

Abstract
Introduction: Agalsidase alfa and agalsidase beta, recombinant enzyme preparations for treatment of Fabry disease (FD), have different approved dosing schedules: 0.2 mg/kg and 1.0 mg/kg every other week (EOW), respectively.
Methods: This open-label, multicenter, exploratory phase 4 study evaluated plasma globotriaosylsphingosine (lyso-GL-3) and plasma and urine globotriaosylceramide (GL-3) levels at baseline and 2, 4, and 6 months after the switch from agalsidase alfa (0.2 mg/kg EOW for ≥12 months) to agalsidase beta (1.0 mg/kg EOW) in 15 male patients with FD. Immunoglobulin (Ig)G antidrug antibody titers were assessed, and safety was monitored throughout the study.
Results: Plasma lyso-GL-3 concentrations decreased significantly within 2 months after switch and reductions continued through month 6 (mean absolute changes, −12.8, −16.1, and −16.7 ng/mL at 2, 4, and 6 months, respectively; all P < 0.001). The mean percentage reduction from baseline was 39.5% (P < 0.001) at month 6. For plasma GL-3, the mean absolute change from baseline (−0.9 μg/mL) and percentage reduction (17.9%) at month 6 were both significant (P < 0.05). Urine GL-3 measurements showed intra-patient variability and changes from baseline were not significant. No clinical outcomes were assessed in this 6-month study, and, therefore, no conclusions can be drawn regarding the correlation of observed reductions in glycosphingolipid concentrations with clinically relevant outcomes. There were no differences in IgG antidrug antibody titers between the two enzymes. The switch from agalsidase alfa to agalsidase beta was well tolerated.
Conclusion: Plasma lyso-GL-3 and GL-3 levels reduced after switching from agalsidase alfa to agalsidase beta, indicating that agalsidase beta has a greater pharmacodynamic effect on these markers at the recommended dose. These data further support the use of agalsidase beta 1.0 mg/kg EOW as enzyme replacement therapy in FD.
Introduction
Fabry disease (FD) is an X-linked disorder that affects both males and females and is caused by deficient activity of the lysosomal enzyme α-galactosidase A (α-Gal A) (Germain 2010). This leads to the accumulation of globotriaosylceramide (GL-3, Gb3), predominantly in the lysosomes of multiple cell types, and the elevation of globotriaosylsphingosine (lyso-GL-3, lyso-Gb3), the more water-soluble deacylated form of GL-3, in the plasma (Aerts et al. 2008). In vitro studies suggest that these elevations trigger a cascade of pathological processes, including inflammatory and fibrotic responses that cause progressive damage to multiple organs (Germain 2010). In males with classic FD, early signs and symptoms, including neuropathic pain, hypohidrosis, and gastrointestinal dysmotility, usually appear in early childhood (Hopkin et al. 2008), and life-threatening renal, cardiac, and cerebrovascular complications typically develop by the fourth or fifth decade of life (Germain 2010).
FD is treated with recombinant α-Gal A enzyme replacement therapy (ERT). In the USA, agalsidase beta (Fabrazyme®; Genzyme, a Sanofi company) is the only ERT approved to treat FD. Two ERTs are licensed in the European Union: agalsidase beta and agalsidase alfa (Replagal®; Shire Human Genetic Therapies) (Schaefer et al. 2009). Agalsidase alfa and agalsidase beta have identical amino acid sequences and are functionally equivalent (Lee et al. 2003). However, agalsidase beta contains more fully sialylated oligosaccharides and more mannose-6-phosphate than agalsidase alfa (Lee et al. 2003; Togawa et al. 2014); cellular uptake of recombinant agalsidases is mediated by mannose-6-phosphate receptors. Greater uptake of agalsidase beta than of agalsidase alfa has been suggested in vitro (in skin fibroblasts from patients with FD) (Keslová-Veselíková et al. 2008; Lee et al. 2003) and in vivo (in the kidney and heart after administration of the same dose of the two enzymes in a mouse model of FD) (Desnick and Schuchman 2012; Sakuraba et al. 2006). Although not adequately powered, the results from a study in patients with FD suggest that the two agents have similar clinical effects when administered at the same dose of 0.2 mg/kg every other week (EOW) (Vedder et al. 2007).
However, based on the doses used in the clinical development programs for these agents, the recommended doses of the two agents differ by a factor of 5 – agalsidase alfa is 0.2 mg/kg EOW (Replagal® Summary of product characteristics, last updated September 2014), whereas agalsidase beta is 1.0 mg/kg EOW (Fabrazyme® Summary of product characteristics, last updated October 2014; Fabrazyme® Prescribing information, last updated May 2010). The optimal dose of agalsidase has long been a matter of debate (Desnick 2004), and identification of the biochemically and clinically most effective dose has been hampered by the slowly progressive nature of FD that lacks reliable, predictive biochemical or clinical surrogate end points. Additionally, the clinical presentation of FD is highly heterogeneous and the timing of symptom onset highly variable (Eng et al. 2007; Hopkin et al. 2008). There have been no randomized, double-blind trials of patients matched for sex, age, and disease severity to allow a direct comparison of the effectiveness of agalsidase beta and agalsidase alfa at their approved doses (Desnick and Schuchman 2012). Recent data from the Canadian Fabry Disease Initiative, where agalsidase alfa and beta have been used head-to-head, have shown no difference in end points between patients treated with agalsidase alfa (0.2 mg/kg) and patients receiving agalsidase beta (1.0 mg/kg), although patients were not randomized for variables known to affect prognosis, and GL-3 and lyso-GL-3 data were not reported (Sirrs et al. 2014). In addition, as this study was underpowered, a definite conclusion about comparability of enzymes at equal or licensed dose is still unresolved.
GL-3, a substrate for α-Gal A, is routinely measured in blood and urine and occasionally in biopsy samples. The level of GL-3 is used as a surrogate marker to assess treatment efficacy, and clearance of GL-3 from the tissues, mainly the liver and kidneys, has been used in several studies as an outcome measure (reported as relative to normal and/or percentage decrease from baseline) (Eng et al. 2001; Schiffmann et al. 2000). Short-term therapy has been shown to rapidly decrease plasma GL-3 levels in adult and pediatric male patients with FD (Clarke et al. 2007; Schiffmann et al. 2010). Recently, lyso-GL-3 has emerged as a more sensitive biomarker that may correlate with disease severity and/or organ involvement in FD (Rombach et al. 2010). Untreated patients with classic FD typically have highly elevated plasma levels of lyso-GL-3 (Aerts et al. 2008; Togawa et al. 2010; Niemann et al. 2014), and plasma lyso-GL-3 decreased within 3 months of initiation of treatment in naive FD patients treated with either agalsidase alfa or agalsidase beta, with significantly larger reductions with agalsidase beta 1.0 mg/kg versus agalsidase alfa and agalsidase beta 0.2 mg/kg (van Breemen et al. 2011). Recurrent elevations in plasma lyso-GL-3 have been reported in patients whose agalsidase dose was reduced. These patients either changed to a lower dose of agalsidase beta (from 1.0 to 0.5 mg/kg EOW or 0.5 mg/kg per month) or switched from agalsidase beta to agalsidase alfa (0.2 mg/kg EOW) (Smid et al. 2011), leading to the hypothesis that ERT using agalsidase reduces plasma lyso-GL-3 concentration in a dose-dependent manner regardless of the type of agalsidase (beta or alfa) administered.
