

Abstract
The molecular basis of gastrointestinal intolerances in a severe case of Niemann-Pick type C disease was analyzed in an intestinal biopsy specimen. The enzyme activities of intestinal sucrase-isomaltase and maltase-glucoamylase are reduced in the patient, while that of lactase is comparable to the control. The association of SI with lipid rafts is reduced in the patient’s biopsy as a consequence of altered composition of membrane microdomains. As association with lipid rafts influences the intracellular transport and the enzyme activities of sucrase-isomaltase and maltase-glucoamylase, these data explain reduced carbohydrate digestion in the intestinal lumen and delineate the effect of deficient cholesterol and sphingolipid homeostasis in development of gastrointestinal symptoms in NPC patients.
Introduction
Niemann-Pick type C (NPC; OMIM#257220) is a lysosomal storage disease (LSD) that is characterized by intracellular accumulation of unesterified cholesterol in the lysosomes and late endosomes (Patterson et al. 2001). The disease is caused by autosomal recessive mutations in the genes encoding the proteins NPC1 and NPC2, which act together in transporting cholesterol out of the lysosome into cellular membranes including ER, Golgi, and the plasma membrane. Cholesterol exerts many of its functions via membrane microdomains known as lipid rafts, which are enriched in cholesterol and sphingolipids (Lindner and Naim 2009). Accumulation of these lipids in the endosomes/lysosomes may elicit altered lipid raft composition and function in diverse cells in which these membrane structures play pivotal roles.
In many LSDs including NPC, gastrointestinal (GI) symptoms are early and common manifestations where the cellular homeostasis of lipids especially cholesterol and different sphingolipids is affected (Banikazemi et al. 2005; Bernstein et al. 2010). The GI symptoms include diarrhea, nausea, bloating, abdominal pain, and weight loss which resemble the symptoms in disaccharidase deficiencies (Jacob et al. 2000; Amiri and Naim 2012). We have previously shown that cholesterol- and sphingolipid-enriched lipid rafts are essential for polarized sorting of the GI enzymes sucrase-isomaltase (SI; EC 3.2.1.10/48; SUC for sucrase and IM for isomaltase) and dipeptidyl peptidase 4 (DPPIV or CD26; EC 3.4.14.5) to the apical surface in intestinal epithelium (Jacob et al. 2000; Alfalah et al. 2002). Noteworthy, the catalytic activities of SUC and IM within SI are substantially increased upon association of SI with lipid rafts (Jacob and Naim 2001).
This strongly suggests that an unbalanced lipid homeostasis in NPC or other LSDs can be associated with digestive and subsequent absorptive malfunction in the intestine due to impaired trafficking and reduced lipid raft association of SI and possibly other disaccharidases, such as maltase-glucoamylase (MGAM; EC 3.2.1.20/3). These effects could additionally increase the severity of the overall symptoms observed in patients who are subjected to treatment with miglustat or N-butyldeoxynojirimycin, an iminosugar analogue of glucose used in treatment of Gaucher disease and NPC. Miglustat inhibits glucosylceramide synthase and thus prevents glycosphingolipid (GSL) accumulation in lysosomes via substrate reduction therapy (Cox et al. 2000; Rosenbaum and Maxfield 2011). Due to its chemical structure, miglustat binds directly to the catalytic sites of intestinal α-glucosidases, such as SI and MGAM, and inhibits their activity at μM concentrations (Amiri and Naim 2014).
Materials and Methods
Materials
Disaccharides sucrose, isomaltose, maltose, and lactose; proteinase inhibitors pepstatin, leupeptin, antipain, and phenylmethanesulfonyl fluoride; and trypsin inhibitor aprotinin were purchased from Sigma (Steinheim, Germany) or Carl Roth GmbH (Karlsruhe, Germany). PVDF membrane and DTT were purchased from Carl Roth GmbH (Karlsruhe, Germany). Lubrol WX was obtained from MP Biomedicals (Eschwege, Germany). Molecular weight standards for SDS-PAGE and SuperSignal® West Femto Maximum Sensitivity Substrate ECL reagents were purchased from Thermo Scientific GmbH (Schwerte, Germany). Glucose oxidase-peroxidase reagent for colorimetric glucose detection was purchased from Axiom (Worms, Germany).
Antibodies
The monoclonal mouse antibodies against sucrase (HBB2/614/88) and isomaltase (HBB3/705/60) (Hauri et al. 1985) were generously provided by Prof. Dr. H. P. Hauri (Biocenter, Basel, Switzerland) and Prof. Dr. E. Sterchi (University of Bern, Bern, Switzerland). Monoclonal antibodies mlac6 and mlac10 (Maiuri et al. 1991) for immunoblotting of lactase-phlorizin hydrolase were generously provided by Prof. Dr. D. Swallow (University College London, United Kingdom). The secondary horseradish peroxidase-conjugated anti-mouse antibody was purchased from Thermo Fisher Scientific (Bonn, Germany).
