

Abstract
ACAD9 (acyl-CoA dehydrogenase 9) is an essential factor for the mitochondrial respiratory chain complex I assembly. ACAD9, a member of acyl-CoA dehydrogenase family, has high homology with VLCAD (very long-chain acyl-CoA dehydrogenase) and harbors a homodimer structure. Recently, patients with ACAD9 deficiency have been described with a wide clinical spectrum ranging from severe lethal form to moderate form with exercise intolerance.
We report here a prenatal presentation with intrauterine growth retardation and cardiomegaly, with a fatal outcome shortly after birth. Compound heterozygous mutations, a splice-site mutation – c.1030-1G>T and a missense mutation – c.1249C>T; p.Arg417Cys, were identified in the ACAD9 gene. Their effect on protein structure and expression level was investigated. Protein modeling suggested a functional effect of the c.1030-1G>T mutation generating a non-degraded truncated protein and the p.Arg417Cys, creating an aberrant dimer. Our results underscore the crucial role of ACAD9 protein for cardiac function.
Electronic supplementary material
The online version of this chapter (doi:10.1007/8904_2015_499) contains supplementary material, which is available to authorized users.
Keywords: ACAD9, Fetal cardiomegaly, Mitochondrial respiratory chain, β-Oxidation
Introduction
The mitochondrial flavoenzymes acyl-CoA dehydrogenases (ACADs) are involved in the β-oxidation of acyl-CoA and amino acid catabolisms. Acyl-CoA dehydrogenase catalyzes the first step of fatty acid β-oxidation in the mitochondria (Swigonova et al. 2009).
Acyl-CoA dehydrogenase 9 (ACAD9) is a 621-amino-acid protein encoded by the ACAD9 gene. The expression of ACAD9 mRNA is ubiquitous with high levels of expression in the heart, skeletal muscle, brain, kidney, and liver. In contrast to the other ACADs, which are homotetrameric flavoenzymes in the mitochondrial matrix, ACAD9 and very long-chain acyl-CoA dehydrogenase (VLCAD) are homodimers associated with the inner mitochondrial membrane (Ensenauer et al. 2005). ACAD9 shares 47% amino acid identity and 65% amino acid similarity with VLCAD, which is encoded by the gene ACADVL (Zhang et al. 2002); this could be explained by ACADVL gene duplication. The main difference is a 35-amino-acid stretch within the ACAD9 sequence, which is predicted to form an α helix at the surface of the molecule (Nouws et al. 2010). After dimer assembly, these helices could form a hydrophobic cleft interacting with mitochondrial partners different from VLCAD ones. This structural difference may underlie the distinct roles of these enzymes. In fact, although ACAD9 conserves its β-oxidation activity (Ensenauer et al. 2005), VLCAD has a significant higher activity level in degrading unsaturated long-chain fatty acids. It has been shown recently that ACAD9 plays a pivotal role in the assembly of chain respiratory complex I (Nouws et al. 2014a, b). It’s worth noting that the use of catalytically inactive ACAD9 allowed a partial-to-complete rescue of ACAD9 deficient cells (Nouws et al. 2014a, b). Complex I consists of 45 subunits derived from both mitochondrial and nuclear genome (Efremov et al. 2010; Mimaki et al. 2012). Importantly, defects in complex I assembly underlie 50% of the isolated complex I deficiencies (Nouws et al. 2012). The assembly of its different subunits requires specific factors which are involved in complex I biogenesis and stability without taking any part in the final mature complex I. Models have been put forward to explain the assembly process which include several steps. ACAD9 is linked to two other factors, NDUFAF1 and Ecsit, within an assembly intermediate.
ACAD9 deficiency causes decreased levels of NDUFAF1, Ecsit, and mature complex I (Nouws et al. 2010; Mimaki et al. 2012). Clinical presentation of ACAD9 deficiency consists either in a severe presentation including Reye-like episodes with acute liver dysfunction, hypertrophic cardiomyopathy, rapidly progressive encephalomyopathy with rhabdomyolysis, and sudden infant death syndrome, or in a milder childhood-onset form consisting in fatigue and vomiting episodes with cognitive dysfunction, or in a pure myopathy with exercise intolerance and lactic acidosis (He et al. 2007; Haack et al. 2010; Gerards et al. 2011; Nouws et al. 2014a, b). The prognosis for severe form of ACAD9 deficiency is poor with death reported from 46 days of age to adulthood (He et al. 2007; Haack et al. 2010). Riboflavin treatment has been reported to improve clinical condition in some patients. Riboflavin may increase mitochondrial FAD concentration, favors FAD binding to ACAD9, and, thus, improve catalytic activity, folding, and assembly of ACAD9 (Henriques et al. 2010). Accordingly, riboflavin treatment has been reported to ameliorate clinical condition in some patients. However, depending on the position of the causative mutation, riboflavin treatment may be ineffective (Nouws et al. 2014a, b).
