

Abstract
Krabbe disease is an autosomal recessive neurodegenerative lysosomal storage disease caused by the deficiency of β-galactocerebrosidase. This deficiency results in the impaired degradation of β-galactocerebroside, a major myelin lipid, and of galactosylsphingosine. Based on the age of onset of neurological symptoms, an infantile form (90% patients) and late-onset forms (10% patients) of the disease are recognized. Over 130 disease-causing mutations have been identified in the β-galactocerebrosidase gene. We present the biochemical and molecular findings in 19 cases of Krabbe disease, 17 of them unrelated, diagnosed in Greece over the last 30 years. β-Galactocerebrosidase activity assayed in leukocyte homogenates using either the tritium-labeled or the fluorescent substrate was diagnostic for all. Increased plasma chitotriosidase activity was found in 11/15 patients.
Mutational analysis, carried out in 11 unrelated cases, identified seven different mutations, four previously described (p.I250T, c.1161+6532_polyA+9kbdel, p.K139del, p.D187V) and three novel mutations (p.D610A, c.583-1 G>C, p.W132X), and seven distinct genotypes. The most prevalent mutation was mutation p.I250T, first described in a patient of Greek origin. It accounted for 36.4% (8/22) of the mutant alleles. The second most frequent mutation was c.1161+6532_polyA+9kbdel that accounted for 22.7% (5/22) of the mutant alleles. The observed frequency was lower than that described in Northern European countries and closer to that described in Italian patients.
Introduction
Krabbe disease, also known as globoid leukodystrophy (GLD, OMIM # 245200), is a rare autosomal recessive demyelinating disorder that belongs to the group of lysosomal storage diseases. It is caused by the deficiency of β-galactocerebrosidase (GALC, E.C. 3.2.146) which impairs the degradation of galactocerebroside, a major myelin lipid, and of galactosylsphingosine or psychosine (Suzuki 1998; Wenger et al. 2013). Accumulation of galactocerebroside elicits the formation of the characteristic multinucleated macrophages known as globoid cells. Psychosine is cytotoxic to oligodendrocytes and Schwann cells, arresting myelination and contributing to the progressive demyelination observed. According to the “psychosine hypothesis,” it is the main contributor to the disease pathology (Suzuki 1998; Svennerholm et al. 1980; Tanaka et al. 1989).
Based on the age of onset of neurological symptoms, an infantile form and late-onset forms of the disease are recognized. It has been reported that approximately 90% of the patients have the former and 10% the latter forms of the disease (Wenger 2008); however, a higher incidence for the later-onset forms is suggested in a recent report (Duffner et al. 2012). The galactocerebrosidase gene (GALC; MIM # 606890) was cloned by two groups more than 20 years ago (Chen and Wenger 1993; Sakai et al. 1994). It is localized on chromosome 14q31, has 17 exons and 16 introns, and spreads over 58 kb (Luzi et al. 1995). It encodes a 669-amino-acid (80 kDa) mature protein containing six potential glycosylation sites, which is proteolytically cleaved into 30 and 50 kDa forms that comprise the active form of the enzyme (Chen and Wenger 1993).
Up-to-date 131 mutations have been identified in the GALC gene, 128 of which have been reported to be the cause of Krabbe disease (Human Gene Mutation Database, HGMD professional 2015.1; Graziano and Cardile 2015; Wenger et al. 2001) that include missense, nonsense, deletion, and insertion mutations. Although genotype-phenotype correlations have been reported (Tappino et al. 2010 ; Wenger et al. 1997; Xu et al. 2006), the interpretation of the effects of mutations is complicated by the concomitant presence of polymorphisms (Harzer et al. 2002; Luzi et al. 1996; Wenger et al. 2001).
In Greece, the Institute of Child Health is the only center offering laboratory diagnosis for lysosomal storage diseases, including Krabbe disease. We report the biochemical and molecular findings in 19 cases of Krabbe disease diagnosed in our center over the last 30 years.
Patients and Methods
Patients
The present paper includes 19 patients with Krabbe disease, originating from all over Greece. They were referred for diagnosis to the Institute of Child Health on the basis of their clinical evaluation and neuroimaging studies.