Patient and Processing of Intestinal Biopsy Specimens
The NPC patient is a girl, born in 1999, with severe gastrointestinal symptoms. Using Sanger sequencing, compound heterozygosity for the mutations P1007A (c.3019C>G) and L1244P (c.3731T>C) in the gene encoding NPC1 was already identified. Control tissue was obtained from a non-NPC patient who underwent biopsy procedure for diagnostic purposes. The biopsy specimens (>10 mg) were taken under anesthesia and were frozen at −80°C immediately.
Protein Analysis
Biopsy specimens were homogenized in 0.55% Lubrol WX in PBS on ice using a Potter-Elvehjem homogenizer followed by passing through 26G needle for 20 times. The lysis was then continued by gentle rotation at 4°C for 1 h. After removing the cell debris by centrifugation at 1,000×g, a part of the supernatant was aliquoted as the total lysate, and the rest was subjected to ultracentrifugation at 100,000×g for 1 h at 4°C to isolated lipid rafts (pellet) from the non-lipid rafts (supernatant) fractions. Total lysates and raft/non-raft fractions were resolved by SDS-PAGE and the different disaccharidases were detected by immunoblotting.
The protein bands of the Western blots were quantified using the Quantity One® software from Bio-Rad Laboratories GmbH (Munich, Germany).
Enzymatic Activity Measurement
The biopsy specimens were processed according to Dahlqvist (Dahlqvist 1968) using sucrose, isomaltose, maltose, and lactose as substrates. Briefly, samples were incubated 1:1 with 0.056 M of the respective disaccharide for 1 h at 37°C. The amount of the glucose generated by disaccharidase activity was assessed by colorimetric method using glucose oxidase-peroxidase reagent from Axiom.
Results and Discussion
In this report we addressed the correlation between GI symptoms and the association of disaccharidases with lipid rafts in a biopsy specimen from an NPC patient. The patient is a 15-year-old girl, who is compound heterozygous for the mutations c.3019C>G and c.3731T>C within the NPC1 gene (NM_000271) that result in the amino acid exchanges P1007A and L1244P. She shows the late infantile type of the disease. Her development was normal until the age of 3.5 years, when she started stuttering and stumbling. Her motoric and cognitive skills regressed and she developed cataplexy and epilepsy at the age of 9. NPC was diagnosed 2 years later and she was directly treated with miglustat. The progression of the disease slowed down, but she developed severe GI symptoms that resemble those in carbohydrate malabsorption resulting in a strong weight loss. A duodenal biopsy specimen was taken to assess the basis of the GI symptoms. The activities of the enzymes SUC, IM, and MGAM were below the normal range, while that of lactase (lactase-phlorizin hydrolase; EC 3.2.1.68/108) was normal (Table 1). Western blotting revealed protein bands corresponding to SUC and IM similar to those in the control counterparts (Fig. 1a), while their enzymatic activities were 30% and 57%, respectively, below the activity range in the controls (Table 1, Fig. 1a). This inconsistency suggests that a posttranslational processing event in the NPC patient may account for these variations.
Table 1.
Enzymatic activities and protein expression levels of major intestinal disaccharidases
| Enzyme | Activity (IU/g) | Protein expression in NPC (% of control) | |
|---|---|---|---|
| Control range | NPC | ||
| Sucrase | 40–136 | 28 | 90 |
| Isomaltase | 35–123 | 15 | 80 |
| Maltase-glucoamylase | 140–298 | 94 | – |
| Lactase | 25–64 | 26 | 96 |
An intestinal biopsy specimen of the NPC patient was homogenized and used for determination of enzyme activities of sucrase, isomaltase, maltase-glucoamylase, and lactase. Expression levels of these enzymes were detected by immunoblotting (Fig. 1a) and quantified densitometrically
Fig. 1.
