

Abstract
Lysinuric protein intolerance (LPI) is a rare autosomal recessive metabolic disorder, caused by defective transport of cationic amino acids at the basolateral membrane of epithelial cells, typically in intestines and kidneys. The SLC7A7 gene, mutated in LPI patients, encodes the light subunit (y+LAT1) of a member of the heterodimeric amino acid transporter family.
The diagnosis of LPI is difficult due to unspecific clinical features: protein intolerance, failure to thrive and vomiting after weaning. Later on, patients may present delayed growth osteoporosis, hepatosplenomegaly, muscle hypotonia and life-threatening complications such as alveolar proteinosis, haemophagocytic lymphohistiocytosis and macrophage activation syndrome. Renal involvement is also a serious complication with tubular and more rarely, glomerular lesions that may lead to end-stage kidney disease (ESKD). We report six cases of LPI followed in three different French paediatric centres who presented LPI-related nephropathy during childhood. Four of them developed chronic kidney disease during follow-up, including one with ESKD. Five developed chronic tubulopathies and one a chronic glomerulonephritis. A histological pattern of membranoproliferative glomerulonephritis was first associated with a polyclonal immunoglobulin deposition, treated by immunosuppressive therapy. He then required a second kidney biopsy after a relapse of the nephrotic syndrome; the immunoglobulin deposition was then monoclonal (IgG1 kappa). This is the first observation of an evolution from a polyclonal to a monotypic immune glomerulonephritis. Immune dysfunction potentially attributable to nitric oxide overproduction secondary to arginine intracellular trapping is a debated complication in LPI. Our results suggest all LPI patients should be monitored for renal disease regularly.
Keywords: Children, Glomerulonephritis, Lysinuric protein intolerance, Nephrocalcinosis
Introduction
Lysinuric protein intolerance (LPI) (MIM 222700) is a rare autosomal recessive metabolic disorder with an estimated incidence of 1.7 cases for every 100,000 live births in France. It is caused by defective sodium-independent transport of cationic amino acids (CAA, i.e. l-arginine, l-lysine, l-ornithine) at the basolateral membrane of epithelial cells, mostly of the intestine and kidney (Camargo et al. 2008). The SLC7A7 gene, mutated in LPI patients, encodes the light subunit (y+LAT1) of a member of the heterodimeric amino acid transporter (HAT) family. y+LAT-1 is associated with 4F2hc, the heavy subunit required for normal expression of the transporter at the basolateral membrane (Palacin et al. 2004).
The highest prevalence of LPI (1/60,000 live births) is found in Finland, due to a founder mutation (c.895-2A>T) (Thelle et al. 2006). There is also a high prevalence in Southern Italy and Japan (Sebastio et al. 2011). More than 50 mutations have been described to date (Ogier de Baulny et al. 2012). The mutations result in defective transport of CAA and subsequently low blood concentrations of CAA due to poor intestine absorption and high renal clearance. The reduced availability of arginine and ornithine for the urea cycle is responsible for hyperammonaemia after rich protein intakes (Ogier de Baulny et al. 2012).
The diagnosis of LPI is often difficult due to unspecific and sometimes subtle clinical features. Protein intolerance and failure to thrive are common findings in infants after weaning. Later on, patients may present growth delay, osteoporosis, hepatosplenomegaly, muscle hypotonia, pancreatitis (Parenti et al. 1995) and life-threatening complications such as alveolar proteinosis, haemophagocytic lymphohistiocytosis with macrophage activation syndrome and autoimmune disorders (Ogier de Baulny et al. 2012). Renal involvement is also a serious complication with tubular and more rarely, glomerular involvement that may lead to end-stage kidney disease (ESKD) (Tanner et al. 2007).
We report herein six LPI patients followed in three different French paediatric centres who presented LPI-related nephropathy during childhood: five chronic tubulopathies and one chronic glomerulonephritis. Four of them developed chronic kidney disease (CKD) during follow-up, including one with ESKD at last follow-up.
Materials and Methods
Patients
After reviewing the last 30 years of archives (1984–2013) of all the French paediatric metabolic disease departments and paediatric nephrology departments, we identified six LPI patients who developed a renal disease before 18 years of age in an estimated 25 patients diagnosed during this period. The mean follow-up duration was 13 years (6 to 17 years). Data were collected from clinical and biological files. Renal biopsy reports were all reviewed.