Patients 8 and 9 and 15 and 16 are siblings. It is also reported that the family of patient 6 had lost a boy at the age of 10 months with psychomotor retardation, regression, and neurological disease.
A brief description of the patients is shown in Table 1. The study was approved by the Ethics Committee of the Institute of Child Health.
Table 1.
The clinical signs on referral along with the results of the biochemical investigations and DNA studies
| Patient | Sex | Age of onset | Age at diagnosis | Symptoms on referral | GALC activity (fluorescent substrate; nmol/mg protein/h) normal: 0.4–3.8 units | GALC activity (radioactive substrate; nmol/mg protein/h) normal: 0.1–0.97 units | Chitotriosidase activity (nmol/ml/h) normal: 0–150 units | Genetic variations: mutations/polymorphisms |
|---|---|---|---|---|---|---|---|---|
| 1 | F | 6 months | 9 months | Psychomotor regression, muscular hypertonia, myoclonic jerks, irritability, megalencephaly, ↑CSF protein level | – | 0.0 | 434 | c.1161+6532_polyA+9kbdel/c.1161+6532_polyA+9kbdel hom p.R184C(c.550C>T) |
| 2 | M | Unknown | 4 months | Seizures, irritability, feeding difficulties ↑CSF protein level |
– | 0.03 | 23 | – |
| 3 | F | 6 months | 10 months | Psychomotor retardation, axial hypotonia, ↑CSF protein level | – | 0.0 | – | – |
| 4 | M | 6 months | 8 months | Psychomotor regression, hypotonia, ↑CSF protein level, normal brain CT scan | – | 0.0 | 34 | – |
| 5 | M | 5 months | 5.5 months | Psychomotor retardation, cerebral palsy, hypertonia, ↑CSF protein level, normal brain CT scan | – | 0.0 | – | – |
| 6 | M | 26 months | 26.5 months | Spastic tetraplegia, loss of vision, abnormal MRI | – | 0.04 | 0.0 | – |
| 7 | M | 3 years 1 month | 3 years 5 months | Psychomotor regression, speech, walking and vision impairment, abnormal MRI | – | 0.01 | 800 | – |
| 8 | M | 3 months | 6.5 months | Infantile seizures, irritability, hypotonia, megalencephaly, psychomotor retardation, loss of vision, abnormal MRI | – | 0.03 | 75 | p.I250T (c.749T>C)/p.I250T (c.749T>C) |
| 9 | F | 4.5 months | 6.5 months | Irritability, hypertonia, opisthotonus, loss of vision, abnormal MRI | – | 0.03 | 398 | p.I250T (c.749T>C)/p.I250T (c.749T>C) |
| 10 | M | Unknown | 10 months | Psychomotor retardation, hypertonia, ↑CSF protein level, abnormal MRI | – | 0.05 | 276 | p.I250T (c.749T>C)/c. 583-1G>Ca
het p.I562T (c.1685T>C) |
| 11 | M | 5 months | 7 months | Irritability, regression, feeding difficulties ↑CSF protein level, abnormal MRI, CT scan, NCV |
– | 0.04 | 404 | p. W132X (c.396G>A)a/p. W132X (c.396G>A)a |
| 12 | M | 6 months | 8 months | Psychomotor retardation, hyperpyrexia, spastic tetraplegia, ↑ CSF protein peripheral hypertonia, abnormal MRI | – | 0.0 | 994 | c.1161+6532_polyA+9kbdel /p.D187V (c.560A>T) het p. R184C (c.550C>T) |
| 13 | F | 4 months | 11 months | Regression, mild axial hypotonia, hypertonia of the extremities, opisthotonus, ↑ CSF protein, abnormal MRI | – | 0.0 | 380 | p.I250T (c.749T>C)/p.I250T (c.749T>C) |
| 14 | M | 6 months | 1.5 years | Psychomotor regression, abnormal MRI, abnormal NCVs | – | 0.0 | 217 | p.I250T (c.749T>C)/p.I250T (c.749T>C) |
| 15 | M | 7 years | 37 years | Spastic paraparesis, optic atrophy, demyelinating peripheral neuropathy, abnormal MRI | 0.0 | 0.015 | 11 | p. D610A (c.1829A>C)a/p. K139del (c.411-413delTAA) |
| 16 | F | Unknown | 42 years | Abnormal MRI, behavioral problems | 0.0 | 0.01 | 33 | p. D610A (c.1829A>C)a/p. K139del (c.411-413delTAA) |
| 17 | M | 3.5 months | 6 months | Generalized hypertonia, developmental delay, abnormal MRI, decreased NCVs | 0.0 | 0.003 | 1,556 | p. K139del (c.411-413delTAA)/p. K139del(c.411-413delTAA) |
