFP-tagged ETB (rETB-GFP) were lysed and subjected to immunoprecipitation (IP) with rendomab-B4 (upper panel) or GFP-Trap beads (lower panel). Immunoprecipitated proteins were analyzed by protein gel blot with an anti-GFP antibody (IB). Position of molecular mass standards is indicated.
When hETB-GFP was subjected to immunoprecipitation with rendomab-B4 followed by western blot analysis with an anti-GFP antibody, only one immunoreactive band was detected (Fig. 5 upper panel). This band corresponds to the higher mass band visible in the lower panel. This result shows that rendomab-B4 is able to interact and precipitate the full length ETB, but not its N-terminally truncated forms. This result is consistent with the finding of the epitope sequence in its N-terminal domain. When the same experiment was performed on rETB-GFP instead of hETB-GFP, no immunoreactive band was detected, confirming that rendomab-B4 does not cross-react with rETB and strengthening the hypothesis that the amino acids that differ between human and rat ETB are important for rendomab-B4 binding.
Rendomab-B4 specifically recognizes ETB in UACC-257 melanoma cell line: binding properties
Considering that rendomab-B4 was able to bind human ETB in CHO cells with high affinity and specificity, its binding on human melanoma cell lines and on non-malignant cell lines expressing ETB was further investigated. Results in Fig. 6A show that rendomab-B4 clearly displays saturable binding curves on UACC-257 cells. Binding curves obtained with WM-266-4 and SLM8 also tend to saturate and could be easily fitted to sigmoid curves. Analysis of the binding curves gave half-maximal binding concentration values of 0.14 ± 0.03 nM, 1.5 ± 0.32 nM and ∼10 nM for UACC-257, WM-266-4 and SLM8, respectively. In contrast, binding curves obtained with HEK293T and HUVEC clearly did not tend to saturate, and these curves could not be fitted to sigmoid curves, indicating that no specific binding of rendomab-B4 was observed in these non-tumoral, yet ETB-expressing cells (Fig. 6A). This result further supports our previous assumption of the occurrence of tumor-specific conformations of ETB.25
Figure 6.
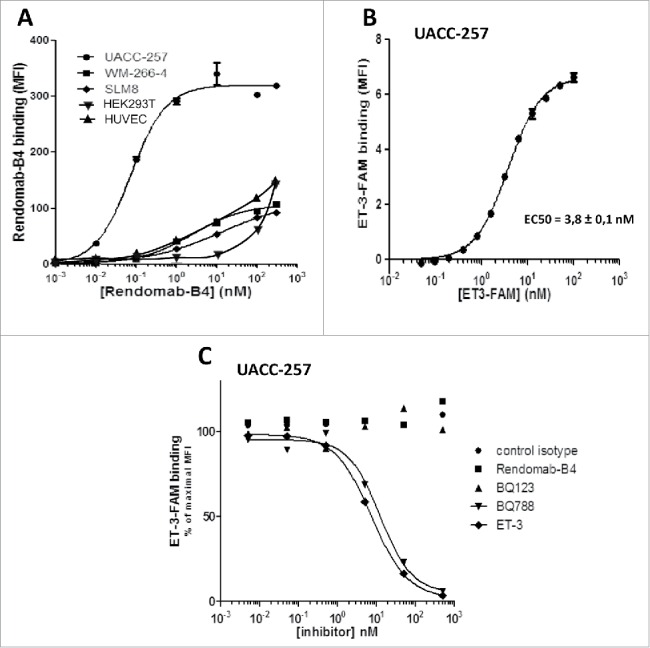
Rendomab-B4 specifically recognizes ETB in melanoma: Binding properties. (A) Human melanoma cell lines (UACC-257, WM-266-4 and SLM8), HEK293T and HUVEC were incubated with increasing concentrations of rendomab-B4. Rendomab-B4 binding was evaluated using R-PE-labeled anti-mouse antibody followed by flow cytometry analysis. The results are representative of 3 independent experiments. (B) To control human ETB expression at the surface of UACC-257 cells, the cells were incubated with varying concentrations of ET-3-FAM and fluorescence was measured by flow cytometry. Three independent experiments were performed. (C) UACC-257 melanoma cells were simultaneously incubated with 10 nM of ET-3-FAM and increasing concentrations of isotype control antibody, rendomab-B4, selective antagonists of ETA (BQ123) or ETB (BQ788) or unlabelled ligand ET-3. The fluorescence was quantified by flow cytometry. All these curves were plotted and fitted using GraphPad Prism software.
Since its apparent affinity was also higher in UACC-257 than in the 2 other melanoma cell lines and, in addition, among the 9 selected mAbs, rendomab-B4 displayed the best apparent affinity for UACC-257 (Fig. 1C), this cell line was chosen for further characterization of the fine binding properties and pharmacological effects of the antibody on tumor cells. Fig. 6B shows the saturable binding on UACC-257 cells of labeled ET-3, which is a specific agonist of ETB, known to play a leading role in melanoma development. This binding is characterized by an EC50 value of 3.8 ± 0.1 nM, which is in the same order of magnitude as that obtained in CHO-ETB cells (Fig. 2B). Competition experiments revealed that, as expected, ET-3 and BQ788 were able to fully inhibit ET-3-FAM binding, while control antibody and BQ123 were without any effect (Fig. 6C). Moreover, as already observed in CHO-ETB cells, rendomab-B4 was not able to reduce ET-3-FAM in UACC-257 cells, confirming that rendomab-B4 is not a competitor of ET-3 binding on hETB.
Rendomab-B4 is internalized in UACC-257 cells
Given that ETB-targeting-rendomab-B4 binds UACC-257 cells with high affinity, and that ETB is known to have high internalization and turnover rates,27,28 the internalization of the rendomab-B4 was studied. Internalization of a mAb is a crucial property because it allows uptake of a cytotoxic drug coupled to the mAb into cancer cells expressing the target.17,29 Cytometry experiments were carried out to study the internalization of ETB in UACC-257 cells (Fig. 7A). Because no competition was observed between ET-3 and rendomab-B4 binding on UACC-257 (Fig. 6C), rendomab-B4 was used to quantify receptors present at the surface of the cells after incubation of living adherent cells with ET-3 in different conditions. The maximal binding was determined by incubating the cells with ET-3 for 1 h at 4°C, a condition where internalization is blocked. Incubation of the cells with ET-3 for 1 h at 37°C, a temperature for which internalization can occur, led to a decrease of the fluorescent signal by about 70%. This signal decrease reflects a massive ET-3-mediated internalization of ETB.
Figure 7.
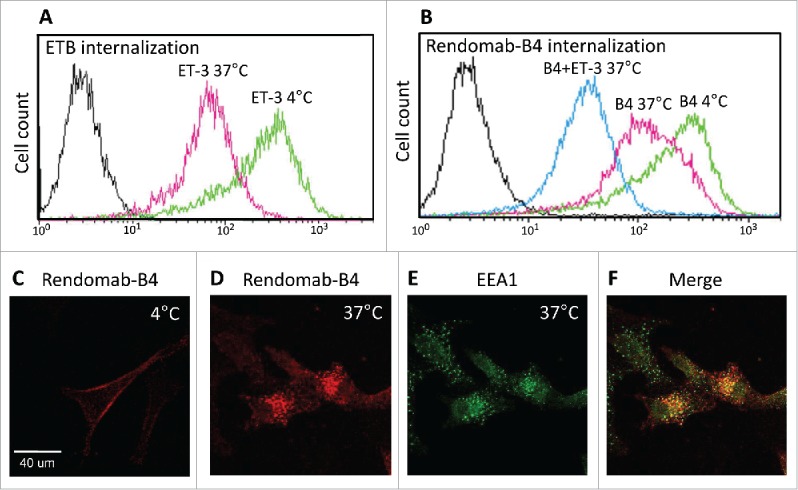
Rendomab-B4 is internalized in UACC-257 cells. Flow cytometry experiments were performed to study the internalization of ETB (A) and of rendomab-B4 itself (B). (A) Living, adherent UACC-257 cells were incubated at 4°C (green curve) or 37°C (pink curve) in medium in the presence of 50 nM ET-3. After washing, cells were detached and incubated for 2 hours at 4°C with rendomab-B4 in order to evaluate ETB amount at the surface of the cells. Cells were then incubated with a fluorescent secondary antibody and the fluorescence was measured using a FACS calibur cytometer. Black curve corresponds to basal fluorescence. (B) UACC-257 living cells were incubated for 3 hours at 4°C (green curve) with 100 nM rendomab-B4. Cells were incubated for 3 hours at 37°C with 100 nM rendomab-B4 prior to 1 additional hour incubation at 37°C with 50 nM ET-3 (blue curve) or not (pink curve). Cells were then detached and remaining antibodies at cell surface were detected using secondary antibody. The fluorescence was quantified by flow cytometry. (C and D) Living UACC-257 cells were incubated for 2 h at 4°C (C) or 37°C (D) with rendomab-B4, fixed and incubated in Cy3-anti-mouse antibody (red). Green labeling in (E) corresponds to early endosomal antigen 1 (EEA1) staining. Merged image is represented in (F).
