

ABSTRACT
We generated an anti-albumin antibody, CA645, to link its Fv domain to an antigen-binding fragment (Fab), thereby extending the serum half-life of the Fab. CA645 was demonstrated to bind human, cynomolgus, and mouse serum albumin with similar affinity (1–7 nM), and to bind human serum albumin (HSA) when it is in complex with common known ligands. Importantly for half-life extension, CA645 binds HSA with similar affinity within the physiologically relevant range of pH 5.0 – pH 7.4, and does not have a deleterious effect on the binding of HSA to neonatal Fc receptor (FcRn). A crystal structure of humanized CA645 Fab in complex with HSA was solved and showed that CA645 Fab binds to domain II of HSA. Superimposition with the crystal structure of FcRn bound to HSA confirmed that CA645 does not block HSA binding to FcRn. In mice, the serum half-life of humanized CA645 Fab is 84.2 h. This is a significant extension in comparison with < 1 h for a non-HSA binding CA645 Fab variant. The Fab-HSA structure was used to design a series of mutants with reduced affinity to investigate the correlation between the affinity for albumin and serum half-life. Reduction in the affinity for MSA by 144-fold from 2.2 nM to 316 nM had no effect on serum half-life. Strikingly, despite a reduction in affinity to 62 µM, an extension in serum half-life of 26.4 h was still obtained. CA645 Fab and the CA645 Fab-HSA complex have been deposited in the Protein Data Bank (PDB) with accession codes, 5FUZ and 5FUO, respectively.
KEYWORDS: Anti-albumin, Fab fragment, FcRn, human serum albumin, serum half-life
Introduction
Increasing the serum half-life of biological drugs by targeting human serum albumin (HSA) is now well established.1,2 HSA is utilized because it is the most abundant protein in blood serum (34–54 g/L), it is widely distributed in tissues and has a non-acute function, and its half-life is 19 d.3 Therefore, HSA is a target that is readily available and safe for binding, as such a small percentage is utilized in this approach.
Of the serum proteins, only IgG has a similarly long half-life (21 days). The long serum half-lives of HSA and IgG are primarily due to protection from intracellular lysosomal degradation by the neonatal Fc receptor (FcRn).4,5 FcRn recycles HSA and IgG back to the cell surface following non-specific pinocytosis of the plasma into vesicles by endothelial cells and haematopoietic cells lining the vascular space. The pinocytotic vesicles acidify by fusion with the early endosome enabling HSA and IgG to bind to FcRn in a pH-dependent manner. Vesicles bearing membrane receptors, including FcRn, and in turn bound HSA and IgG, are recycled back to the cell surface while the remaining unbound material is channeled to the lysosome for degradation. HSA and IgG bind weakly to FcRn at neutral pH, and so are released back into the circulation when the recycled vesicles are exposed to the neutral pH of the blood.2
HSA can be exploited in one of 2 ways. One approach is to directly couple the therapeutic protein to HSA, either genetically or chemically.6,7 A second approach is to use an albumin-binding domain. Examples of binding domains used to date include fatty acids (myristic acid),8 organic molecules (Albutag),9 synthetic peptides,10,11 bacterial albumin-binding domains (AlbumodTm),12,13 single domain antibodies (NanobodyTm, AlbudAbTm)14-17 and a Fab.18
Using a fatty acid or an organic molecule to bind HSA takes advantage of the principal function of HSA, which is to act as a transporter of fatty acids, hormones, metal ions, waste products, and its additional capability to transport exogenous compounds such as ibuprofen and warfarin. Novo Nordisk has extended the serum half-life in man of an insulin analog from 4–6 min to 5–7 h by fusing it to myristic acid.8 Similarly, Trussel et al., used an organic molecule, 6-(4-(4-iodophenyl)butanamido)hexanoate, otherwise named AlbuTag, to extend the half-life of a scFv in mice from 20 min to 40 h.9
Ablynx and GlaxoSmithKline (GSK) have developed single domain antibodies to bind albumin. NanobodiesTm are VH-like domains, termed VHH, which are derived from heavy chain only camel, llama and dromedary antibodies. Ablynx have linked a NanobodyTm with a 22 nM affinity for HSA to an anti-interleukin-6 receptor (IL-6R) NanobodyTm, and achieved a serum half-life of ∼6 days in cynomolgus monkeys. This is equivalent to the serum half-life of cynomolgus serum albumin (CSA). Ablynx predict that a NanobodyTm will have a half-life close to that of HSA in humans.15 GSK has utilized phage libraries to isolate albumin-binding single VH and VL domain antibodies, named AlbudAbTm. The AlbudAbTm technology has recently been studied in humans. A clinical trial was conducted with a 39-residue peptide, extendin-4, fused to the N-terminus of a VL AlbudAbTm. The serum half-lives of the peptide and the AlbudAbTm portions were 6 and 14 days, respectively, indicating that the peptide was vulnerable to degradation.17
In contrast to the long serum half-life of IgG, an antibody Fab fragment has a half-life of only 12–20 h when administered to humans.18 We hypothesized that binding HSA using an Fv domain, or VH-VL pair, from an antibody raised against HSA to extend the serum half-life of a Fab would be an attractive alternative to the methods documented to date. As a proof of concept, we previously demonstrated that an anti-rat serum albumin (RSA)/anti-tumor necrosis factor (TNF) bispecific F(ab′)2 had a serum half-life of 42.5 h in rats.19 This is close to the 48 h half-life of endogenous RSA. An Fv domain should be less vulnerable to proteolysis and cleavage than fatty acid, organic molecule and peptide albumin-binding domains. Moreover, an Fv domain more closely resembles an endogenous domain of IgG than single VH, VL or VHH domains, and so may be more likely to be tolerated by the immune system. Potential immunogenicity can be further mitigated by humanization of the framework sequences.
Here, we describe the generation and characterization of a humanized anti-HSA antibody. We have identified its binding site on HSA by solving a crystal structure of the Fab-HSA complex. We also investigated the association between affinity and serum half-life by making a panel of reduced affinity mutants and assessing their half-lives in mice.
Results
Generation and characterization of a mAb to serum albumin across species
To generate a panel of anti-HSA antibodies with cross-species reactivity, 2 rabbits were immunized with HSA. B cells were harvested from the sera, cultured, stimulated to secrete IgG and screened using fluorescent microvolume assay technology (FMAT) to identify antigen-specific wells.20-22 Further FMAT screens assessed binding to RSA and binding to HSA in the presence or absence of known albumin-binding compounds warfarin, ibuprofen, myristic acid, and copper chloride. Data for the 5 top-ranked antibodies is shown in Table 1. The fluorescence intensity signal for binding to HSA was lowest for CA645. However, CA645 did retain 80% of binding activity in the presence of the compounds, whereas CA646, CA647, CA648 and CA649 retained only 40%. The levels of binding to HSA and RSA were most closely matched for CA645 and CA646, being within 5-fold. In contrast, the levels of binding to RSA by CA647, CA648 and CA649 were 9-to 18-fold lower than for HSA.
Table 1.