The INFORM study was designed to confirm this hypothesis by monitoring the changes in plasma lyso-GL-3 levels in patients with FD whose agalsidase dose was increased by a switch from agalsidase alfa administered at 0.2 mg/kg EOW to agalsidase beta administered at 1.0 mg/kg EOW (when investigational agalsidase alfa treatment became unavailable in the USA). Fifteen male FD patients were recruited to the study, and the effects of the dose change were followed for 6 months, during which patients’ plasma and urine concentrations of glycosphingolipids (plasma lyso-GL-3 and urinary and plasma GL-3) were evaluated. Antibody titers to both agalsidase preparations were also investigated.
Methods
Patients
Adult and pediatric male patients with a diagnosis of FD, confirmed by α-Gal A activity assay and/or GLA gene mutation analysis in accordance with local standards, were eligible for the study if they had received agalsidase alfa (0.2 mg/kg EOW) for ≥12 months (Fig. 1). Patients who had switched to agalsidase beta before the start of the study who met the eligibility criteria and had provided blood and urine samples at protocol-specified time points (at baseline and 2 and 4 months after switching to agalsidase beta) were also included.
Fig. 1.
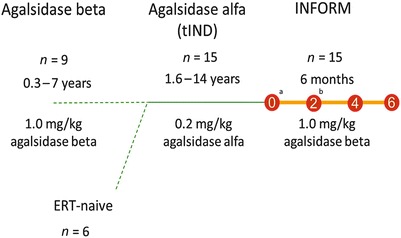
Enzyme replacement therapy before and during INFORM study. aSixteen patients screened for 15 eligible patients. bOne patient voluntarily discontinued on day 64 of treatment as he no longer wished to participate in the study. tIND treatment Investigational New Drug
Patients who were on dialysis or had undergone renal transplantation, patients with end-stage cardiac disease, and patients (or their guardians or parents) who were unable to fully comply with study requirements were excluded from the study.
Study Design
INFORM was an open-label, multicenter, exploratory phase 4 study conducted at six sites in the USA between April 2012 and March 2013. After baseline samples were collected, eligible patients received agalsidase beta at 1.0 mg/kg EOW via intravenous infusion (Fig. 1). No patient had a treatment hiatus between his last agalsidase alfa infusion and his first agalsidase beta infusion.
Assessments
Assessments were conducted at baseline and months 2, 4, and 6 after the switch from agalsidase alfa to agalsidase beta. The end points were the mean percentage change and absolute change from baseline in plasma lyso-GL-3, plasma GL-3, and urine GL-3 at months 2, 4, and 6.
Concentrations of plasma GL-3, plasma lyso-GL-3, and urine GL-3 were assessed in samples obtained at baseline and months 2, 4, and 6. Plasma GL-3 concentrations were quantified using a validated liquid chromatography–tandem mass spectrometry (LC/MS/MS) method as previously described (Nelson et al. 2004; Roddy et al. 2005; Wilcox et al. 2004). Urine GL-3 was extracted from whole urine with chloroform/methanol and measured by LC/MS/MS. Urine GL-3 levels were normalized to creatinine and expressed as micrograms of GL-3 per 1 mmol creatinine. Plasma lyso-GL-3 was measured using a validated LC/MS/MS method. Briefly, 100 μL of plasma was treated with acidified acetonitrile containing dimethyl psychosine (Avanti Polar Lipids Inc.; Alabaster, AL, USA) as internal standard and filtered in a 96-well lipid-removal filtration plate. The eluent was dried down and reconstituted for LC/MS/MS analysis. The lower limit of quantification (LLOQ) was defined as the lowest concentration of the analyte that could be measured with accuracy (80–120% recovery) and precision (percentage coefficient of variation ≤20%). The LLOQ was 2.0 μg/mL for plasma GL-3, 0.2 μg/mL for urine GL-3, and 5.0 ng/mL for plasma lyso-GL-3. All assays were validated and performed in a clinically compliant laboratory environment. Normal values for plasma GL-3 and plasma lyso-GL-3 were <7.0 μg/mL (n = 203, from 104 males and 101 females) and <5 ng/mL (n = 100, from 50 females and 50 males), respectively. The normal range for urine GL-3 was <81 μg/mmol creatinine (n = 50 normal subjects).
Antidrug IgG antibodies against agalsidase beta and agalsidase alfa were measured in serum samples obtained at baseline, and serum IgG antibodies against agalsidase beta were measured at months 2, 4, and 6. A specific enzyme-linked immunosorbent assay (ELISA) method to assess IgG antibodies to agalsidase beta or agalsidase alfa was used. The method for agalsidase beta was adapted for agalsidase alfa, and modifications were made to achieve similar assay sensitivity. Briefly, IgG antibody to agalsidase beta or agalsidase alfa was screened and confirmed. Both assays were used for baseline sample analysis, and only agalsidase beta ELISA was used for posttreatment sample analysis. Confirmed positive samples were further analyzed to determine titers for all of the posttreatment samples. Serum was serially diluted twofold starting at an initial 1/100 dilution. The titer was reported as the reciprocal of the last dilution above the assay cutoff point.
A potential neutralizing effect of IgG antibody formation was assessed in a post hoc analysis. The neutralizing capacity of enzyme activity was expressed as micrograms of recombinant human α-Gal A inhibited by 1 mL of patient sera. In addition, inhibition of enzyme uptake was assessed in all IgG antibody-positive patients.
Safety was assessed by recording the incidence and type of adverse events (AEs) associated with protocol-related procedures and other AEs not considered associated with agalsidase beta treatment. AEs associated with agalsidase beta treatment were also recorded. AEs were classified according to the Medical Dictionary for Regulatory Activities, version 16.0, and grouped by preferred term and system organ class. Each AE or serious AE (SAE) was judged, in the opinion of the study investigators, to be not related, unlikely related, possibly related, or related to agalsidase beta treatment; the investigators graded the severity of the AE or SAE as mild, moderate, or severe. No clinical efficacy outcomes were assessed.
Statistical Methods
Sample size was chosen on the basis of a study that reported a statistically significant median increase in lyso-GL-3 concentration in a group of male patients switched from agalsidase beta to agalsidase alfa or a lower dose of agalsidase beta (Smid et al. 2011). Based on the assumption that a reverse effect on lyso-GL-3 concentrations in patients who switched from agalsidase alfa (0.2 mg/kg EOW) to agalsidase beta (1.0 mg/kg EOW) would be observed, a cohort of 20 patients would provide >90% power to detect changes in lyso-GL-3 concentration of the same magnitude. Plasma lyso-GL-3, rather than plasma GL-3, was chosen to power the study, as levels are more pronouncedly increased in untreated classically affected males (Aerts et al. 2008), which makes it a more suitable discriminator of a dose effect.