| 18 | M | Neonatal period | 12 months | Psychomotor retardation, infantile seizures, ↑ CSF protein level, abnormal MRI | 0.0 | 0.0 | 209 | p. I250T (c.749T>C) c.1161+6532_polyA+9kbdel het p. R184 C (c.550C>T) |
| 19 | M | 3 months | 5 months | Psychomotor retardation, regression, hypotonia, seizures, abnormal MRI | 0.0 | 0.007 | 653 |
c.1161+6532_polyA+9kbdel/unknown het p. R184C (c. 550C >T) hem p. I562T (c. 1685T>C) |
aNovel mutation
↑increased
NCV nerve conduction velocity. Patients 8 and 9 and 15 and 16 are siblings
Methods
β-Galactocerebrosidase activity was assayed in leukocyte homogenates using tritium-labeled [H3] galactosylceramide (ARC; American Radiolabeled Chemicals, Inc.) or 6-hexadecanoyl-4-methylumbelliferyl-β-d-galactopyranoside (Glycosynth) as substrates (Wiederschain et al. 1992; Young et al. 1972).
Chitotriosidase activity in plasma was measured using 4-methylumbelliferyl-β-d-N,N′,N′-triacetylchitotriose as substrate (Michelakakis et al. 2004).
Molecular Analysis
DNA analysis was carried out by PCR amplification of all the exons and flanking regions of the GALC gene followed by automate sequencing. Mutations were confirmed by restriction enzyme analysis when appropriate or by resequencing. RNA extraction and cDNA analysis were performed as previously described (Rodriguez-Pascau et al. 2009). Primer sequences are available on demand. The novel missense mutation p.D610A was confirmed by restriction analysis with the HphI enzyme. It was analyzed in samples from 50 control individuals and was not found. All mutations are described according to the current mutation nomenclature guidelines (http://www.hgvs.org/mutnomen), ascribing the A of the first ATG translational codon as nucleotide +1.
Results: to – Discussion
Krabbe disease is a panethnic rare neurodegenerative autosomal recessive disorder. Its estimated overall prevalence is 1:100,000 births, while a very high prevalence is found in two separate inbred Druze and Arab Muslim communities in Israel (Foss et al. 2013; Rafi et al. 1996). Assuming a birth rate of 100,000/year, a rough incidence estimate for Krabbe disease in Greece would be 0.63/100,000 births. However, this is most probably an underestimate since underdiagnosis, especially of adult-onset forms, is highly probable.
The Institute of Child Health is the only center in Greece providing the diagnosis of lysosomal storage diseases. This is the first study on a large number of Greek Krabbe patients. The cases presented here account for 4.4% of the total number of patients diagnosed with a lysosomal storage disorder in our center. The patients originated from different parts of Greece including the Ionian and Aegean Sea islands (7/19) and Northern and Central Greece (11/19), whereas one patient was of Gypsy origin (#11).
The clinical phenotypes of Krabbe patients range from the classical infantile form, typically with onset ≤6 months of age and rapid progression, to late-onset forms (6 months–≥9 years) with varying age of onset and rate of progression (Graziano and Cardile 2015).
According to the information available to us, at least 13/19 patients reported had onset of symptoms ≤6 months of age (Table 1). A much later onset was clearly observed in three patients (#7, #15, #16). The two latter are siblings, and they are still alive at the ages of 37 and 42 years, respectively, ages at which the diagnosis was established. The time between onset and diagnosis in our cohort ranged from 15 days to 30 years, the longer delay being observed for patient 15.