Written informed consent for genetic analysis was obtained from participants or their parents. Patient care and study conduct complied with good clinical practice and the Declaration of Helsinki Principles.
The cohort consisted of six patients (four boys) (Table 1). The age at LPI diagnosis ranged from few days of life to 11 years of age. Patient 1 and patient 2 were siblings (brother and sister) from a non consanguineous Moroccan family. Patient 3 and patient 4 were from Turkish families; they were not related, but both of them came from a consanguineous family. Patient 5 was Caucasian; his parents were not related. Patient 6 had consanguineous Moroccan parents; his older brother had LPI without renal impairment.
Table 1.
Characteristics and treatments of this study's patients
| Patient 1 | Patient 2 | Patient 3 | Patient 4 | Patient 5 | Patient 6 | |
|---|---|---|---|---|---|---|
| Birth/gender | 1976/F (sister of patient 2) | 1977/M (brother of patient 1) | 1991/F | 1997/M | 1997/M | 2005/M |
| Ethnic background | Morocco | Morocco | Turkey | Turkey | Caucasian | Morocco |
| Age at the diagnosis of LPI | 3 months old | 10 months old | 19 months old | 2 months old | 11 years old | Few days old |
| Presenting symptoms | Coma, metabolic acidosis | Failure to thrive | Vomiting, failure to thrive | Failure to thrive | Proteinuria | Neonatal hypotonia |
| Remarkable symptoms | Interstitial pulmonary syndrome, severe growth retardation | Local emphysema on pulmonary tomodensitometry | Interstitial pulmonary syndrome on tomodensitometry | |||
| Treatment | Citrulline (dose not known) | Citrulline (3 g/day) | Citrulline (3 g/day), lysine, l-carnitine | Citrulline (dose not known), l-carnitine | Citrulline (3 g/day), arginine (250 mg/kg/day) | Citrulline (1.6 g/day), l-carnitine, lysine |
| Age(yo) at renal impairment diagnosis | 8 | 7 | 3 | 6 | 12 | 2 |
| Renal dysfunction | Tubular | Tubular | Tubular | Tubular | Tubular, glomerular | Tubular |
| Kidney biopsy | Normal | No | No | No | Membranoproliferative glomerulopathy | No |
| Nephrocalcinosis | Yes | No | Yes | No | No | Yes |
| CKD at the end of study (clearance mL/min/1.73 m2) | No | Yes (70) | ESKD | Yes (44) | No | Yes (72) |
| Supplementation | Bicarbonate, phosphate | Bicarbonate | Bicarbonate, phosphate | Bicarbonate, phosphate | Bicarbonate, phosphate | No |
| Age (years) at the end of study | Death at 10 | 36 | 21 | 16 | 16 | 8 |
M male, F female, CKD chronic kidney disease, ESKD end-stage kidney disease
Biochemical and Molecular Analyses
The diagnosis of LPI was confirmed for all patients if they had low lysine (<71 μmol/L), arginine (<23 μmol/L) and ornithine (<27 μmol/L) blood concentrations associated with high urinary concentrations of these following cationic amino acids: lysine (>150 μmol/mmol creatinine), arginine (>5 μmol/mmol creatinine) and ornithine (>5 μmol/mmol creatinine). They all had high orotic aciduria (>1.2 μmol/mmol creatinine). LPI was confirmed by Sanger sequencing in patient 5, bearing the already-described homozygous c.106-108delGAG SLC7A7 mutation (Sperandeo et al. 2005).
Results
General LPI Features
The main presenting symptom was significant growth retardation detected between 2 and 19 months of age (patients 2, 3, 4). Patient 1 presented with coma with metabolic acidosis at 3 months of age. Patient 5 was diagnosed after investigations for proteinuria discovered at 11 years of age, and patient 6, who had an affected sibling and neonatal hypotonia, was diagnosed soon after birth. All of the six patients had biochemical features characteristic of LPI.