Activity profiles of anti-human serum albumin (HSA) antibodies. Fluorescent microvolume assay technology (FMAT) screening of secreted anti-HSA antibodies in B cell supernatants for binding to 100ng/ml HSA in the presence or absence of 25 µM albumin binding compounds (warfarin, ibuprofen, myristic acid, and copper chloride) and for binding to 100ng/ml rat serum albumin (RSA). FL = fluorescence intensity. Equilibrium binding constants (KD) of anti-HSA rabbit Fab fragments for human and mouse serum albumin (MSA), and of equivalent humanized IgG antibodies for HSA, MSA and RSA determined by surface plasmon resonance (SPR).
| FMAT |
SPR |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| B cell sup |
Rabbit Fab |
Humanized |
|||||||
| mAb CA# | HSA (FL) | HSA + compounds (FL) | RSA (FL) | HSA KD x10−9 (M) | MSA KD x10−9 (M) | HSA KD x10−9 (M) | MSA KD x10−9 (M) | IgGRSA KD x10−9 (M) | Yield (mg/ml) |
| 645 | 272 | 220 | 114 | 0.31 | 2.6 | 0.82 | 2.9 | 7.9 | 161 |
| 646 | 2310 | 964 | 484 | 0.14 | 1.6 | 0.57 | 1.7 | 4.5 | 35 |
| 647 | 1213 | 520 | 69 | 0.60 | 36.0 | 1.30 | 26 | 10 | 23 |
| 648 | 1048 | 465 | 72 | 0.33 | 12.0 | 0.13 | 23 | 54 | 312 |
| 649 | 1338 | 534 | 142 | 0.54 | 13.0 | 0.32 | 17 | 44 | 188 |
To recover the heavy and light chain variable regions of the 5 antibodies, fluorescent foci method was used to isolate single B cells and then RT-PCR was performed. The variable regions were cloned into expression vectors containing rabbit heavy CH1 and light chain constant regions, and then the DNA was sequenced. This revealed that the antibody sequences were unique with the exception of CA645 and CA646, which had identical heavy chain sequences. Given CA645 and CA646 must bind to the same epitope through the heavy chain, it is unclear why CA646 binding was more affected by the presence of ligands. The sequencing also revealed that the complementarity-determining regions (CDRs) of all of the antibodies lacked histidine residues. This was important for further progression of these antibodies because histidine residues protonate at acidic pH, and this can potentially disrupt antigen binding.
Following transfection of HEK293 cells, the recombinant Fab molecules were analyzed by surface plasmon resonance (SPR) for affinity for HSA and mouse serum albumin (MSA). CA645 and CA646 both exhibited the strongest affinities for HSA, at 0.31 nM and 0.14 nM, respectively, and for MSA at 2.6 nM and 1.6 nM, respectively (Table 1). Furthermore, their affinities for HSA and MSA were the most closely matched of all of the antibodies. This was in line with the B cell supernatant screening data against HSA and RSA.
Humanization and selection of lead candidate
All five antibodies were humanized by grafting the CDRs onto human Vκ1 and VH3 frameworks and back-mutating framework residues in positions considered important for retention of binding activity.23 The humanization scheme for CA645 is shown in Fig. 1. Of note is that the framework 3 regions (residues 66–94) of the rabbit donor heavy chains were shorter than that of the human acceptor framework sequences. CA647 and CA649 were shorter by one residue whereas CA645 and CA646 (identical sequences) and CA648 were shorter by 2 residues. In all cases, the gap was retained in the initial conservative gL1gH1 graft.
Figure 1.
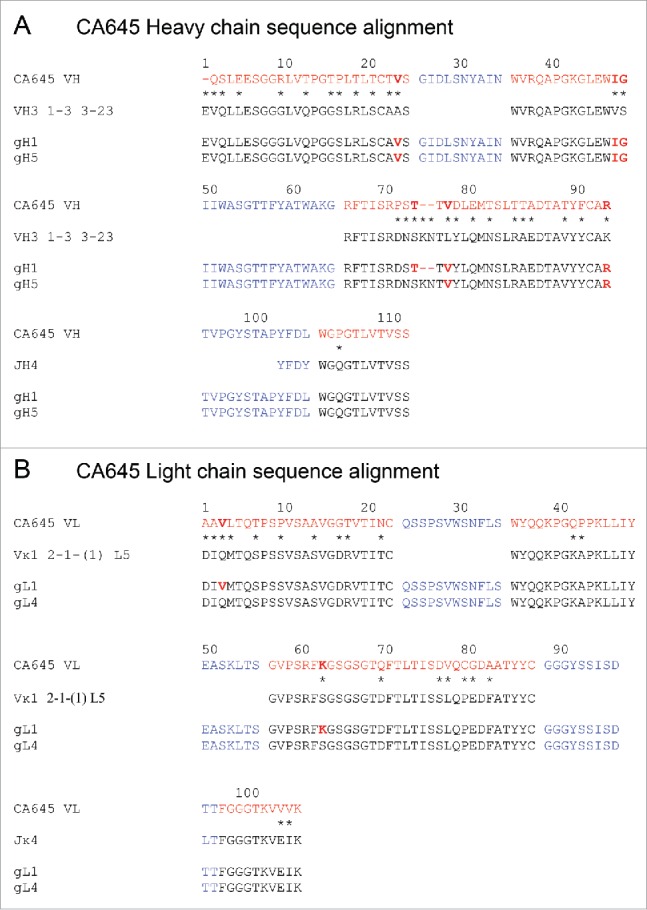
Humanization and affinity reduction of antibody CA645. The heavy (A) and light (B) chain sequences of antibody CA645 are aligned with human germline acceptor framework sequences VH3 1-3 3-23/JH4 and Vκ1 2-1-(1) L5/Jκ4. Rabbit residues are in red, human residues are in black and CDRs are in blue (J-region CDR residues are shown but acceptor V-region CDRs are not). The grafted VH (gH) and VL (gL) sequences are shown below their corresponding human acceptor germline frameworks. Framework sequence differences between the rabbit and human framework sequences are shown with asterisks. Rabbit framework residues retained in the humanized grafts are highlighted in bold.
The conservative grafts were expressed as human IgG1 antibodies and analyzed by SPR for binding to HSA, MSA and RSA. The humanized IgGs displayed the same trend in binding to HSA and MSA as observed with the recombinant parental rabbit Fabs (Table 1). The affinities for HSA of CA647, CA648 and CA649 were similar to those of CA645 and CA646, but they showed a 6- to 10-fold reduction in affinity for MSA by comparison. The affinities for RSA of CA648 and CA649 also showed a 5- to 10-fold reduction compared with CA645 and CA646. CA646 exhibited marginally stronger affinities for HSA, MSA and RSA than CA645, but the transient expression yields were 4-fold lower at 35 mg/ml compared with 161 mg/ml (Table 1). Based on the near maximal retention of binding to HSA in the presence of known albumin binders, the consistent binding activity for albumin across multiple species and good yields in transient expression, CA645 was selected as our lead candidate for further progression. Further graft variants of CA645 gL1gH1 were generated by replacing rabbit donor residues with human acceptor residues and filling the gap in framework 3 of the heavy chain with the equivalent human residues. The graft variants were assessed on affinity for HSA and transient yield expression (data not shown). The final graft pairing selected was gL4 and gH5 (Fig. 1).
The affinities of CA645 gL4gH5 Fab for HSA, MSA, RSA and rabbit serum albumin (RbSA) were shown to be 4.6, 7.1, 54 and 162 nM, respectively (Table 2). Significantly for utility of CA645 gL4gH5 Fab in cynomolgus monkey toxicology studies and disease models, the affinity for cynomolgus serum albumin (CSA) was very similar to that of HSA at 3.3 nM. In addition, CA645 Fab failed to bind to bovine serum albumin.
Table 2.
Affinity of CA645 gL4gH5 Fab for serum albumin from different species. Association (ka) and dissociation (kd) rate constants and equilibrium binding constants (KD) determined by SPR.
| Albumin species | ka x104 (1/Ms) | kd x10−4 (1/s) | KD x10−9 (M) |
|---|---|---|---|
| Human | 9.0 | 4.1 | 4.6 |
| Mouse | 4.8 | 3.4 | 7.1 |
| Rat | 2.4 | 13 | 54 |
| Cynomolgus | 10 | 3.5 | 3.3 |
| Rabbit | 0.2 | 2.9 | 162 |
| Bovine | – | – | No binding |
To determine whether CA645 gL4gH5 Fab is likely to remain bound to albumin in the acidic environment of the early endosome and be recycled to the cell surface, the affinity was measured at pH 5.0−7.0 (Table S1). The affinities of CA645 gL4gH5 Fab for HSA at pH 5.0, pH 5.5, pH 6.0, and pH 7.0 were 7.1, 10.7, 12.5 and 13.3 nM, indicating that binding is largely unaffected within this physiologically relevant pH range.
To determine whether HSA can bind to FcRn in the presence of CA645 gL4gH5 Fab, SPR was used. The kinetic assays were conducted at pH5.5 to ensure optimal binding by FcRn to albumin. HSA or MSA was bound directly onto the sensor chip, then either CA645 gL4gH5 Fab, to saturate CA645 albumin binding sites, or running buffer was injected into the flow cell. This was followed by an injection of FcRn plus CA645 Fab, or FcRn alone. CA645 Fab was included in the co-injection with FcRn to maintain CA645 binding site saturation. Fig. 2 shows the levels of FcRn binding to both HSA and MSA following subtraction of the signals for CA645. Binding by FcRn to both albumins was unaffected by the presence of CA645 gL4gH5 Fab.