This delay is in agreement with previous reports, suggesting a substantial difficulty in the diagnosis of the late-onset forms of the disease due to their variable phenotype (Duffner et al. 2012 ; Kolodny et al. 1991). Assaying of β-galactocerebrosidase, irrespective of the substrate used, was diagnostic for all cases. As previously reported, assaying of residual GALC activity could not reliably differentiate earlier and later-onset patients (Wenger et al. 2014).
Chitotriosidase is an enzyme produced by activated macrophages. Increased plasma activity, which is at least 100-fold elevated compared to controls, is observed in Gaucher disease (Hollak et al. 1994), whereas more modest increases have been reported in other lysosomal storage diseases (Michelakakis et al. 2004).
Plasma chitotriosidase activity was assayed in 16/19 patients. One patient (#6) had zero activity, apparently belonging to the approximately 6% of the general population that does not synthesize enzymatically active enzyme (Boot et al. 1998). In 11 of the remaining 15 patients, an increase that ranged from 1.4× to 10.4×, the upper normal limit, was observed. It is possible that the increase in chitotriosidase activity observed in Krabbe disease patients reflects the psychosine-induced activation of peripheral immune cells described in the disease (Parsqui et al. 2007; Formici et al. 2007). Although further studies will be needed to investigate the mechanism and significance of the observed increase in the plasma chitotriosidase activity, our results are in agreement with previous observations and strongly suggest that assaying this enzyme activity can be useful in the diagnosis of Krabbe disease (Michelakakis et al. 2004; Wajner et al. 2006).
DNA was available from 11 of the 17 unrelated patients (and for two siblings). Mutational analysis resulted in the identification of 21/22 mutant alleles. They included four previously described mutations and three novel ones (Table 1).
The most frequent mutation was the missense mutation p.I250T (c.749T>C). It was first described in homozygosity in a Greek female patient, born to consanguineous parents, with disease onset at 28 months of age (De Gasperi et al. 1996). Recent data confirm that the mutation is associated with a negligible level of residual activity and strongly suggest that it leads to altered processing of the 80 kDa precursor resulting in an undetectable 50–54 kDa N-terminal GALC fragment (Lee et al. 2010). In our cohort it was identified either in homozygosity (three unrelated patients – and the sib of one of them) or heterozygosity (two patients). It accounts for 36.4% (8/22 alleles, considering only unrelated individuals). No information regarding possible consanguinity of the parents of the homozygous patients was available. However, the parents of the siblings #8 and #9, homozygous for the mutation, originated from the same village of an Ionian island. Furthermore, no common geographical origin for this mutation was identified since the patients came from Northern, Central, and Western Greece. All the patients of our series bearing this allele had a much earlier onset of the disease than the described for the original patient. Two of the patients bearing the p.I250T mutation in heterozygosity are also carriers of either the p.I562T (c1685T>C) or the p. R184C (c.550C>T) polymorphisms.
The second most frequent mutation in our cohort was the large deletion c.1161+6532_polyA+9kbdel. The mutation has probably originated in Sweden where Krabbe disease has recently been reported to be the most frequent lysosomal storage disorder (Hult et al. 2014). It is found all over Europe but also in other countries like Mexico and India. It has been described to account for 44% of the alleles of American patients with Northern European ancestry and for 52% of unrelated Dutch cases. An overall 35% frequency is described in patients from different European countries. Recently it has been reported to account for 18% of the disease alleles in an Italian cohort of patients. In this study it accounted for 22.7% of the disease alleles, a frequency lower than that described in Northern European countries and closer to that described in Italian patients. It is a severe mutation that in the homozygous state results in the classic infantile form or, depending on the nature of the second mutation, in later-onset disease when in compound heterozygosity. It is invariably found to be associated with the transition c.550C>T (p.R184C) which is present on the same allele (Debs et al. 2013; Kleijer et al. 1997; Tappino et al. 2010; Luzi et al. 1995; Rafi et al. 1995).