They all received a protein-restricted diet and citrulline supplement, associated with arginine supplement in patient 5 or lysine supplement in patients 3 and 6, and three patients (patients 3, 4 and 6) received also a temporary l-carnitine supplement. All had hyperferritinaemia (ferritin >80 μg/L) (data not found for patient 2) and enlarged liver and spleen. Patient 1 had serious lung involvement with severe interstitial syndrome at 8 years of age. The lung biopsy showed extended fibrosis and fatty acid crystals. She died 2 years later from refractory hypoxemia. Two other patients had asymptomatic pulmonary impairment discovered on systematic tomodensitometry (patients 3 and 6) (Table 1).
Renal Involvement
All patients had renal tubular impairment with increased β2 microglobulinuria discovered between 2 and 12 years of age (Table 1). Four had phosphate leaks (patients 1, 3, 4 and 5), and five had renal tubular acidosis (patients 1, 2, 3, 4, 5) requiring bicarbonate supplements. Three patients developed nephrocalcinosis (patients 1, 3 and 6), secondary to hypercalciuria (ranging from 1.3 to 1.8 mmol ca/mmol creatinine), detected between 2 and 8 years of age. In two of them, nephrocalcinosis was associated with CKD that was diagnosed, respectively, at 7 and 14 years of age (patients 3 and 6); the third one (patient 1) died at 10 years of age with preserved renal function.
Four patients had CKD diagnosed between 6 and 14 years of age (Table 1). Three had CKD diagnosed before 10 years old (patients 2 and 6 had CKD stage 2 and patient 4 had CKD stage 3 at the end of the study). Patient 3 presented CKD at 14 years of age that eventually progressed to ESKD at 21 years of age. Concurrently, he developed a non-nephrotic range proteinuria (0.71 g/L) and hypoalbuminaemia (29 g/L) but did not undergo kidney biopsy.
Patient 3 had high blood pressure, revealing his renal impairment at three years old; patients 4, 5 and 6 had normal blood pressure (data not found for patients 1 and 2). Patient 5 had microscopic haematuria when the renal impairment was diagnosed (no data for the other patients).
Kidney biopsy was performed in two patients (Table 1). Patient 1 underwent kidney biopsy at 9 years of age because of nephrocalcinosis: kidney histology was normal. Patient 5 had presented hyperferritinaemia and hepatosplenomegaly since infancy, and a tubular proteinuria appeared at 11 years of age, etiological investigation of which finally led to the diagnosis of LPI. He developed a nephrotic syndrome two years later (hypoalbuminaemia 9 g/L, hypoproteinaemia 40 g/L, proteinuria> 3 g/L). Renal biopsy, performed at the onset of the nephrotic syndrome, showed a membranoproliferative glomerulonephritis with polyclonal immunoglobulin deposits associated with C3 and C1q deposits (Fig. 1). He received corticosteroids (60 mg/day) associated with mycophenolate mofetil (1,600 mg/day) and achieved partial remission within 6 months (normalization of albuminaemia, but persistence of a non-nephrotic proteinuria (0.6 g/L)). Steroid was tapered off after 11 months, and the patient was treated with mycophenolate mofetil alone. The nephrotic syndrome relapsed 10 months after. A second kidney biopsy was performed and showed the same membranoproliferative glomerulonephritis pattern but with a surprising evolution of the immunoglobulin deposits that were monoclonal IgG1 kappa (Fig. 2). Corticosteroid was started again (30 mg/day) and resulted in a complete remission, but proteinuria reappeared after tapering corticosteroid doses. Immunologic investigations found anti-nuclear antibodies (1/400) in plasma with anti-SSA (Sjögren syndrome A) antibodies (49 mUA/L); C3 nephritic factor was inconsistently detectable. No monoclonal proliferation was detected after myelogram, plasma and urinary protein electrophoresis and immunoelectrophoresis.
Fig. 1.