In the present report, we describe the earliest fatal cardiac presentation of ACAD9 deficiency associated with two deleterious mutations. The effect of these mutations on protein structure and expression level is investigated.
Case Report
A male newborn was delivered at 36 weeks’ gestation (WG) by cesarean section due to intrauterine growth retardation (UIGR) detected at 22 WG during ultrasound (US) examination. At 33 WG, US confirmed UIGR and revealed cardiomegaly. This newborn was the fourth child of a non-consanguineous healthy couple. Two other healthy children were born at term 6 and 1 years before. The second child, a boy born at 38 WG, who also presented with intrauterine growth retardation, died 6 h after birth from hypoglycemia with no definite diagnosis. Considering the family background, the patient was investigated soon after birth; lactate concentration in the cord blood was within the reference range (5 mmol/L N < 6). One hour after birth, the patient presented with severe recurrent hypoglycemic events (1.6 mmol/L). Despite oral milk intake, he remained hypoglycemic (0.8 mmol/L), and admixture of premature milk and 10% glucose solution was introduced by means of a nasogastric tube, with normalization of glycemia within 3 h. But he rapidly presented with cardiac failure and metabolic acidosis. Clinical examination revealed global hypotonia and systolic murmurs and no hepatomegaly. Heart US examination confirmed hypokinetic myocardiopathy and cardiomegaly. Biological tests showed metabolic acidosis with a pH measured at 6.85. Lactates were at 28 mmol/L with a lactate/pyruvate ratio at 91 (N < 20), alkaline reserve at 5 mmol/L, and hyperammonemia at 197 μmol/L. Total and free carnitine were low 4 h after birth, but acylcarnitine analysis using tandem mass spectrometry was normal.
Despite appropriate and intensive treatments, the child died within 10 h. Immediately after death, muscular and liver biopsies were performed for histological and electron microscopy analyses, and a complete autopsy was carried out with the informed consent of both parents in accordance with the French law and following standardized protocols.
Material and Methods
Respiratory Chain Analysis
Spectrophotometric assays of respiratory chain (RC) enzymes were performed on muscle, as described by Rustin et al. (1994). Mitochondrial proteins from patient and control were analyzed by blue native polyacrylamide gel electrophoresis (BN-PAGE). The gel was subsequently blotted and incubated with five antibodies specific of each respiratory chain complex [GRIM19 (complex I subunit), 70 kDa (complex II subunit), core 2 (complex III subunit), COX1 (complex IV subunit), and beta complex V subunit (MitoSciences)] as previously described (Assouline et al. 2012).
Genomic Amplification and Sequencing
Genomic DNA was amplified in vitro by PCR. Primers used to amplify exonic regions, including intron/exon boundaries and promoter of ACAD9 gene, were designed according to the sequence NM_014049.4: PCR consisted of one cycle of 96°C for 5 min, followed by 30 cycles of 96°C for 30 s, 55°C for 30 s, and 72°C for 30 s; PCR was terminated after a final cycle at 72°C for 10 min. Direct DNA fragment sequencing of ACAD9 gene was performed with the PRISM Ready Reaction Sequencing Kit (Perkin-Elmer, Oak Brook, IL) on an automatic sequencer (ABI 3130xl; PE Applied Biosystems, Foster City, CA).
In Silico Analysis of Splicing Effect
The analysis was performed by the integrated software Alamut v.2.3 (http://www.interactive-biosoftware.com) using default settings in all predictions of five splice-site prediction programs: SpliceSiteFinder-like (http://www.interactive-biosoftware.com), GeneSplicer (http://www.cbcb.umd.edu/software/GeneSplicer), Splice Site Prediction by Neural Network (http://www.fruitfly.org/seq_tools/splice.html), MaxEntScan (http://genes.mit.edu/burgelab/maxent/Xmaxentscan_scoreseq.html), and Human Splicing Finder (http://www.umd.be/HSF/).
cDNA Amplification and Sequencing
Total RNA was extracted from fibroblasts using NucleoSpin® RNA (Macherey NAGEL, EURL) according to the manufacturer’s recommendations. The cDNAs were obtained by reverse transcription of 1 μg of total RNA using reverse transcription system (Promega, Charbonnières les Bains, France).
Amplification of the ACAD9 transcript was performed using a forward primer within exon 10 [5′TCGGAGATGGGTTTAAGGTG3′] and a reverse primer within exon 15 [5′GGCCTGTTTAAGCTCATGGA3′].
To assay for the presence of a short intronic insertion in patient cDNA, we used a forward primer within this putative additional intronic sequence [5′GATTTGGCTCTCAGCACATG3′] in combination with a primer located in exon 15 [5′GGCCTGTTTAAGCTCATGGA3′] to produce a 448 bp band if the inserted fragment is present in the cDNA sequence.
