

Abstract
Misfolded α-synuclein is a key factor in the pathogenesis of Parkinson's disease (PD). However, knowledge about a physiological role for the native, unfolded α-synuclein is limited. Using brains of mice lacking α-, β-, and γ-synuclein, we report that extracellular monomeric α-synuclein enters neurons and localizes to mitochondria, interacts with ATP synthase subunit α, and modulates ATP synthase function. Using a combination of biochemical, live-cell imaging and mitochondrial respiration analysis, we found that brain mitochondria of α-, β-, and γ-synuclein knock-out mice are uncoupled, as characterized by increased mitochondrial respiration and reduced mitochondrial membrane potential. Furthermore, synuclein deficiency results in reduced ATP synthase efficiency and lower ATP levels. Exogenous application of low unfolded α-synuclein concentrations is able to increase the ATP synthase activity that rescues the mitochondrial phenotypes observed in synuclein deficiency. Overall, the data suggest that α-synuclein is a previously unrecognized physiological regulator of mitochondrial bioenergetics through its ability to interact with ATP synthase and increase its efficiency. This may be of particular importance in times of stress or PD mutations leading to energy depletion and neuronal cell toxicity.
SIGNIFICANCE STATEMENT Misfolded α-synuclein aggregations in the form of Lewy bodies have been shown to be a pathological hallmark in histological staining of Parkinson's disease (PD) patient brains. It is known that misfolded α-synuclein is a key driver in PD pathogenesis, but the physiological role of unfolded monomeric α-synuclein remains unclear. Using neuronal cocultures and isolated brain mitochondria of α-, β-, and γ-synuclein knock-out mice and monomeric α-synuclein, this current study shows that α-synuclein in its unfolded monomeric form improves ATP synthase efficiency and mitochondrial function. The ability of monomeric α-synuclein to enhance ATP synthase efficiency under physiological conditions may be of importance when α-synuclein undergoes the misfolding and aggregation reported in PD.
Keywords: alpha-synuclein, astrocytes, ATP synthase, bioenergetics, mitochondria, neurons
Introduction
Mitochondria have multiple crucial roles in cell homeostasis. The major function of this organelle is the production of energy in form of ATP, but it is also linked to other cellular functions such as calcium signaling and cell proliferation and differentiation (Griffiths and Rutter, 2009). Mitochondrial dysfunction has been associated with a number of disorders ranging from cancer to neurodegeneration. A strong pathogenic link to mitochondrial health has been established for Parkinson's disease (PD), one of the most common disabling and incurable neurodegenerative conditions. The major histopathological hallmarks in PD are Lewy bodies, which are intracellular deposits of misfolded, aggregated α-synuclein fibrils (Spillantini et al., 1998). In order for these fibrillar structures to form, monomeric α-helical α-synuclein must undergo a series of conformational changes and oligomeric intermediates, a process that has been reported to be toxic to the cell (Cremades et al., 2012; Angelova et al., 2016; Deas et al., 2016).
Many studies have overexpressed α-synuclein in a variety of models to mimic α-synuclein-induced pathogenesis. These studies have reported α-synuclein-induced calcium homeostasis dysregulation, compromised synaptic transmission, dopaminergic imbalances, and mitochondrial abnormalities (Hettiarachchi et al., 2009; Venda et al., 2010; Luth et al., 2014). However, overexpression studies are unlikely to provide a clear insight into the physiological roles of native unfolded α-synuclein and its implications in disease-causing processes. Studies of a physiological function of α-synuclein in the nervous system are often confounded by the presence of two other members of the synuclein family, β-synuclein and γ-synuclein. The three synucleins possess a high degree of amino acid similarity, significantly overlapping expression patterns, and intraneuronal localization. The generation of synuclein triple knock-out (TKO) mice has made it possible to investigate the physiological role(s) of α-synuclein in a synuclein-null background while avoiding a potential compensatory effect by endogenous β- or γ-synuclein (Greten-Harrison et al., 2010). These TKO mice do not display a strong phenotype except for some age-dependent neuronal dysfunction, making them a useful model with which to study the physiological function of exogenously applied monomeric α-synuclein (Chandra et al., 2004; Robertson et al., 2004).