We also examined the internalization of the rendomab-B4 itself, and the results are shown in Fig. 7B. To avoid internalization of the receptor and to obtain the maximal binding, adherent UACC-257 cells were first incubated for 3 hours at 4°C with the rendomab-B4. To allow the internalization, the cells were incubated for 3 hours at 37°C in the presence of the antibody. This resulted in a signal decrease by about 40%. Moreover, when ET-3 was added to the cells and incubated for one additional hour at 37°C, still in the presence of rendomab-B4, the signal was further reduced by about 80%. In control experiments, no binding was observed with an isotypic control antibody (data not shown). Thus, rendomab-B4 is internalized by UACC-257 cells, and this effect is increased in the presence of the agonist ET-3, reaching a percentage of internalization higher than that obtained with ET-3 alone, suggesting that rendomab-B4 by itself is able to promote ETB endocytosis to some extent. Immunofluorescence analysis of rendomab-B4 binding on UACC-257 living cells at 4°C is shown in Fig. 7C. The result clearly shows that rendomab-B4 bound to the UACC-257 cell surface. After 2 hours of incubation at 37°C, the labeling disappeared from the cell membrane (Fig. 7D) and appeared as a punctuate labeling in the cytoplasm, reflecting the internalization of the mAb. As expected, this labeling obtained with rendomab-B4 mainly colocalized with Early Endosome Antigen 1 (EEA1) staining (Fig. 7E and F), indicating that after 2 h ET-3 treatment, a large proportion of rendomab-B4 is located in early endosomes.
Rendomab-B4 inhibits PLC but not ERK response due to ETB receptor activation in UACC
Although rendomab-B4 does not compete with ET binding on hETB, we cannot exclude the possibility that it might exert allosteric modulation on receptor activity or might reduce the number of receptors at the cell surface, as suggested by the internalization results described above. Therefore, the effect of rendomab-B4 on signaling pathways coupled to hETB in UACC-257 cells was investigated. It is well established that ETB is coupled to Gq and/or Gi families of G proteins, leading to the activation of phospholipase C (PLC) and ERK1/2 MAP kinases pathways. Data shown in Fig. 8A showed that incubation of UACC-257 cells with ET-1, but also with ET-3, stimulated the PLC activity by about 5 times, as reflected by the increased production of inositol phosphates (InsPs). Incubation of the cells for 2 hours in the presence of control isotype antibody had no effect. In contrast, a similar treatment with the rendomab-B4 strongly reduced (by about 75%) the production of InsPs stimulated both by ET-1 and ET-3. These data indicate that rendomab-B4 is able to reduce the PLC transduction pathway without interfering with the binding of endogenous receptor ligands.
Figure 8.
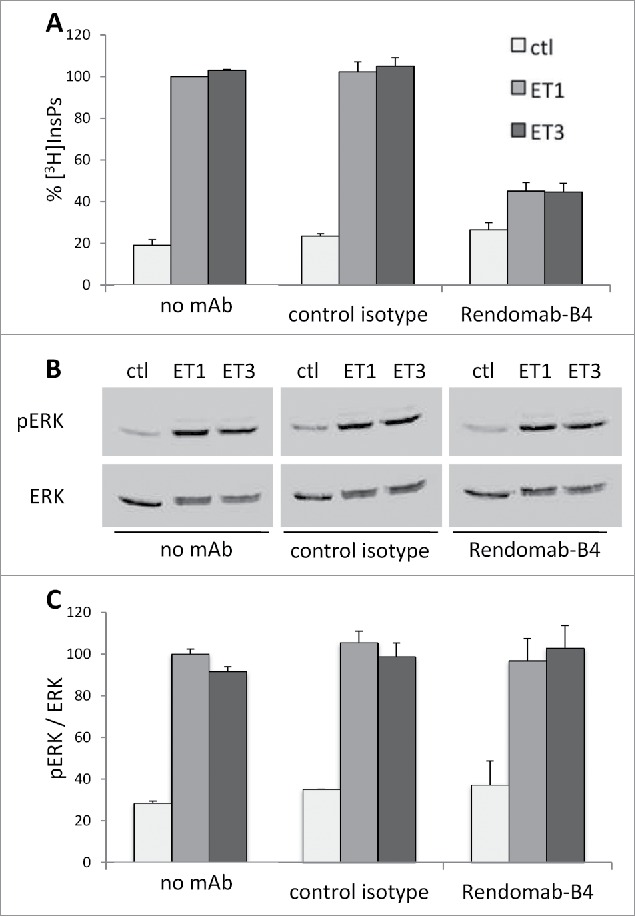
Rendomab-B4 inhibits PLC but not ERK response due to ETB receptor activation in UACC. (A) [3H]inositol-labeled UACC-257 cells were treated or not for 2 h with 150 nM of control isotype antibody or Rendomab-B4 before stimulation for 30 min in the presence or the absence of 50 nM ET-1 or ET-3. Total [3H]InsPs amount was determined as described in materials and methods. Results are expressed as percent of InsPs production induced by ET-1 in the absence of antibody and are means ± SEM of 6 independent experiments, each performed in duplicate. (B) Cells were treated for 2 h with or without 150 nM of control isotype antibody or rendomab-B4 before the addition of 10 nM ET-1 or ET-3. After a 10 min incubation, cells were lysed and total proteins were analyzed by 10% SDS/PAGE followed by immunoblotting with anti-active phosphorylated ERK1/2 (pERK) antibody and anti-total ERK2 (ERK). pERK and ERK signals were quantified (C), and the levels of phosphorylated ERK1/2 were normalized with respect to total ERK2 amount in the corresponding sample. Results were expressed as percent of ET-1 stimulation without antibody treatment (100%). Values are the means ± SEM of 3 independent experiments performed in duplicate.
The MAP kinases pathway is known to be involved in tumor progression in melanoma. Therefore, we also investigated the potential effect of rendomab-B4 on this pathway. Our data confirmed that in UACC-257, both ET-1 and ET-3 increased ERK1/2 phosphorylation and activation, as previously shown.17 Indeed, in UACC-257, the 2 ETs induced a rapid increase of ERK1/2 phosphorylation, with a maximal response obtained at 5 to 10 min, followed by a progressive decrease (data not shown). The effect of rendomab-B4 was then tested on the activation of ERK1/2 by ET-1 and ET-3 (Fig. 8B and C). Data depicted in Fig. 8C clearly indicate that the activation of ERK1/2 was unaffected when UACC-257 were treated in the presence of rendomab-B4, as it was observed using control mAb. These results show that rendomab-B4 exhibits differential inhibitory effects on PLC and ERK pathways activated by ETs.
Rendomab-B4 inhibits melanoma cell migration induced by endothelin
It is now well established that the ET axis plays an important role in cancer progression, triggering cellular events involved in cancer cells invasion. Given this crucial role, we next studied the effect of rendomab-B4 on ET-1-induced migration of UACC-257. Data shown in Fig. 9 demonstrated that the migration of the melanoma cell line was increased by 3-fold in the presence of ET-1. Incubation of the cells with control antibody failed to modify the cell migration due to ET-1. By contrast, rendomab-B4 completely abolished UACC-257 cells migration induced by ET-1. We verified that under similar conditions, the viability and the number of cells were not affected by ET-1 and rendomab-B4 (data not shown). Altogether, these results show that, although rendomab-B4 does not prevent ET binding on ETB, it is able to inhibit several of its effects on melanoma cells.
Figure 9.