In agreement with the above, in our cohort, the mutation was identified either in homozygosity (one patient) or heterozygosity (three patients) in four patients with severe early-onset disease. In all the cases, the genotyping results are consistent with its association with the c.550C>T transition.
p.K139 del (c.411–413 del TAA) was the third most frequently identified mutation. It was found in homozygosity in a patient (#17) with severe early-onset disease and in heterozygosity in siblings #15 and #16, both with late-onset slowly progressing disease. The sibs and the homozygous patient originated from Northern Greece pointing to a possible common origin. The mutation leads to the deletion of a lysine residue that is highly conserved in eukaryotes (http://www.ensebml.org). It has recently been reported at the heterozygous state in a Turkish patient with onset of disease reported to occur at the age of 25 years (Debs et al. 2013). It appears then that in homozygosity the mutation results in a severe early-onset form, whereas in heterozygosity, apparently depending on the second mutation, it can be associated with later-onset forms of the disease. Interestingly, deletion of the previous, equally conserved, lysine residue (p.K138del) has been reported in a Greek patient; however, no details regarding the phenotype or the origin of the patient are available (Wenger et al. 2001).
The novel mutation p.D610A (c.1829A>C) was identified in heterozygosity with the p.K139del in two siblings, patients #15 and #16, with a late-onset form. The missense mutation in exon 16 changes aspartate, an acidic amino acid, to valine, a neutral branched-chain amino acid. Its impact on the enzyme structure is not clear; however, since it was found in patients with late-onset forms in heterozygosity with the apparently severe mutation p.K139del, it could be argued that it predisposes to later-onset forms of the disease.
Two other novel mutations were found in two patients with early-onset disease. The first one, c.583-1G>C, observed in heterozygosity with p.I250T, is a splicing mutation. We were able to show that it results in the skipping of exon 6 (Fig. 1) and the synthesis of a shorter protein (p.I179- K191del). The second is the nonsense mutation p.W132X (c.396G>A) leading to premature termination of synthesis of the enzyme. It was found in homozygosity in a patient of Gypsy origin whose parents were third cousins.
Fig. 1.
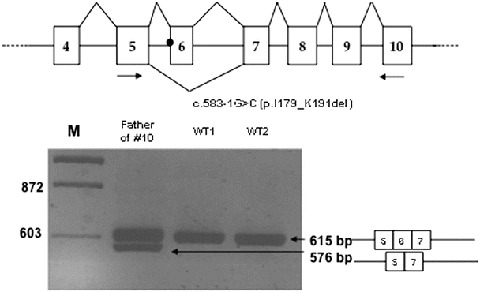
Top: scheme of the splicing pattern of exons 4–10 of the GALC gene in the wt allele (above the figure) and in the allele bearing the c.583-1G>C mutation (below the figure, only the skipping of exon 6 is indicated). Arrows correspond to primers used for the PCR shown below. Bottom: PCR amplification of cDNAs from the father of patient #10 and from two wild-type individuals. A scheme of exons 5–7 is indicated. M molecular weight markers
Finally, the p.D187V (c.560A>T) mutation, previously described in an adult patient (Luzi et al. 1996), was found in heterozygosity with the c.1161+6532_polyA+9kbdel in an early-onset case.
Only one allele of this series remained unidentified. It is the case of patient 19, who bears the c.1161+6532_polyA+9kbdel mutation in the paternal allele. We performed the analysis of the maternal cDNA and gDNA, since no sample from the patient was available. No mutation was detected, neither at gDNA nor at cDNA level. However, her cDNA was homozygous for several polymorphisms that were heterozygous at genomic level. These polymorphisms were p.Q328Q, c.984G>A (exon 9); p.S450S, c.1350C>T (exon 13); and p.I562T, c.1685C>T (exon 15). Some level of recovery of the heterozygosity was observed upon cycloheximide (CHX) treatment. This suggests that the mother is transmitting an allele that suffers nonsense-mediated decay, but the causing mutation was not found. Alternatively, it could be a problem of expression (in spite of the CHX results). We have analyzed part of the promoter and found no change. A deletion of part of the gene could be the causing mutation. This deletion should include one or more exons and generate a frameshift. The exons involved should not be the last 11–17 because the patient bears these exons and his paternal allele does not. Moreover, exons 5 and 9 should not be deleted because of heterozygosity of at least one SNP in the mother (exon 9) or in the patient (exon 5). Southern blot was performed, but no clear results were obtained. Further studies will be performed to characterize this allele.