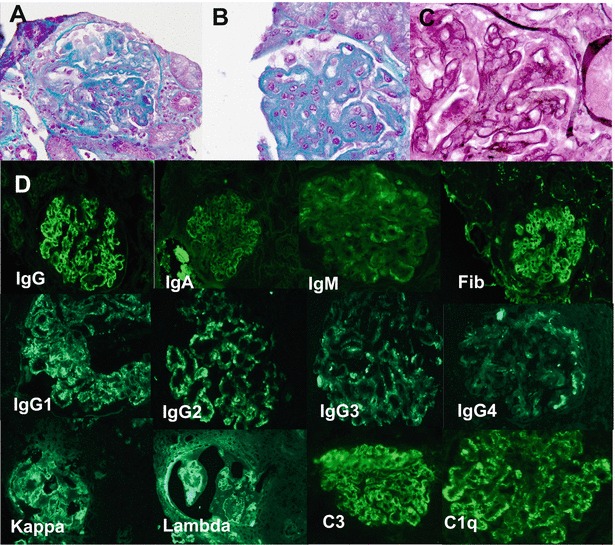
Patient 5, first kidney biopsy: membranoproliferative glomerulonephritis: (a) mesangial proliferation, accumulation of mesangial matrix (trichrome Masson, ×100). (b) Intramembranous, extramembranous and subendothelial deposits (trichrome Masson, ×250). (c) Diffuse doubles contours (Marinozzi staining, ×250). (d) Immunofluorescence study (×100): Ig G, Ig A, Ig M, fibrin (Fib), IgG1, IgG2, IgG3, IgG4, C3 and C1q positivity: granular deposits along glomerular capillary walls, kappa and lambda light chains positivity
Fig. 2.
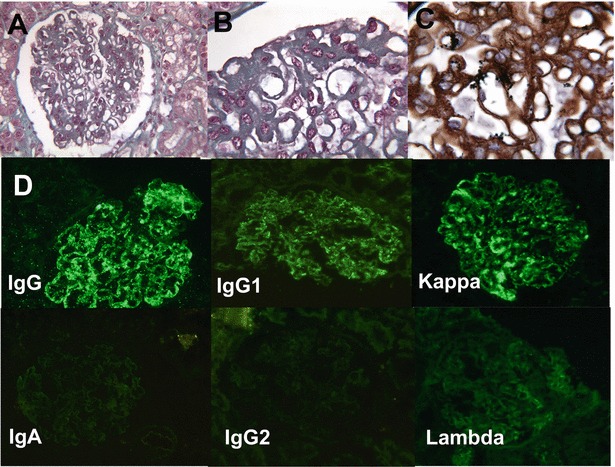
Patient 5, second kidney biopsy, membranoproliferative glomerulonephritis: (a) mesangial proliferation, extramembranous deposits (trichrome Masson, ×100). (b) Intramembranous and subendothelial deposits (trichrome Masson, ×250). (c) Irregular thickening of basal membranes, double contours (Marinozzi staining, ×250). (d) Immunofluorescence study (×100): Ig G, IgG1 and kappa light chain: intramembranous and extramembranous deposits. IgA, IgG2 and lambda light chain: no deposit; IgM, Ig3, Ig4 and fibrinogen: no deposit (data not shown)
Discussion
We report six patients with LPI and renal involvement, consisting of either tubular or glomerular features, which appeared before 18 years of age. In a Finnish series including mostly adults, 74% of the 39 patients were reported to have had proteinuria, 38% haematuria and 15% an elevated serum creatinine (Torrents et al. 1999). Few other cases of glomerulonephritis have been reported (Parto et al. 1994).
In our series, five out of six patients were diagnosed with LPI before 2 years of age because of characteristic symptoms such as failure to thrive, vomiting, hepatomegaly, hyperammonaemia. The renal impairment was detected during the follow-up between 2 and 8 years of age. In patient 5, however, the work-up of kidney disease led to the diagnosis of LPI at 11 years of age. It is well known that LPI can be diagnosed late in life (Dionisi-Vici et al. 1998), and complications may reveal the disease because LPI severity is rather dependent on complications (renal, pulmonary) than on moderate metabolic disorders (hyperammonaemia, low cationic amino acids in blood and high in urine) (Ogier de Baulny et al. 2012; Tanner et al. 2007).