Real-Time PCR
Real-time PCR was performed on different concentrations of cDNA (1, 1/10, 1/100) samples in triplicate using iQ™ SYBR® Green Supermix according to the manufacturer’s protocol and CFX96™ real-time PCR detection system (Bio-Rad, Hercules, CA). Amplification of specific transcripts was confirmed by melting curve profiles generated at the end of the PCR program. The primers were designed with Primer Express software and are ACAD9-F: 5′GTACGCCTGCACAAGGAAAC3′; ACAD9-R:5′TGCTGAGAGCCAAATCTGAA3′ (Applied Biosystem).
For quantitative data analysis, CT values were normalized to those of GAPDH using ΔΔCT method.
Immunoblotting
Control human or patient fibroblasts were homogenized in 250 μL of lysis buffer (50 μM Hepes, pH 7.5, 150 mM NaCl, 10 mM EDTA, 10 mM glycerophosphate, 100 mM natrium fluoride, 1% triton X100, 1 mM PMSF, and phosphatase inhibitor cocktail). After centrifugation of the homogenates (20,000 g × 15 min), the supernatants were used for Western blotting. Fifty microgram of protein extracts was suspended in Laemmli buffer (100 mM Hepes, pH 6.8, 10% β-mercaptoethanol, 20% SDS) and boiled for 5 min. They were then loaded on 15% SDS-polyacrylamide gel. After separation, proteins were electrically transferred to nitrocellulose membrane. The membranes were then incubated with the blocking solution (1X TBS, 0.05% TWEEN 20, 5% bovine serum albumin (BSA)) at room temperature for 1 h and incubated overnight with the primary antibodies raised against ACAD9 (1/1,000; sc-135148, Santa Cruz Biotechnology) or β-actin (1/1,000. A5441, Sigma Aldrich). After incubation with the corresponding secondary antibody coupled to peroxidase (Santa Cruz Biotechnology), proteins were visualized using an enhanced chemiluminescence ECL Plus immunoblotting detection system (Amersham Biosciences Europe GmbH, Freiburg, Germany). Commercial markers (SeeBlue Pre-Stained Standard, Invitrogen) were used as molecular weight standards.
Immunohistochemistry
Control or patient-cultured fibroblasts were fixed with 4% PFA in PBS and were incubated overnight at 4°C with ACAD9 antibody diluted in incubation buffer (PBS containing 1% BSA and 3% Triton X-100). Then, the slices were rinsed twice with PBS for 20 min and incubated with the same incubation buffer containing the adequate secondary antibody. Cell nuclei were visualized by incubating the slices for 5 min with 1 μg/mL Hoechst 33258 in PBS. Fluorescent signals were observed with a Leica DMI 6000B microscope. Substituting the primary antibodies by PBS controlled the specificity of the immunoreactions.
Protein Structure Prediction
The protein resulting from the c.1030-1G>T mutation was generated using the protein fold recognition server PHYRE (Kelley and Sternberg 2009). Atom contacts and structure validation were checked using the MolProbity server (Davis et al. 2007; Chen et al. 2010). The p.Arg417Cys mutation was built using the ACAD9 dimer homology model as a template (Nouws et al. 2010). The best score rotamer calculated by the mutation tool of SPDBV software (Guex and Peitsch 1997) (http://www.expasy.org/spdbv/) was chosen. The structures were analyzed with SYBYL package (Tripos Associates, St. Louis, MO) and displayed with MOLSCRIPT (Kraulis 1991). Sequence alignment rendering was performed with the ENDscript server (Robert and Gouet 2014) (ESPript – http://espript.ibcp.fr)
Results
Pathological Findings
The neonate weighed 1,919 g (<3rd percentile). No craniofacial dysmorphism was observed. The heart was globular and enlarged, its weight reaching the 95th percentile (22.1 g, N = 18.21 ± 3.69 g). On cut sections, the cardiac walls were hypertrophic and pale (Fig. 1a). The only associated anomaly consisted in lung hypoplasia. The myocardium was histologically normal. Brain examination revealed diffuse edema due to agonic status.
Fig. 1.
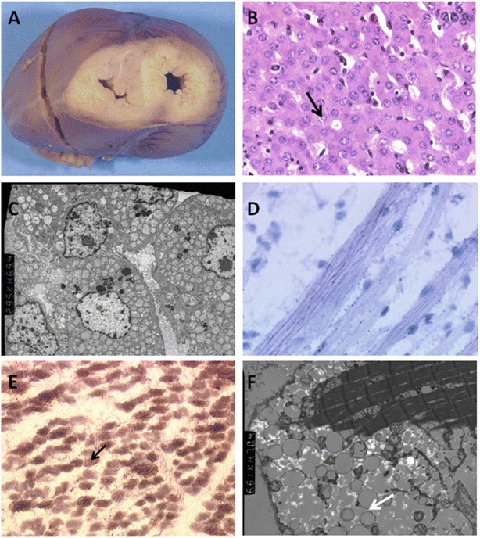
Morphological results obtained from autopsy. (a) Cross section of the heart showing enlarged and pale ventricular walls. (b) Acinar arrangements (arrow) and mild steatosis in the liver parenchyma (G × 400). (c) Accumulation of mitochondria in the cytoplasm of hepatocytes and of dispersed neutral lipid droplets (arrow) (G × 2,200). (d) Mild muscular microvacuolar lipidosis (Oil Red O staining; G × 1,000). (e) Reduced cytochrome-oxidase activity on combined succinate dehydrogenase-cytochrome-oxidase histoenzymology and blue muscle fibers (arrow) lacking cytochrome-oxidase activity (G × 400). (f) Moderately electron dense neutral lipids dispersed between muscle fibers (arrows) (G × 6,100)
Histological examination of the liver biopsy displayed microvacuolar steatosis and pseudo-acinous arrangements of hepatocytes (Fig. 1b). Ultrastructural analysis confirmed mild steatosis, with an excessive accumulation of mitochondria in the cytoplasms (Fig. 1c). Oil Red O staining performed on the quadriceps revealed microvacuolar lipidosis, along with a reduction of cytochrome-oxidase activity visualized by histoenzymological methods (Fig. 1d). At the ultrastructural level, muscle fibers contained multiple neutral lipid droplets, but no structural anomaly of the mitochondria (Fig. 1e, f).