Demographic data and baseline characteristics were summarized using descriptive statistics. Patient-level plasma lyso-GL-3, plasma GL-3, and urine GL-3 concentrations at baseline and months 2, 4, and 6 after switching to agalsidase beta are presented graphically. Changes from baseline were summarized as mean percentage change and absolute change by using descriptive statistics for each visit. Changes in plasma lyso-GL-3 and plasma GL-3 concentrations were tested using one-sample t-tests. Urine GL-3 concentration showed considerable fluctuations between visits, and changes were not normally distributed. Therefore, changes were tested using a one-sample test of medians (sign test) and described as median changes from baseline. Statistical tests were performed and the corresponding P-values are reported. Statistical reports were generated by using SAS® version 9.2 in a secure and validated environment. AE data were also summarized descriptively.
Results
Patient Demographics
Patient demographics and GLA mutations are summarized in Table 1. Fourteen of the 15 male patients (93.3%) who enrolled completed the study. One patient (patient 5) voluntarily withdrew consent from the study on day 64 of treatment for reasons unrelated to safety.
Table 1.
Characteristics and individual GLA mutations at baseline for the 15 patients enrolled in the study
| Patient number (random) | GLA gene mutation | Associated phenotype, as reported in the literature | Reference | Age categorya | Duration of Fabry disease (years)b | Leukocyte α-Gal A activity (U/mg)/plasma α-Gal A activity (U/mL) |
|---|---|---|---|---|---|---|
| 1 | 358Edel | Classic | Blanch et al. (1996) | B | 11.5 | 0.9/1.0 |
| 2 | c.256delT | Classic | Topaloglu et al. (1999) | C | 14.1 | NA/1.0 |
| 3 | c.717_718delAA | Classic | Blanch et al. (1996) | E | NA | 0.1/0.0 |
| 4 | R220X | Classic | Meaney et al. (1994) | C | 10.0 | 1.4c/NA |
| 5 | D322E | Classic | Lee et al. (2010) | E | 2.7 | NA/NA |
| 6 | NA | – | – | D | 18.8 | 3.5c/NA |
| 7 | R227Q | Classic | Eng et al. (1993) | C | 11.6 | 6.8c/0.1 |
| 8 | C56X | Classic | Shabbeer et al. (2006) | A | 8.1 | NA/NA |
| 9 | g.IVS4+919G>A | Late onset | Chien et al. (2013) and Eng et al. (1994) | A | 5.9 | 5.1c/NA |
| 10 | R112C | Classic | Shabbeer et al. (2006) | C | 3.1 | 0.4/0.2 |
| 11 | C202Y | Classic | Eng et al. (1997) | E | 1.9 | NA/0.1 |
| 12 | R227Q | Classic | Eng et al. (1993) | D | 8.6 | 0.1/0.1 |
| 13 | R112H | Late onset | Chien et al. (2013) and Eng et al. (1994) | B | 10.7 | 1.7c/0.1 |
| 14 | R227Q | Classic | Eng et al. (1993) | B | 8.3 | 0.1/0.1 |
| 15 | R301X | Classic | Eng et al. (1994) | F | 13.2 | ND/0.4 |
| Mean (SD) | 28.5 (16.1) | 9.2 (4.7)d | ||||
| Median (range) | 24.0 (5–61) | 9.3 (1.9–18.8) | ||||
α-Gal A α-galactosidase A, NA not available, ND not detectable, SD standard deviation
aAge was calculated from the date of birth to the date of the first study infusion of agalsidase beta. To protect patients’ data privacy, age categories are used: A, >0 to ≤10 years; B, >10 to ≤20 years; C, >20 to ≤30 years; D, >30 to ≤40 years; E, >40 to ≤50 years; F, >50 years
bDuration of Fabry disease was calculated from the date of initial diagnosis of Fabry disease to the date of the first study infusion
cIn nmol/h/mg protein
d n = 14; the date of diagnosis was not available for one patient
The majority (86.7%) of subjects included in the study was Caucasian, with a mean age (standard deviation [SD]) at enrollment of 28.5 (16.1) years. The median duration of FD at baseline (from initial diagnosis to first study treatment) was 9.3 years (range 2–19 years). Prior to agalsidase alfa treatment, nine patients received agalsidase beta treatment for a mean of 3.5 years (range 0.3–7 years), and six were ERT-naive. Patients had received agalsidase alfa (0.2 mg/kg EOW) for a mean of 3.7 years (range 1.6–14 years) before switching to agalsidase beta (1.0 mg/kg EOW) (Fig. 1).
Among the patients with known GLA gene mutations, 12 patients had mutations previously reported to be associated with a classical presentation of FD, and two had mutations reported to be associated with later-onset FD (patients 9 and 13) (references are presented in Table 1).
Plasma Lyso-GL-3
The patient-level plasma lyso-GL-3 concentrations according to Clinical visit after the switch to agalsidase beta at 1.0 mg/kg EOW are presented in Fig. 2.
Fig. 2.
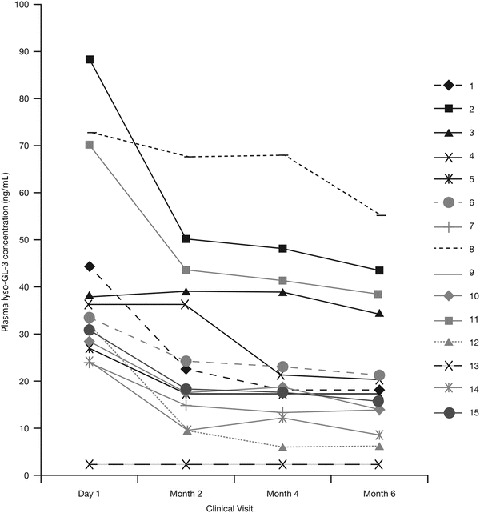
Patient-level plasma lyso-GL-3 concentrations according to Clinical visit after the switch to agalsidase beta at 1.0 mg/kg every other week. Figure includes three of the four patients (patients 2, 3, 8, and 11) who had the highest agalsidase antibody titers before switch. Symbols overlap for two patients (9 and 13) whose lyso-GL-3 concentrations were below quantitative limits at baseline and throughout the study. These patients have GLA gene mutations reported to be associated with a later-onset form of Fabry disease. Lyso-GL-3 globotriaosylsphingosine
Plasma lyso-GL-3 concentration decreased significantly within 2 months of the switch to agalsidase beta 1.0 mg/kg EOW, and the reduction continued at months 4 and 6. The mean absolute change in plasma lyso-GL-3 concentration was −12.8 ng/mL (P < 0.001) at month 2, −16.1 ng/mL (P < 0.001) at month 4, and −16.7 ng/mL (P < 0.001) at month 6. The mean (SD) percentage reduction from baseline in plasma lyso-GL-3 concentration was 39.5% (23.57) (P < 0.001) at month 6.
In most cases, the plasma lyso-GL-3 concentration decreased consistently at each successive assessment for each patient (Fig. 2). The two patients with a GLA mutation associated with later-onset FD (patients 9 and 13) had lyso-GL-3 concentrations below quantitative limits at study baseline and throughout the study.