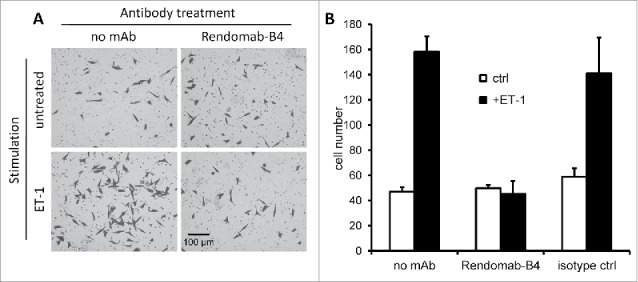
Rendomab-B4 inhibits melanoma cell migration induced by endothelin. UACC-257 (150 nM) seeded in culture inserts were treated for 2 h with or without 150 nM of rendomab-B4 or control isotype antibody before the addition of 10 nM ET-1. After 20 h of incubation, cells were stained with crystal violet and counted from 3 randomly selected images per insert. (A) Images of UACC-257 cells on the lower side of the insert membranes. (B) Quantification of the number of cells present on the lower side of the insert membranes. Values are the means ± SEM of 3 independent experiments.
Discussion
The development of mAbs for the treatment of cancers is an alternative to conventional therapeutic agents and is currently a rapidly expanding sector. In the particular case of metastatic melanoma treatment, the anti-CTLA-4 blocking antibody ipilimumab and the anti-PD-1 antibodies pembrolizumab and nivolumab that modulate the immune checkpoints have been recently approved.30-32 However, despite the improvement of overall survival observed with these mAbs, they do not lead to remission of the disease. Several studies clearly established that ETB plays a pivotal role in melanoma and represents a good candidate for completing the therapeutic strategies.12,20,33-35 Despite the promising results obtained with the dual receptor antagonist bosentan in melanoma cell lines,36 the 2 Phase 2 studies in patients with stage IV metastatic melanoma were disappointing.22,23 Used as a monotherapy, bosentan stabilized less than 20% of the patients, and no additional effect on temporal progression of tumors was observed when bosentan was combined with a chemotherapeutic agent (dacarbazine).23 Hence new therapeutic molecules, like antibodies, targeting ETB, need to be developed in order to inhibit the pleiotropic effects on tumor cells induced by the ET axis.
In a previous study, we reported the in vitro characterization of rendomab-B1, a mAb directed against human ETB. Rendomab-B1 was the first-reported mAb behaving as a potent antagonist of human ETB, blocking ET-1-induced signaling in CHO overexpressing ETB and recognizing ETB on HUVEC. Nevertheless, rendomab-B1 recognizes only with a poor affinity ETB overexpressed at the surface of melanoma cells, strongly suggesting a structural heterogeneity among ETB receptors.25 In the study reported here, we selected and characterized another anti-ETB mAb, rendomab-B4, on the criterion of melanoma recognition.
We first characterized the rendomab-B4 pharmacological properties in CHO cells stably expressing human ETB. Experiments using flow cytometry demonstrated that rendomab-B4 recognized human, but not rat, ETB with high affinity and without any cross-reactivity with ETA in living cells.
Data obtained from epitope mapping experiments demonstrated that rendomab-B4 recognizes 2 noncontiguous sequences present in the N-terminal domain of the receptor. This interpretation was strengthened by complementary results showing that rendomab-B4 does not recognize human ETB when CHO-ETB cell lysates were analyzed by protein gel blot, a condition where the receptor is in a denatured form, but is able to immunoprecipitate it from a similar cell lysate, a condition where the receptor is under its native form. Altogether, these observations indicate that rendomab-B4 binds with a high affinity a conformational epitope formed by the 3-dimensional arrangement and juxtaposition of 2 peptidic sequences in the structure of the N-terminal domain of ETB. Furthermore, pepscan analysis results indicate that rendomab-B4 is also able to bind the 2 sequences separately, while negative results of western blotting experiments tend to indicate the opposite. This apparent discrepancy indicates that rendomab-B4 binding to the individual epitopic peptides is a low affinity binding that can only be revealed by pepscan analysis, which is a more sensitive assays than protein gel blot. All these rendomab-B4 epitope mapping data constitute a clue concerning the conformation of the receptor, since it provides information about the proximity of the 2 distinct sequences of ETB recognized by rendomab-B4. Epitopes formed by discontinuous sequences were already described such as for the rendomab-B1, which recognized 3 discontinuous regions in extracellular domains of the receptor; more precisely, 2 other sequences in the N-terminal domain of ETB and one in the extracellular loop E2.25 Other mAbs recognizing discontinuous epitopes have already been described. Indeed, mAbs directed against epithelial cell adhesion molecule37 or against glycoprotein B of herpes simplex virus38 were also shown to bind discontinuous epitopes.
The crystal structure of the human ETB has still not been solved and the precise binding site for ET-1 and ET-3 has not been clearly defined, either by site-directed mutagenesis or molecular modeling approaches. However, TMI-III, VII and extracellular loops appear to contain an agonist binding motif,39,40 and the E1 loop seems to be more particularly involved in the interaction with ET-1.41 The observation that the epitope sequences recognized by rendomab-B4 are located in a region that has not been implicated in ligand binding is consistent with our binding data showing that there was no competition between the antibody and ETs.
Although rendomab-B4 did recognize ETB on UACC-257 melanoma cells with high affinity, it is striking to observe that it was unable to bind ETB on HEK293T and HUVEC, thus displaying opposite binding properties compared to those of rendomab-B1.25 These diverging properties may be explained by differential levels of ETB expression, specific cellular context such as receptor interacting proteins or post-translational modifications in the various studied cell lines. Data from literature indicate that CHO-ETB and UACC-257 express ETB at higher level (> 1000 fmol/106 cells and 56 fmol/106 cells, respectively17) than non-cancer cells, i.e., HUVEC that express only 4.8 fmol/106 cells.42 Although this low amount of receptor is detected by rendomab-B1, rendomab-B4 is unable to bind HUVEC. To explain this difference it can be speculated that in CHO-ETB cells, due to its very high expression level, ETB would tend to form dimers (or oligomers), which would constitute the real target of rendomab-B4. According to this reasoning, in cells expressing low levels of ETB, the receptor would be rather monomeric and recognized by rendomab-B1. Binding of rendomab-B4 on ETB dimers is compatible with the epitope mapping results. Indeed, we observed that rendomab-B4 epitope is conformational and formed by 2 sequences which have to be in close proximity to bind the antibody. One possibility, as stated above, is that the proximity of the 2 sequences is just the result of the intrinsic 3 dimensional structure of the receptor, but it can also be envisaged that the 2 peptides are not in proximity in one receptor molecule, but that dimerization of the receptor would bring the 2 peptides in proximity allowing rendomab-B4 to bind on. This last hypothesis is also compatible with the finding that rendomab-B4 does not recognizes denatured ETB on western blots.
Alternatively, as stated above, post-translational modifications, particularly glycosylation that occurs at the level of the N-terminal tail of,43 may affect the binding of antibodies. Indeed, a glycosylation site in the N-terminal region of ETB (N59) is located between the 2 sequences constituting the epitope. Then, it is tempting to speculate that modifications in the glycosylation status of this site may influence binding properties of the rendomab-B4 and may contribute to the different binding properties of rendomab-B1 and -B4. Moreover, it is now well demonstrated that the glycosylation of many cell surface proteins is altered in cancer cells.44 However, many data in the literature tend to make this hypothesis rather unlikely. Indeed, although the N 59 glycosylation site has been predicted by analysis of the primary structure of the receptor, the role of ETB glycosylation remains unclear.43 Several reports show that treatment of cell membranes with deglycosylating enzymes did not change the binding of ET-1 on ETB, while it strongly reduced ET-1 binding on ETA receptor.45 Other studies, where the putative N-glycosylation site of ETB was mutated, did not reveal any modification of the expression of ETB at cell surface, binding to ET1 or coupling to signaling pathways.46,47 Moreover, Roos et al.48 found, by mass spectrometry approach, that ETB, extracted from bovine lung, was not glycosylated in its N-terminal domain. These results indicate that the glycosylation of ETB does not influence the structure and function of the receptor, and has very few chances to play a role in the discriminating properties of rendomab-B4 toward CHO-ETB and UACC-257 cells. However, to fully reject this hypothesis, further investigations, such as site-directed mutagenesis experiments at the glycosylation site followed by immunoprecipitation of ETB by rendomab-B4, would be necessary.