Figure 2.
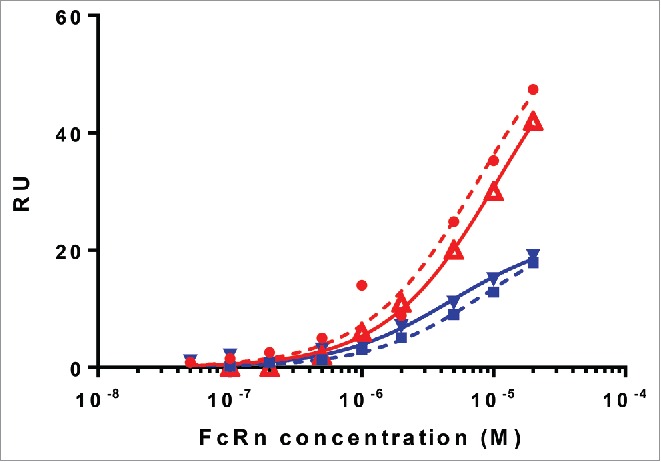
Binding of FcRn to HSA and MSA in the presence or absence of CA645 gL4gH5 Fab. Binding to HSA in absence of Fab (red circle), binding to HSA in presence of Fab (red triangle), binding to MSA in absence of Fab (blue square), binding to MSA in presence of Fab (blue triangle).
Crystallography
To identify where CA645 binds to HSA, the crystal structure of CA645 Fab-HSA complex was determined. The CA645 Fab-HSA complex protein preparation was concentrated to 70mg/ml,24 and crystallized using ethanol and PEG1000 as precipitants. To aid solving the structure of the complex with molecular replacement, we also determined the structure of unbound CA645 Fab. We observed single copies of both the Fab and Fab-HSA complex in the asymmetric units of their respective crystals (Table 3). The structure of free Fab was refined to 2.68 Å with a final Rwork value of 21.14% and Rfree value of 25.13%. The structure of the complex was refined to 3.6 Å with a final Rwork value of 21.38% and Rfree value of 25.23%.
Table 3.
X-ray data collection and refinement statistics. Values in parentheses are for highest-resolution shell.
| CA645 Fab | CA645 Fab-HSA | |
|---|---|---|
| Data collection | ||
| Space group | P 31 2 1 | P 31 2 1 |
| Cell dimensions | ||
| a, b, c (Å) | 111.21, 111.21, 89.20 | 217.68, 217.68, 78.68 |
| α, β, γ (°) | 90.00, 90.00, 120.00 | 90.00, 90.00, 120.00 |
| Resolution (Å) | 30.0–2.68 (2.82–2.68) * | 30.0–3.58 (3.79–3.58) * |
|
Rmeas CC1/2 |
0.120(0.365) 99.7(98.2) |
0.108(0.439) 99.5(78.5) |
| I / σI | 23.0(10.1) | 7.57(1.64) |
| Completeness (%) Redundancy |
99.5(99.4) 21.5(22.2) |
93.9(86.1) 2.4(1.9) |
| Refinement | ||
| Resolution (Å) | 30.00–2.68 | 30.00–3.6 |
| No. reflections | 18,142 | 24,409 |
| Rwork / Rfree | 0.2119 / 0.2509 | 0.2151 / 0.2536 |
| No. atoms | ||
| Protein | 3299 (excluding H) | 7869 (ex H) |
| Water | 37 | – |
| B-factors | ||
| Protein | 31.26 | 115.00 |
| Water | 24.24 | – |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.007 | 0.010 |
| Bond angles (°) | 1.421 | 1.442 |
The crystal structure of the CA645 Fab-HSA complex showed that CA645 binds to domain II of HSA (Fig. 3A; PDB code 5FUO). Superimposition of the crystal structure of FcRn in complex with HSA (PDB code 4N0F),25 showed that CA645 does not block binding of HSA to FcRn (Fig. 3B). HSA contains 7 fatty acid (FA) binding sites. Sites FA7 and FA3/FA4 are the 2 main drug binding sites.26 Drugs also bind at sites FA1, FA5 and FA6, but with weaker affinity. Metal ion binding sites are located between domains I and II and at a site at the N-terminus.27 Superimposition of the complex with the crystal structures of HSA in complex with warfarin (PDB code 2BXD),28 ibuprofen (PDB code 2BXG)28 and myristic acid (PDB code 1BJ5)29 showed that CA645 binds close to site FA6 and does not occlude the main drug (FA7 and FA3/FA4), fatty acid or metal ion binding sites (Fig. 3B).
Figure 3.
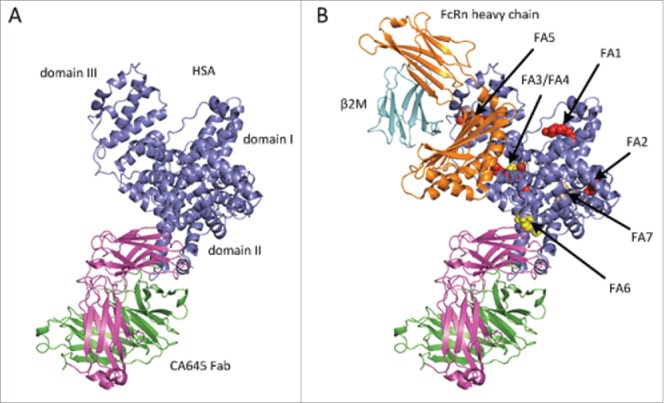
(A) Crystal structure of CA645 gL4gH5 Fab in complex with HSA (B) Superimposition of CA645 Fab-HSA with the crystal structures of HSA in complex with myristic acid, PDB code 1BJ5, shown in red, ibuprofen, PDB code 2BXG, shown in yellow, and warfarin, PDB code 2BXD, shown in wheat, and FcRn, PDB code 4N0F. FcRn is composed of heavy chain, shown in orange, and common β2-microglobulin (β2M), shown in cyan. The seven fatty acid (FA) binding sites in albumin are also labeled.
The binding kinetics of CA645 gL4gH5 Fab to HSA in comparison with those for MSA, CSA, RSA and RbSA (Table 2) may be explained by close visual inspection of the crystal structure. The epitope on HSA is formed by residues F206, G207, R209, C316, K317, AEAKD 320-324, K351, E354, E358, K359, C361, A362 and A364. The affinities of CA645 for CSA (3.3 nM) and MSA (7.1 nM) are very similar to the affinity for HSA (4.6 nM). This is likely due to the presence in CSA and MSA of the same residues that form the epitope in HSA. RSA shares all of these residues except for position 364, which is glycine. Position 364 is located at the tip of a short loop (positions 362–365) that links 2 α-helices (positions 366–398 and 342–361) together (Fig. 4A). This short loop is bound by CDRs 1 and 2 of the CA645 heavy chain. The affinity of CA645 for RSA is ∼10-fold lower than for HSA. It is possible that the absence of the alanine side chain increases the flexibility of the loop, compared with that of HSA, and alters the binding kinetics.
Figure 4.
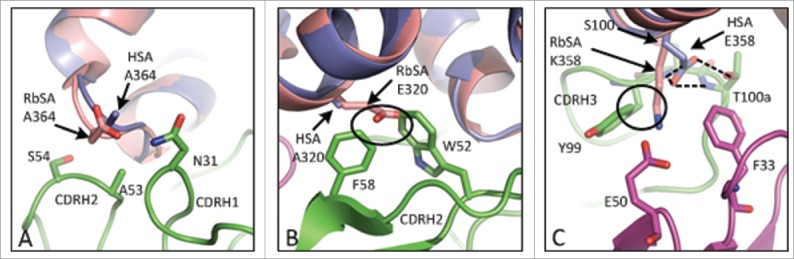
Superimposition of CA645-HSA with RbSA. Close up views of regions around albumin residues at positions (A) 364, (B) 320 and (C) 358. CA645 heavy chain shown in green; CA645 light chain shown in magenta; HSA shown in blue; RbSA shown in pink; hydrogen bonds shown as dashed lines. Clashes are defined as 2 heavy atoms from different residues being within 2 Å of each other and are denoted by a black circle.