Plasma GL-3
The patient-level plasma GL-3 concentrations according to Clinical visit after the switch to agalsidase beta at 1.0 mg/kg EOW are presented in Fig. 3.
Fig. 3.
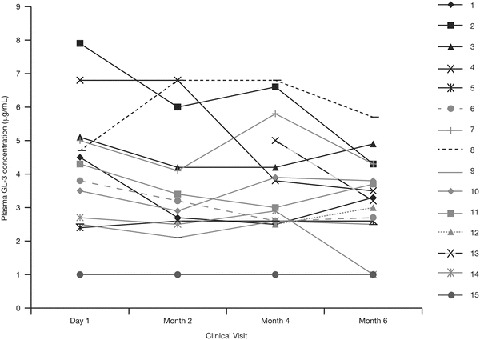
Patient-level plasma GL-3 concentrations according to Clinical visit after the switch to agalsidase beta at 1.0 mg/kg every other week. For two patients (patients 12 and 13), baseline and month 2 values were not available. GL-3 globotriaosylceramide
The mean absolute change from baseline in plasma GL-3 concentration was −0.5 μg/mL at month 2, −0.7 μg/mL at month 4, and −0.9 μg/mL at month 6. The mean absolute change from baseline at month 6 was statistically significant (P < 0.05). The mean percentage change in plasma GL-3 concentration at month 6 was −17.9% (P < 0.05).
Urine GL-3
The patient-level urine GL-3 concentrations according to Clinical visit after the switch to agalsidase beta at 1.0 mg/kg EOW are presented in Fig. 4. The two patients with a GLA mutation associated with later-onset FD (patients 9 and 13) had urine GL-3 concentrations below quantitative limits at study baseline and throughout the study.
Fig. 4.
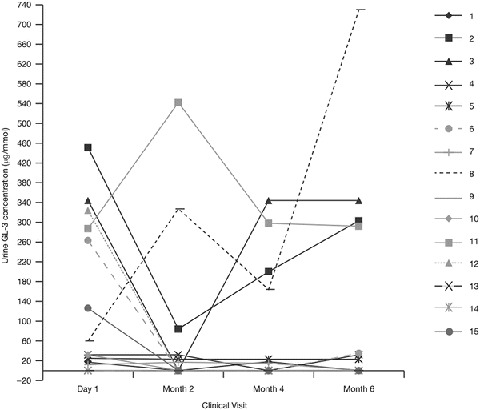
Patient-level urine GL-3 concentrations according to clinical visit after the switch to agalsidase beta at 1.0 mg/kg every other week. GL-3 globotriaosylceramide
The median absolute change from baseline in urine GL-3 concentration was −7.6 μg/mmol at month 2, −9.9 μg/mmol at month 4, and −11.1 μg/mmol at month 6. None of these median changes from baseline were statistically significant.
The median percentage change from baseline in median urine GL-3 concentration was −33.8% at month 6. None of the median percentage changes from baseline in urine GL-3 were statistically significant.
Serum Anti-agalsidase Beta IgG Antibody Titers
Prior to first agalsidase beta infusion, eight patients were positive for serum IgG antibodies to both agalsidase alfa and agalsidase beta when the same assay format was used (patients 1, 2, 3, 4, 6, 8, 11, and 15); patients 1, 3, and 8 had never been treated with agalsidase beta (patient 8 had the highest pre-agalsidase beta antibody titer) (Fig. 5). Seven patients were seronegative for IgG antibodies to both agalsidase beta and agalsidase alfa, four of whom had been treated with both agalsidase alfa and agalsidase beta (patients 7, 9, 10, and 12). No patient was positive for antibodies against one ERT and negative for the other ERT. Titers of antibodies to agalsidase alfa and agalsidase beta at baseline were not significantly different (no difference of >1 dilution), even in the six patients who had not yet been exposed to agalsidase beta (patients 1, 3, 5, 8, 13, and 14). Patient 14 was the only seronegative patient who seroconverted during the agalsidase beta study treatment period. He seroconverted by month 2 but had low antibody titers (maximum antibody titer 1:400).
Fig. 5.
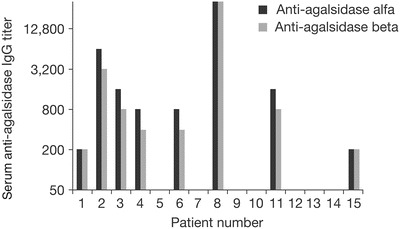
Serum anti-agalsidase IgG titers at baseline. Titers of antibodies to agalsidase alfa and agalsidase beta at baseline were not significantly different even in patients who had not yet been exposed to agalsidase beta (patients 1, 3, 5, 8, 13, and 14). Ig immunoglobulin
In patients who were positive for agalsidase beta antibodies, there was no discernible pattern between the titer measured at any time point during the study and concentration of lyso-GL-3, plasma GL-3, or urine GL-3; patients who were negative for antibodies at baseline were among those with the lowest plasma concentrations of lyso-GL-3 and GL-3.
Patient 8, who had a baseline antibody titer of 102,000, was found to be positive in the neutralizing activity and enzyme uptake inhibition assays. Prior to enrollment into the trial, he had received agalsidase alfa for 1.6 years and had not been exposed to agalsidase beta. His enzyme activity inhibition was >100% and the uptake assay was positive. The level of neutralizing activity (expressed in micrograms of recombinant human α-Gal inhibited by 1 mL of patient sera) decreased from 538 to 389 at 6 months, and lyso-GL-3 decreased by 23% over the course of 6 months.
None of the other patients demonstrated uptake inhibition, and only one additional patient (patient 11) had some inhibition of enzyme activity at 2 months, which subsequently decreased. This patient’s lyso-GL-3 level decreased significantly at 2 months of agalsidase beta treatment (37%) and continued to further decrease at 4 months (41%) and 6 months (45%).
Adverse Events
There were no SAEs reported during the study. Only one of the 15 patients (7%) experienced a non-SAE (an infusion-associated reaction) following completion of the first agalsidase beta infusion after switching. The AE resolved after 2 days and the patient continued treatment without experiencing any further AEs. The serum sample drawn prior to the first agalsidase beta infusion was positive for agalsidase IgG antibodies (titer 1:3,200).