It has been reported that, compared to ETA, ETB is rapidly internalized and degraded.27,28 Interestingly, we observed that rendomab-B4 was internalized in UACC-257 cells and the internalization was increased in the presence of ET-3, known to play a crucial role in melanoma invasiveness.49,50 These results were strengthened by immunofluorescence experiments showing the internalization of rendomab-B4 in the presence of ET-3 in the melanoma cell line. Furthermore, the internalized complexes were detected in early endosomes. Finding rendomab-B4 in early endosomes is consistent with previously published data showing that, after activation by ET, ETB is rapidly internalized and then targeted to lysosomes.28,51,52
In this study, we observed that after internalization rendomab-B4 staining mainly colocalized with an early endosome specific protein, but some mAb-positive dots were not positive for the endosomal marker. These mAb-positive dots may reflect the presence of rendomab-B4 having reached lysosomes with ETB. This property of internalization of rendomab-B4, associated with the high expression level of ETB in melanoma cells, is particularly important because the mAb could be used for the specific delivery of cytotoxic molecules into melanoma overexpressing the ET-3/ ETB axis, increasing antibody-mediated cell killing. In line with this interpretation, it has been demonstrated that a relationship exists between ETB density and efficacy of mAb 5E9 raised against ETB.17,53 Although the 5E9 mAb had no inhibitory effect by itself, its use as an ADC (5E9 conjugated with the potent cytotoxic compound monomethyl auristatin E) showed good efficacy against human melanoma cell lines and xenograft tumor models.17 Since rendomab-B4, by itself, has the ability to inhibit PLC and migration in melanoma cells, its use as an ADC may lead to greater efficacy. Moreover, since rendomab-B4 does not inhibit ERK1/2 pathway and this pathway is crucial for melanoma growth, the efficacy of rendomab-B4-based ADC would be further enhanced by co-treatment with ERK1/2 pathway inhibitor, a combination treatment previously reported to highly efficient,53 or by directly coupling the antibody to such inhibitor.
Although we observed that rendomab-B4 does not compete with ETs for binding on ETB, the possibility that this mAb may modulate ETB-dependent signaling processes was investigated. Indeed, increasing data indicate that GPCR signaling may be regulated by allosteric modulators that do not bind to the orthosteric site of the receptor.54-56 Interestingly, we found that rendomab-B4 was able to inhibit PLC activation, which is a G protein dependent process, but had no effect on ERK1/2 activation induced by ET-1 or ET-3. Comparable results were obtained in HEK293 cells expressing angiotensin I receptors, where a synthetic peptide mimicking the second intracellular loop of the receptor inhibits angiotensin II-mediated PLC activation and Ca2+ influx, but does not alter ERK1/2 activation.57 Furthermore, ORG27569, an allosteric ligand for the CB1 cannabinoid receptor, was found to inhibit G protein signaling but induced ERK1/2 activation.58 Protease-activated receptor (PAR)2 cleaved by trypsin induces the activation of both PLC/calcium and ERK1/2 signaling pathways. However, when cleaved with Elastase-2, PAR2 lost its coupling to Gq/calcium but retains its capacity to activate ERK1/2,59 indicating that N-terminal domain of this GPCR is able to control receptor-mediated selective signaling pathway.
Also, rendomab-B4 does not inhibit ETB endocytosis, but rather tends to increase it. Since ETB endocytosis has been shown to be a β-arrestin-dependent process,51 it can be assumed that rendomab-B4 does not inhibit β-arrestin-dependent signaling. Interestingly, it is now admitted that ERK1/2 pathway can be activated through β-arrestin recruitment by various GPCR.60 Such a mechanism occurring in UACC-257 cells may explain why rendomab-B4 does not inhibit ERK1/2 activation in these cells, but no information is available on the signaling pathway linking ETB to ERK1/2 in UACC-257 cells. To conclude on this point, to our knowledge, rendomab-B4 is the first reported mAb to possess the ability to differentially affect ETB-coupled signaling pathways, behaving as a biased allosteric modulator inhibiting G protein-dependent processes but not β-arrestin-dependent ones. These particular properties of rendomab-B4 merit further investigations.
As mentioned, ETB plays a crucial role in metastatic spread of melanoma, which is the critical step of the disease. Taking into account this property of the ET axis, migration assays were performed, and they demonstrated that rendomab-B4 is able to completely inhibit the migration induced by ET-1. Although the demonstration has not been made, this effect is likely linked to the potentiality of rendomab-B4 to inhibit G-protein signaling. This inhibition of melanoma cell migration suggests that rendomab-B4 may represent a valuable starting point for the development of new tools for treatment of melanoma. Future development of these tools will require the demonstration that rendomab-B4, alone or as an ADC, is able to inhibit melanoma growth and/or spreading in an in vivo model consisting of human melanoma xenografts implanted in nude mice. Then, the mAb must be humanized and its toxicity evaluated in non-rodent models since it does not cross react with rat and mice ETB.
In conclusion, anti-human ETB rendomab-B4 displays features that are unique, and not observed with the other anti ETB mAbs described until now.17 Rendomab-B4 specifically binds to ETB expressed on cancer cells with a very high affinity, and it is internalized by these cells and blocks their migration. These particular properties make rendomab-B4 an interesting tool to analyze ETB-structure/function, and a promising starting point for the development of future immunological tools in the field of melanoma therapeutics.
Materials and methods
Animals
Six-week-old female C57BL/6 mice (ElevageJanvier) were kept in a specific pathogen-free animal facility. All animal experiments complied with French animal experimentation regulations.
Plasmids
The cDNA clone of the human ETB (generous gift from Dr. M.J. Brownstein, Bethesda, MD, USA) was subcloned in pcDNA3.1 vector (Life Technologies). The integrity of the construct was confirmed by sequencing.
Cell culture and transfection
CHO-K1 cells (ECACC) were cultured in Ham-F12 medium, HEK293T (EACC) and human umbilical vascular endothelial cells (HUVEC, ATCC) in DMEM medium. The melanoma cell line UACC-257 (NCI-60 cell collection) was cultured in RPMI-1640. Melanoma cell lines WM-266-4 (ECACC) and SLM8 (kind gift from Dr M. Viguier, Hôpital Saint-Louis, Paris, France) were cultured in DMEM: Ham-F12 medium (1:1). All media were supplemented with 10% fetal calf serum, 1 mM pyruvate, 1% nonessential amino acids, 2 mM glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin. Eker rat uterine leiomyoma cells (ELT-3, kindly provided by Dr. C. Walker, Anderson Cancer Center, University of Texas, Smithville, TX, USA) were cultured in DF8 medium supplemented with 10% fetal calf serum. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2. All media and cell culture supplements were from Life Technologies. The ETB expression vector was transfected and stably overexpressed in CHO and HEK cells using FUGENE HD reagent (Roche Diagnostics). Human ETA stably transfected CHO cells were kindly provided by Dr. M. Iglarz (Actelion). Stably transfected cells expressing human ETB or ETA were termed CHO-ETB, CHO-ETA or HEK-ETB, and they were always cultured in the presence of 1 mg/mL G1418.
DNA immunization protocol and production of monoclonal antibodies
MAb production, by DNA immunizations, was performed as previously described;25,61 collected splenocytes of the 2 best-responding mice were fused to NS1 mouse myeloma cells. Hybridoma supernatants were screened for production of anti-ETB specific antibodies by a living cell-based ELISA test, using untransfected CHO cells and CHO-ETB cells as targets, prior to confirmation of specificity and reactivity of antibodies by flow cytometry. After subcloning by limiting dilutions, antibodies were isotyped using a mouse immunoglobulin isotyping kit according to the manufacturer (Pierce) instructions and purified by affinity chromatography on Protein A-Sepharose (Millipore).
Flow cytometry analysis
Affinity determination
For affinity measurements, saturation binding experiments were performed with stably transfected CHO-ETB, melanoma cell lines (UACC-257, WM266-4 and SLM8), HEK293T and HUVEC. Collected cells were seeded (100,000 cells/well) in V-shaped 96-well plates. Plates were centrifuged, supernatant was discarded and cells were incubated overnight at 4°C with 100 µL of D-phosphate-buffered saline (PBS) supplemented with 0.1% BSA and 5% normal goat serum (NGS, Life Technologies) and containing increasing concentrations of rendomab-B4. After two washes with 150 µL of ice-cold D-PBS - 0.1% BSA - 1% NGS, cells were incubated for 2 h at 4°C in the dark with R-phycoerythrin (R-PE)-conjugated AffiniPure goat anti-mouse IgG (H+L) (Jackson ImmunoResearch). After two washes, cells were re-suspended in 100 µL of D-PBS and their fluorescence was measured using a GUAVA flow cytometer (Guava Easycyte Plus, Millipore). Mean fluorescence intensity (MFI) of samples was then calculated. To control ETB expression at the cell surface, increasing concentrations of fluorescein-labeled ET-1 or ET-3 (ET-1-FAM or ET-3-FAM, Phoenix Pharmaceuticals) were used.