The renal disease varies from tubular dysfunction to glomerulonephritis, with or without CKD (Tanner et al. 2007; Verzola et al. 2012). Tubular impairment is often the first manifestation of kidney involvement. All of our six patients had rather severe tubular dysfunction that ranged from isolated metabolic acidosis with normal anion gap (patient 2) to complete Fanconi syndrome (patient 3) and preceded glomerular features for patient 5. It was associated with hypercalciuria and nephrocalcinosis in three out of six patients probably responsible, at least in part, for CKD. Tanner et al. did not report any nephrocalcinosis in the Finnish LPI cohort (Tanner et al. 2007). We could not find any previous report of nephrocalcinosis in LPI in the literature. However, our results are not surprising since nephrocalcinosis is a classical complication of proximal or distal tubular dysfunction (Ogier de Baulny et al. 2012).
Four out of six patients developed CKD between the age of 6 years and adulthood (before 10 years of age for three of them) attributed to chronic tubular damage. However, since no kidney biopsy was performed in these patients, an associated glomerulopathy cannot be excluded. Patient 3 had severe renal impairment with ESKD at 20 years of age. The proteinuria and hypoalbuminuria attributed to nephron reduction may have been caused by other glomerular lesions. Patient 1 died early at 10 years of age from lung disease: her renal function was normal at that time. Patient 5 developed glomerulonephritis with normal renal function at the last follow-up at 15 years of age. Our results suggest that when LPI is complicated with renal disease during childhood, the evolution of renal impairment is severe.
Pathophysiological mechanisms of renal impairment in LPI are not fully elucidated, and several explanations have been suggested. The first is a direct toxicity of intracellular trapped lysine on proximal tubular cells (Thelle et al. 2006; Verzola et al. 2012). Indeed oral lysine supplementation in rats promotes proteinuria and inhibits albumin reabsorption by proximal tubular cells (Thelle et al. 2006). Lysine can also trigger apoptosis through an NADPH (nicotinamide adenine dinucleotide phosphate) oxidase-dependent mechanism that increases ROS (reactive oxygen species) production (Verzola et al. 2012). And the renal oxidative stress plays a central role in hypertension, CKD and diabetic nephropathy (28). Moreover, patients with cystinuria (another amino acid transport defect) have a high urinary concentration of cysteine, a natural anti-oxidant, but do not develop any tubular leaks (they typically develop microcalculi). The anti-oxidative properties of cysteine may have a protective effect on the tubular cells (Verzola et al. 2012). Finally, one patient with LPI has been described with renal Fanconi syndrome, and the kidney biopsy showed a sloughing of brush border and vacuolization of proximal tubular cells. But the author did not consider the lysine accumulation as the toxic agent since patients with hyperlysinaemia do not develop renal Fanconi syndrome (Benninga et al. 2007).
The second hypothesis is a dysregulation of the immune system, in particular locally in renal tissue through the nitric oxide (NO) pathway. Arginine which accumulates in LPI cells due to its defective efflux (Mannucci et al. 2005) is the substrate of inducible nitric oxide synthase (iNOS), which synthesizes NO and citrulline (Nagasaka et al. 2009). Three LPI patients have been reported with increased plasma NO (Mannucci et al. 2005). iNOS is naturally present in epithelial tubular and renal mesangial cells and also plays a key role in fine regulation of vascular resistance; NO plays a role in inflammation and triggers mechanisms of immune defence (Nagasaka et al. 2009). iNOS synthesis is stimulated in LPI so the increased NO production may further contribute to the development of a multisystemic inflammation status (Mannucci et al. 2005). This overstimulation of the immune system could lead to autoimmune diseases. Patients with LPI often show disturbed immune functions and lupus erythematosus is the major autoimmune disease observed in patients with LPI (Aoki et al. 2001; Di Rocco et al. 1998; Kamoda et al. 1998; Parto et al. 1994). Several cases of lupus erythematosus diagnosed before LPI have been reported (Di Rocco et al. 1998; Parto et al. 1994). Patient 5 illustrates a new involvement of the immune system in LPI, through a very unique immune glomerulopathy. In this case, renal impairment revealed the diagnosis of LPI with a proximal tubular dysfunction (beta2 microglobulinuria) but unexpectedly evolved towards a glomerular disease with nephrotic syndrome. On initial kidney biopsy, a membranoproliferative pattern and immunoglobulin deposits associated with C3 and C1q deposits led to the diagnosis of lupus-like kidney disease. Plasma analyses found anti-SSA antibodies. Immunosuppressive treatment efficacy is an additional argument for the involvement of the immune system in LPI. For patient 5, an immunosuppressive therapy with corticosteroid and mycophenolate mofetil led to the remission of the nephrotic syndrome. Several other LPI cases have been treated successfully with various immunosuppressive therapies (steroid, polyvalent immunoglobulin or azathioprine) (Aoki et al. 2001; Dionisi-Vici et al. 1998; Di Rocco et al. 1998). These treatments were introduced because of a systemic disorder with fever, skin rash or erythroblasto-phagocytosis without macrophagic activation syndrome and showed an improvement of clinical symptoms. Patient 5’s kidney biopsy immunofluorescence pattern also supports the hypothesis of the immune system involvement in LPI glomerular injuries, with a very unique evolution from polyclonal Ig deposition at onset to a monoclonal IgG1 kappa deposition 18 months later. The unsuccessful search for a systemic immune abnormality with monoclonal peak of Ig or plasmocyte abnormalities supports the hypothesis of a local dysregulation. Increased immunoglobulin concentration has already been reported but never as a monoclonal disease (Yoshida et al. 1995).