Discussion
This study demonstrates that, in male patients with FD, increasing the dose of ERT by switching from agalsidase alfa at 0.2 mg/kg EOW to agalsidase beta at 1.0 mg/kg EOW can further reduce plasma lyso-GL-3 and GL-3 concentrations beyond reductions previously achieved with agalsidase alfa. These reductions were evident 2 months after increasing the dose of agalsidase. The reductions provide further evidence that the in vivo activity of agalsidase alfa at its recommended dose is below that of agalsidase beta at its recommended dose. These results underscore the importance of dose in the treatment of FD and corroborate findings by other groups (Smid et al. 2011; Tøndel et al. 2013; van Breemen et al. 2011; Weidemann et al. 2014). In previously untreated patients, plasma lyso-GL-3 decreased, reaching almost stable levels within 3 months of initiation of treatment with either agalsidase alfa or agalsidase beta administered at 0.2 mg/kg, and with 1.0 mg/kg of agalsidase beta (van Breemen et al. 2011). However, the reduction of plasma lyso-GL-3 was significantly greater for patients treated with agalsidase beta 1.0 mg/kg versus patients who received agalsidase alfa or agalsidase beta at a dose of 0.2 mg/kg. Plasma lyso-GL-3 concentrations tended to be higher in antibody-positive males when treated with agalsidase alfa or agalsidase beta at 0.2 mg/kg, compared with antibody-negative males. This was not the case after 12 months of agalsidase beta 1.0 mg/kg; the lyso-GL-3 reduction at this dose was similar for antibody-positive and antibody-negative patients (van Breemen et al. 2011), suggesting that a dose increase can compensate for potentially reduced efficacy in patients with anti-agalsidase antibodies. Another study reported a significant increase in lyso-GL-3 concentration in adult male patients who switched from agalsidase beta 1.0 mg/kg to agalsidase alfa 0.2 mg/kg or received a reduced agalsidase beta dose (one group subsequently switched to agalsidase alfa), suggesting recurrence of disease activity (Smid et al. 2011). The observed relapse in plasma lyso-GL-3 elevation in almost all patients within a few months after agalsidase dose reduction in that cohort led experts to conclude that there is an ERT dose effect on plasma lyso-GL-3 levels and that the biochemical response to ERT is suboptimal at an agalsidase dose of 0.2 mg/kg EOW in the majority of classic FD patients (Smid et al. 2011; Ferraz et al. 2014). Systematic and complete data collections from 105 FD patients showed that patients receiving agalsidase beta at 1.0 mg/kg EOW had a stable disease course, whereas patients whose agalsidase beta dose was reduced and patients who were switched to agalsidase alfa experienced worsening of renal function and deterioration of Fabry-related symptoms (Warnock and Mauer 2014; Weidemann et al. 2014). Moreover, the magnitude of GL-3 clearance in podocytes (terminally differentiated cells) in young patients (median age 16.5 years) was dependent on the cumulative dose of agalsidase beta or alfa administered over time (Tøndel et al. 2013).
An important consideration of ERT with agalsidase is the patient’s immunological response to the enzyme. In this study, titers of serum IgG antibodies to both agalsidase alfa and agalsidase beta were measured using the same assay format, and the findings suggest that agalsidase alfa and agalsidase beta have epitopes in common; over half of the patients who had never been exposed to agalsidase beta were positive for agalsidase beta antibodies at baseline. Importantly, no relationship was apparent between IgG antibody titers and concentration of plasma lyso-GL-3, plasma GL-3, or urine GL-3 in patients receiving agalsidase beta 1.0 mg/kg EOW. In contrast, as noted above, patients with antibodies receiving 0.2 mg/kg of either agalsidase alfa or agalsidase beta tended toward smaller correction in plasma lyso-GL-3 concentrations compared with patients receiving agalsidase beta 1.0 mg/kg, who experienced a reduction similar to patients without antibodies (van Breemen et al. 2011). It is important to note that, to date, a comparison of antibody test results obtained from different laboratories and different studies has been hampered by the lack of a standardized assay and reference standard (Schellekens 2008). The number of samples that test positive for neutralizing activity and/or inhibition of uptake is dependent on the sensitivity and specificity of the method (Fabrazyme® Prescribing information, last updated May 2010). Thus, it is not surprising that neutralizing antibody activity results from this study differ from results reported by other groups (Linthorst et al. 2004; Lenders et al. 2015). Studies have demonstrated that both agalsidase alfa and agalsidase beta are structurally equivalent and, on a milligram basis, induce similar, fully cross-reactive, antibody responses in vivo (Blom et al. 2003; Lee et al. 2003; Linthorst et al. 2004), refuting the suggestion that variation in glycosylation patterns between agalsidase beta and agalsidase alfa may have implications for the long-term safety of ERT (Barbey et al. 2008). The perceived difference in immunogenicity of the two enzymes could be based on differences in analytical testing.
Our observations further support the suggestion that plasma lyso-GL-3 could be a sensitive pharmacodynamic biomarker for dose–response effects of ERT in FD. Of the glycosphingolipids examined in this study, lyso-GL-3 showed more consistent changes in concentration than did plasma or urine GL-3. Lyso-GL-3 concentration was significantly reduced from baseline at each successive Clinical assessment, and individual patient values of plasma lyso-GL-3 concentration fluctuated less than did those of plasma and urine GL-3 concentrations (data not shown). Although during the past decade attention has largely focused on the pathogenic role of GL-3 in FD (DeGraba et al. 2000; Park et al. 2011; Shen et al. 2008), the importance of lyso-GL-3 in the FD process has become evident. For example, exposure of smooth muscle cells in vitro to lyso-GL-3 at concentrations similar to those seen in patients with FD (e.g., in the patients treated for 12 months with agalsidase alfa or beta at 0.2 mg/kg [van Breemen et al. 2011]) stimulates proliferation of the smooth muscle cells (Aerts et al. 2008), a finding consistent with the increased intima–media thickness observed in patients with FD (Barbey et al. 2006).
Lyso-GL-3 has also been proposed to have a role in renal injury by causing podocyte injury and glomerular accumulation of extracellular matrix – key characteristics of glomerulosclerosis (Sanchez-Niño et al. 2011). In vitro, human podocytes exposed to lyso-GL-3 increased their expression of mediators of glomerular injury (transforming growth factor [TGF]-β1, the extracellular matrix proteins fibronectin and type IV collagen, and CD74) in a dose- and time-dependent manner, and the effect of lyso-GL-3 on extracellular matrix production was mediated by TGF-β1 (Sanchez-Niño et al. 2011).
It has not yet been unequivocally demonstrated that lyso-GL-3 contributes to manifestations of FD in vivo. However, studies in patients with classic FD have explored correlations between levels of plasma lyso-GL-3 (a potential pathogenic factor inducing vascular dysfunction [Rombach et al. 2012]) and disease manifestations (Rombach et al. 2010; Rombach et al. 2012). One study found that lifetime exposure to plasma lyso-GL-3 correlates with severity of FD manifestations (assessed by Mainz severity scoring index), and plasma lyso-GL-3 was reported to be an independent risk factor for development of cerebrovascular white matter lesions in male patients and left ventricular hypertrophy in female patients with classic FD. No correlation was observed between plasma lyso-GL-3 and renal function (Rombach et al. 2010). Other studies found significant correlations between carotid intima–media thickness (previously reported to be predictive of stroke [Chambless et al. 2000]) and plasma lyso-GL-3 levels in female patients with classic FD, and between exposure to lyso-GL-3 and cold detection threshold and thermal sensory limen at the upper limb (Biegstraaten et al. 2012).
No clinical outcomes were assessed in our 6-month study because FD is a chronic disease and progresses slowly, and, therefore, no conclusions can be drawn from this short study regarding the correlation of observed reductions in plasma lyso-GL-3 with clinically relevant outcomes. As acknowledged by others, following the biochemical response may prove to be a valuable tool in clinical management of FD patients if treatment is started early, before irreversible damage has occurred (van Breemen et al. 2011), and is probably the most sensitive way to evaluate the effects of a change in agalsidase dose in short-term studies (Smid et al. 2011). The switch from agalsidase alfa to agalsidase beta in our study was safe and well tolerated.