Competition experiments
For competition tests, CHO-ETB or melanoma cells (UACC-257) were treated according to 2 different protocols: (i) cells were co-incubated with 10 nM ET-1-FAM or ET-3-FAM and varying concentrations of different competitors (rendomab-B4, control isotype antibody, ET-1, ET-3, selective ETA receptor antagonist (BQ123) or selective ETB receptor antagonist (BQ788); and (ii) cells were co-incubated with fixed rendomab-B4 concentration (10 nM) and varying concentrations of competitor peptides ET-1, ET-3. To reach equilibrium, cells were incubated overnight at 4°C. For type (i) experiments, cells were washed twice before measuring the fluorescence. For type (ii) experiments, after 2 washes, cells were incubated for 2 h with (R-PE)-conjugated secondary antibody. The MFI of the cells was measured using a GUAVA cytometer.
Receptor and rendomab-B4 internalization
To study receptor and antibody internalization in UACC-257 cell line, 300,000 cells were used per assay. The internalization of the receptor due to ET was evaluated by incubating living adherent cells for 1 h at 4°C or 37°C with 50 nM ET-3. After detachment, cells were incubated for 2 h at 4°C in D-PBS - 0.1% BSA - 5%NGS with 100 nM rendomab-B4, washed twice and were incubated for 2 h at 4°C with (R-PE)-conjugated AffiniPure goat anti-mouse IgG (H+L). Cells were washed twice and the fluorescence was analyzed.
For antibody internalization experiments, living adherent cells were incubated for 3 h at 4°C or 37°C with either 100 nM rendomab-B4 or control isotype antibody. Cells were incubated for one additional hour with or without 50 nM ET-3. Then, cells were detached and incubated for 2 h at 4°C with (R-PE)-conjugated AffiniPure goat anti-mouse IgG (H+L) and were washed twice again.
For these 2 kinds of experiments, cell fluorescence was finally assayed using a FACSCalibur flow cytometer (FACSCalibur Flow Cytometry System, BD Biosciences) and the MFI determined.
Microscopy analysis
Wide-field microscopy analysis
Wide-field microscopy analysis was performed on CHO and CHO-ETB cell lines seeded on glass coverslips at the density of 15,000 cells per cm2. After 48 h, cells were washed in PBS and fixed for 15 min in 4% paraformaldehyde (PFA) at room temperature. Cells were then incubated for 10 min in 50 mM NH4Cl and washed once with PBS. Cells were then incubated at room temperature for 1 hour in the presence of 100 nM of rendomab-B4 in PBS-1% BSA. This incubation is performed in the absence of detergent in order to only detect membrane receptors at the cell surface. After three successive washes with PBS, cells were incubated for 1 h with PBS - 1% BSA containing Cy3-conjugated anti-mouse IgG antibody (Jackson ImmunoResearch). After three washes, cells were mounted on slides with Fluoroshield containing DAPI (Sigma-Aldrich). Images were taken using a fluorescence microscope (Zeiss Axiophot II), at 40x magnification.
Confocal microscopy analysis
Confocal microscopy analysis was performed on UACC-257 cells seeded on glass coverslips at a density of 30,000 cells per cm2. For internalization experiments, living cells were incubated at 4 or 37°C for 1 h with 100 nM of rendomab-B4 before fixation with 4% PFA. To stain the endosomal compartment, cells were permeabilized by treatment for 15 min with PBS - 1% BSA - 0.2% Triton X-100 before incubation with an anti-EEA1 antibody (Millipore, France). After three washes, cells were incubated in PBS - 1% BSA - 0.2% Triton X-100 containing Cy3-conjugated anti-mouse IgG antibody and FITC-conjugated anti-rabbit antibody. Images were taken using a confocal microscope (Zeiss LSM510), at 63x magnification.
Peptide synthesis and ETB-binding epitope mapping
The entire extracellular amino acid sequence of the ETB receptor was synthesized on a cellulose membrane using the previously described SPOT technique,62 laying down overlapping 12-mer peptides, frameshifted by one residue. Rendomab-B4 epitope mapping was performed according to a protocol previously described,61 incubating the membrane in 1 µg/mL of rendomab-B4 for 90 min at room temperature. The hybridization was revealed using an anti-mouse IgG labeled with alkaline phosphatase (Sigma-Aldrich). ImageJ software was used to quantify the signal obtained for each spot. Peptides were considered as antigenically relevant if they were part of a consecutive series of reactive spots, presenting at least one signal peak 3 times higher than the background.62
Cloning of GFP-tagged rat and human ETB
Rat ETB cDNA was cloned by RT-PCR technique from rat ELT3 cells, which express high level of ETB.26 Total RNA from these cells was prepared using NucleoSpin® RNAII (Macherey-Nagel) according to the manufacture's protocol. The reverse transcription reaction was performed with 5 μg of total RNA using 200 units of Moloney Murine Leukemia Virus (M-MLV)-reverse transcriptase, 200 μM deoxynucleoside triphosphates (dNTPs), and 10 μM random hexamer primers. PCR primers for amplification of rat EBT (rETB) were based on the GenBank sequence (NM_017333). The primers span the start codon and introduced a modified codon in place of the stop codon and contain HindIII and BamHI sites (italics): 5′-GCAAGCTTATGCAATCGTCCGCAAGCC-3′ (Forward primer) and 5′-GCGGATCCACAGATGAGCTGTATTTATTGCTGG -3′ (Reverse primer).
For hETB cloning, hETB cDNA was amplified by PCR from pCDNA3.1-ETB plasmid (described above) with PCR primers based on the GenBank sequence (NM_000115.3). As for rETB cloning, the primers span the start codon and introduced a modified codon in place of the stop codon and contain HindIII and BamHI sites (italics): 5′-GCAAGCTTATGC AGCCGCCTCCAAGTC-3′ (Forward primer) and 5′-GCGGATCCACAGATGAGCTGT ATTTATTACTGG-3′ (Reverse primer).
The PCR reactions were performed, in a thermal cycler (icycler; Bio-Rad) with 3 μl of the RT reaction (rETB) or 10 ng pCDNA-ETB plasmid (hETB) using the Phusion® High-Fidelity DNA Polymerase according to the manufacturer's protocol. The amplified condition was 98°C for 30 s followed by 30 cycles of 98°C for 10 s, 70°C for 30 s, 72°C for 45 s, and a final extension at 72°C for 10 min. The PCR products were digested by BamHI and HindIII restriction enzymes and ligated into pEGFP-N1 (Clonetech), using T4 DNA ligase according to the manufacturer's indication. Resulting plasmids were named phETB-GFP and prETB-GFP, and their sequences were verified by sequencing (Eurofins MWG Operon).
Immunoprecipitation of ETB
CHO cells seeded in 24-well plates were transfected with phETB-GFP or prETB-GFP plasmids using Lipofectamine LTX (Life Technologies) according to the manufacturer's instructions. 24 h after transfection, cells were washed with PBS and lysed in 200 µL of ice cold lysis buffer containing 10 mM Tris/Cl pH 7.5; 150 mM NaCl; 0.5 mM EDTA; 1% Triton X-100. Lysates were centrifuged for 5 min at 15,000 × g and the supernatants diluted to 1 mL with lysis buffer without Triton X-100. Antibodies, 4 µg of rendomab-B4 or 4 µL of GFP-Trap beads (Chromotek), were added and the samples were incubated on a wheel for 4 h at 4°C. When rendomab-B4 was used, 20 µL G-protein conjugated beads (Santa Cruz) were added after the 2 first hours of incubation. At the end of the incubations, samples were washed 4 times with 1 mL ice cold lysis buffer without detergent. Immunoprecipitated samples were then analyzed by protein gel blot using an anti-GFP antibody (Cell Signaling Technologies) and a secondary anti-rabbit IgG antibody conjugated to HRP (Cell Signaling Technologies).