In conclusion, in our cohort of patients, a considerable delay in the diagnosis of late-onset cases was observed. Sequencing analysis identified seven different mutations, three of which were novel, which accounted for 25/26 alleles studied (21/22, if only unrelated patients are considered), and seven distinct genotypes. The prevailing mutation was the missense mutation p.I250T (c.749T>C) that accounted for about 36% of the mutated alleles. Both p.I250T and the c.1161+6532_polyA+9kbdel were identified either in homozygosity or in heterozygosity in severe early-onset forms of the disease.
Acknowledgments
We are grateful to Prof. Zafeiriou, Dr. Garoufi, Dr. Skiadas, Dr. Lourbopoulos, and all the clinicians that collaborated with us in the diagnosis of the Krabbe patients. We are also grateful to A. Bayona for technical help. This work was partially supported by grants SAF2011-25431 from the Spanish Ministry of Science and Innovation and 2009SGR971 and 2014SGR932, from the Catalan Government.
Take Home Message
In 11 Krabbe patients we identified seven mutations (three novel) and seven genotypes; chitotriosidase activity can be of value in diagnosing Krabbe disease.
Compliance with Ethics Guidelines
Conflict of Interest
Evangelia Dimitriou, Mοnica Cozar, Irene Mavridou, Daniel Grinberg, Lluïsa Vilageliu, and Helen Michelakakis declare that they have no conflict of interest.
Contribution of Authors
Monica Cozar performed the molecular analysis and Lluïsa Vilageliu and Daniel Grinberg supervised the work and revised the MS. Evangelia Dimitriou and Irene Mavridou were involved in the diagnosis of patients and the collection of data. Helen Michelakakis planned the study, supervised the diagnostic work, and wrote the manuscript.
Footnotes
Competing interests: None declared
References
- Boot RG, Renkema GH, Verhoek M, et al. The human chitotriosidase gene. Nature of inherited enzyme deficiency. J Biol Chem. 1998;273:25680–25685. doi: 10.1074/jbc.273.40.25680. [DOI] [PubMed] [Google Scholar]
- Chen YQ, Wenger DA. Galactocerebrosidase from human urine: purification and partial characterization. Biochim Biophys Acta. 1993;1170:53–61. doi: 10.1016/0005-2760(93)90175-9. [DOI] [PubMed] [Google Scholar]
- De Gasperi R, Gama Sosa MA, Sartorato EL, et al. Molecular heterogeneity of late-onset forms of globoid-cell leukodystrophy. Am J Hum Genet. 1996;59:1233–1242. [PMC free article] [PubMed] [Google Scholar]
- Debs R, Froissart R, Aubourg P, et al. Krabbe disease in adults: phenotypic and genotypic update from a series of 11 cases and a review. J Inherit Metab Dis. 2013;36:859–868. doi: 10.1007/s10545-012-9560-4. [DOI] [PubMed] [Google Scholar]
- Duffner PK, Barczykowski A, Kay DM, et al. Later onset phenotypes of Krabbe disease: results of the worldwide registry. Pediatr Neurol. 2012;46:298–306. doi: 10.1016/j.pediatrneurol.2012.02.023. [DOI] [PubMed] [Google Scholar]
- Formici P, Radi E, Battisti C, et al. Psychosine induced apoptosis and cytokine activation in immune peripheral cells of Krabbe patients. J Cell Physiol. 2007;212:737–743. doi: 10.1002/jcp.21070. [DOI] [PubMed] [Google Scholar]
- Foss AH, Duffner PK, Carter RL (2013) Lifetime risk estimators in epidemiological studies of Krabbe disease: review and Monte Carlo comparison. Rare Dis 1:e25212 [DOI] [PMC free article] [PubMed]
- Graziano ACE, Cardile V. History, genetic, and recent advances on Krabbe disease. Gene. 2015;555:2–13. doi: 10.1016/j.gene.2014.09.046. [DOI] [PubMed] [Google Scholar]