Of note, the methods used to measure lyso-GL-3 in the literature vary. Standardization of these methods would allow comparison of lyso-GL-3 data from different studies. Moreover, discussing our results in the context of results of recently published agalsidase beta – agalsidase alfa switch studies is hampered by the fact that these studies did not report plasma lyso-GL-3 or GL-3 data (Pisani et al. 2013), included Fabry-variant patients with normal or low lyso-GL-3 levels at baseline (six out of nine patients) (Lin et al. 2014), and included many female patients with low lyso-GL-3 at baseline (seven out of 11 patients) (Tsuboi and Yamamoto 2014).
The small number of patients and the lack of long-term clinical follow-up are limitations of our study. However, although fewer patients were included in the analysis than required by the statistical power calculation, a statistically significant difference in lyso-GL-3 clearance was still detected. Although a two-arm crossover design with a switch from agalsidase alfa to agalsidase beta followed by a crossover to alternate ERT product would have been a more robust design than our open-label, single-arm study, this would have been challenging to conduct after termination of the US treatment Investigational New Drug permit for agalsidase alfa in June 2012. Given that the long-term clinical benefits of ERT for patients with FD are still unclear, the study was not intended to assess clinical outcomes related to dose differences and only measured GL-3 and lyso-GL-3 concentrations. The lack of pre-study data is another limitation of this study. However, as already discussed, the lack of a standardized assay and reference standard precludes comparisons of glycosphingolipid and antibody test results across studies. With regard to the impact of ERT on plasma lyso-GL-3 levels, it is important to note that lyso-GL-3 levels in classic FD patients have been reported to decrease rapidly after start of ERT in a dose-dependent manner, reaching almost stable levels within 3 months of treatment (van Breemen et al. 2011; Ferraz et al. 2014). Therefore, it was expected that the patients in the current study should have reached a maximal reduction in plasma lyso-GL-3 levels during the agalsidase alfa (0.2 mg/kg EOW) treatment period (mean 3.7 years, range 1.6–14 years) prior to switching to agalsidase beta (1.0 mg/kg EOW).
In conclusion, the reduction in plasma lyso-GL-3 and GL-3 observed in patients after switch from agalsidase alfa to agalsidase beta indicates that the recommended dose of agalsidase beta at 1.0 mg/kg EOW has a greater pharmacodynamic effect on these markers. Plasma lyso-GL-3 appears to be a biomarker to assess the initial response to ERT in patients with FD. Structural similarities between the two ERTs may suggest similar immunogenic profiles. Additionally, despite a fivefold increase in ERT dose, switching to a higher-dose ERT (agalsidase beta) is safe and well tolerated. Further studies with a larger sample size are needed to determine the clinical implications of lyso-GL-3 as an indicator of disease severity and progression and the clinical response to treatment.
Acknowledgments
The authors received editorial/writing support in the preparation of this manuscript provided by Alessia Piazza, PhD, of Excerpta Medica, funded by Genzyme. The authors were responsible for all content and editorial decisions and have not received honoraria related to the development of this publication.
The authors would like to acknowledge the staff at the Genzyme Clinical Specialty Laboratory (Framingham, MA, USA) for performing all laboratory assays.
We dedicate this manuscript to the memory of John A. Barranger who acted as the principal study investigator but sadly passed away.
Synopsis
Agalsidase beta at a dose of 1.0 mg/kg administered EOW may further reduce plasma and urine glycosphingolipid concentrations beyond reductions previously achieved with agalsidase alfa in patients with Fabry disease.
Compliance with Ethics Guidelines
Conflict of Interest
Ozlem Goker-Alpan has received research support (Actelion, Shire HGT, Genzyme, Amicus, Pfizer-Protalix Biotherapeutics), payments for consultancy (Actelion, Shire HGT, Pfizer-Protalix Biotherapeutics), and speaker bureaus (Actelion, Shire HGT, Genzyme). Daniel J. Gruskin and Larry Blankstein are Genzyme employees. Neal J. Weinreb receives travel reimbursements and/or honoraria and/or research support from Shire HGT, Genzyme, Pfizer Corporation, and Actelion Corporation. Michael J. Gambello, Gustavo H.B. Maegawa, and Khan J. Nedd declare that they have no conflict of interest.
The study is registered at www.ClinicalTrials.gov under the identifier NCT01650779 and was sponsored by Genzyme, a Sanofi company.
Patient Consent Statement
All procedures followed were in accordance with the ethical standards of the responsible committee on human experimentation (institutional and national) and with the Helsinki Declaration of 1975, as revised in 2000. Informed consent was obtained from all patients, or their parents, or legal guardians, for being included in the study.
Details of the Contributions of Individual Authors
Daniel J. Gruskin and Larry Blankstein were involved in the study planning and coordination of statistical analyses. Ozlem Goker-Alpan, Michael J. Gambello, Gustavo H.B. Maegawa, Khan J. Nedd, and Neal J. Weinreb were involved in the study conduct. All authors contributed to the first draft of the manuscript, were involved in the critical review and revision of subsequent drafts, and approved the final draft for submission.