Measurement of PLC activity
Confluent UACC-257 cells seeded in 12-well plates were serum starved for one night in MEM in the presence of 10 µCi/mL Myo-[2-3H]inositol (16 Ci/mmol; PerkinElmer). The cells were washed 3 times with MEM and then incubated at 37°C for 2 h in 800 µl of MEM in the presence of 150 nM rendomab-B4 or control isotype antibody. Cells were then exposed to the agonists ET-1 or ET-3 (50 nM) in the presence of 10 mM LiCl (added 20 min before the agonists). Total inositol phosphates (InsPs) produced by PLC were quantified as previously described.26 Results were expressed as the mean ± SEM of at least 3 independent experiments performed in duplicate.
Western blot analysis of phosphorylated ERK1/2
Confluent UACC-257 cells seeded in 12-well plates were serum starved for one night in RPMI and then incubated at 37°C for 2 h in 800 µl of MEM in the presence of 150 nM rendomab-B4 or control isotype antibody. Cells were then exposed to the agonists ET-1 or ET-3 (50 nM) for 5 min and then lysed in 50 µl lysis buffer (50 mM HEPES (pH 7.4), 150 mM NaCl, 100 mM NaF, 10% glycerol, 10 mM Na4P2O7, 200 µM Na3VO4, 10 mM EDTA, 1% Triton X-100, 10 µg/mL aprotinin and leupeptin). Detergent-extracted proteins were analyzed by western blot technique using mouse monoclonal anti-active phosphorylated ERK1/2 (Cell Signaling Technology) and rabbit polyclonal anti-ERK2 (Santa Cruz Biotechnology) antibodies (1:5000 each). The membranes were then incubated with antirabbit IgG antibody conjugated to IRDye 800CW (LICOR Biosciences) and antimouse IgG antibody conjugated to AlexaFluor 680 (Life technologies) for 1 h at 37˚C. Protein bands were detected and quantified on a 2color Odyssey Infrared Imaging System (LICOR Biosciences).
Migration assay
UACC-257 cells were seeded within cell culture inserts (Falcon PET membrane with 8 µm porosity) placed in a 24-well plate (30,000 cells/insert). After 8 h of culture, media were removed and the cells were incubated for 2 h in RPMI medium without FCS, in presence or absence of 150 nM rendomab-B4 or 150 nM antibody control. Cells were then stimulated or not by addition of 50 nM ET-1 in the 2 compartments. After 20 h, inserts were removed and the cells were stained with crystal violet (0.1% in 20% ethanol). After scrapping off non-migrated cells, the inserts were observed on an Axiophot II Zeiss microscope and images were taken at low magnification. The mean number of cells present on each insert was determined from 3 randomly selected images.
Disclosure of potential conflicts of interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Karine Moreau and Marc Plaisance for their expert assistance with monoclonal antibody production, and Jean-Charles Robillard for animal experiments. This work was supported by grants from Université Paris Diderot, Université Paris Sud-11, the Center National de la Recherche Scientifique (CNRS, France) and the Commissariat à l'Energie Atomique et aux Energies Alternatives (CEA, France). A.B was the recipient of a fellowship from the Ministère de l'Education Nationale, de l'Enseignement Supérieur et de la Recherche.
References
- 1.Horinouchi T, Terada T, Higashi T, Miwa S. Endothelin Receptor Signaling: New Insight Into Its Regulatory Mechanisms. J Pharmacol Sci 2013; 123:85-101; PMID:24077109; http://dx.doi.org/ 10.1254/jphs.13R02CR [DOI] [PubMed] [Google Scholar]
- 2.Yanagisawa M, Kurihara H, Kimura S, Tomobe Y, Kobayashi M, Mitsui Y, Yazaki Y, Goto K, Masaki T. A novel potent vasoconstrictor peptide produced by vascular endothelial cells. Nature 1988; 332:411-5; PMID:2451132; http://dx.doi.org/ 10.1038/332411a0 [DOI] [PubMed] [Google Scholar]
- 3.Tanfin Z, Leiber D, Robin P, Oyeniran C, Breuiller-Fouche M. Endothelin-1: physiological and pathological roles in myometrium. Int J Biochem Cell Biol 2011; 43:299-302; PMID:20974279; http://dx.doi.org/ 10.1016/j.biocel.2010.10.009 [DOI] [PubMed] [Google Scholar]
- 4.Freeman BD, Machado FS, Tanowitz HB, Desruisseaux MS. Endothelin-1 and its role in the pathogenesis of infectious diseases. Life Sci 2014; PMID:24780317; http://dx.doi.org/ 10.1016/j.lfs.2014.04.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kohan DE, Inscho EW, Wesson D, Pollock DM. Physiology of endothelin and the kidney. Compr Physiol 2011; 1:883-919; PMID:23737206; http://dx.doi.org/ 10.1002/cphy.c100039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dhaun N, Goddard J, Kohan DE, Pollock DM, Schiffrin EL, Webb DJ. Role of endothelin-1 in clinical hypertension: 20 years on. Hypertension 2008; 52:452-9; PMID:18678788; http://dx.doi.org/ 10.1161/HYPERTENSIONAHA.108.117366 [DOI] [PubMed] [Google Scholar]
- 7.Rosano L, Spinella F, Bagnato A. Endothelin 1 in cancer: biological implications and therapeutic opportunities. Nat Rev Cancer 2013; 13:637-51; PMID:23884378; http://dx.doi.org/ 10.1038/nrc3546 [DOI] [PubMed] [Google Scholar]
- 8.Nelson J, Bagnato A, Battistini B, Nisen P. The endothelin axis: emerging role in cancer. Nat Rev Cancer 2003; 3:110-6; PMID:12563310; http://dx.doi.org/ 10.1038/nrc990 [DOI] [PubMed] [Google Scholar]
- 9.Carducci MA, Jimeno A. Targeting bone metastasis in prostate cancer with endothelin receptor antagonists. Clin Cancer Res 2006; 12:6296s-300s; PMID:17062717; http://dx.doi.org/ 10.1158/1078-0432.CCR-06-0929 [DOI] [PubMed] [Google Scholar]
- 10.Rosano L, Di Castro V, Spinella F, Nicotra MR, Natali PG, Bagnato A. ZD4054, a specific antagonist of the endothelin A receptor, inhibits tumor growth and enhances paclitaxel activity in human ovarian carcinoma in vitro and in vivo. Mol Cancer Ther 2007; 6:2003-11; PMID:17620430; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-0151 [DOI] [PubMed] [Google Scholar]
- 11.Yohn JJ, Smith C, Stevens T, Hoffman TA, Morelli JG, Hurt DL, Yanagisawa M, Kane MA, Zamora MR. Human melanoma cells express functional endothelin-1 receptors. Biochem Biophys Res Commun 1994; 201:449-57; PMID:8198607; http://dx.doi.org/ 10.1006/bbrc.1994.1722 [DOI] [PubMed] [Google Scholar]
- 12.Bittner M, Meltzer P, Chen Y, Jiang Y, Seftor E, Hendrix M, Radmacher M, Simon R, Yakhini Z, Ben-Dor A, et al.. Molecular classification of cutaneous malignant melanoma by gene expression profiling. Nature 2000; 406:536-40; PMID:10952317; http://dx.doi.org/ 10.1038/35020115 [DOI] [PubMed] [Google Scholar]
- 13.Demunter A, De Wolf-Peeters C, Degreef H, Stas M, van den Oord JJ. Expression of the endothelin-B receptor in pigment cell lesions of the skin. Evidence for its role as tumor progression marker in malignant melanoma. Virchows Arch 2001; 438:485-91; PMID:11407477; http://dx.doi.org/ 10.1007/s004280000362 [DOI] [PubMed] [Google Scholar]
- 14.Garbe C, Leiter U. Melanoma epidemiology and trends. Clin Dermatol 2009; 27:3-9; PMID:19095149; http://dx.doi.org/ 10.1016/j.clindermatol.2008.09.001 [DOI] [PubMed] [Google Scholar]
- 15.Thompson JF, Scolyer RA, Kefford RF. Cutaneous melanoma in the era of molecular profiling. Lancet 2009; 374:362-5; PMID:19647595; http://dx.doi.org/ 10.1016/S0140-6736(09)61397-0 [DOI] [PubMed] [Google Scholar]