Alpha-helical α-synuclein has been reported to bind the inner mitochondrial membrane, making a physiological role for α-synuclein in mitochondrial metabolism possible (Guardia-Laguarta et al., 2014; Robotta et al., 2014). Therefore, this study presents an in-depth analysis of monomeric α-synuclein-induced effects on mitochondrial metabolism in the TKO model and provides a physiological role of α-synuclein in ATP synthase function.
Materials and Methods
Animals.
Synuclein TKO, α-synuclein knock-out (AKO), β-synuclein knock-out (BKO), γ-knock-out (GKO), α/γknock-out (A/GKO), and matching wild-type (WT) mice on a pure C57BL/6J genetic background (RRID:IMSR_JAX:000664) of either sex were produced and maintained as described previously (Anwar et al., 2011). Sprague Dawley rat pups 1–3 d postpartum (University College London breeding colony; RRID:RGD_734476) of either sex were used for neuronal cocultures and experimental procedures were performed in full compliance with the United Kingdom Animal (Scientific Procedures) Act of 1986.
Cell culture and synuclein species.
Mixed cultures of cortical or midbrain neurons and glial cells were prepared from postnatal pups (day 1–3). The preparation, maintenance, and characterization of primary neuronal cultures were performed according to the method published by Gandhi et al. (2009). Neurons were easily distinguishable from glia: they appeared phase bright, had smooth rounded somata and distinct processes, and lay just above the focal plane of the glial layer.
Untagged recombinant human synuclein proteins were expressed in E. coli and purified as described previously by us and others (Jakes et al., 1994; Ninkina et al., 2012). Purified proteins were immediately aliquoted, snap-frozen, and kept at −80°C. An absence of oligomers and higher-order aggregates in defrosted aliquots was confirmed by gel electrophoresis. A protein sample was used for assays immediately after defrosting of an aliquot; 100 nm were applied unless otherwise stated.
Oxygen consumption measurements.
Intact mitochondria were isolated from the brains of WT, TKO, and AKO mice a method of differential centrifugation (Plun-Favreau et al., 2012) and resuspended in medium containing 250 mm sucrose, 1 mm EDTA, and 19 mm Tris-HCl, pH 7.1. Oxygen consumption was measured in a Clark-type oxygen electrode (Hansatech) thermostatically maintained at 25°C containing the following (in mm): 135 KCl, 10 NaCl, 20 HEPES, 0.5 KH2PO4, 1 MgCl2, and 5 EGTA, pH 7.1. Glutamate (5 mm), malate (5 mm), and sodium succinate (5 mm) were added to allow basal respiration (V2). Data were obtained using an Oxygraph Plus system with Chart recording software. Protein levels were established using a Pierce BCA protein assay kit (recordings were adjusted accordingly).
Live-cell imaging.
The mitochondrial membrane potential (ΔΨm) was measured by loading cells with 25 nm tetramethyl rhodamine methyl ester (TMRM) in a HEPES-buffered salt solution (Invitrogen) for 40 min at room temperature. Measurements were obtained with a Zeiss 710 VIS CLSM equipped with a META detection system and a 40× oil-immersion objective while keeping 40 nm TMRM in the imaging solution. TMRM was excited using the 560 nm laser line and fluorescence was measured >580 nm. Z-stack images were obtained by confocal microscopy and the basal Δψm was measured using Zen software (Zeiss). Assessments of the mitochondrial membrane potential maintenance through application of oligomycin, rotenone, and the uncoupler carbonylcyanide-p-trifluoromethoxyphenylhydrazone (FCCP) were performed through recordings from a single focal plane. The TMRM was used in the redistribution mode to assess the Δψm, so a reduction in TMRM fluorescence represents mitochondrial depolarization.
Kinetic ATP measurements.