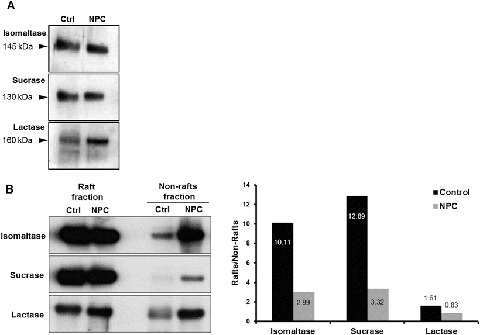
Protein expression and lipid raft association of major intestinal disaccharidases. Immunoblot analysis of (a) total lysate and (b) lipid raft and non-raft fractions prepared from biopsy specimens of an NPC patient in comparison to a non-NPC control patient using Lubrol WX detergent. Lipid rafts were isolated by centrifugation at 100,000g for 1 h at 4°C. The ratio of raft/non-raft association of the analyzed disaccharidases in the NPC patient versus the control indicates a more substantial decrease in lipid raft association for sucrase and isomaltase than lactase
We therefore examined whether the accumulation of cholesterol and sphingolipids in NPC influences the association of SI with lipid rafts. Strikingly, these associations were substantially reduced by 3.9-fold for SUC and 3.4-fold for IM in comparison to the healthy control (Fig. 1b, c). Since the trafficking and sorting of SI to the apical membrane in epithelial cells depend on its association with cholesterol- and sphingolipid-enriched lipid rafts (Jacob and Naim 2001; Alfalah et al. 2002), the results shown here clearly indicate that sorting of SI is markedly impaired and its expression at the apical or microvillus membrane is greatly diminished. Unfortunately, the limited availability of biopsy material has hampered further investigation on the expression levels of SUC and IM in the brush border in the biopsy sample. Another role for the lipid rafts in conjunction with SI is regulatory relative to the activities of SUC and IM, which increase approximately threefold when SI is associated with lipid rafts (Wetzel et al. 2009). As shown here, the association of SUC and IM is markedly reduced, compatible therefore with a decrease in their activities. Altogether, we propose that the reduced delivery of a less-active SI to the microvillus membrane in the patient’s enterocytes elicits carbohydrate malabsorption and appearance of GI symptoms in the NPC patient.
In biological membranes, cholesterol and sphingolipids are often associated with each other, and it has been suggested that metabolism of either one of these lipids in different LSDs results in an impaired localization and function of both of them (Aguilera-Romero et al. 2014). NPC disease offers a support to this view, since the impaired cholesterol transport due to mutations in the NPC1 gene leads to accumulation of cholesterol and subsequently sphingolipids in late endosomes and lysosomes (Parkinson-Lawrence et al. 2010). An imbalance in cholesterol and sphingolipid composition affects the overall function and transport of lipid raft-associated proteins such as SI and possibly also MGAM. Although MGAM association with lipid rafts is not demonstrated yet with certainty, its high level of structural, functional, and trafficking similarities with SI (Nichols et al. 2003) suggests that its function could be impaired as well due to aberrant lipid raft composition.
Finally, the association of lactase with lipid rafts was also reduced, but to a lesser extent than SUC and IM (2.0-fold) (Fig. 1b, c). However, since lactase does not depend on lipid rafts in its trafficking (Jacob and Naim 2001) or function, any alterations in the association of lactase with lipid rafts remain without consequences on enzymatic levels as shown here and on the patient’s tolerance toward lactose.
In conclusion, the current study has delineated the pathobiochemical rationale behind occurrence of GI symptoms in LSDs like NPC, relative to the unbalanced lipid homeostasis in the intestinal epithelial cells. Based on these data, complementary assessments of intestinal disaccharidases in addition to low-carbohydrate diets for managing the GI intolerances in similar cases can be recommended. By virtue of our results, the patient described here was subjected to low-carbohydrate diet that resulted in a substantial improvement of her quality of life.
It should be noted that the GI side effects vary between different patients and variations in the responsiveness of LSD patients to treatment with miglustat have been observed. Therefore, future studies should focus on the elucidation of genotype/phenotype relationships and the extent of alterations in the intracellular lipid composition elicited by a particular phenotype in order to understand the basis for the severity of GI symptoms in NPC patients.
Acknowledgements
The authors would like to thank the patient and her family as well as the Clinic of Pediatrics of the University Hospital Bonn, Germany, for donating the biopsy.
During the course of this work Hadeel Shammas was recipient of a Ph.D. scholarship from the German Academic Exchange Service (DAAD), Bonn, Germany.
Take-Home Message
Gastrointestinal symptoms observed in NPC patients can be due to reduced enzyme activities and lipid raft association of major intestinal disaccharidases.
Compliance with Ethics Guidelines
Conflict of Interest
Mahdi Amiri, Eva-Maria Kuech, Hadeel Shammas, Gabi Wetzel and Hassan Y. Naim declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from the patient for being included in the study.
Details on the Contributions of Individual Authors
Mahdi Amiri, Eva-Maria Kuech, Hadeel Shammas and Gabi Wetzel have equally contributed to designing and performing the experiments, analyzing the data and drafting the manuscript. Hassan Y. Naim has designed the concept of the study and written the final manuscript.