Conclusion
We report six paediatric cases of LPI with renal involvement. All of them had tubular disease, while three were also diagnosed with nephrocalcinosis and another had a glomerular disease. The renal evolution was severe in three cases with CKD during childhood. The occurrence of glomerulopathy in LPI supports the role of an immune system dysregulation in LPI renal disease. All LPI patients should therefore be regularly monitored for renal disease.
Take-Home Message
Kidney involvement can complicate lysinuric protein intolerance, and it is therefore essential to monitor patients for the appearance of kidney diseases such as tubulopathy or glomerulopathy.
Compliance with Ethics Guidelines
Conflict of Interest
Camille Nicolas, Nathalie Bednarek, Vincent Vuiblet, Olivia Boyer, Anais Brassier, Pascale De Lonlay, Louise Galmiche, Pauline Krug, Véronique Baudouin, Samia Pichard, Manuel Schiff, Christine Pietrement declare that they have no conflict of interest.
All authors have nothing to declare.
The authors declare that the content of the article has not been influenced by the sponsors.
This article does not contain any studies with animal performed by the any of the authors.
Patient care and study conduct complied with good clinical practice and the Declaration of Helsinki Principles.
Camille Nicolas wrote the paper and gathered patients’ data. Vincent Vuiblet and Louise Galmiche interpreted the biopsy pictures, and all authors contributed to the interpretation and final manuscript preparation.
Footnotes
Competing interests: None declared
Contributor Information
C. Nicolas, Email: cnicolas@chu-reims.fr
Collaborators: Matthias R. Baumgartner, Marc Patterson, Shamima Rahman, Verena Peters, Eva Morava, and Johannes Zschocke
References
- Aoki M, Fukao T, Fujita Y, et al. Lysinuric protein intolerance in siblings: complication of systemic lupus erythematosus in the elder sister. Eur J Pediatr. 2001;160(8):522–523. doi: 10.1007/PL00008455. [DOI] [PubMed] [Google Scholar]
- Benninga M, Lilien M, De Koning T, et al. Renal Fanconi syndrome with ultrastructural defects in lysinuric protein intolerance. J Inherit Metab Dis. 2007;30(3):402–403. doi: 10.1007/s10545-007-0446-9. [DOI] [PubMed] [Google Scholar]
- Camargo S, Bockenhauer D, Kleta R. Aminoacidurias: clinical and molecular aspects. Kidney Int. 2008;73(8):918–925. doi: 10.1038/sj.ki.5002790. [DOI] [PubMed] [Google Scholar]
- Di Rocco M, Buoncompagni A, Gatton M, et al. Complications of lysinuric protein intolerance must be treated with immunosuppressive drugs. J Inher Metab Dis. 1998;21(6):675–676. doi: 10.1023/A:1005440802688. [DOI] [PubMed] [Google Scholar]
- Dionisi-Vici D, De Felice L, el Hachem M, et al. Intravenous immune globulin in lysinuric protein intolerance. J Inherit Metab Dis. 1998;21(2):95–102. doi: 10.1023/A:1005383307100. [DOI] [PubMed] [Google Scholar]
- Kamoda T, Nagai Y, Shigeta M. Lysinuric protein intolerance and systemic lupus erythematosus. Eur J Pediatr. 1998;157(2):130–131. doi: 10.1007/s004310050784. [DOI] [PubMed] [Google Scholar]