To determine the ATP levels, primary cocultures were transfected with a mitochondrial ATP probe (mitAT1.03) generated by Imamura et al. (2009) using Effectene (Qiagen) according to the manufacturer's instructions. The FRET was quantified by the 527:475 nm ratio with an excitation of 405 nm and a filter from 515 to 580 nm (Imamura et al., 2009; Holmström et al., 2013). ATP kinetics were assessed in permeabilized primary cocultures. To permeabilize the transfected cells, a buffer (0.137 m NaCl, 5 mm KCl, 0.7 mm NaH2PO4, 25 mm Tris-HCl, pH 7.1) containing low concentration of digitonin (20 μm) was added to the cocultures. Upon permeabilization, the buffer and digitonin were replaced by fresh buffer containing mitochondrial substrates (5 mm glutamate and 5 mm malate). Cells were allowed to rest for at least 5 min before measurements were taken.
Biochemical ATP measurements.
Total ATP levels were measured using the ATP Assay Kit (Abcam, ab83355) and following the manufacturer's protocol. Tissue was sampled and immediately frozen using liquid N2 until experimental procedures were started. Quantification was performed by colorimetric measurements (ODmax = 570 nm).
NADH measurements.
NADH autofluorescence was measured using an epifluorescence inverted microscope equipped with a 40× fluorite objective. Excitation light at a wavelength of 350 nm was provided by a xenon arc lamp, with the beam passing through a monochromator (Cairn Research). Emitted fluorescence light was reflected through a 455 nm long-pass filter to a cooled CCD camera (Retiga; QImaging) and digitized to 12 bit resolution. Imaging data were collected and analyzed using software from Andor (RRID:SCR_014461).
Mag-Fura2 measurements.
To assess the ATP levels, which correlate with Mg2+ changes, [Mg2+] was imaged using Mag-Fura2 AM. Fluorescence images were acquired (30 s interval) on an epifluorescence inverted microscope equipped with a 20× fluorite objective (excitation at 340 and 380 nm). The emitted light was reflected through a 515 nm long-pass filter to a cooled CCD camera (Retiga; QImaging) and digitized to 12-bit resolution (Cairn Research). Andor iQ (RRID:SCR_014461) was used to collect and analyze data.
Enzyme activity measurements.
ATP synthase enzyme activity was measured using an ATP synthase microplate kit (Abcam) according to the manufacturer's protocol. Briefly, brain mitochondria were isolated, lysed, and the F0F1-ATPase was immunocaptured in the wells of the 96-well microplate supplied with the kit. The hydrolysis of ATP to ADP is coupled to the oxidation of NADH to NAD+, which can be detected by a decrease in absorbance at 340 nm (30°C). Monomeric α-synuclein (100 nm) was added immediately before measurements were taken and results were compared against control wells containing F0F1-ATPase with or without oligomycin.
Proximity ligation assay.
The α-synuclein proximity ligation assay was performed using the Duolink in situ red kit (mouse/rabbit; Sigma-Aldrich; DUO92101) supplied by Sigma-Aldrich according to the manufacturer's instructions. Human neuroblastoma cells (SH-SY5Y catalog #94030304, RRID:CVCL_0019) and rat neuronal cocultures were used in this assay because the most robust commercially available monoclonal antibodies against ATP synthase subunits are raised in mice, which is preventing the use of TKO primary neuronal cultures. Cells were exposed to α-synuclein for 1 h at 37°C before fixation. ATP synthase α-subunit antibody (Abcam catalog #ab14748, RRID:AB_301447) was omitted for the control proximity ligation assay (PLA). Please note that the antibody raised against α-synuclein (Abcam catalog #ab138501, RRID:AB_2537217) does not detect endogenous rat α-synuclein.
Coimmunoprecipitation (co-IP) and Western blotting.
For co-IP, rat brain mitochondria were isolated (Plun-Favreau et al., 2012). The mitochondrial sample was divided and one part was exposed to monomeric α-synuclein for 15 min. Immunoprecipitating antibodies (anti-α-synuclein antibody; Abcam catalog #ab138501, RRID:AB_2537217) were coupled to Dynabeads Protein A (10006D) according to the manufacturer's protocol (Thermo Fisher Scientific). It should be noted that the anti-α-synuclein antibody binds human α-synuclein only (e.g., the exogenous applied monomeric α-synuclein; Abcam) and not the endogenous rat α-synuclein. The coupled beads were exposed to the mitochondrial samples and the eluents (and inputs were separated by SDS-PAGE, transferred to nitrocellulose, and probed for ATP synthase subunit α (53 kDa; Abcam catalog #ab14748, RRID:AB_301447) and α-synuclein (predicted 14 kDa; Abcam catalog #ab138501, RRID:AB_2537217).
