

Abstract
Acute intermittent porphyria (AIP) is an autosomal dominant disorder of heme biosynthesis due to a mutation in the porphobilinogen deaminase gene. The mutation causes a deficiency in the porphobilinogen deaminase enzyme, thereby causing an accumulation of heme precursors (δ-aminolevulinic acid and porphobilinogen). These neurotoxic heme precursors elicit acute neurovisceral attacks, which can be treated with heme-arginate infusions. Some patients require heme-arginate infusions on a regular basis for many years, which ultimately leads to an iron accumulation (increased serum ferritin and iron accumulation in the liver, spleen, and bone marrow on MRI). We report three AIP patients, who developed iron accumulation (with serum ferritin up to 7,850 microgram/liter) due to multiple heme-arginate infusions. We report for the first time that the iron accumulation in these patients was associated with fibrosis on liver histology.
Conclusion: Regular heme-arginate treatment in AIP does not only lead to increased serum ferritin but may also induce liver fibrosis. This should be taken into account, when weighing the risks and benefits of repeated heme-arginate treatment against the risk and benefits of treating refractory AIP by liver transplantation.
Introduction
Acute intermittent porphyria (AIP) is an autosomal dominant disorder of the heme biosynthesis pathway, resulting from mutations in the gene of the porphobilinogen deaminase (PBGD), also called hydroxymethylbilane synthase (HMBS; OMIM #609806). Patients suffer from acute neurovisceral attacks with abdominal pain, nausea, vomiting, and neuropsychiatric symptoms. These acute attacks, which can be life-threatening, are commonly treated with carbohydrate solutions and intravenous administration of heme-arginate preparations (Puy et al. 2010). Heme-arginate molecules contain the central iron molecule, the reactive center of the heme molecule. Less than 10% of patients develop recurrent attacks (Puy et al. 2010) and therefore require heme-arginate preparations on a regular basis for several years, which can result in progressive iron overload. We present three AIP cases developing a distinct iron overload secondary to the heme-arginate treatment, complicated with the development of liver fibrosis, confirmed on pathology.
Cases
Case 1
In 2000, a 42-year-old male patient was diagnosed with AIP, based on clinical signs and increased urinary δ-aminolevulinic acid (ALA) and porphobilinogen (PBG), later on confirmed by genetic analysis (HMBS mutations c.517 C>T, p.173 P>W). Initially he suffered from recurrent acute attacks every few months. In the following years, he experienced a gradual increase of attack frequency, with attacks occurring every 2–3 weeks by the year 2006. Acute attacks were treated with glucose and heme-arginate infusions (3 mg/kg/day for 4 days). In May 2007 prophylactic treatment with heme-arginate infusions was introduced on a weekly basis, which initially led to an improvement of symptom control. Despite this regular medical treatment, he experienced a further increase in frequency of attacks the following years, severely affecting his quality of life. As a result of the chronic treatment with heme-arginate infusions, he developed a progressive iron overload, with increasing ferritin levels (Fig. 1) and on MRI reduced signal intensity of the spleen and liver on T1- and T2-weighted images as sign of secondary hemochromatosis (Fig. 2). Echocardiography showed a borderline left ventricular function, but without evidence of myocardial iron accumulation on MRI. Liver transaminases were slightly elevated (<2× ULN) and liver synthetic capacity remained normal (normal albumin and INR). Underlying mutations in the HFE1 gene (C282Y, H63D) on chromosome 6p21 were excluded, pointing to the absence of a genetic predisposition to hereditary hemochromatosis.
Fig. 1.
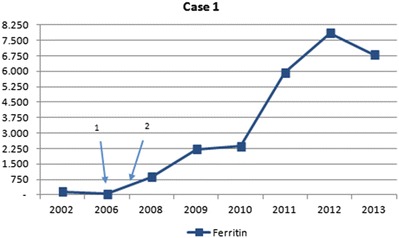
Evolution of serum ferritin levels in case 1 (expressed in μg/L – normal values: 13–150 μg/L). Arrow 1 introduction of heme-arginate infusions. Arrow 2 introduction of weekly infusions
Fig. 2.
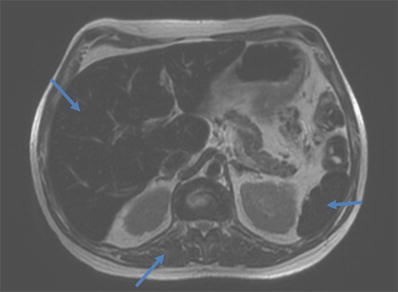
T2-weighted image with signal intensity of the liver and spleen lower than the signal in the paravertebral muscle, pointing to iron accumulation in the liver and spleen
Treatment with phlebotomies was introduced in April 2009 in between acute attacks but was poorly tolerated (recurrence of attacks immediately after the phlebotomies). Eventually the patient was referred to our hospital for orthotopic liver transplantation because of poor quality of life, based on the frequent recurrence of acute attacks despite maximal medical treatment and progressive hyperferritinemia. In January 2014 he received a liver transplant. Histological review of the explant liver showed diffuse deposition of iron in the Kupffer cells and the hepatocytes but surprisingly also showed a septal stage of fibrosis. Currently the patient is doing well, without significant posttransplantation complications.