Footnotes
Competing interests: None declared
References
- Aerts JM, Groener JE, Kuiper S, et al. Elevated globotriaosylsphingosine is a hallmark of Fabry disease. Proc Natl Acad Sci U S A. 2008;105:2812–2817. doi: 10.1073/pnas.0712309105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbey F, Brakch N, Linhart A, et al. Cardiac and vascular hypertrophy in Fabry disease: evidence for a new mechanism independent of blood pressure and glycosphingolipid deposition. Arterioscler Thromb Vasc Biol. 2006;26:839–844. doi: 10.1161/01.ATV.0000209649.60409.38. [DOI] [PubMed] [Google Scholar]
- Barbey F, Lidove O, Schwarting A. Fabry nephropathy: 5 years of enzyme replacement therapy – a short review. NDT Plus. 2008;1:11–19. doi: 10.1093/ndtplus/sfm022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biegstraaten M, Hollak CE, Bakkers M, Faber CG, Aerts JM, van Schaik IN. Small fiber neuropathy in Fabry disease. Mol Genet Metab. 2012;106:135–141. doi: 10.1016/j.ymgme.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Blanch LC, Meaney C, Morris CP. A sensitive mutation screening strategy for Fabry disease: detection of nine mutations in the alpha-galactosidase A gene. Hum Mutat. 1996;8:38–43. doi: 10.1002/(SICI)1098-1004(1996)8:1<38::AID-HUMU5>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- Blom D, Speijer D, Linthorst GE, Donker-Koopman WG, Strijland A, Aerts JM. Recombinant enzyme therapy for Fabry disease: absence of editing of human alpha-galactosidase A mRNA. Am J Hum Genet. 2003;72:23–31. doi: 10.1086/345309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambless LE, Folsom AR, Clegg LX, et al. Carotid wall thickness is predictive of incident clinical stroke: the Atherosclerosis Risk in Communities (ARIC) study. Am J Epidemiol. 2000;151:478–487. doi: 10.1093/oxfordjournals.aje.a010233. [DOI] [PubMed] [Google Scholar]
- Chien YH, Bodamer OA, Chiang SC, Mascher H, Hung C, Hwu WL. Lyso-globotriaosylsphingosine (lyso-Gb3) levels in neonates and adults with the Fabry disease later-onset GLA IVS4+919G>A mutation. J Inherit Metab Dis. 2013;36:881–885. doi: 10.1007/s10545-012-9547-1. [DOI] [PubMed] [Google Scholar]
- Clarke JT, West ML, Bultas J, Schiffmann R. The pharmacology of multiple regimens of agalsidase alfa enzyme replacement therapy for Fabry disease. Genet Med. 2007;9:504–509. doi: 10.1097/GIM.0b013e318133fb1b. [DOI] [PubMed] [Google Scholar]
- DeGraba T, Azhar S, Dignat-George F, et al. Profile of endothelial and leukocyte activation in Fabry patients. Ann Neurol. 2000;47:229–233. doi: 10.1002/1531-8249(200002)47:2<229::AID-ANA13>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Desnick RJ. Enzyme replacement therapy for Fabry disease: lessons from two alpha-galactosidase A orphan products and one FDA approval. Expert Opin Biol Ther. 2004;4:1167–1176. doi: 10.1517/14712598.4.7.1167. [DOI] [PubMed] [Google Scholar]
- Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307–335. doi: 10.1146/annurev-genom-090711-163739. [DOI] [PubMed] [Google Scholar]
- Eng CM, Resnick-Silverman LA, Niehaus DJ, Astrin KH, Desnick RJ. Nature and frequency of mutations in the alpha-galactosidase A gene that cause Fabry disease. Am J Hum Genet. 1993;53:1186–1197. [PMC free article] [PubMed] [Google Scholar]
- Eng CM, Niehaus DJ, Enriquez AL, Burgert TS, Ludman MD, Desnick RJ. Fabry disease: twenty-three mutations including sense and antisense CpG alterations and identification of a deletional hot-spot in the alpha-galactosidase A gene. Hum Mol Genet. 1994;3:1795–1799. doi: 10.1093/hmg/3.10.1795. [DOI] [PubMed] [Google Scholar]
- Eng CM, Ashley GA, Burgert TS, Enriquez AL, D’Souza M, Desnick RJ. Fabry disease: thirty-five mutations in the alpha-galactosidase A gene in patients with classic and variant phenotypes. Mol Med. 1997;3:174–182. [PMC free article] [PubMed] [Google Scholar]
- Eng CM, Guffon N, Wilcox WR, et al. Safety and efficacy of recombinant human alpha-galactosidase A – replacement therapy in Fabry’s disease. N Engl J Med. 2001;345:9–16. doi: 10.1056/NEJM200107053450102. [DOI] [PubMed] [Google Scholar]
- Eng CM, Fletcher J, Wilcox WR, et al. Fabry disease: baseline characteristics of a cohort of 1765 males and females in the Fabry Registry. J Inherit Metab Dis. 2007;30:184–192. doi: 10.1007/s10545-007-0521-2. [DOI] [PubMed] [Google Scholar]
- Fabrazyme® Summary of product characteristics, last updated October 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000370/WC500020547.pdf. Accessed 23 Jul 2015
- Fabrazyme® Prescribing information, last updated May 2010. http://www.fabrazyme.com/hcp/pi/fz_us_hc_pi.pdf. Accessed 23 Jul 2015
- Ferraz MJ, Kallemeijn WW, Mirzaian M, et al. Gaucher disease and Fabry disease: new markers and insights in pathophysiology for two distinct glycosphingolipidoses. Biochim Biophys Acta. 2014;1841:811–825. doi: 10.1016/j.bbalip.2013.11.004. [DOI] [PubMed] [Google Scholar]
- Germain DP. Fabry disease. Orphanet J Rare Dis. 2010;5:30. doi: 10.1186/1750-1172-5-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hopkin RJ, Bissler J, Banikazemi M, et al. Characterization of Fabry disease in 352 pediatric patients in the Fabry Registry. Pediatr Res. 2008;64:550–555. doi: 10.1203/PDR.0b013e318183f132. [DOI] [PubMed] [Google Scholar]
- Keslová-Veselíková J, Hůlková H, Dobrovolný R, et al. Replacement of alpha-galactosidase A in Fabry disease: effect on fibroblast cultures compared with biopsied tissues of treated patients. Virchows Arch. 2008;452:651–665. doi: 10.1007/s00428-008-0586-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee K, Jin X, Zhang K, et al. A biochemical and pharmacological comparison of enzyme replacement therapies for the glycolipid storage disorder Fabry disease. Glycobiology. 2003;13:305–313. doi: 10.1093/glycob/cwg034. [DOI] [PubMed] [Google Scholar]
- Lee BH, Heo SH, Kim GH, et al. Mutations of the GLA gene in Korean patients with Fabry disease and frequency of the E66Q allele as a functional variant in Korean newborns. J Hum Genet. 2010;55:512–517. doi: 10.1038/jhg.2010.58. [DOI] [PubMed] [Google Scholar]
- Lenders M, Stypmann J, Duning T, Schmitz B, Brand SM, Brand E (2015) Serum-mediated inhibition of enzyme replacement therapy in Fabry disease. J Am Soc Nephrol. pii: ASN.2014121226 (Epub ahead of print) [DOI] [PMC free article] [PubMed]
- Lin HY, Huang YH, Liao HC, et al. Clinical observations on enzyme replacement therapy in patients with Fabry disease and the switch from agalsidase beta to agalsidase alfa. J Chin Med Assoc. 2014;77:190–197. doi: 10.1016/j.jcma.2013.11.006. [DOI] [PubMed] [Google Scholar]
- Linthorst GE, Hollak CE, Donker-Koopman WE, Strijland A, Aerts JM. Enzyme therapy for Fabry disease: neutralizing antibodies toward agalsidase alpha and beta. Kidney Int. 2004;66:1589–1595. doi: 10.1111/j.1523-1755.2004.00924.x. [DOI] [PubMed] [Google Scholar]