Statistical analysis.
Statistical analysis and exponential curve fitting were performed using Origin 9 (Microcal Software; RRID:SCR_014212) software. Experimental data are shown as means ± SEM. Statistical analysis between samples was performed using a one-way ANOVA with Bonferroni correction. Differences were considered to be significantly different if p < 0.05.
Results
Lack of α,β,γ-synuclein leads to a decreased mitochondrial membrane potential
Primary neuron/glia cocultures were prepared from the cerebral cortex of WT, TKO, and AKO mice (Anwar et al., 2011) and the ΔΨm was assessed as an indicator of mitochondrial health and metabolic function. The ΔΨm of neurons and astrocytes were analyzed separately because these cell types have different metabolisms and could exhibit different responses to unfolded proteins (Abramov et al., 2004). Using TMRM as a fluorescent indicator of ΔΨm, it was found that the basal ΔΨm was significantly lower in both AKO astrocytes (70 ± 3%, n = 20 cells; p < 0.05) and TKO astrocytes (63 ± 3%, n = 50 cells; p < 0.001) compared with the basal ΔΨm values for these cells in cultures prepared from WT mice (normalized to 100%, n ≥ 39 cells; Fig. 1A). Similar results were obtained in neurons of AKO (70 ± 11%, n = 22, 3 independent experiments, p < 0.05) and TKO (85 ± 3%, n = 72 cells, 3 independent experiments; p < 0.05) mice compared with the basal ΔΨm values for these cells in cultures prepared from WT mice (normalized to 100%, n ≥ 39 cells; 3 independent experiments). Impaired mitochondrial respiration can induce a switch of the F0F1-ATPsynthase to work in reverse mode (ATPase) to maintain the ΔΨm (Yao et al., 2011). The F0F1-ATPsynthase inhibitor oligomycin was used to test whether synuclein deficiency affects the mechanism of ΔΨm maintenance. Both, WT and TKO cells showed no decrease of ΔΨm in response to the F0F1-ATPsynthase inhibitor oligomycin (2 μg/ml), whereas subsequent inhibition of complex I by rotenone (5 μm) caused a rapid loss of potential (Fig. 1B). Complete depolarization was achieved in all TKO and WT cells by the addition of the mitochondrial uncoupler FCCP (1 μm). Therefore, although synuclein deficiency results in a lower mitochondrial membrane potential, it does not alter the mechanism of ΔΨm maintenance.
Figure 1.

Mitochondrial membrane potential is lower in synuclein-deficient cells and can be rescued by exogenous monomeric α-synuclein. A, Basal Δψm was quantified in WT, AKO, and TKO cells. B, Representative traces of TMRM fluorescence in WT and TKO primary cells after addition of oligomycin (2 μg/ml), rotenone (1 μm), and FCCP (1 μm). C, Representative images of cells loaded with TMRM before and after α-synuclein addition. *p < 0.05; ***p < 0.001; n = 3 independent experiments. Scale bar, 20 μm.
To investigate whether α-synuclein affects the ΔΨm, exogenous monomeric α-synuclein (100 nm) was applied to both cocultures before ΔΨm was measured. The cultures were preincubated for 10 min and analysis of the ΔΨm revealed that low concentrations of α-synuclein raises the basal ΔΨm significantly in TKO cultures (astrocytes by 58 ± 5%; n = 20 cells, p < 0.001, neurons by 50 ± 14%, n = 10 cells, p < 0.001), as well as in WT (astrocytes by 51 ± 9%; n = 15 cells, neurons by 46 ± 8%; n = 15 cells, p < 0.001) compared with basal ΔΨm levels without exogenous α-synuclein (Fig. 1A,C).