RbSA shares all of the HSA epitope residues except positions 320, 358 and 364. Superimposition of the crystal structure of RbSA (PDB code 3V09)30 showed clear clashes with CA645 Fab at positions 320 and 358, and a potential clash at position 364. In RbSA, position 364 is aspartic acid, and, while there was no clear clash, this position is a contact residue and therefore likely to influence binding by CA645. In HSA, position 320 is an alanine that forms a hydrophobic interaction with F58 of CDRH2 (Fig. 4B). In RbSA, position 320 is glutamic acid, and it clashes with CDRH2 residues W52 and F58. Residue E358 in HSA forms a hydrogen bond network with S100 and T100a of CDRH3 (Fig. 4C). Position 358 in RbSA is lysine, and it clashes with Y99 of CDRH3. The weaker affinity of CA645 for RbSA compared with HSA is entirely due to an 18-fold reduction in the association rate (Table 2). This is likely to be caused by the presence in RbSA of the larger side chains at positions 320 and 358, and possibly 364.
Pharmacokinetics of reduced affinity variants
To investigate the correlation between the half-life of CA645 and its affinity for albumin, we generated a panel of mutants of CA645 gL4gH5 Fab with a broad range of reduced affinities and then analyzed their pharmacokinetic properties in mice. The mutations were designed using the crystal structure of the CA645 gL4gH5 Fab-HSA complex as a guide. Oligonucleotide-directed mutagenesis was used to generate 20 graft variants across 6 residue positions of the heavy chain, and 27 graft variants across 6 residue positions of the light chain (Tables 4, S2A, S2B and S2C). BALB/c mice were dosed by a single intravenous injection at 10 mg/kg with CA645 gL4gH5 Fab and a subset of 4 of the graft variants, gL5gH5, gL4gH37, gL5gH37 and gL5gH47. Blood sera were serially sampled and the level of Fab quantified by liquid chromatography – mass spectrometry (LC-MS).Variability in the concentrations of the graft variants at the first time point was observed (Fig. 5). This is in part explained by the collection of data across 3 experiments. However, the relevant PK parameter calculated, t1/2, is concentration independent and thus enables a comparison of the panel of antibodies administered.
Table 4.
Binding kinetics and pharmacokinetics of CA645 Fab graft variants. Association (ka) and dissociation (kd) rate constants and equilibrium binding constants (KD) determined by SPR. Three mice (M1-3)/group were dosed intravenously at 10 mg/kg with a single CA645 graft variant. Mean and standard deviation (SD) of each group is shown. *measured by steady-state.
| mutation |
SPR |
Half-life (h) |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| CA645 graft variants | Light chain | Heavy chain | ka x105 (1/Ms) | kd x10−4 (1/s) | KD x10−9 (M) | M1 | M2 | M3 | Mean | SD |
| gL4gH5 | – | – | 1.39 | 1.72 | 1.23 | 85 | 88 | 79 | 84 | 4.6 |
| gL5gH5 | W30A | – | 1.26 | 571 | 453 | 120 | 82 | 88 | 96.7 | 20.4 |
| gL4gH37 | – | F58E | 0.61 | 583 | 955 | 55 | 48 | 80 | 61 | 16.8 |
| gL5gH47 | W30A | T100aS | – | – | 52 µM* | 27 | 23 | 29 | 26.3 | 3.1 |
| gL5gH37 | W30A | F58E | – | – | NB | 0.54 | 0.42 | 0.47 | 0.48 | 0.06 |
Figure 5.
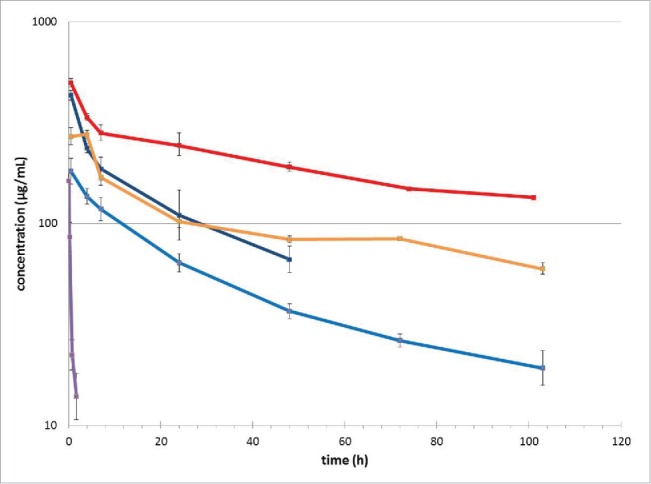
Pharmacokinetics. CA645 Fab graft variants were intravenously injected into mice at 10mg/kg and serum concentrations of the Fabs were determined at various time points by LC-MS. The lower level of quantification is 5 µg/ml. CA645 graft variants are colored as follows gL4gH5 (red), gL5gH5 (orange), gL4gH37 (cyan), gL5gH47 (dark blue) and gL5gH37 (purple). Mean concentration and standard deviation of each group (3 mice per variant) is shown.
CA645 gL5gH37 showed no detectable binding to HSA by SPR and was cleared rapidly with a serum half-life of only 0.48 ± 0.06 h (Table 4). This is in line with the short half-life (0.7 h) of an anti-TNF Fab observed in rats.19 In contrast, gL4gH5 exhibited a significantly extended half-life of 84 ± 4.6 h. The variant with the weakest affinity for which there was no difference in pharmacokinetic profile from gL4gH5 was gL5gH5 (Fig. 5). gL5gH5 contained a single mutation in the light chain, W30A, and its affinity was 453 nM. This affinity was 368-fold weaker than that of gL4gH5 (1.23 nM), but its half-life (96.7 ± 20.4 h) was equivalent to that of gL4gH5. A change in the pharmacokinetic profile was observed for gL4gH37. It has a single mutation in the heavy chain, F58E, and its affinity was 955 nM. This affinity was 776-fold lower than that of gL4gH5, but the half-life still extended to 61 ± 16.8 h. gL5gH47 contained one mutation in the light chain, W30A, and one mutation in the heavy chain, T100aS, and had an affinity of 52 µM, as measured by steady state SPR. This affinity was 42,276-fold weaker than that of gL4gH5, and yet the pharmacokinetic profile did not differ dramatically from gL4gH37 and the half-life increased to 26.3 ± 3.1 h.
The mutants were designed and selected on the basis of affinity for HSA, but the pharmacokinetic model was murine. Therefore, to confirm that the affinities of the mutants for HSA reflected their affinities for albumin in a mouse, SPR was repeated (Table 5). The affinities of gL4gH5 and gL5gH5 were 1.8 and 254 nM for HSA, and similarly 2.2 and 316 nM for MSA. These data were in line with the previously determined affinities of gL4gH5 and gL5gH5 for HSA of 1.23 and 453 nM, respectively.
Table 5.
Affinities of CA645 Fab graft variants gL4gH5 and gL5gH5 for HSA and MSA. Association (ka) and dissociation (kd) rate constants and equilibrium binding constants (KD) determined by SPR.
| CA645 graft variants | Albumin species | ka x104 (1/Ms) | kd x10−4 (1/s) | KD x10−9 (M) |
|---|---|---|---|---|
| gL4gH5 | HSA | 22 | 4.0 | 1.8 |
| gL4gH5 | MSA | 31 | 6.8 | 2.2 |
| gL5gH5 | HSA | 8.5 | 220 | 254 |
| gL5gH5 | MSA | 1.2 | 38 | 316 |
Discussion
This study describes the generation of anti-HSA antibody CA645 for the purpose of using the Fv domain as an albumin-binding domain to extend the serum half-life of a Fab fragment. Raising an antibody through immunization of rabbits combined with rational screening was a very effective means by which to isolate an Fv domain with the required binding characteristics. Inclusion of known albumin ligands in our early screens ensured we would isolate a candidate that would not occlude the ligand binding sites, and that was able to recognize albumin when a fatty acid, metal ion or drug was bound. CA645 was by far the strongest candidate as it only showed a 20% reduction in binding activity to HSA that had been pre-incubated with ligands compared with a reduction of 60% for the other lead candidates. We also screened against MSA and RSA, as well as HSA, to select a candidate with consistent cross-species reactivity. CA645, along with CA646, which had an identical heavy chain sequence, exhibited affinities that were the most similar for the 3 species of albumin. The affinity of CA645 gL4gH5 Fab for CSA was subsequently measured and shown to be within 1.4-fold of that for HSA. This is a desirable property for an albumin-binding domain because CA645 can be utilized in disease and toxicology models of rodents and cynomolgus monkeys, as well as in man therapeutically, without the need to make species-specific domains.