Footnotes
Competing interests: None declared
References
- Aguilera-Romero A, Gehin C, Riezman H. Sphingolipid homeostasis in the web of metabolic routes. Biochim Biophys Acta. 2014;1841:647–656. doi: 10.1016/j.bbalip.2013.10.014. [DOI] [PubMed] [Google Scholar]
- Alfalah M, Jacob R, Naim HY. Intestinal dipeptidyl peptidase IV is efficiently sorted to the apical membrane through the concerted action of N- and O-glycans as well as association with lipid microdomains. J Biol Chem. 2002;277:10683–10690. doi: 10.1074/jbc.M109357200. [DOI] [PubMed] [Google Scholar]
- Amiri M, Naim HY. Miglustat-induced intestinal carbohydrate malabsorption is due to the inhibition of alpha-glucosidases, but not beta-galactosidases. J Inherit Metab Dis. 2012;35:949–954. doi: 10.1007/s10545-012-9523-9. [DOI] [PubMed] [Google Scholar]
- Amiri M, Naim HY. Long term differential consequences of miglustat therapy on intestinal disaccharidases. J Inherit Metab Dis. 2014;37:929–937. doi: 10.1007/s10545-014-9725-4. [DOI] [PubMed] [Google Scholar]
- Banikazemi M, Ullman T, Desnick RJ. Gastrointestinal manifestations of Fabry disease: clinical response to enzyme replacement therapy. Mol Genet Metab. 2005;85:255–259. doi: 10.1016/j.ymgme.2005.04.009. [DOI] [PubMed] [Google Scholar]
- Bernstein DL, Bialer MG, Mehta L, Desnick RJ. Pompe disease: dramatic improvement in gastrointestinal function following enzyme replacement therapy. A report of three later-onset patients. Mol Genet Metab. 2010;101:130–133. doi: 10.1016/j.ymgme.2010.06.003. [DOI] [PubMed] [Google Scholar]
- Cox T, Lachmann R, Hollak C, et al. Novel oral treatment of Gaucher’s disease with N-butyldeoxynojirimycin (OGT 918) to decrease substrate biosynthesis. Lancet. 2000;355:1481–1485. doi: 10.1016/S0140-6736(00)02161-9. [DOI] [PubMed] [Google Scholar]
- Dahlqvist A. Assay of intestinal disaccharidases. Anal Biochem. 1968;22:99–107. doi: 10.1016/0003-2697(68)90263-7. [DOI] [PubMed] [Google Scholar]
- Hauri HP, Sterchi EE, Bienz D, Fransen JA, Marxer A. Expression and intracellular transport of microvillus membrane hydrolases in human intestinal epithelial cells. J Cell Biol. 1985;101:838–851. doi: 10.1083/jcb.101.3.838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob R, Naim HY. Apical membrane proteins are transported in distinct vesicular carriers. Curr Biol. 2001;11:1444–1450. doi: 10.1016/S0960-9822(01)00446-8. [DOI] [PubMed] [Google Scholar]
- Jacob R, Zimmer KP, Schmitz J, Naim HY. Congenital sucrase-isomaltase deficiency arising from cleavage and secretion of a mutant form of the enzyme. J Clin Invest. 2000;106:281–287. doi: 10.1172/JCI9677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner R, Naim HY. Domains in biological membranes. Exp Cell Res. 2009;315:2871–2878. doi: 10.1016/j.yexcr.2009.07.020. [DOI] [PubMed] [Google Scholar]
- Maiuri L, Raia V, Potter J, et al. Mosaic pattern of lactase expression by villous enterocytes in human adult-type hypolactasia. Gastroenterology. 1991;100:359–369. doi: 10.1016/0016-5085(91)90203-W. [DOI] [PubMed] [Google Scholar]
- Nichols BL, Avery S, Sen P, Swallow DM, Hahn D, Sterchi E. The maltase-glucoamylase gene: common ancestry to sucrase-isomaltase with complementary starch digestion activities. Proc Natl Acad Sci U S A. 2003;100:1432–1437. doi: 10.1073/pnas.0237170100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkinson-Lawrence EJ, Shandala T, Prodoehl M, Plew R, Borlace GN, Brooks DA. Lysosomal storage disease: revealing lysosomal function and physiology. Physiology (Bethesda) 2010;25:102–115. doi: 10.1152/physiol.00041.2009. [DOI] [PubMed] [Google Scholar]
- Patterson MC, Vanier MT, Suzuki K, et al. et al. Niemann-Pick disease type C: a lipid trafficking disorder. In: Scriver CR, Beaudet AL, Sly WS, et al.et al., editors. The metabolic and molecular bases of inherited disease. New York: McGraw-Hill; 2001. pp. 3611–3634. [Google Scholar]
- Rosenbaum AI, Maxfield FR. Niemann-Pick type C disease: molecular mechanisms and potential therapeutic approaches. J Neurochem. 2011;116:789–795. doi: 10.1111/j.1471-4159.2010.06976.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wetzel G, Heine M, Rohwedder A, Naim HY. Impact of glycosylation and detergent-resistant membranes on the function of intestinal sucrase-isomaltase. Biol Chem. 2009;390:545–549. doi: 10.1515/BC.2009.077. [DOI] [PubMed] [Google Scholar]