

Abstract
GM2-gangliosidosis, AB variant is a very rare form of GM2 gangliosidosis due to a deficiency of GM2 activator protein, associated with autosomal recessive mutations in GM2A. Less than ten patients, confirmed by molecular analysis, have been described in the literature.
A 12-month-old Hmong girl presented to the neurometabolic clinic for evaluation of global developmental delay, hypotonia, and cherry red spots. The parents were not known to be consanguineous. Her examination was remarkable for hypotonia with hyperreflexia and excessive startling. The head circumference was normal. An extensive neurometabolic evaluation was negative.
Developmental regression began at 14 months of age. Retinal examination at 16 months of age disclosed 4+ cherry red/black spots with “heaped up” ring of whitish infiltrate surrounding both foveae but no evidence of optic atrophy or peripheral retinal abnormalities. Repeat magnetic resonance imaging (MRI) scan at 17 months of age revealed delayed but interval myelination associated with abnormal signal intensity of the bilateral thalami presenting as T2 hyperintensity of the posterior thalami in the region of the pulvinar nuclei and T2 hypointensity in the anterior thalami. Sequencing of the GM2A gene revealed a homozygous c.160 G>T mutation, predicted to result in a premature protein termination p. Glu54*.
Introduction
The GM2 gangliosidoses are a group of disorders characterized by progressive neurological deterioration associated with the excessive accumulation of GM2 ganglioside and related glycolipids in the lysosomes, primarily of neuronal cells. GM2-gangliosidosis, AB variant (OMIM # 272750) is a very rare form of GM2 gangliosidosis due to a deficiency of GM2 activator protein, associated with autosomal recessive mutations in GM2A. The clinical phenotype of the AB variant is very similar to the classic infantile form of Tay-Sachs disease. Hexosaminidase A (Hex A) and B (Hex B) levels are normal (Gravel et al. 2014). Less than ten patients with AB variant, confirmed by molecular analysis, have been described in the literature.
We describe the clinical, ophthalmological, magnetic resonance imaging (MRI), and molecular findings in a patient of Hmong descent. The finding of a homozygous mutation present in a previous patient of Hmong descent suggests a founder mutation.
Case Report
A 12-month-old Hmong girl presented to the neurometabolic clinic for evaluation of global developmental delay, hypotonia, and cherry red spots.
She was the product of her mother’s fifth pregnancy. Her mother had experienced three early miscarriages. She was born at term by spontaneous vaginal delivery following an uncomplicated pregnancy apart from mild gestational diabetes requiring dietary management. The first concern was at 3–4 months of age, when she developed roving eye movements and decreased visual fixation. By 6 months of age, hypotonia was noted and global developmental delay was noted at 10–11 months. She started to crawl just before 12 months of age and was still gaining skills at the time of her first assessment. Excessive startling and irritability were noted and myoclonic jerks at sleep onset also developed. She was otherwise healthy. The family history was negative and the parents were not known to be consanguineous. Her examination was remarkable for global developmental delay and hypotonia with hyperreflexia and excessive startling. The head circumference was normal. Bilateral cherry red spots were present. An extensive neurometabolic evaluation including beta-hexosaminidase A activity and evaluation for other causes of cherry red spots including GM1-gangliosidosis, Krabbe disease, cherry red spot myoclonus, and Niemann-Pick type C were negative.
Developmental regression began at 14 months of age. Progressive hypotonia developed over the subsequent 2 months. By 16 months of age, she was no longer able to crawl, roll, or sit independently. Her grasp became weaker and her fine motor skills declined. She started to have difficulty with swallowing and choking on solids and liquids. Retinal examination at 16 months of age disclosed 4+ cherry red spots with a ring of “heaped up” whitish infiltrate surrounding both foveae but no evidence of optic atrophy or peripheral retinal abnormalities (Fig. 1).
Fig. 1.
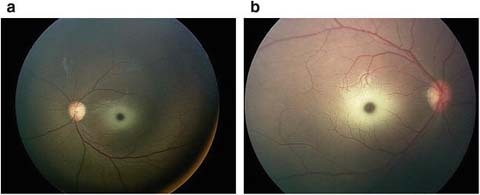
Retinal examination disclosed 4+ cherry red with a ring of “heaped up” whitish infiltrate surrounding both fovea with no optic atrophy or peripheral retinal abnormalities
Repeat magnetic resonance imaging (MRI) scan at 17 months of age revealed delayed but interval myelination associated with abnormal signal intensity of the bilateral thalami presenting as T2 hyperintensity of the posterior thalami in the region of the pulvinar nuclei and T2 hypointensity in the anterior thalami (Fig. 2)
Fig. 2.
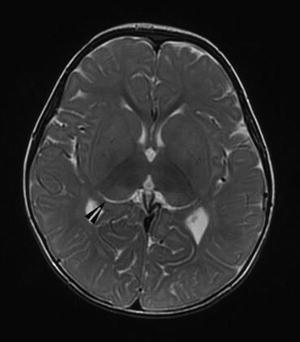
Magnetic resonance imaging (MRI) scan at 17 months of age revealed delayed but interval myelination associated with abnormal signal intensity of the bilateral thalami presenting as T2 hyperintensity of the posterior thalami in the region of the pulvinar nuclei (arrowhead) and T2 hypointensity in the anterior thalami
Molecular analysis of the GM2A gene revealed a homozygous c.160 G>T mutation, predicted to result in a premature protein termination p. Glu54*.