The long half-life of albumin is substantially due to FcRn-mediated protection from intracellular degradation. HSA binds to FcRn through domains I and III when exposed to low pH in the early endosome compartment following pinocytosis.31 To benefit from this mechanism, it was essential to choose a candidate that could bind HSA at low pH, and at a site that did not prevent HSA from binding to FcRn. CA645 met both those criteria. CA645 showed no loss in binding activity when the pH was reduced to pH 5.0 and saturation of CA645 binding sites on HSA had no effect on FcRn-HSA complex formation. Determination of the crystal structure of CA645 Fab-HSA revealed that CA645 bound in domain II with no apparent steric inhibition to FcRn-binding.
In mice, CA645 gL4gH5 Fab exhibited a half-life of 84.2 ± 4.6 h. This was a significant extension in comparison with a non-albumin binding variant that had a half-life of just 0.48 ± 0.1 h. The half-life of MSA is reportedly 24 h.11 This suggests that CA645 Fab has gained the maximal possible benefit from binding albumin to extend serum half-life. Since the affinity of CA645 for MSA is similar to that of HSA then there is good reason to believe that this significant extension in half-life would translate to man.
One striking aspect of this study has been the degree to which the affinity for albumin needed to be reduced, at least for CA645, to observe a change in the pharmacokinetic profile and the serum half-life. gL5gH5 was generated by mutating light chain residue W29 of gL4gH5 to alanine. The resulting affinity of 316 nM for MSA was 144-fold weaker than that of the parent, and yet there was no change in the pharmacokinetic profile and half-life. In contrast, mutagenesis of heavy chain residue F58 to glutamic acid to generate gL4gH37 did alter the pharmacokinetic profile. The extension in half-life was reduced to 60 ± 16.8 h and the affinity for MSA weakened to a predicted 1146 nM. Remarkably, gL5gH47, generated by combining W30A of the light chain and T100aS of the heavy chain, with a predicted affinity for MSA of 62.4 µM, still achieved an extension in half-life of 26.3 h.
Nguyen et al.,11 have also investigated the correlation between affinity and half-life. They generated variants of a short albumin-binding peptide fused to a Fab and measured the pharmacokinetics in mice, rats and rabbits. In mice, a Fab-peptide fusion with an affinity of 41 nM for MSA, as measured by solution binding ELISA, showed a serum half-life of just 14 h. This is in stark contrast to gL5gH5, which has a much weaker affinity for MSA of 316 nM and yet showed a half-life of 95 ± 20.4 h. In rats, Fab-peptide fusions with affinities for RSA of 92, 493 and 2429 nM exhibited serum half-lives of 26.9, 10.9 and 4.21 h. Here, 5- and 26-fold reductions in affinity reduced half-lives by 2- and 6-fold. The fold reductions in affinity to achieve a reduced half-life are far less than we observed for CA645. These data suggest that the type of albumin-binding domain and the kinetics of the interaction between the albumin-binding domain and albumin are more important than the overall KD value. The mutations we generated could be used to tune the half-life of a Fab or another antibody format that utilizes CA645.
In summary, we generated a humanized anti-HSA antibody, CA645, which binds to albumin from multiple species with similar affinity. CA645 binds in domain II of albumin, distal to the main fatty acid and drug binding sites, and importantly, distal to the FcRn binding site, thereby ensuring FcRn-mediated protection from lysosomal degradation. As a Fab, CA645 attained a serum half-life of 84.2 h in mice. We have subsequently linked CA645 Fv to a humanized Fab and fully characterized its biophysical and functional properties. The Fab-Fv fusion showed good stability and attained a serum half-life equivalent to that of endogenous IgG in mice and in cynomolgus monkeys,32 We predict that the Fv domain of CA645 will confer a long, but tunable, serum half-life to a therapeutic Fab fragment in man.
Materials and methods
Antibody discovery
Two Half Lop rabbits were immunized subcutaneously with 200 µg HSA (Jackson ImmunoResearch). Complete Freund's adjuvant (Sigma Aldrich) was co-administered with the first dose and subsequent doses included incomplete Freund's adjuvant. B cells were harvested from the rabbit sera and cultured for 7 d to induce clonal expansion and antibody secretion. Fluorescence microvolume assay technology (FMAT) was used to screen the supernatants for binding to HSA.20-22 The supernatants were mixed with streptavidin beads (Bangs Laboratories, Inc.) coated with biotinylated goat anti-rabbit Fc and Alexa Fluor 647 Chrompure Human Albumin (Jackson ImmunoResearch). Plates were read on an Applied Biosystems 8200 Cellular Detection System. The 48-wells with the highest fluorescence intensity (FL) signal were transferred to a single master plate and the screening repeated as before, but with 2 additional screens. In one screen, Alexa Fluor 647 Chrompure Human Albumin was pre-incubated for 1 hour with a 25 µM solution of albumin binders; warfarin, ibuprofen, myristic acid, and copper chloride (all individually sourced from Sigma Aldrich). In the second screen, HSA was replaced with rat serum albumin (Sigma Aldrich) that had been labeled using Alexa Fluor 647 ® monoclonal antibody labeling kit (Molecular Probes).
Individual HSA-specific B cells were isolated by fluorescent foci method.20-22 B cells from positive wells were mixed with streptavidin beads (Bangs Laboratories, Inc.) coated with biotinylated-HSA (Jackson ImmunoResearch) and goat anti-rabbit Fc fragment fluorescein isothiocynate conjugate (Chemicon). Following 1 hour incubation at 37°C, antigen-specific B cells could be identified due to the presence of a fluorescent halo surrounding that B cell. An Olympus IX70 microscope and an Eppendorf micromanipulator were used to identify and transfer the individual B cells to PCR tubes. The heavy and light chain immunoglobulin variable (V) region genes of single cells were amplified by RT-PCR and cloned into UCB mammalian expression vectors containing rabbit heavy CH1 and rabbit light Cκ regions, respectively. Following transient expression in HEK293 cells, anti-HSA recombinant Fabs were further screened in SPR binding assays against HSA and MSA.
Humanization
Albumin specific antibodies were humanized in silico by grafting the CDRs from antibody V-regions onto the Vκ1 and VH3 human germline antibody V-region frameworks. The CDRs grafted from the donor to the acceptor sequence were as defined by Kabat et al.,33 with the exception of CDR-H1 (residues 26–35) where the combined definitions of Kabat et al., and loop structure was used.23 Where a framework residue differed between the donor rabbit sequence and the acceptor human sequence in a position that was considered to be important for retention of antigen binding, then the donor residue was included in the initial conservative graft.21 The conservative graft genes were chemically synthesized by Entelechon, GmbH. Heavy chain graft genes (gH1) were cloned into 2 UCB expression vectors, one containing human γ1 CH1 domain and another containing the full human γ1 constant region. Light chain graft genes (gL1) were cloned into a UCB expression containing human kappa constant region (Km3 allotype). These constructs were subsequently modified by oligonucleotide-directed mutagenesis to create a number of different variants of both the heavy and light chain grafts. Heavy and light chain vectors were co-transfected into HEK293 cells and the recombinant Fab or IgG molecules screened using a SPR binding assay to measure affinity for HSA, MSA, RSA, CSA, RbSA and bovine serum albumin.