- Mannucci L, Emma F, Markert M, et al. Increased NO production in lysinuric protein intolerance. J Inherit Metab Dis. 2005;28(2):123–129. doi: 10.1007/s10545-005-5954-x. [DOI] [PubMed] [Google Scholar]
- Nagasaka H, Tsukahara H, Yorifuji T, et al. Evaluation of endogenous nitric oxide synthesis in congenital urea cycle enzyme defects. Metab Clin Exp. 2009;58(3):278–282. doi: 10.1016/j.metabol.2008.09.025. [DOI] [PubMed] [Google Scholar]
- Ogier de Baulny H, Schiff M, Dionisi-Vici C. Lysinuric protein intolerance (LPI): a multi organ disease by far more complex than a classic urea cycle disorder. Mol Genet Metab. 2012;106(1):12–17. doi: 10.1016/j.ymgme.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Palacin M, Bertran J, Chillaron J (2004) Lysinuric protein intolerance: mechanisms of pathophysiology. Mol Genet Metab 81(Suppl 1):S27–S37 [DOI] [PubMed]
- Parenti G, Sebastio G, Strisciuglio P. Lysinuric protein intolerance characterized by bone marrow abnormalities and severe clinical course. J Pediatr. 1995;126(2):246–251. doi: 10.1016/S0022-3476(95)70552-X. [DOI] [PubMed] [Google Scholar]
- Parto K, Kallajoki M, Aho H, et al. Pulmonary alveolar proteinosis and glomerulonephritis in lysinuric protein intolerance: case reports and autopsy findings of four pediatric patients. Hum pathol. 1994;25(4):400–407. doi: 10.1016/0046-8177(94)90150-3. [DOI] [PubMed] [Google Scholar]
- Sebastio G, Sperandeo MP, Andria G. Lysinuric protein intolerance: reviewing concepts on a multisystem disease. Am J Med Genet C Semin Med Genet. 2011;157C(1):54–62. doi: 10.1002/ajmg.c.30287. [DOI] [PubMed] [Google Scholar]
- Sperandeo MP, Paladino S, Maiuri L, et al. A y(+)LAT-1 mutant protein interferes with y(+) LAT-2 activity: implications for the molecular pathogenesis of lysinuric protein intolerance. Eur J Hum Genet. 2005;13(5):628–634. doi: 10.1038/sj.ejhg.5201376. [DOI] [PubMed] [Google Scholar]
- Tanner L, Näntö-Salonen K, Niinikoski H, et al. Nephropathy advancing to end-stage renal disease: a novel complication of lysinuric protein intolerance. J Pediatr. 2007;150(6):631–634. doi: 10.1016/j.jpeds.2007.01.043. [DOI] [PubMed] [Google Scholar]
- Thelle K, Christensen E, Vorum H. Characterization of proteinuria and tubular protein uptake in a new model of oral L-Lysine administration in rats. Kidney Int. 2006;69(8):1333–1340. doi: 10.1038/sj.ki.5000272. [DOI] [PubMed] [Google Scholar]
- Torrents D, Mykkänen J, Pineda M, et al. Identification of SLC7A7, encoding y+LAT-1 as the lysinuric protein intolerance gene. Nat Genet. 1999;21(3):293–296. doi: 10.1038/6809. [DOI] [PubMed] [Google Scholar]
- Verzola D, Fama A, Villaggio B, et al. Lysine triggers apoptosis through a NADPH oxidase-dependent mechanism in human renal tubular cells. J Inherit Metab Dis. 2012;35(6):1011–1019. doi: 10.1007/s10545-012-9468-z. [DOI] [PubMed] [Google Scholar]
- Yoshida Y, Machigashira K, Suehara M, et al. Immunological abnormality in patients with lysinuric protein intolerance. J Neurol Sci. 1995;134(1-2):178–182. doi: 10.1016/0022-510X(95)00237-1. [DOI] [PubMed] [Google Scholar]