- Harzer K, Knoblich R, Rolfs BP, Eggers J. Residual galactosylsphingosine (psychosine) beta-galactosidase activities and associated GALC mutations in late and very late onset Krabbe disease. Clin Chim Acta. 2002;317:77–84. doi: 10.1016/S0009-8981(01)00791-4. [DOI] [PubMed] [Google Scholar]
- Hollak CE, van Weely S, van Oers MH, Aerts JM. Marked elevation of plasma chitotriosidase activity. A novel hallmark of Gaucher disease. J Clin Invest. 1994;93:1288–1292. doi: 10.1172/JCI117084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hult M, Darin N, von Doblen MJE. Epidemiology of lysosomal storage diseases in Sweden. Acta Paediatr. 2014;103:1258–1263. doi: 10.1111/apa.12807. [DOI] [PubMed] [Google Scholar]
- Kleijer WJ, Keulemans JLM, van Der Kraan M, et al. Prevalent mutations in the GALC gene of patients with Krabbe disease of Dutch and other European origin. JInher Metab Dis. 1997;20:587–594. doi: 10.1023/A:1005315311165. [DOI] [PubMed] [Google Scholar]
- Kolodny EH, Raghavan S, Krivit W. Late-onset Krabbe disease (globoid cell leukodystrophy): clinical and biochemical features of 15 cases. Dev Neurosci. 1991;13:232–239. doi: 10.1159/000112166. [DOI] [PubMed] [Google Scholar]
- Lee WC, Kang D, Causevic E, Herdt AR, Eckman EA, Ekman CB. Molecular characterization of mutations that cause globoid cell leukodystrophy and pharmacological rescue using small molecule chemical chaperones. J Neurosci. 2010;30:5489–5497. doi: 10.1523/JNEUROSCI.6383-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Wenger DA. Characterization of the large deletion in the GALC gene found in patients with Krabbe disease. Hum Mol Genet. 1995;4:2335–2338. doi: 10.1093/hmg/4.12.2335. [DOI] [PubMed] [Google Scholar]
- Luzi P, Rafi MA, Wenger DA. Multiple mutations in the GALC gene in a patient with adult-onset Krabbe disease. Ann Neurol. 1996;40:116–119. doi: 10.1002/ana.410400119. [DOI] [PubMed] [Google Scholar]
- Michelakakis H, Dimitriou E, Labadaridis I. The expanding spectrum of disorders with elevated plasma chitotriosidase activity: an update. J Inher Metab Dis. 2004;27:705–706. doi: 10.1023/B:BOLI.0000043025.17721.fc. [DOI] [PubMed] [Google Scholar]
- Parsqui AL, Di Renzo M, Auteri A, Federico G, Puccelti L. Increased TNF-alpha production by peripheral blood mononuclear cells in patients with Krabbe’s disease: effect of psychosine. Eur J Clin Invest. 2007;37:742–745. doi: 10.1111/j.1365-2362.2007.01850.x. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Chen YQ, Wenger DA. A large deletion together with a point mutation in the GALC gene is a common mutant allele in patients with infantile Krabbe disease. Hum Mol Genet. 1995;4:1285–1289. doi: 10.1093/hmg/4.8.1285. [DOI] [PubMed] [Google Scholar]
- Rafi MA, Luzi P, Zlotogora J, Wenger DA. Two different mutations are responsible for Krabbe disease in Druze and Moslem Arab populations in Israel. Hum Genet. 1996;97:304–308. doi: 10.1007/BF02185759. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Pascau L, Gort L, Schuchman EH, Vilageliu L, Gringberg D, Chabas A. Identification and characterization of SMPD1 mutations causing Niemann-Pick types A and B in Spanish patients. Hum Mutat. 2009;30:1117–1122. doi: 10.1002/humu.21018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai N, Inui K, Fujii N, et al. Krabbe disease: isolation and characterization of a full-length cDNA for human galactocerebrosidase. Biochem Biophys Res Commun. 1994;198:485–491. doi: 10.1006/bbrc.1994.1071. [DOI] [PubMed] [Google Scholar]