Case 2
A female patient was diagnosed with AIP in 1973, at the age of 38 years. In 2001 her medical history was complicated with a breast carcinoma, which was treated with a mastectomy followed by chemotherapy. Diagnosis of AIP was made when she presented with severe back pain and dark-colored urine. The diagnosis was confirmed by genetic analysis (HMBS mutations c.181 ins G). Because of a high frequency of acute attacks, she was started on weekly prophylactic heme-arginate infusions in 1996 (200 mg/day). Despite this intensive treatment, severe acute attacks continued at high frequency. In addition, there was a progressive increase in ferritin levels (Fig. 3) and transferrin saturation, with signs of bone marrow iron deposition on MRI of the spine. The latter was performed because of predominant back pain in a patient with previous breast carcinoma. No evidence of metastasis was found, but a clear iron deposition was detected. Liver function remained normal. Underlying mutations in the HFE1 gene (C282Y, H63D, S65C) on chromosome 6p21 were excluded. Heme-arginate administration was reduced and monthly phlebotomies were started in 2010. Because of persisting disabling attacks, the patient was eventually transplanted in August 2012. Pathological review of the explant liver showed excessive iron deposition in the hepatocytes and Kupffer cells as well as advanced fibrosis with portoportal fibrous septa (Fig. 4). The posttransplantation period was complicated by a thrombosis of the hepatic artery, which was treated with an immediate surgical thrombectomy, but eventually she required a second transplant in November 2013. The patient is currently doing well.
Fig. 3.
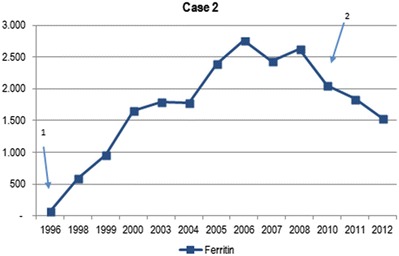
Evolution of serum ferritin levels in case 2 (expressed in μg/L – normal values: women 10–140 μg/L). Arrow 1 introduction of heme-arginate infusions. Arrow 2 start of phlebotomies
Fig. 4.
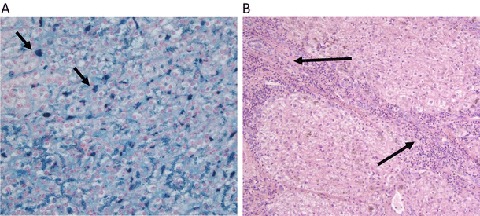
(a) Iron staining on liver resection specimen from case 2 (arrows: iron deposits in Kupffer cells). (b) Fibrosis on liver resection specimen from case 2 (arrows: fibrous septum running from the upper left portal tract to the lower right portal tract)
Case 3
Another female patient was diagnosed with AIP in 1987 at the age of 45 years, confirmed by urinary and genetic analysis (HMBS mutations c.cgg-346-tgg, p.arg-116-trp, R116W). Treatment with heme arginate was administered on a regular basis for recurrent neurovisceral attacks from 2005, at 3 mg/kg/day (lifelong dose estimated at 40,000 mg). Biochemical follow-up revealed an asymptomatic elevation of ferritin levels up to 1904 μg/L. Mutations in the HFE1 gene (C282Y, H63D, S65C) were excluded. In 2010, the patient was diagnosed with hepatocellular carcinoma and treated with chemoembolization, followed by a partial hepatectomy (segments IV, V, and VIII) in 2011. Liver function was normal up until this complication. Histological review of the resection specimen showed excessive iron deposition in the hepatocytes and advanced fibrosis. Eventually the patient died in 2013, at the age of 70 years, of metastatic disease.
Discussion
We collected data of three illustrated cases of AIP patients presenting with acute neurovisceral attacks on an almost weekly basis and in need of frequent medical treatment. All patients were treated during attacks with the classical dose of 3–4 mg/kg of heme-arginate infusions per day and also periodically received preventive treatment, for many years. Secondary to this iron-loaded treatment, they developed secondary hemochromatosis, both biochemically and on imaging. For the first time, we report here that these ferritin increases were associated with advanced fibrosis on liver histology.