- 16.Ponti G, Ruini C, Girolomoni G, Pellacani G, Farnetani F, Pastorino L, Ghiorzo P, Witkowski AM, Bianchi-Scarra G, Tomasi A, et al.. Brooke-Spiegler syndrome tumor spectrum beyond the skin: a patient carrying germline R936X CYLD mutation and a somatic CYLD mutation in Brenner tumor. Future Oncol 2014; 10:345-50; PMID:24559443; http://dx.doi.org/ 10.2217/fon.13.198 [DOI] [PubMed] [Google Scholar]
- 17.Asundi J, Reed C, Arca J, McCutcheon K, Ferrando R, Clark S, Luis E, Tien J, Firestein R, Polakis P. An antibody-drug conjugate targeting the endothelin B receptor for the treatment of melanoma. Clin Cancer Res 2011; 17:965-75; PMID:21245091; http://dx.doi.org/ 10.1158/1078-0432.CCR-10-2340 [DOI] [PubMed] [Google Scholar]
- 18.Spinella F, Caprara V, Di Castro V, Rosano L, Cianfrocca R, Natali PG, Bagnato A. Endothelin-1 induces the transactivation of vascular endothelial growth factor receptor-3 and modulates cell migration and vasculogenic mimicry in melanoma cells. J Mol Med (Berl) 2013; 91:395-405; PMID:22965194; http://dx.doi.org/ 10.1007/s00109-012-0956-2 [DOI] [PubMed] [Google Scholar]
- 19.Lahav R, Heffner G, Patterson PH. An endothelin receptor B antagonist inhibits growth and induces cell death in human melanoma cells in vitro and in vivo. Proc Natl Acad Sci U S A 1999; 96:11496-500; PMID:10500205; http://dx.doi.org/ 10.1073/pnas.96.20.11496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bagnato A, Rosano L, Spinella F, Di Castro V, Tecce R, Natali PG. Endothelin B receptor blockade inhibits dynamics of cell interactions and communications in melanoma cell progression. Cancer Res 2004; 64:1436-43; PMID:14973117; http://dx.doi.org/ 10.1158/0008-5472.CAN-03-2344 [DOI] [PubMed] [Google Scholar]
- 21.Lahav R. Endothelin receptor B is required for the expansion of melanocyte precursors and malignant melanoma. Int J Dev Biol 2005; 49:173-80; PMID:15906230; http://dx.doi.org/ 10.1387/ijdb.041951rl [DOI] [PubMed] [Google Scholar]
- 22.Maguire JJ, Davenport AP. Endothelin receptors and their antagonists. Semin Nephrol 2015; 35:125-36; PMID:25966344; http://dx.doi.org/ 10.1016/j.semnephrol.2015.02.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kefford RF, Clingan PR, Brady B, Ballmer A, Morganti A, Hersey P. A randomized, double-blind, placebo-controlled study of high-dose bosentan in patients with stage IV metastatic melanoma receiving first-line dacarbazine chemotherapy. Mol Cancer 2010; 9:69; PMID:20350333; http://dx.doi.org/ 10.1186/1476-4598-9-69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Recondo G, Diaz Canton E, de la Vega M, Greco M, Valsecchi ME. Therapeutic options for HER-2 positive breast cancer: Perspectives and future directions. World J Clin Oncol 2014; 5:440-54; PMID:25114858; http://dx.doi.org/ 10.5306/wjco.v5.i3.440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Allard B, Wijkhuisen A, Borrull A, Deshayes F, Priam F, Lamourette P, Ducancel F, Boquet D, Couraud JY. Generation and characterization of rendomab-B1, a monoclonal antibody displaying potent and specific antagonism of the human endothelin B receptor. MAbs 2013; 5:56-69; PMID:23221682; http://dx.doi.org/ 10.4161/mabs.22696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Raymond MN, Robin P, De Zen F, Vilain G, Tanfin Z. Differential endothelin receptor expression and function in rat myometrial cells and leiomyoma ELT3 cells. Endocrinology 2009; 150:4766-76; PMID:19628575; http://dx.doi.org/ 10.1210/en.2009-0118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Oksche A, Boese G, Horstmeyer A, Papsdorf G, Furkert J, Beyermann M, Bienert M, Rosenthal W. Evidence for downregulation of the endothelin-B-receptor by the use of fluorescent endothelin-1 and a fusion protein consisting of the endothelin-B-receptor and the green fluorescent protein. J Cardiovasc Pharmacol 2000; 36:S44-7; PMID:11078332; http://dx.doi.org/ 10.1097/00005344-200036051-00016 [DOI] [PubMed] [Google Scholar]
- 28.Bremnes T, Paasche JD, Mehlum A, Sandberg C, Bremnes B, Attramadal H. Regulation and intracellular trafficking pathways of the endothelin receptors. J Biol Chem 2000; 275:17596-604; PMID:10747877; http://dx.doi.org/ 10.1074/jbc.M000142200 [DOI] [PubMed] [Google Scholar]
- 29.Sievers EL, Appelbaum FR, Spielberger RT, Forman SJ, Flowers D, Smith FO, Shannon-Dorcy K, Berger MS, Bernstein ID. Selective ablation of acute myeloid leukemia using antibody-targeted chemotherapy: a phase I study of an anti-CD33 calicheamicin immunoconjugate. Blood 1999; 93:3678-84; PMID:10339474 [PubMed] [Google Scholar]
- 30.Russo A, Ficili B, Candido S, Pezzino FM, Guarneri C, Biondi A, Travali S, McCubrey JA, Spandidos DA, Libra M. Emerging targeted therapies for melanoma treatment (review). Int J Oncol 2014; 45:516-24; PMID:24899250; http://dx.doi.org/ 10.3892/ijo.2014.2481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, Chmielowski B, Spasic M, Henry G, Ciobanu V, et al.. PD-1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014; 515:568-71; PMID:25428505; http://dx.doi.org/ 10.1038/nature13954 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Topalian SL, Sznol M, McDermott DF, Kluger HM, Carvajal RD, Sharfman WH, Brahmer JR, Lawrence DP, Atkins MB, Powderly JD, et al.. Survival, durable tumor remission, and long-term safety in patients with advanced melanoma receiving nivolumab. J Clin Oncol 2014; 32:1020-30; PMID:24590637; http://dx.doi.org/ 10.1200/JCO.2013.53.0105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cruz-Munoz W, Jaramillo ML, Man S, Xu P, Banville M, Collins C, Nantel A, Francia G, Morgan SS, Cranmer LD, et al.. Roles for endothelin receptor B and BCL2A1 in spontaneous CNS metastasis of melanoma. Cancer Res 2012; 72:4909-19; PMID:22865454; http://dx.doi.org/ 10.1158/0008-5472.CAN-12-2194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rosano L, Spinella F, Genovesi G, Di Castro V, Natali PG, Bagnato A. Endothelin-B receptor blockade inhibits molecular effectors of melanoma cell progression. J Cardiovasc Pharmacol 2004; 44(Suppl 1):S136-9; PMID:15838263; http://dx.doi.org/ 10.1097/01.fjc.0000166247.35992.dd [DOI] [PubMed] [Google Scholar]
- 35.Spinella F, Caprara V, Cianfrocca R, Rosano L, Di Castro V, Garrafa E, Natali PG, Bagnato A. The interplay between hypoxia, endothelial and melanoma cells regulates vascularization and cell motility through endothelin-1 and vascular endothelial growth factor. Carcinogenesis 2014; 35:840-8; PMID:24473118; http://dx.doi.org/ 10.1093/carcin/bgu018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Berger Y, Bernasconi CC, Juillerat-Jeanneret L. Targeting the endothelin axis in human melanoma: combination of endothelin receptor antagonism and alkylating agents. Exp Biol Med (Maywood) 2006; 231:1111-9; PMID:16741059 [PubMed] [Google Scholar]
- 37.Ruf P, Gires O, Jager M, Fellinger K, Atz J, Lindhofer H. Characterisation of the new EpCAM-specific antibody HO-3: implications for trifunctional antibody immunotherapy of cancer. Br J Cancer 2007; 97:315-21; PMID:17622246; http://dx.doi.org/ 10.1038/sj.bjc.6603881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Daumer MP, Schneider B, Giesen DM, Aziz S, Kaiser R, Kupfer B, Schneweis KE, Schneider-Mergener J, Reineke U, Matz B, et al.. Characterisation of the epitope for a herpes simplex virus glycoprotein B-specific monoclonal antibody with high protective capacity. Med Microbiol Immunol 2011; 200:85-97; PMID:20931340; http://dx.doi.org/ 10.1007/s00430-010-0174-x [DOI] [PubMed] [Google Scholar]