- Meaney C, Blanch LC, Morris CP. A nonsense mutation (R220X) in the alpha-galactosidase A gene detected in a female carrier of Fabry disease. Hum Mol Genet. 1994;3:1019–1020. doi: 10.1093/hmg/3.6.1019. [DOI] [PubMed] [Google Scholar]
- Nelson BC, Roddy T, Araghi S, et al. Globotriaosylceramide isoform profiles in human plasma by liquid chromatography-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 2004;805:127–134. doi: 10.1016/j.jchromb.2004.02.032. [DOI] [PubMed] [Google Scholar]
- Niemann M, Rolfs A, Störk S, et al. Gene mutations versus clinically relevant phenotypes: lyso-Gb3 defines Fabry disease. Circ Cardiovasc Genet. 2014;7:8–16. doi: 10.1161/CIRCGENETICS.113.000249. [DOI] [PubMed] [Google Scholar]
- Park S, Kim JA, Joo KY, et al. Globotriaosylceramide leads to K(Ca)3.1 channel dysfunction: a new insight into endothelial dysfunction in Fabry disease. Cardiovasc Res. 2011;89:290–299. doi: 10.1093/cvr/cvq333. [DOI] [PubMed] [Google Scholar]
- Pisani A, Spinelli L, Visciano B, et al. Effects of switching from agalsidase beta to agalsidase alfa in 10 patients with Anderson-Fabry disease. JIMD Rep. 2013;9:41–48. doi: 10.1007/8904_2012_177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Replagal® Summary of product characteristics, last updated September 2014. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/000369/WC500053612.pdf. Accessed 23 Jul 2015
- Roddy TP, Nelson BC, Sung CC, et al. Liquid chromatography-tandem mass spectrometry quantification of globotriaosylceramide in plasma for long-term monitoring of Fabry patients treated with enzyme replacement therapy. Clin Chem. 2005;51:237–240. doi: 10.1373/clinchem.2004.038323. [DOI] [PubMed] [Google Scholar]
- Rombach SM, Dekker N, Bouwman MG, et al. Plasma globotriaosylsphingosine: diagnostic value and relation to clinical manifestations of Fabry disease. Biochim Biophys Acta. 2010;1802:741–748. doi: 10.1016/j.bbadis.2010.05.003. [DOI] [PubMed] [Google Scholar]
- Rombach SM, van den Bogaard B, de Groot E, et al. Vascular aspects of Fabry disease in relation to clinical manifestations and elevations in plasma globotriaosylsphingosine. Hypertension. 2012;60:998–1005. doi: 10.1161/HYPERTENSIONAHA.112.195685. [DOI] [PubMed] [Google Scholar]
- Sakuraba H, Murata-Ohsawa M, Kawashima I, et al. Comparison of the effects of agalsidase alfa and agalsidase beta on cultured human Fabry fibroblasts and Fabry mice. J Hum Genet. 2006;51:180–188. doi: 10.1007/s10038-005-0342-9. [DOI] [PubMed] [Google Scholar]
- Sanchez-Niño MD, Sanz AB, Carrasco S, et al. Globotriaosylsphingosine actions on human glomerular podocytes: implications for Fabry nephropathy. Nephrol Dial Transplant. 2011;26:1797–1802. doi: 10.1093/ndt/gfq306. [DOI] [PubMed] [Google Scholar]
- Schaefer RM, Tylki-Szymańska A, Hilz MJ. Enzyme replacement therapy for Fabry disease: a systematic review of available evidence. Drugs. 2009;69:2179–2205. doi: 10.2165/11318300-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Schellekens H. The immunogenicity of therapeutic proteins and the Fabry antibody standardization initiative. Clin Ther. 2008;30(Suppl B):S50–S51. doi: 10.1016/S0149-2918(08)80041-0. [DOI] [PubMed] [Google Scholar]
- Schiffmann R, Murray GJ, Treco D, et al. Infusion of alpha-galactosidase A reduces tissue globotriaosylceramide storage in patients with Fabry disease. Proc Natl Acad Sci U S A. 2000;97:365–370. doi: 10.1073/pnas.97.1.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiffmann R, Martin RA, Reimschisel T, et al. Four-year prospective clinical trial of agalsidase alfa in children with Fabry disease. J Pediatr. 2010;156:832–837. doi: 10.1016/j.jpeds.2009.11.007. [DOI] [PubMed] [Google Scholar]
- Shabbeer J, Yasuda M, Benson SD, Desnick RJ. Fabry disease: identification of 50 novel alpha-galactosidase A mutations causing the classic phenotype and three-dimensional structural analysis of 29 missense mutations. Hum Genomics. 2006;2:297–309. doi: 10.1186/1479-7364-2-5-297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen JS, Meng XL, Moore DF, et al. Globotriaosylceramide induces oxidative stress and up-regulates cell adhesion molecule expression in Fabry disease endothelial cells. Mol Genet Metab. 2008;95:163–168. doi: 10.1016/j.ymgme.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirrs SM, Bichet DG, Casey R, et al. Outcomes of patients treated through the Canadian Fabry disease initiative. Mol Genet Metab. 2014;111:499–506. doi: 10.1016/j.ymgme.2014.01.014. [DOI] [PubMed] [Google Scholar]
- Smid BE, Rombach SM, Aerts JM, et al. Consequences of a global enzyme shortage of agalsidase beta in adult Dutch Fabry patients. Orphanet J Rare Dis. 2011;6:69. doi: 10.1186/1750-1172-6-69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Togawa T, Kodama T, Suzuki T, et al. Plasma globotriaosylsphingosine as a biomarker of Fabry disease. Mol Genet Metab. 2010;100:257–261. doi: 10.1016/j.ymgme.2010.03.020. [DOI] [PubMed] [Google Scholar]
- Togawa T, Takada M, Aizawa Y, Tsukimura T, Chiba Y, Sakuraba H. Comparative study on mannose 6-phosphate residue contents of recombinant lysosomal enzymes. Mol Genet Metab. 2014;111:369–373. doi: 10.1016/j.ymgme.2013.12.296. [DOI] [PubMed] [Google Scholar]
- Tøndel C, Bostad L, Larsen KK, et al. Agalsidase benefits renal histology in young patients with Fabry disease. J Am Soc Nephrol. 2013;24:137–148. doi: 10.1681/ASN.2012030316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topaloglu AK, Ashley GA, Tong B, et al. Twenty novel mutations in the alpha-galactosidase A gene causing Fabry disease. Mol Med. 1999;5:806–811. [PMC free article] [PubMed] [Google Scholar]
- Tsuboi K, Yamamoto H. Clinical course of patients with Fabry disease who were switched from agalsidase-β to agalsidase-α. Genet Med. 2014;16:766–772. doi: 10.1038/gim.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Breemen MJ, Rombach SM, Dekker N, et al. Reduction of elevated plasma globotriaosylsphingosine in patients with classic Fabry disease following enzyme replacement therapy. Biochim Biophys Acta. 2011;1812:70–76. doi: 10.1016/j.bbadis.2010.09.007. [DOI] [PubMed] [Google Scholar]
- Vedder AC, Linthorst GE, Houge G, et al. Treatment of Fabry disease: outcome of a comparative trial with agalsidase alfa or beta at a dose of 0.2 mg/kg. PLoS One. 2007;2 doi: 10.1371/journal.pone.0000598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnock DG, Mauer M. Fabry disease: dose matters. J Am Soc Nephrol. 2014;25:653–655. doi: 10.1681/ASN.2013121322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weidemann F, Krämer J, Duning T, et al. Patients with Fabry disease after enzyme replacement therapy dose reduction versus treatment switch. J Am Soc Nephrol. 2014;25:837–849. doi: 10.1681/ASN.2013060585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox WR, Banikazemi M, Guffon N, et al. Long-term safety and efficacy of enzyme replacement therapy for Fabry disease. Am J Hum Genet. 2004;75:65–74. doi: 10.1086/422366. [DOI] [PMC free article] [PubMed] [Google Scholar]