AIP is an autosomal dominant disorder of the heme biosynthesis pathway. Mutations result in an enzyme activity of approximately 50% of normal. More than 200 different mutations have been described in the gene encoding porphobilinogen deaminase (PBGD), also called hydroxymethylbilane synthase (HMBS) (Siegesmund et al. 2010). Prevalence of a mutant AIP gene is estimated at 1 per 500, but the penetrance of the disorder is incomplete and the prevalence of symptomatic disease is estimated at only 1–2 per 100,000 in Europe (Elder et al. 2013). The reduced PBGD enzyme activity causes accumulation of heme precursors ALA and PBG, which are neurotoxic and cause the acute neurovisceral attacks. These acute attacks are debilitating and may become life-threatening if unrecognized or left untreated. Most commonly patients present with abdominal pain, but pain can also occur in the back or thighs. Gastrointestinal dysmotility symptoms such as nausea, vomiting, and constipation are also highly prevalent. In addition, a variety of neurological and psychiatric symptoms can be associated with the attacks.
Diagnosis of AIP is based on clinical suspicion, with first-line confirmation through high urinary ALA and PBG during or shortly after acute attacks. Direct DNA sequencing has become the gold standard for diagnostic confirmation and defining the underlying genetic defects. This also allows counseling of family members, thereby preventing attacks in those with latent disease (Puy et al. 2010; Herrick and McColl 2005).
Treatment is primarily based on avoidance of precipitating factors that either induce production of ALA and PBG or increase the demand for heme synthesis in the liver and bone marrow. These precipitants include hormonal fluctuations in women, porphyrinogenic drugs, infections, alcohol use, smoking, and caloric deprivation (Badminton and Elder 2002; Herrick and McColl 2005). In case of an acute attack, treatment consists of intravenous administration of carbohydrates (minimum of 300 g/day), heme-arginate preparations (3 mg/kg heme arginate once daily for 4 days), and further supportive measures (e.g., pain treatment with safe drugs). The intravenously administrated heme inhibits the upregulation of ALA synthase in the liver resulting in a dramatic reduction of urinary and plasma ALA and PBG (Puy et al. 2010). Liver transplantation has become a valid treatment option in selected patients with recurrent attacks and significant disabling symptoms with poor quality of life (Seth et al. 2007; Singal et al. 2014).
Less than 10% of patients show frequently recurring attacks requiring medical treatment. In some of these patients, prophylactic use of heme arginate – e.g., every few weeks – is applied to prevent further attacks and improve symptom control and therefore also quality of life. In the literature, few complications of treatment with heme-arginate administration have been reported. Aside from anaphylactic reactions and local irritation of blood vessels (with thrombophlebitis and progressive loss of venous access), there are a few rare complications reported such as renal failure and acute liver failure due to overdose (Anderson et al. 2005; Frei et al. 2012). However, knowing that a single dose of human heme arginate contains 22.7 mg of iron (Puy et al. 2010; Siegesmund et al. 2010), there is also a high risk of developing iron overload and secondary hemochromatosis with related complications in the subgroup of patients receiving repeated administration of heme arginate over several years. Current literature is limited on this matter, but AIP seems not to be directly associated with fibrosis or cirrhosis in itself. Even in AIP patients developing HCC, cirrhosis is rarely reported (Innala and Andersson 2011; Stewart 2012). Based on the current literature, it is even suggested that patients diagnosed with HCC in a non-cirrhotic liver should be screened for acute intermittent porphyria (Deybach and Puy 2011). In the patients we report here, the liver fibrosis we observed is most likely due to the secondary hemochromatosis induced by years of treatment with heme-arginate preparations. Other underlying causes of liver fibrosis, such as chronic viral hepatitis, were excluded on serology and on biopsies. To our knowledge, these are the first patients described with fibrosis due to medical treatment of acute AIP attacks with heme arginate. This implies the necessity for close monitoring of ferritin levels in patients receiving regular heme arginate and the need to screen for related complications such as liver fibrosis/cirrhosis as well as for extrahepatic complications (cardiac involvement, diabetes, etc.). It remains unclear when treatment and screening for complications should be started, although applying the same guidelines as for hereditary hemochromatosis and other forms of iron overload seems the most appropriate (Siddique and Kowdley 2012; European Association for the Study of the Liver 2010).
Conclusion
Chronic administration of heme arginate over many years in AIP patients can result in secondary hemochromatosis with liver fibrosis. Recurrence of acute neurovisceral attacks in patients with AIP necessitates regular heme-arginate infusion in less than 10% of AIP patients. This subgroup appears to be at risk for evolution to secondary hemochromatosis with associated complications, such as liver fibrosis. Close monitoring of ferritin levels is needed during follow-up, as well as screening for complications of iron overload. Increasing ferritin levels are to be taken into account when evaluating these patients for liver transplantation, in addition to the standard considerations as are the presence of repeated life-threatening acute attacks, failure of medical therapy, and poor quality of life.