Biochemical Studies
Pyruvate dehydrogenase and pyruvate carboxylase deficiency were ruled out by enzyme assay carried out on fibroblasts. Spectrophotometric assays of respiratory chain enzymes performed on muscle showed a very low complex I activity (9% of lower control range) and a partially decreased complex V activity (64% of lower control range) (Table 1).
Table 1.
Spectrophotometric dosage of respiratory chain complexes in skeletal muscle
| RCC | Absolute values (nmol/min/mg protein) | Normal range (nmol/min/mg protein) | % of lower control range (%) |
|---|---|---|---|
| Complex I | 1 | 11–28 | 9 |
| Complex II | 33 | 20–49 | 165 |
| Complex III | 250 | 224–478 | 112 |
| Complex IV | 120 | 90–223 | 133 |
| Complex V | 28 | 44–120 | 64 |
| Normalized activity/CS | |||
| Citrate synthase (CS) | 92 | 69–144 | |
| Complex I | 0.01 | 0.12–0.22 | 8 |
| Complex II | 0.36 | 0.24–0.38 | 150 |
| Complex III | 2.72 | 2.29–3.62 | 119 |
| Complex IV | 1.31 | 1.23–1.96 | 107 |
| Complex V | 0.30 | 0.46–0.98 | 65 |
| Activity ratio/RCC total activity | Values (%) | Normal range (%) | % of lower control range (%) |
| Complex I/RCC | 0.10 | 2.50–3.70 | 4 |
| Complex II/RCC | 7.70 | 4.20–6.70 | 183 |
| Complex III/RCC | 58.00 | 47.90–57.20 | 121 |
| Complex IV/RCC | 27.80 | 22.20–28.20 | 125 |
| Complex V/RCC | 6.40 | 10.60–17.50 | 60 |
RCC respiratory chain complex, CS citrate synthase
Further experiments were performed on cultured skin fibroblasts since additional muscle was not available. The respiratory chain complexes were studied by BN-PAGE analysis. The gel was subsequently blotted and incubated with five antibodies specific of each RC complex. BN-PAGE profile was suggestive of quantitative and qualitative complex I assembly anomalies showing low amount of the fully assembled complex I in contrast to control cells (~1 MDa) and the presence of a lower molecular weight subcomplex between 350 and 450 KDa. Other RC complexes including complex V remained normal as compared to control (Supplementary Fig. 1).
These results in combination with clinical presentation led to the investigation of complex I assembly deficiency factors such as ACAD9.
Molecular Studies
Molecular study of the ACAD9 gene revealed two heterozygous mutations, a splice mutation in intron 10, c.1030-1G>T and a missense mutation in exon 12 c.1249C>T; p.Arg417Cys (Fig. 2). Parents were also investigated and were both heterozygous, the mother carrying the c.1030-1G>T mutation and the father carrying the c.1249C>T substitution.
Fig. 2.