- 39.Mazzuca MQ, Khalil RA. Vascular endothelin receptor type B: structure, function and dysregulation in vascular disease. Biochem Pharmacol 2012; 84:147-62; PMID:22484314; http://dx.doi.org/ 10.1016/j.bcp.2012.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sakamoto A, Yanagisawa M, Sawamura T, Enoki T, Ohtani T, Sakurai T, Nakao K, Toyo-oka T, Masaki T. Distinct subdomains of human endothelin receptors determine their selectivity to endothelinA-selective antagonist and endothelinB-selective agonists. J Biol Chem 1993; 268:8547-53; PMID:8473300 [PubMed] [Google Scholar]
- 41.Lattig J, Oksche A, Beyermann M, Rosenthal W, Krause G. Structural determinants for selective recognition of peptide ligands for endothelin receptor subtypes ETA and ETB. J Pept Sci 2009; 15:479-91; PMID:19466696; http://dx.doi.org/ 10.1002/psc.1146 [DOI] [PubMed] [Google Scholar]
- 42.Fujitani Y, Oda K, Takimoto M, Inui T, Okada T, Urade Y. Autocrine receptors for endothelins in the primary culture of endothelial cells of human umbilical vein. FEBS Lett 1992; 29:79-83; PMID:1312017; http://dx.doi.org/ 10.1016/0014-5793(92)80026-D [DOI] [PubMed] [Google Scholar]
- 43.Okamoto Y, Ninomiya H, Masaki T. Posttranslational modifications of endothelin receptor type B. Trends Cardiovasc Med 1998; 8:327-9; PMID:14987546; http://dx.doi.org/ 10.1016/S1050-1738(98)00027-9 [DOI] [PubMed] [Google Scholar]
- 44.Christiansen MN, Chik J, Lee L, Anugraham M, Abrahams JL, Packer NH. Cell surface protein glycosylation in cancer. Proteomics 2014; 14:525-46; PMID:24339177; http://dx.doi.org/ 10.1002/pmic.201300387 [DOI] [PubMed] [Google Scholar]
- 45.Shraga-Levine Z, Sokolovsky M. Functional role for glycosylated subtypes of rat endothelin receptors. Biochem Biophys Res Commun 1998; 246:495-500; PMID:9610390; http://dx.doi.org/ 10.1006/bbrc.1998.8646 [DOI] [PubMed] [Google Scholar]
- 46.Grossmann S, Higashiyama S, Oksche A, Schaefer M, Tannert A. Localisation of endothelin B receptor variants to plasma membrane microdomains and its effects on downstream signaling. Mol Membr Biol 2009; 26:279-92; PMID:19757321; http://dx.doi.org/ 10.1080/09687680903191682 [DOI] [PubMed] [Google Scholar]
- 47.Grantcharova E, Furkert J, Reusch HP, Krell HW, Papsdorf G, Beyermann M, Schulein R, Rosenthal W, Oksche A. The extracellular N terminus of the endothelin B (ETB) receptor is cleaved by a metalloprotease in an agonist-dependent process. J Biol Chem 2002; 277:43933-41; PMID:12226103; http://dx.doi.org/ 10.1074/jbc.M208407200 [DOI] [PubMed] [Google Scholar]
- 48.Roos M, Soskic V, Poznanovic S, Godovac-Zimmermann J. Post-translational modifications of endothelin receptor B from bovine lungs analyzed by mass spectrometry. J Biol Chem 1998; 273:924-31; PMID:9422751; http://dx.doi.org/ 10.1074/jbc.273.2.924 [DOI] [PubMed] [Google Scholar]
- 49.Tang L, Su M, Zhang Y, Ip W, Martinka M, Huang C, Zhou Y. Endothelin-3 is produced by metastatic melanoma cells and promotes melanoma cell survival. J Cutan Med Surg 2008; 12:64-70; PMID:18346402; http://dx.doi.org/ 10.2310/7750.2008.06164 [DOI] [PubMed] [Google Scholar]
- 50.Lin N, Huang C, Tian J, Tao J, Zhang J, Yang L, Li Y, Liu Y, Chen S, Shen G, et al.. The expression of endothelin receptor B in melanoma cells A375 and Sk-mel-1 and the proliferative effects of endothelin 3 on A375 cells. J Huazhong Univ Sci Technolog Med Sci 2007; 27:611-3; PMID:18060649; http://dx.doi.org/ 10.1007/s11596-007-0535-x [DOI] [PubMed] [Google Scholar]
- 51.Paasche JD, Attramadal T, Sandberg C, Johansen HK, Attramadal H. Mechanisms of endothelin receptor subtype-specific targeting to distinct intracellular trafficking pathways. J Biol Chem 2001; 276:34041-50; PMID:11382773; http://dx.doi.org/ 10.1074/jbc.M103243200 [DOI] [PubMed] [Google Scholar]
- 52.Oksche A, Boese G, Horstmeyer A, Furkert J, Beyermann M, Bienert M, Rosenthal W. Late endosomal/lysosomal targeting and lack of recycling of the ligand-occupied endothelin B receptor. Mol Pharmacol 2000; 57:1104-13; PMID:10825380 [PubMed] [Google Scholar]
- 53.Asundi J, Lacap JA, Clark S, Nannini M, Roth L, Polakis P. MAPK pathway inhibition enhances the efficacy of an anti-endothelin B receptor drug conjugate by inducing target expression in melanoma. Mol Cancer Ther 2014; 13:1599-610; PMID:24651527; http://dx.doi.org/ 10.1158/1535-7163.MCT-13-0446 [DOI] [PubMed] [Google Scholar]
- 54.Kenakin T. A holistic view of GPCR signaling. Nat Biotechnol 2010; 28:928-9; PMID:20829829; http://dx.doi.org/ 10.1038/nbt0910-928 [DOI] [PubMed] [Google Scholar]
- 55.May LT, Leach K, Sexton PM, Christopoulos A. Allosteric modulation of G protein-coupled receptors. Annu Rev Pharmacol Toxicol 2007; 47:1-51; PMID:17009927; http://dx.doi.org/ 10.1146/annurev.pharmtox.47.120505.105159 [DOI] [PubMed] [Google Scholar]
- 56.Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov 2013; 12:205-16; PMID:23411724; http://dx.doi.org/ 10.1038/nrd3954 [DOI] [PubMed] [Google Scholar]
- 57.Yu J, Taylor L, Mierke D, Berg E, Shia M, Fishman J, Sallum C, Polgar P. Limiting angiotensin II signaling with a cell-penetrating peptide mimicking the second intracellular loop of the angiotensin II type-I receptor. Chem Biol Drug Des 2010; 76:70-6; PMID:20492449; http://dx.doi.org/ 10.1111/j.1747-0285.2010.00985.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ahn KH, Mahmoud MM, Kendall DA. Allosteric modulator ORG27569 induces CB1 cannabinoid receptor high affinity agonist binding state, receptor internalization, and Gi protein-independent ERK1/2 kinase activation. J Biol Chem 2012; 287:12070-82; PMID:22343625; http://dx.doi.org/ 10.1074/jbc.M111.316463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ramachandran R1, Mihara K, Chung H, Renaux B, Lau CS, Muruve DA, DeFea KA, Bouvier M, Hollenberg MD. Neutrophil elastase acts as a biased agonist for proteinase-activated receptor-2 (PAR2). J Biol Chem 2011; 286:24638-48; PMID:21576245; http://dx.doi.org/ 10.1074/jbc.M110.201988 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.DeWire SM1, Ahn S, Lefkowitz RJ, Shenoy SK. Beta-arrestins and cell signaling. Annu Rev Physiol 2007; 69:483-510; PMID:17305471; http://dx.doi.org/ 10.1146/annurev.physiol.69.022405.154749 [DOI] [PubMed] [Google Scholar]
- 61.Allard B, Priam F, Deshayes F, Ducancel F, Boquet D, Wijkhuisen A, Couraud JY. Electroporation-aided DNA immunization generates polyclonal antibodies against the native conformation of human endothelin B receptor. DNA Cell Biol 2011; 30:727-37; PMID:21688998; http://dx.doi.org/ 10.1089/dna.2011.1239 [DOI] [PubMed] [Google Scholar]
- 62.Laune D, Molina F, Ferrieres G, Villard S, Bes C, Rieunier F, Chardes T, Granier C. Application of the Spot method to the identification of peptides and amino acids from the antibody paratope that contribute to antigen binding. J Immunol Methods 2002; 267:53-70; PMID:12135800; http://dx.doi.org/ 10.1016/S0022-1759(02)00140-0 [DOI] [PubMed] [Google Scholar]