Synopsis
Regular treatment with heme arginate in AIP may lead to iron overload and liver fibrosis.
Compliance with Ethics Guidelines
Conflict of Interest
Barbara Willandt, Janneke G. Langendonk, Katharina Biermann, Wouter Meersseman, François D’Heygere, Christophe George, Chris Verslype, Diethard Monbaliu, and David Cassiman declare that they have no conflict of interest.
Informed Consent
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000 (5). Informed consent was obtained from all patients for being included in the study.
Details of the Contributions of Individual Authors
Barbara Willandt collected the data and wrote the first report. Janneke G. Langendonk, Katharina Biermann, Wouter Meersseman, François D’Heygere, Christophe George, Chris Verslype, Diethard Monbaliu, and David Cassiman were all involved in planning and executing the clinical follow-up and hence data collection. All authors contributed in writing or correcting/improving the final manuscript.
Footnotes
Competing interests: None declared
References
- Anderson KE, Bloomer JR, Bonkovsky HL, et al. Recommendations for the diagnosis and treatment of the acute porphyrias. Ann Intern Med. 2005;14:439–450. doi: 10.7326/0003-4819-142-6-200503150-00010. [DOI] [PubMed] [Google Scholar]
- Badminton MN, Elder GH. Management of acute and cutaneous porphyrias. Int J Clin Pract. 2002;56:272–278. [PubMed] [Google Scholar]
- Deybach JC, Puy H. Hepatocellular carcinoma without cirrhosis: think acute hepatic porphyrias and vice versa. J Intern Med. 2011;269:521–524. doi: 10.1111/j.1365-2796.2011.02358.x. [DOI] [PubMed] [Google Scholar]
- Elder G, Harper P, Badminton M, Sandberg S, Deybach JC. The Incidence of inherited porphyrias in Europe. JIMD. 2013;36(5):849–857. doi: 10.1007/s10545-012-9544-4. [DOI] [PubMed] [Google Scholar]
- European Association for the Study of the Liver EASL clinical practice guidelines for HFE hemochromatosis. J Hepatol. 2010;53(1):3–22. doi: 10.1016/j.jhep.2010.03.001. [DOI] [PubMed] [Google Scholar]
- Frei P, Minder EI, Corti N, et al. Liver transplantation because of acute liver failure due to heme arginate overdose in a patient with acute intermittent porphyria. Case Rep Gastroenterol. 2012;6:190–196. doi: 10.1159/000338354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrick AL, McColl KEL. Acute intermittent porphyria. Best Pract Res Clin Gastroenterol. 2005;19(2):235–249. doi: 10.1016/j.bpg.2004.10.006. [DOI] [PubMed] [Google Scholar]
- Innala E, Andersson C. Screening for hepatocellular carcinoma in acute intermittent porphyria: a 15-year follow-up in northern Sweden. J Intern Med. 2011;269:538–545. doi: 10.1111/j.1365-2796.2010.02335.x. [DOI] [PubMed] [Google Scholar]
- Puy H, Gouya L, Deybach JC. Porphyrias. Lancet. 2010;375:924–937. doi: 10.1016/S0140-6736(09)61925-5. [DOI] [PubMed] [Google Scholar]
- Seth AK, Badminton MN, Mirza D, Russell S, Elias E. Liver transplantation for porphyria: who, when, and how? Liver Transpl. 2007;13:1219–1227. doi: 10.1002/lt.21261. [DOI] [PubMed] [Google Scholar]
- Siddique A, Kowdley KV. Review article: the iron overload syndromes. Aliment Pharmacol Ther. 2012;35(8):876–893. doi: 10.1111/j.1365-2036.2012.05051.x. [DOI] [PubMed] [Google Scholar]
- Siegesmund M, Van Serooskerken T, Poblete-Gutiérrez P, Frank J. The acute hepatic porphyrias: current status and future challenges. Best Pract Res Clin Gastroenterol. 2010;24(5):593–605. doi: 10.1016/j.bpg.2010.08.010. [DOI] [PubMed] [Google Scholar]
- Singal A, Parker C, Bowden C, Thapar M, Liu L, McGuire BM. Liver transplantation in the management of porphyria. Hepatology. 2014;60:1082–1089. doi: 10.1002/hep.27086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart FM. Review of hepatocellular cancer, hypertension and renal impairment as late complications of acute porphyria and recommendations for patient follow-up. J Clin Pathol. 2012;65:976–980. doi: 10.1136/jclinpath-2012-200791. [DOI] [PubMed] [Google Scholar]