Discussion
The GM2 gangliosidoses are a group of disorders characterized by the excessive accumulation of GM2 ganglioside and related glycolipids in the lysosomes, primarily of neuronal cells. Three forms of GM2 gangliosidosis have been described. Tay-Sachs disease and its variants result from mutations in the HEXA gene and are associated with deficient hex A activity but normal hex B activity. Sandhoff disease and its variants result from mutations in HEXB and are associated with a deficiency of both Hex A and Hex B activity. GM2-gangliosidosis, AB variant is a very rare form of GM2 gangliosidosis due to a deficiency of GM2 activator protein, associated with autosomal recessive mutations in GM2A. Hexosaminidase A and B levels are normal. The clinical phenotype of the classic infantile form of GM2 gangliosidosis is characterized by normal early development followed by developmental regression, progressive weakness, exaggerated startle, vision loss with cherry red spots, and seizures (Gravel et al. 2014). All patients with GM2-gangliosidosis, AB variant to date have presented with the classic infantile clinical phenotype.
The MRI findings in patients with GM2-gangliosidosis, AB variant, have not been well characterized. Chen et al. (1999) describe abnormal MRI signal intensity in the periventricular white matter and basal ganglia in a patient with AB variant. The MRI in Tay-Sachs demonstrates diffuse dysmyelination of hemispheric white matter and bilateral symmetric signal change in the thalami with hyperintensity on T1 and hypointensity on T2 and FLAIR images (Sharma et al. 2008). Magnetic resonance imaging of our patient at 17 months of age revealed delayed but interval myelination associated with abnormal signal intensity of the bilateral thalami presenting as T2 hyperintensity of the posterior thalami in the region of the pulvinar nuclei and T2 hypointensity in the anterior thalami (Fig. 1).
The first case confirmed by molecular analysis was a black female diagnosed on the basis of autopsy findings suggestive of GM2 gangliosidosis in the setting of normal hexosaminidase A enzyme activity. A homozygous c.412T>C missense mutation was found in the GM2AB gene resulting in a single amino acid substitution, p.Arg169Pro (Schroder et al. 1991; Xie et al. 1992). Another missense mutation, c.506G>C, described by Schroder et al. (1993) leads to a substitution of proline for Arg169 resulting in premature degradation of the mutant GM2 activator protein. Schepers et al. (1996) described two small homozygous intragenic deletions in patients presenting with a family history of known consanguinity. The first in a patient of Saudi origin had a three base pair in frame deletion (AAG 262-264) with the deletion of lysine 88, and the other in a patient of Spanish origin had a single base deletion (A410) resulting in a frame shift beginning at codon 137 and leading to a stop codon at 170. A recent review of 73 cases of GM2 gangliosidosis in the United Kingdom included 2 cases of AB variant (one of Caucasian and the other of Pakistani origin); however, molecular findings were not described (Smith et al. 2012).
Chen et al. (1999) described a boy of Laotian Hmong ancestry with no known consanguinity. He was normal until 5 months of age when he developed delayed motor milestones and increasing weakness associated with cherry red spots. His hexosaminidase A activity was normal. Molecular analysis of the GM2A gene revealed a homozygous c.160 G>T mutation, predicted to result in a premature protein termination p. Glu54*. This mutation is the same as our patient who is also the product of non-consanguineous parents of Hmong ancestry, suggesting a founder mutation.
Take-Home Message
Typical clinical symptoms and MRI findings associated with cherry red spots in the setting of normal beta-hexosaminidase A activity should lead to sequencing of GM2A for AB variant, a rare, but underdiagnosed, form of GM2 gangliosidosis.
Compliance with Ethics Guidelines
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Author Contributions
Deborah L. Renaud, M.D., is the primary author and corresponding author.
Michael Brodsky, M.D., contributed to the writing and editing of the manuscript and provided Fig. 1.
Footnotes
Competing interests: None declared
References
- Chen B, Rigat B, et al. Structure of the GM2A gene: identification of an exon 2 nonsense mutation and a naturally occurring transcript with an in-frame deletion of exon 2. Am J Hum Genet. 1999;65(1):77–87. doi: 10.1086/302463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gravel R, Kaback MM et al (2014) The GM2 Gangliosidoses. In: Valle D, Vogelstein B, Kinzler KW, Antonarakis SE, Ballabio A, Gibson K, Mitchell G (eds) OMMBID. McGraw-Hill, New York
- Schepers U, Glombitza G, et al. Molecular analysis of a GM2-activator deficiency in two patients with GM2-gangliosidosis AB variant. Am J Hum Genet. 1996;59(5):1048–1056. [PMC free article] [PubMed] [Google Scholar]
- Schroder M, Schnabel D, et al. A mutation in the gene of a glycolipid-binding protein (GM2 activator) that causes GM2-gangliosidosis variant AB. FEBS Lett. 1991;290(1-2):1–3. doi: 10.1016/0014-5793(91)81211-P. [DOI] [PubMed] [Google Scholar]
- Schroder M, Schnabel D, et al. Molecular genetics of GM2-gangliosidosis AB variant: a novel mutation and expression in BHK cells. Hum Genet. 1993;92(5):437–440. doi: 10.1007/BF00216446. [DOI] [PubMed] [Google Scholar]
- Sharma S, Sankhyan N, et al. Thalamic changes in Tay-Sachs’ disease. Arch Neurol. 2008;65(12):1669. doi: 10.1001/archneur.65.12.1669. [DOI] [PubMed] [Google Scholar]
- Smith NJ, Winstone AM, et al. GM2 gangliosidosis in a UK study of children with progressive neurodegeneration: 73 cases reviewed. Dev Med Child Neurol. 2012;54(2):176–182. doi: 10.1111/j.1469-8749.2011.04160.x. [DOI] [PubMed] [Google Scholar]
- Xie B, Wang W, et al. A Cys138-to-Arg substitution in the GM2 activator protein is associated with the AB variant form of GM2 gangliosidosis. Am J Hum Genet. 1992;50(5):1046–1052. [PMC free article] [PubMed] [Google Scholar]