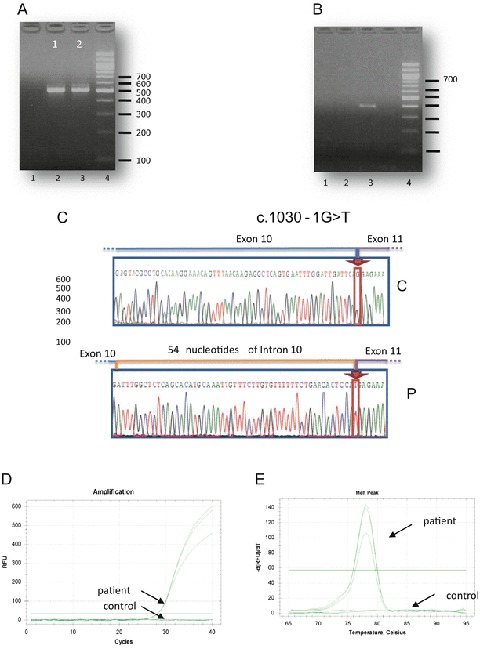
Characterization of the mutation NM_014049.4: c.1030-1G>T in the intron 10 of the ACAD9 gene. (a) Amplification of the ACAD9 transcript using a forward primer within exon 10 and a reverse primer within exon 15. Lane 1 control; Lane 2 patient; Lane 3 DNA 100-bp increment ladder. The amplification showed 509 and 563 bp products in the patient sample while only the 509 bp product was present in the control DNA sample. (b) Amplification of the ACAD9 transcript using a forward primer within the putative inserted intronic sequence and a reverse primer within exon 15. Lane 1 control; Lane 2 patient; Lane 3: DNA 100-bp increment ladder. The corresponding fragment (448 bp) was amplified using patient sample while no amplification was obtained with control sample. (c) Sequence of the 563 bp PCR product showing the 54 intronic nucleotide in the patient DNA sample. C control, P patient. (d) qPCR amplification of ACAD9 cDNA in proband and control fibroblasts. No amplification was observed with control sample. (e) Dissociation curves for ACAD9 qPCR. A single dissociation temperature suggests a single product
The c.1249C>T (p.Arg417Cys) mutation has already been reported as deleterious by Haack et al. (2010), while the second mutation (c.1030-1G>T) has been identified in this patient. The substitution of G to T abolishes the splice acceptor site at the 3′ end of intron 10.
The impact of this mutation was evaluated at the cDNA level and amplification using primers surrounding exon 11 allowed to reveal the presence of two different bands (509; 563 bp) in patient sample while the 563 bp product was very faint in the control sample (Fig. 2a). This result goes along with the enhanced use of a cryptic splice site located at 54 nucleotides upstream exon 11. In order to amplify the abnormal fragment alone, we designed a forward primer within the putative inserted intronic sequence. The corresponding fragment (448 bp) was amplified using patient sample while no amplification was obtained with control sample (Fig. 2b). The PCR product sequence harbored an insertion of 54 intronic nucleotides between exon 10 and exon 11, confirming the use of this cryptic site in the patient (Fig. 2c).
Real-time RT-PCR analysis, using primer within the inserted region, confirmed that the mRNA with intronic insertion is present in the index case while no amplification was observed with cDNA obtained with one control (Fig. 2d, e).
This insertion is predicted to result in a truncated protein product of 351AA in which the 278 C-terminal amino acids are replaced by an amino acid stretch of 18AA.
Western blot analysis, with anti-ACAD9 antibodies against upstream-mutation part of ACAD 9, revealed that an additional form was present in high level in the patient protein extract. As shown in the Fig. 3a, the mitochondrial (66 kDa) and the cytosolic/nuclear protein forms (76 kDa) previously described in fibroblast lysate (He et al. 2007) were observed in the control and in the patient protein extracts. One other form corresponding in size (molecular mass about 50 kDa) to the predicted truncated protein was present at a high level in the patient protein extract.
Fig. 3.
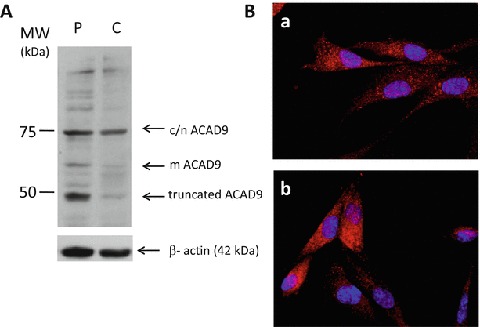
Characterization of c.1030-1G>T mutation at protein level. (a) Immunoblotting. Fifty micrograms of control (C) or patient (P) fibroblast lysate were analyzed by Western blot by using ACAD9 and β-actin antibodies. The mACAD9 band corresponds in size to mitochondrial form (66 kDa), and the c/nACAD9 band corresponds to cytosolic or nuclear ACAD9 species (76 kDa). One other form corresponding in size (about 50 kDa) to the predicted truncated protein is present at a high level in the patient protein extract. (b) Immunocytochemistry analysis from control (a) or proband (b) fibroblasts by using ACAD9 antibody
In an immunochemistry study using anti-ACAD9 antibodies targeting the protein upstream, the mutation made it possible to confirm that ACAD9 protein was expressed at least at the same level in the patient and control fibroblasts (Fig. 3b). These results suggest that c.1030-1G>T mutation may induce the production of a truncated protein, which is probably not degraded.
Protein Structure Prediction
Homology modeling was used to investigate the possible structural implication of both c.1249C>T (p.Arg417Cys) and c.1030-1G>T mutations.