- Suzuki K. Twenty five years of the “psychosine hypothesis”: a personal perspective of its history and present status. Neurochem Res. 1998;23:251–272. doi: 10.1023/A:1022436928925. [DOI] [PubMed] [Google Scholar]
- Svennerholm L, Vanier MT, Mansson JE. Krabbe disease: a galactosylsphingosine (psychosine) lipidosis. J Lipid Res. 1980;21:53–64. [PubMed] [Google Scholar]
- Tanaka K, Nagara H, Kobayashi T, Goto I. The twitcher mouse: accumulation of galactosylsphingosine and pathology of the central nervous system. Brain Res. 1989;482:347–450. doi: 10.1016/0006-8993(89)91198-0. [DOI] [PubMed] [Google Scholar]
- Tappino B, Biancheri R, Mort M, et al. Identification and characterization of 15 novel GALC gene mutations causing Krabbe disease. Hum Mutat. 2010;31:E1894–E1914. doi: 10.1002/humu.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wajner A, Michelin K, Burin MG, et al. Comparison between the biochemical properties of plasma chitotriosidase from normal individuals and from patients with Gaucher disease, GM1-gangliosidosis, Krabbe disease and heterozygotes for Gaucher disease. Clin Biochem. 2006;40:365–369. doi: 10.1016/j.clinbiochem.2006.12.003. [DOI] [PubMed] [Google Scholar]
- Wenger DA (2008) Krabbe disease. In: Gene reviews and gene tests: medical genetics information resource (database on line). Copyright, University Washington, Seattle. 1997–2010. http://www.genetest.org
- Wenger DA, Rafi MA, Luzi P. Molecular genetics of Krabbe disease (globoid cell leukodystrophy): diagnostic and clinical implications. Hum Mutat. 1997;10:268–279. doi: 10.1002/(SICI)1098-1004(1997)10:4<268::AID-HUMU2>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Wenger DA, Suzuki K, Suzuki Y. Galactosylceramide lipidosis: globoid cell leukodystrophy (Krabbe disease) In: Scriver CR, Beaudet AL, Sly WS, Valle D, Chilids B, Vogelstein D, editors. The metabolic and molecular bases of inherited disease. New York: McGraw Hill; 2001. pp. 3669–3694. [Google Scholar]
- Wenger DA, Escolar MI, Luzi P, Rafi MA (2013) Krabbe disease (globoid cell leukodystrophy). In: Valle D, Beaudet AL, Vogelstein D, Kinzler KW, Antonarakis SE, Ballabio A (eds) The online metabolic and molecular bases of inherited disease. McGraw Hill, New York
- Wenger DA, Luzi P, Rafi AM. Krabbe disease: are certain mutations disease-causing only when specific polymorphisms are present or when inherited in trans with specific second mutations? Mol Genet Metab. 2014;111:307–308. doi: 10.1016/j.ymgme.2013.12.009. [DOI] [PubMed] [Google Scholar]
- Wiederschain G, Raghavan S, Kolodny E. Characterization of 6-hexadecanoylamino-4-methylumbelliferyl-beta-D-galactopyranoside as fluorogenic substrate of galactocerebrosidase for the diagnosis of Krabbe disease. Clin Chim Acta. 1992;205:87–96. doi: 10.1016/S0009-8981(05)80003-8. [DOI] [PubMed] [Google Scholar]
- Xu C, Sakai N, Taniike M, Inui K, Ozono K. Six novel mutations detected in the GALC gene in 17 Japanese patients with Krabbe disease, and new genotype-phenotype correlation. J Hum Genet. 2006;15:548–554. doi: 10.1007/s10038-006-0396-3. [DOI] [PubMed] [Google Scholar]
- Young E, Wilson J, Patrick AD, Chrome L. Galactocerebrosidase deficiency in globoid cell leukodystrophy of late onset. Arch Dis Child. 1972;47:449–450. doi: 10.1136/adc.47.253.449. [DOI] [PMC free article] [PubMed] [Google Scholar]