Antibody expression
Antibodies were transiently expressed in either HEK-293 cells using 293Fectin lipid transfection (Life Technologies, catalog #12347-019, according to the manufacturer's instructions) or CHO-S XE cells, a CHO-K1 derived cell line,34 using electroporation. HEK-293 cells were used for small scale expression (< 100 ml) to prepare antibodies for SPR analysis. CHO-S XE cells were used for large scale expression (1 L) to prepare antibodies for crystallography and in vivo pharmacokinetic studies.
Protein purification
Affinity chromatography was used to purify Fab protein from culture supernatants. Supernatants were passed over a HiTrap Protein G column (GE Healthcare) at a flow rate that gave a column contact time of 25 min. Following a washing step with PBS pH 7.4, the bound material was eluted with 0.1 M glycine pH 2.7 and neutralized with 2 m Tris-HCl (pH 8.5). Fractions containing Fab were pooled, quantified by absorbance at 280 nm, and concentrated using Amicon Ultra centrifugal filters (Merck Millipore). To isolate the monomeric fraction, size exclusion chromatography (SEC) over a HiLoad 16/60, Superdex 200 column (GE Healthcare) equilibrated with PBS, pH 7.4, was used. Fractions containing monomeric Fab were pooled, quantified, concentrated and stored at 4°C.
Surface plasmon resonance
The binding affinities and kinetic parameters for the interactions of antibodies were determined by SPR conducted on either a Biacore T200 or Biacore 3000 using CM5 sensor chips (GE Healthcare Bio-Sciences AB) and HBS-EP (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.05% v/v P20, pH7.4) running buffer. For analysis at pH 7.0, 6.0, 5.5 and 5.0, a running buffer of 40 mM citric acid, 80 mM sodium phosphate 50 mM NaCl, 3mM EDTA, 0.05% v/v P20 was used. The required pH was achieved by altering the ratio of citric acid to sodium phosphate. All experiments were performed at 25 °C. The antibody samples were captured to the sensor chip surface using either a human F(ab′)2-specific or human Fc-specific goat Fab (Jackson ImmunoResearch). Covalent immobilization of the capture antibody was achieved by standard amine coupling chemistry to a level of 6000–7000 response units (RU).
Human (Jackson ImmunoResearch, catalog #009-000-051), mouse (Sigma Aldrich, catalog #A3559), rat (Sigma Aldrich, catalog #A6414), rabbit (Sigma Aldrich, catalog #A0764), bovine (Sigma Aldrich, catalog #05470) and cynomolgus (Equitech-Bio, #CMSA-0050) albumin were titrated over the captured antibody at various concentrations from 50 nM to 500 µM. Each assay cycle consisted of firstly capturing the antibody sample using a 1 min injection, before an association phase consisting of a 3 min injection of albumin, after which dissociation was monitored. After each cycle, the capture surface was regenerated with 2 1 min injections of 40 mM HCl followed by 30 s of 5 mM NaOH. The flow rates used were 10 µl/min for capture, 30 µl/min for both the association and dissociation phases, and 10 µl/min for regeneration. A blank flow-cell was used for reference subtraction and buffer-blank injections were included to subtract instrument noise and drift. Kinetic parameters were determined by simultaneous global-fitting of the resulting sensorgrams to a standard 1:1 binding model using Biacore T200 Evaluation software v2.0.1 and BIAEvaluation software v4.1.1, with the exception of CA645 gL5gH47 which was fitted in prism using steady-state affinity model.
To measure the effect of CA645 Fab on the binding potency of FcRn to albumin by SPR, a Biacore3000 instrument was used with a CM5 chip prepared by immobilization of HSA and MSA on separate flow cells to levels of 270 RU and 247 RU respectively. FcRn samples were prepared over the range 50 nM to 50 µM in running buffer, (100 mM MES, 150 mM NaCl, 0.05% v/v P20, pH 5.5) and they also contained either zero or 100 nM CA645 Fab. Each assay cycle was run at a flow rate of 10 µl / min and consisted of either a 5 min injection of 100 nM CA645 Fab to pre-saturate immobilized albumin, followed by a 5 min injection of one of the above FcRn solutions prepared in the presence of CA645 Fab, or a 5 min injection of running buffer followed by a 5 min injection of one of the above FcRn solutions in the absence of CA645 Fab. In either case, a third 5 min injection followed immediately at the end of the second injection, using the ‘coinject’ mode, comprising, respectively, buffer or 100 nM CA645 Fab. A blank flow-cell was used for reference subtraction and blank cycles, where FcRn was replaced with buffer, were included to subtract drift and noise. Cycle regeneration was as above. Blank corrected plateau binding levels of FcRn were plotted in Prism and fitted to a steady state model.
Binding kinetics of wild type and mutant CA645 Fabs at pH 5.5 were also investigated in reverse format on the Biacore3000 using the immobilized albumin chip. In this case, cycles were run where Fab solutions over the range 5 to 5000 nM were injected with 5 min association and dissociation phases. Buffer blank cycles were also included to correct for drift.
Crystallography
To prepare the complex, purified CA645 Fab and fatty acid-free HSA (Sigma Aldrich, catalog #A3782) were mixed in a molar ratio of 1:1 and incubated overnight at 4 °C. Both CA645 Fab and the complex were purified by SEC over a HiLoad 16/60, Superdex 200 column (GE Healthcare) equilibrated with 50 mM NaCl, 25 mM Tris, 5% (v/v) glycerol. Fractions containing either CA645 Fab or the complex were pooled and concentrated to 10 mg/ml and 70 mg/ml, respectively. Conditions suitable for crystal growth were identified by the sitting drop vapor diffusion method using commercially available crystallization screens (Qiagen).
To generate diffraction quality crystals, hanging drop vapor diffusion method was used where 1 µl of protein solution was mixed with 1 µl of reservoir solution. For CA645 Fab, the reservoir contained 500 µl 2 M DL-Malic acid. Crystals were harvested and flash frozen in liquid nitrogen without additional cryoprotectant. Diffraction data to 2.68 Å was collected from a single crystal on the I04 beamline at Diamond Light Source, Oxford, UK and processed using MOSFLM and SCALA.35-37 The structure of CA645 Fab was solved by molecular replacement with Phaser,38 using coordinates of an in-house Fab structure as a search model. For the complex, the reservoir contained 500 µl 0.1 M citric acid pH 4.4, 0.1 M di-sodium hydrogen phosphate, 38% v/v ethanol and 5% v/v polyethylene glycol 1000 (PEG1000). The crystals were cryoprotected by multiple additions to the drop of 1 µl reservoir buffer containing 25% (v/v) PEG1000, until the concentration of PEG1000 in the drop reached 20%. To minimize crystal stress, each addition was spaced at least 1 hour apart. Crystals were harvested and flash frozen in liquid nitrogen. Diffraction data to 3.58 Å was collected from a single crystal on the I02 beamline at Diamond Light Source, Oxford, UK and processed using XDS.39 The structure of the complex was solved by molecular replacement with Phaser using coordinates of CA645 Fab structure and HSA (PDB code 4G03)40 as search models.
Both initial structures were refined with iterative cycles of simulated annealing, energy minimization and manual rebuilding using CNS41,42 and COOT.43 Due to the rather low resolution of the complex, the model was constrained during refinement by using the DEN function of CNS. Model geometry was validated using Molprobity.44 Molecular visualizations were generated with Pymol.45 Data collection and refinement statistics are summarized in Table 3.
Accession codes
Coordinates and structure factors of CA645 Fab and the CA645 Fab-HSA complex have been deposited in the Protein Data Bank (PDB) with accession codes, 5FUZ and 5FUO, respectively.