An ACAD9 dimer homology model based on the VLCAD crystallographic structure was available (Nouws et al. 2010). In this model, it has been reported that in each monomer, Arg417 interacts with Glu413 and that the substitution of this arginine by a lysine residue may have deleterious consequences (Nouws et al. 2010). In our case, the substitution of Arg417 by a cysteine residue could also have such consequences, but this substitution in both monomers may have more drastic consequences. This hypothesis was verified by building an ACAD9 dimer with a double Arg417Cys mutation, using the ACAD9 dimer model of Nouws et al. as a template (Fig. 4a). Interestingly, in the obtained dimer model, Cys417 of both monomers were favorably positioned, thus enabling the existence of a disulfide bridge between them.
Fig. 4.
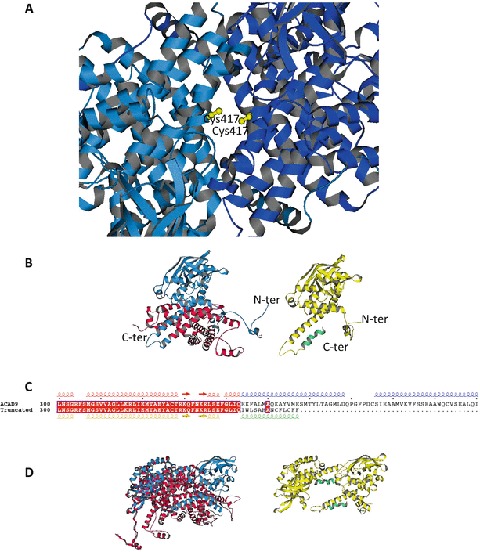
Model of the ACAD9 with the mutations c.1249C>T (p.Arg417Cys) and c.1030-1G>T. (a) ACAD9 dimer with the double c.1249C>T (p.Arg417Cys) mutation (in yellow). The ACAD9 dimer model of Nouws et al. (2010) has been used as a template. (b) Left: monomer of ACAD9 showing the truncated region (in red) resulting from the c.1030-1G>T mutation. The monomer unit is derived from the ACAD9 dimer model of Nouws et al. (2010). Right: monomer of ACAD9 showing the mutated region (in green) resulting from the c.1030-1G>T mutation. The monomer was built using the PHYRE2 server (Kelley and Sternberg 2009). (b) Sequence alignment of the 300–400 range of ACAD9 native and truncated. Secondary structural elements extracted from the models shown in a, using the same colors. (d) Left: dimer model of ACAD9 (Nouws et al. 2010) showing the lost interaction surface (in red) formed by the truncated region resulting from the c.1030-1G>T mutation. Right: dimer model of ACAD9 with the truncated and mutated monomers resulting from the c.1030-1G>T mutation. The monomers are positioned as in the native dimer
The protein resulting from the c.1030-1G>T mutation was generated using the protein fold recognition server PHYRE (Kelley and Sternberg 2009). The obtained model was compared with the monomers constituting the ACAD9 dimer model built by Nouws et al. (2010). The monomers were very close to each other (RMSD = 1.55 Å for monomer 1 and RMSD = 1.49 Å for monomer 2), suggesting that the c.1030-1G>T mutation generates a protein which may be structured. Interestingly, in the model of the truncated ACAD9, the 18 new C-terminal amino acids were assumed to adopt a helix-loop-helix structure and did not perturb the overall structure of the protein (Fig. 4b, c). Nevertheless, the deletion of the 278 C-terminal amino acids and their replacement by a short stretch of 18 amino acids remove a significant interaction surface between the monomers, probably preventing a native dimer formation using this truncated protein (Fig. 4d).
Discussion
We report on the clinical, morphological, and molecular characteristics of an antenatal cardiac presentation of ACAD9 deficiency. UIGR and cardiomegaly were detected antenatally and soon after birth, the patient presented with cardiac failure, dilated cardiomyopathy, and severe lactic acidosis. Besides, severe recurrent episodes of hypoglycemia were observed from 1 h of life; recurrent episodes of hypoglycemia have been reported in only one patient aged 4 months (He et al. 2007). Pathological studies showed an accumulation of mitochondria in hepatocyte cytoplasms with microvacuolar lipidosis in liver and muscle and were consistent with mitochondrial impairment. Autopsy findings have exceptionally been reported, since to our knowledge they were described in a single male case aged 14, who presented with a Reye-like episode, consisting in hepatic steatosis, with moderate to severe chronic neuronal loss on histological examination (He et al. 2007). Actually, fatty acid β-oxidation impairment may be related either to the ACAD9 enzymatic activity decrease or to the lack of mature complex I.
Most reported cases of ACAD9 deficiency present with hypertrophic cardiomyopathy. After birth, the energy required for cardiac function is mainly produced through β-oxidation pathway, Krebs cycle, and respiratory chain. In the present case, both β-oxidation pathway and respiratory chain function may be impaired. If the pivotal role of ACAD9 in complex I assembly has been demonstrated, the physiological function of ACAD9 in β-oxidation still remains controversial. Nouws et al. concluded that ACAD9 seems not necessary for fatty acid oxidation (Nouws et al. 2014a, b), while it has been, recently, demonstrated in cultured cells that ACAD9 plays a physiological role in fatty acid oxidation (Schiff et al. 2015).
Fetal cardiomegaly could be attributed to complex I deficiency since fetal heart metabolism is mainly carbohydrate based. Besides, IUGR has been linked to complex I deficiency. If the decrease of ACAD9 β-oxidation activity has a physiological relevance, it may worsen the prenatal cardiac energetic deficiency by impeding the switch from glucose metabolism to fatty acid metabolism that occurs soon after birth (Onay-Besikci 2006). Fatty acid oxidation defects are mainly diagnosed during the perinatal and infantile period. Only two of them, long-chain 3-hydroxyacyl CoA dehydrogenase deficiency or mitochondrial trifunctional protein deficiencies, are associated with intrauterine growth restriction, prematurity, and maternal pregnancy complications (Oey et al. 2005). Hence, the antenatal presentation of ACAD9 deficiency may be due to its dual role. It is worth noting that one of the identified mutations, c.1249C>T; p.Arg417Cys, has been previously characterized in a compound heterozygous girl who died at the age of 12 (Haack et al. 2010). Therefore, the severity of the function loss could be attributed to the splice-site mutation, since our results indicate clearly that this mutation leads to an activation of an upstream cryptic splice site within intron 10, causing an insertion of 54 bases of intron 10 sequences into the mature RNA. This leads to a frameshift of translation resulting in addition of 18 new amino acids at the carboxyl-terminus, before an in-frame stop translation codon is encountered, truncating the 278 C-terminal amino acids. The 18 new C-terminal amino acids may adopt a helix-loop-helix structure, thus permitting the stabilization of a putatively nonfunctional truncated protein as shown by immunochemistry and Western blotting.
The second mutation involves the residue Arg417 (p.Arg417Cys). The protein structure prediction suggests that the Arg417 of monomer 1 interacts with the Glu143 of monomer 2 to form a homodimer. Substitution of Arg417 by a Cys residue prevents this interaction, a disulfide bridge involving Cys117 of both monomers inducing the formation of an aberrant homodimer. Indeed, mutation causing protein structure alteration is often associated with more severe phenotypes (Fu et al. 2014).
Interestingly, besides the complex I deficiency, a partial defect in complex V was noticed in our patient, whereas other respiratory chain activities were normal. Similarly, Haack et al. reported a profound complex I deficiency with mild reduction of complex V in a patient who presented with respiratory insufficiency, hypertrophic cardiomyopathy, lactic acidosis, severe encephalopathy, and an early death at 46 days (Haack et al. 2010). These findings suggest that ACAD9 may contribute to complex V activity or stability, and the lack of ACAD9 leads to a reduced ATPase activity.
In conclusion, ACAD9 deficiency is a newly recognized pathology with an unknown prevalence and needs to be further documented, especially in severe antenatal presentations.
Electronic Supplementary Material
Below is the link to the electronic supplementary material.
Compliance with Ethics Guidelines
Declaration of Interest
The authors report no conflicts of interest.
Synopsis
A severe ACAD9 deficiency revealed by fetal cardiomegaly with a profound complex I deficiency and a partial complex V deficiency.
Author Contributions
Jennifer Lagoutte-Renosi: data interpretation and drafting the article
Isabelle Ségalas-Milazzo: protein modeling
Marie Crahes: pathological studies
Florian Renosi: data interpretation
Laurence Menu-Bouaouiche: protein modeling
Stéphanie Torre: clinical management of the patient
Caroline Lardennois: clinical management of the patient
Marlène Rio: molecular studies
Stéphane Marret: article editing
Carole Brasse-Lagnel: analysis, data interpretation, and article editing
Annie Laquerrière: interpretation of pathological findings and article editing
Soumeya Bekri: conception and design, data interpretation, and article editing
Guarantor
Soumeya Bekri
Competing Interest Statement
The authors have no competing interests to declare.