Mouse pharmacokinetics
In three separate experiments, BALB/c mice weighing 25-30 g were dosed intravenously with a single CA645 graft variant (gL4gH5, gL4H37, gL5gH5, gL5gH37 or gL5gH47 in PBS, pH 7.4), at 10 mg/kg bodyweight (3 mice/variant). Experiment 1 - gL5gH47 and gL4gH5; experiment 2 - gL4H37, gL5gH5 and gL5gH37; and experiment 3 - gL5gH37. The latter was a repeat that included earlier time points to take into account the very rapid clearance of gL5gH37 that had been observed in experiment 2. Serial blood samples (35 µL) were collected via lateral tail sampling at 0.5, 4, 7, 24, 48, 74 and 101 h (gL4gH5), 0.5, 4, 7, 24, 48, 72 and 103 h (gL4gH37, gL5gH47, gL5gH5) and at 0.033, 0.25, 0.75 and 1.66 h (gL5gH37). To obtain sera, blood samples were centrifuged for 5 min at 10,000 rpm at room temperature. Samples were analyzed for the antibody concentration by LC-MS/MS using an AB SCIEX QTRAP® 5500 system in combination with an Agilent 1290 Infinity Binary UHPLC system. Separation was achieved using an Onyx monolithic C18 column (100×4.6 mm, Phenomenex UK). The methodology is based on quantifying unique “proteotypic” peptides post tryptic digestion by mass spectrometric detection following reverse-phase chromatographic separation.46 Thus, quantitation of total drug is achieved. Ten µl of serum were denatured using acetonitrile, reduced using tris(2-carboxyethyl)phosphine (Sigma Aldrich catalog #C4706), free cysteines were alkylated using iodoacetamide (Sigma Aldrich catalog #I1149) and overnight digestion was performed in ammonium bicarbonate buffer pH 8 using trypsin (Promega catalog #V5111). Samples were analyzed without further sample preparation. Quantitation was achieved by comparison to authentic standard material spiked at known concentrations into control matrix. The limit of quantitation achieved was 5 µg/mL. Pharmacokinetic parameters were calculated from the final dataset using Phoenix WinNonlin 6.2 software (Pharsight).
Supplementary Material
Disclosure of potential conflicts of interest
Ralph Adams, Terry Baker, Tom Ceska, Alastair Lawson, David Humphreys and Sam Heywood hold stock in UCB.
Acknowledgments
The authors would like to thank Simon Carter and Alex Ferecsko for performing the in-life part of the pharmacokinetic studies.
References
- 1.Kontermann RE. Strategies for extended serum half-life of protein therapeutics. Curr Opin Biotechnol 2011; 22:868-76; PMID:21862310; http://dx.doi.org/ 10.1016/j.copbio.2011.06.012 [DOI] [PubMed] [Google Scholar]
- 2.Sleep D, Cameron J, Evans LR. Albumin as a versatile platform for drug half-life extension. Biochim Biophys Acta 2013; 1830:5526-34; PMID:23639804; http://dx.doi.org/ 10.1016/j.bbagen.2013.04.023 [DOI] [PubMed] [Google Scholar]
- 3.Peters TJr. All About Albumin: Biochemistry, Genetics, and Medical Applications 1996; San Diego, CA: Academic Press. [Google Scholar]
- 4.Chaudhury C, Mehnaz S, Robinson JM, Hayton WL, Pearl DK, Roopenian DC, Anderson CL. The major histocompatibility complex-related Fc receptor for IgG (FcRn) binds albumin and prolongs its lifespan. J Exp Med 2003; 197:315-22; PMID:12566415; http://dx.doi.org/ 10.1084/jem.20021829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Junghans RP, Anderson CL. The protection receptor for IgG catabolism is the beta2-microglobulin-containing neonatal intestinal receptor. Proc Natl Acad Sci U S A 1996; 93:5512-6; PMID:8643606; http://dx.doi.org/ 10.1073/pnas.93.11.5512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Müller D, Karle A, Meissburger B, Höfig I, Stork R, Kontermann RE. Improved pharmacokinetics of recombinant bispecific antibody molecules by fusion to human serum albumin. J Biol Chem 2007; 282:12650-60; PMID:17347147; http://dx.doi.org/ 10.1074/jbc.M700820200 [DOI] [PubMed] [Google Scholar]
- 7.Elsadek B, Kratz F. Impact of albumin on drug delivery – New applications on the horizon. J Control Release 2012; 157:4-28; PMID:21959118; http://dx.doi.org/ 10.1016/j.jconrel.2011.09.069 [DOI] [PubMed] [Google Scholar]
- 8.Sørensen AR, Stidsen CE, Ribel U, Nishimura E, Sturis J, Jonassen I, Bouman SD, Kurtzhals P, Brand CL. Insulin detemir is a fully efficacious, low affinity agonist at the insulin receptor. Diabetes Obes Metab 2010; 12:655-73; PMID:20590743; http://dx.doi.org/ 10.1111/j.1463-1326.2010.01206.x [DOI] [PubMed] [Google Scholar]
- 9.Trüssel S, Dumelin C, Frey K, Villa A, Buller F, Neri D. New strategy for the extension of the serum half-life of antibody fragments. Bioconjug Chem 2009; 20:2286-92; PMID:19916518; http://dx.doi.org/ 10.1021/bc9002772 [DOI] [PubMed] [Google Scholar]
- 10.Dennis MS, Zhang M, Meng Y G, Kadkhodayan M, Kirchhofer D, Combs D, Damico LA. Albumin binding as a general strategy for improving the pharmacokinetics of proteins. J Biol Chem 2002; 277:35035-43; PMID:12119302; http://dx.doi.org/ 10.1074/jbc.M205854200 [DOI] [PubMed] [Google Scholar]
- 11.Nguyen A, Reyes AE 2nd, Zhang M, McDonald P, Wong WL, Damico LA, Dennis MS. The pharmacokinetics of an albumin-binding Fab (AB.Fab) can be modulated as a function of affinity for albumin. Protein Eng Des Sel 2006; 9:291-7; PMID:16621915; http://dx.doi.org/ 10.1093/protein/gzl011 [DOI] [PubMed] [Google Scholar]
- 12.Hopp J, Hornig N, Zettlitz KA, Schwarz A, Fuss N, Müller D, Kontermann RE. The effects of affinity and valency of an albumin-binding domain (ABD) on the half-life of a single-chain diabody-ABD fusion protein. Protein Eng Des Sel 2010; 23:827-34; PMID:20817756; http://dx.doi.org/ 10.1093/protein/gzq058 [DOI] [PubMed] [Google Scholar]
- 13.Andersen JT, Pehrson R, Tolmachev V, Daba MB, Abrahmsén L, Ekblad C. Extending half-life by indirect targeting of the neonatal Fc receptor (FcRn) using a minimal albumin binding domain. J Biol Chem 2011; 286:5234-41; PMID:21138843; http://dx.doi.org/ 10.1074/jbc.M110.164848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tijink BM, Laeremans T, Budde M, Stigter-van Walsum M, Dreier T, de Haard HJ, Leemans CR, van Dongen GA. Improved tumor targeting of anti-epidermal growth factor receptor Nanobodies through albumin binding: taking advantage of modular Nanobody technology. Mol Cancer Ther 2008; 8:2288-97; PMID:18723476; http://dx.doi.org/ 10.1158/1535-7163.MCT-07-2384 [DOI] [PubMed] [Google Scholar]
- 15.Van Roy M,Ververken C, Beirnaert E, Hoefman S, Kolkman J, Vierboom M, Breedveld E, Hart B, Poelmans S, Bontinck L, Hemeryck A, et al.. The preclinical pharmacology of the high affinity anti-IL-6R Nanobody® ALX-0061 supports its clinical development in rheumatoid arthritis. Arthritis Res Ther 2015; 17:135; PMID:25994180; http://dx.doi.org/ 10.1186/s13075-015-0651-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Holt LJ, Basran A, Jones K, Chorlton J, Jespers LS, Brewis ND, Tomlinson IM. Anti-serum albumin domain antibodies for extending the half-lives of short lived drugs. Protein Eng Des Sel 2008; 21:283-8; PMID:18387938; http://dx.doi.org/ 10.1093/protein/gzm067 [DOI] [PubMed] [Google Scholar]
- 17.O'Connor-Semmes RL, Lin J, Hodge RJ, Andrews S, Chism J, Choudhury A, Nunez DJ. GSK2374697, a novel albumin-binding domain antibody (AlbudAb), extends systemic exposure of exendin-4: first study in humans-PK/PD and safety. Clin Pharmacol Ther 2014; 96:704-12; PMID:25238251; http://dx.doi.org/ 10.1038/clpt.2014.187 [DOI] [PubMed] [Google Scholar]