Footnotes
Competing interests: None declared
References
- Assouline Z, Jambou M, Rio M, et al. A constant and similar assembly defect of mitochondrial respiratory chain complex I allows rapid identification of NDUFS4 mutations in patients with Leigh syndrome. Biochim Biophys Acta. 2012;1822:1062–1069. doi: 10.1016/j.bbadis.2012.01.013. [DOI] [PubMed] [Google Scholar]
- Chen VB, Arendall WB, 3rd, Headd JJ, et al. MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr D Biol Crystallogr. 2010;66:12–21. doi: 10.1107/S0907444909042073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis IW, Leaver-Fay A, Chen VB, et al. MolProbity: all-atom contacts and structure validation for proteins and nucleic acids. Nucleic Acids Res. 2007;35:W375–W383. doi: 10.1093/nar/gkm216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efremov RG, Baradaran R, Sazanov LA. The architecture of respiratory complex I. Nature. 2010;465:441–445. doi: 10.1038/nature09066. [DOI] [PubMed] [Google Scholar]
- Ensenauer R, He M, Willard JM, et al. Human acyl-CoA dehydrogenase-9 plays a novel role in the mitochondrial beta-oxidation of unsaturated fatty acids. J Biol Chem. 2005;280:32309–32316. doi: 10.1074/jbc.M504460200. [DOI] [PubMed] [Google Scholar]
- Fu R, Ceballos-Picot I, Torres RJ, et al. Genotype-phenotype correlations in neurogenetics: Lesch–Nyhan disease as a model disorder. Brain. 2014;137:1282–1303. doi: 10.1093/brain/awt202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerards M, van den Bosch BJ, Danhauser K, et al. Riboflavin-responsive oxidative phosphorylation complex I deficiency caused by defective ACAD9: new function for an old gene. Brain. 2011;134:210–219. doi: 10.1093/brain/awq273. [DOI] [PubMed] [Google Scholar]
- Guex N, Peitsch MC. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 1997;18:2714–2723. doi: 10.1002/elps.1150181505. [DOI] [PubMed] [Google Scholar]
- Haack TB, Danhauser K, Haberberger B, et al. Exome sequencing identifies ACAD9 mutations as a cause of complex I deficiency. Nat Genet. 2010;42:1131–1134. doi: 10.1038/ng.706. [DOI] [PubMed] [Google Scholar]
- He M, Rutledge SL, Kelly DR, et al. A new genetic disorder in mitochondrial fatty acid beta-oxidation: ACAD9 deficiency. Am J Hum Genet. 2007;81:87–103. doi: 10.1086/519219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henriques BJ, Olsen RK, Bross P, Gomes CM. Emerging roles for riboflavin in functional rescue of mitochondrial beta-oxidation flavoenzymes. Curr Med Chem. 2010;17:3842–3854. doi: 10.2174/092986710793205462. [DOI] [PubMed] [Google Scholar]
- Kelley LA, Sternberg MJ. Protein structure prediction on the Web: a case study using the Phyre server. Nat Protoc. 2009;4:363–371. doi: 10.1038/nprot.2009.2. [DOI] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. doi: 10.1107/S0021889891004399. [DOI] [Google Scholar]
- Mimaki M, Wang X, McKenzie M, Thorburn DR, Ryan MT. Understanding mitochondrial complex I assembly in health and disease. Biochim Biophys Acta. 2012;1817:851–862. doi: 10.1016/j.bbabio.2011.08.010. [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans L, Houten SM, et al. Acyl-CoA dehydrogenase 9 is required for the biogenesis of oxidative phosphorylation complex I. Cell Metab. 2010;12:283–294. doi: 10.1016/j.cmet.2010.08.002. [DOI] [PubMed] [Google Scholar]
- Nouws J, Nijtmans LG, Smeitink JA, Vogel RO. Assembly factors as a new class of disease genes for mitochondrial complex I deficiency: cause, pathology and treatment options. Brain. 2012;135:12–22. doi: 10.1093/brain/awr261. [DOI] [PubMed] [Google Scholar]
- Nouws J, Te Brinke H, Nijtmans LG, Houten SM. ACAD9, a complex I assembly factor with a moonlighting function in fatty acid oxidation deficiencies. Hum Mol Genet. 2014;23:1311–1319. doi: 10.1093/hmg/ddt521. [DOI] [PubMed] [Google Scholar]
- Nouws J, Wibrand F, van den Brand M, et al. A patient with complex I deficiency caused by a novel ACAD9 mutation not responding to riboflavin treatment. JIMD Rep. 2014;12:37–45. doi: 10.1007/8904_2013_242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oey NA, den Boer ME, Wijburg FA, et al. Long-chain fatty acid oxidation during early human development. Pediatr Res. 2005;57:755–759. doi: 10.1203/01.PDR.0000161413.42874.74. [DOI] [PubMed] [Google Scholar]
- Onay-Besikci A. Regulation of cardiac energy metabolism in newborn. Mol Cell Biochem. 2006;287:1–11. doi: 10.1007/s11010-006-9123-9. [DOI] [PubMed] [Google Scholar]
- Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rustin P, Chretien D, Bourgeron T, et al. Biochemical and molecular investigations in respiratory chain deficiencies. Clin Chim Acta. 1994;228:35–51. doi: 10.1016/0009-8981(94)90055-8. [DOI] [PubMed] [Google Scholar]
- Schiff M, Haberberger B, Xia C, et al. Complex I assembly function and fatty acid oxidation enzyme activity of ACAD9 both contribute to disease severity in ACAD9 deficiency. Hum Mol Genet. 2015;24(11):3238–3247. doi: 10.1093/hmg/ddv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swigonova Z, Mohsen AW, Vockley J. Acyl-CoA dehydrogenases: dynamic history of protein family evolution. J Mol Evol. 2009;69:176–193. doi: 10.1007/s00239-009-9263-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Zhang W, Zou D, et al. Cloning and functional characterization of ACAD-9, a novel member of human acyl-CoA dehydrogenase family. Biochem Biophys Res Commun. 2002;297:1033–1042. doi: 10.1016/S0006-291X(02)02336-7. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.