- 18.Flanagan RJ, Jones AL. Fab antibody fragments. Drug Safety 2004; 27:1115-33; PMID:15554746; http://dx.doi.org/ 10.2165/00002018-200427140-00004 [DOI] [PubMed] [Google Scholar]
- 19.Smith BJ, Popplewell A, Athwal D, Chapman AP, Heywood S, West SM, Carrington B, Nesbitt A, Lawson AD, Antoniw P, et al.. Prolonged in vivo residence times of antibody fragments associated with albumin. Bioconjug Chem 2001; 12:750-6; PMID:11562193; http://dx.doi.org/ 10.1021/bc010003g [DOI] [PubMed] [Google Scholar]
- 20.Lightwood DJ, Carrington B, Henry AJ, McKnight AJ, Crook K, Cromie K, Lawson AD. Antibody generation through B cell panning on antigen followed by in situ culture and direct RT-PCR on cells harvested en masse from antigen-positive wells. J Immunol Methods 2006; 316:133-43; PMID:17027850; http://dx.doi.org/ 10.1016/j.jim.2006.08.010 [DOI] [PubMed] [Google Scholar]
- 21.Tickle S, Adams R, Brown D, Griffiths M, Lightwood DJ, Lawson AD. High-throughput screening for high affinity antibodies. J Lab Auto 2009; 14(5):303-07; http://dx.doi.org/ 10.1016/j.jala.2009.05.004 [DOI] [Google Scholar]
- 22.Clargo AM, Hudson AR, Ndlovu W, Wootton RJ, Cremin LA, O'Dowd VL, Nowosad CR, Starkie DO, Shaw SP, Compson JE, et al.. The rapid generation of recombinant functional monoclonal antibodies from individual, antigen-specific bone marrow-derived plasma cells isolated using a novel fluorescence-based method. MAbs 2014; 6:143-59; PMID:24423622; http://dx.doi.org/ 10.4161/mabs.27044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Adair JR, Athwal DS, Emtage JS. Humanised Antibodies International Patent Publication. 1991; WO91/09967. [Google Scholar]
- 24.Curry S. Lessons from the crystallographic analysis of molecule binding to Human Serum Albumin. Drug Metab Pharmacokinet 2009; 24(4):342-57; PMID:19745561; http://dx.doi.org/ 10.2133/dmpk.24.342 [DOI] [PubMed] [Google Scholar]
- 25.Oganesyan V, Damschroder MM, Cook KE, Li Q, Gao C, Wu H, Dall'acqua WF. Structural insights into neonatal Fc receptor-based recycling mechanisms. J Biol Chem 2014; 289:7812-24; PMID:24469444; http://dx.doi.org/ 10.1074/jbc.M113.537563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ascenzi P, Fasano M. Allostery in a monomeric protein: The case of human serum albumin. Biophys Chem 2010; 148:16-22; PMID:20346571; http://dx.doi.org/ 10.1016/j.bpc.2010.03.001 [DOI] [PubMed] [Google Scholar]
- 27.Bal W, Sokolowska M, Kurowska E, Faller P. Binding of transition metal ions to albumin: Sites, affinities and rates. Biochim Et Biophys Act 2013; 1830(12):5444-45; http://dx.doi.org/ 10.1016/j.bbagen.2013.06.018 [DOI] [PubMed] [Google Scholar]
- 28.Ghuman J, Zunszain PA, Petitpas I, Bhattacharya AA, Otagiri M, Curry S. Structural basis of the drug-binding specificity of human serum albumin. J Mol Biol 2005; 353:38-52; PMID:16169013; http://dx.doi.org/ 10.1016/j.jmb.2005.07.075 [DOI] [PubMed] [Google Scholar]
- 29.Curry S, Mandelkow H, Brick P, Franks N. Crystal structure of human serum albumin complexed with fatty acid reveals an asymmetric distribution of binding sites. Nat Struct Biol 1998; 5:827-35; PMID:9731778; http://dx.doi.org/ 10.1038/1869 [DOI] [PubMed] [Google Scholar]
- 30.Majorek KA, Porebski PJ, Dayal A, Zimmerman MD, Jablonska K, Stewart AJ, Chruszcz M, Minor W. Structural and immunologic characterization of bovine, horse, and rabbit serum albumins. Mol Immunol 2012; 52:174-82; PMID:22677715; http://dx.doi.org/ 10.1016/j.molimm.2012.05.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sand KMK, Bern M, Nilsen J, Dalhus B, Gunnarsen KS, Cameron J, Grevys A, Bunting K, Sandlie I, Andersen JT. Interaction with both domain I and III of albumin is required for optimal pH-dependent binding to the neonatal Fc receptor (FcRn). J Biol Chem 2014; 289:34583-94; PMID:25344603; http://dx.doi.org/ 10.1074/jbc.M114.587675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Davé E, Adams R, Griffin L, Zaccheo O, Compson J, Dugdale S, Airey M, Malcolm S, Hailu H, Heywood S, et al.. Fab-dsFv: a bispecific antibody format with extended serum half-life through albumin binding. Manuscript submitted [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kabat EA, Wu TT, Perry HM, Gottesman KS, Foeller C. Sequences of proteins of immunological interest, 1991; 5th edit Public Health Service, National Institutes of Health, Bethesda, MD. [Google Scholar]
- 34.Cain K, Peters S, Hailu H, Sweeney B, Stephens P, Heads J, Sarkar K, Ventom A, Page C, Dickson A. A CHO cell line engineered to express XBP1 and ERO1-Lα has increased levels of transient protein expression. Biotechnol Prog 2013; 29:697-706; PMID:23335490; http://dx.doi.org/ 10.1002/btpr.1693 [DOI] [PubMed] [Google Scholar]
- 35.Leslie AGW. The integration of macromolecular diffraction data. Acta Crysta 2006; D62:48-57; PMID:16369093; http://dx.doi.org/19461840 10.1107/S0907444905039107 [DOI] [PubMed] [Google Scholar]
- 36.Leslie AGW, Powell HR. Evolving Methods for Macromolecular Crystallography 2007; 245:41-51; ISBN 978-1-4020-6314-5; http://dx.doi.org/ 10.1007/978-1-4020-6316-9 [DOI] [Google Scholar]
- 37.Evans P. Scaling and assessment of data. Acta Cryst 2006; D62:72-82; PMID: 16369096; http://dx.doi.org/19461840 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 38.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Cryst 2007; 40:658-74; PMID:19461840; http://dx.doi.org/ 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabsch W. XDS. Acta Cryst 2010; D66:125-32; PMID: 20124692; http://dx.doi.org/9757107 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cao H.L, Yin DC. High-resolution Crystal Structural Variance Analysis between Recombinant and Wild-type Human Serum Albumin. http://dx.doi.org/ 10.2210/pdb4g03/pdb [DOI] [Google Scholar]
- 41.Brünger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al.. Crystallography & NMR system: A new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr 1998; 54(Pt 5):905-21; PMID:9757107; http://dx.doi.org/ 10.1107/S0907444998003254 [DOI] [PubMed] [Google Scholar]
- 42.Brünger AT. Version 1.2 of the Crystallography and NMR system. Nat Protoc 2007; 2:2728-33; PMID:18007608; http://dx.doi.org/ 10.1038/nprot.2007.406 [DOI] [PubMed] [Google Scholar]
- 43.Emsley P, Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr D Biol Crystallogr 2004; D60:2126-32; PMID:15572765; http://dx.doi.org/ 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 44.Chen VB, Arendall III WB, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography; Acta Crystallogr D Biol Crystallogr 2010; D66: 12-21; PMID: 20057044; http://dx.doi.org/18465883 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.DeLano WL. The PyMOL Molecular Graphics System 2002; DeLano Scientific LLC, San Carlos, CA. [Google Scholar]
- 46.Heudi O, Barteau S, Zimmer D, Schmidt J, Bill K, Lehmann N, Bauer C, Kretz O. Towards absolute quantification of therapeutic monoclonal antibody in serum by LC−MS/MS using isotope-labeled antibody standard and protein cleavage isotope dilution mass spectrometry. Anal. Chem 2008; 80:4200-7; PMID:18465883; http://dx.doi.org/ 10.1021/AC800205S [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.