Triple synuclein deficiency decreases NADH redox state
There are several possible explanations for the observed reduction in ΔΨm in the absence of α-, β-, and γ-synuclein. The ΔΨm is maintained by complexes of the respiratory chain, which transfer electrons via redox reactions and transport protons across the membrane, generating an electrical potential. This electrical potential can depolarize in a number of situations, including insufficient respiratory chain activity or uncoupled oxidative phosphorylation. To investigate which of these factors caused the observed lower ΔΨm, respiratory chain activity was measured by NADH autofluorescence (Bartolomé and Abramov, 2015). NADH is the electron donor for complex I, so NADH levels correlate inversely to respiratory chain activity. For measurements of the redox index, FCCP (1 μm) was added to maximize respiration and therefore minimize the mitochondrial NADH pool. The addition of NaCN (1 mm) blocks mitochondrial respiration and therefore maximizes the NADH pool in mitochondria. This allowed the separation of mitochondrial from cytosolic NADH and total NADPH. The initial NADH autofluorescence is then calculated as a percentage of this range (Fig. 2A). In addition, the total mitochondrial pool of NADH (maximum autofluorescence minus minimum) may be taken as an indication of the substrate availability for complex I.
Figure 2.
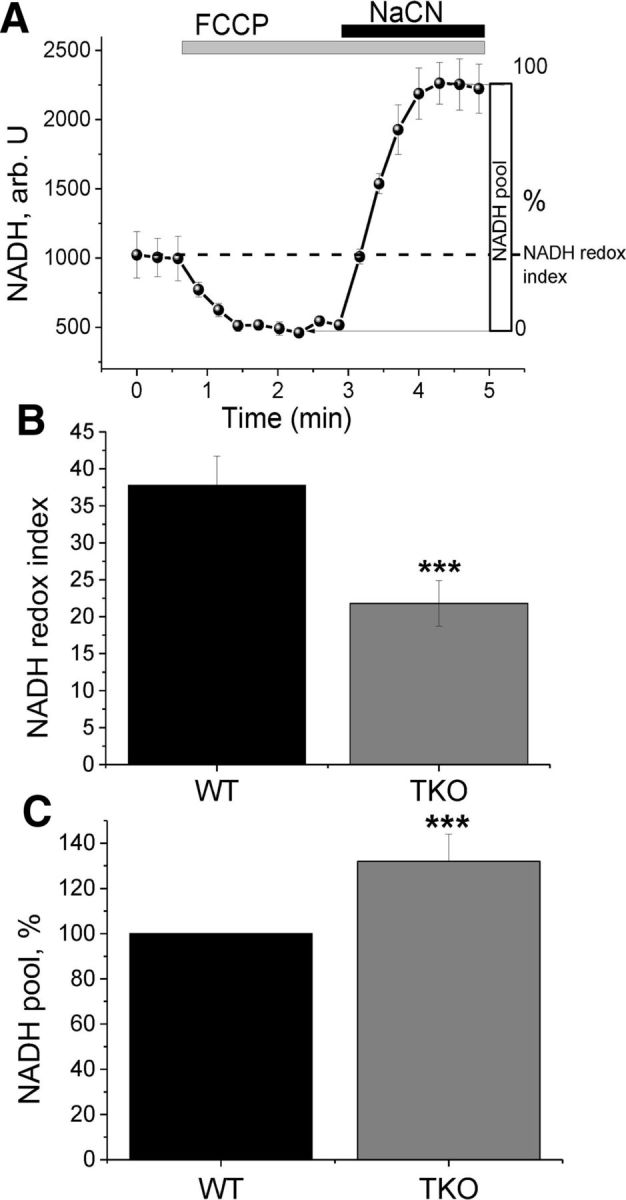
NADH autofluorescence measurements reveal increased respiration in TKO cells. A, Representative traces of NADH in healthy cells. FCCP (1 μm) is applied to maximize respiration and therefore minimize the NADH pool; NaCN (1 mm) is added to block the mitochondrial respiration and therefore maximize the NADH pool. B, Redox index is significantly lower in TKO compared with WT cells. C, Mitochondrial pool of NADH is significantly larger in TKO compared with WT cells, indicating normal substrate supply. *p < 0.05; ***p < 0.001; n = 3 experiments.
We observed a significant decrease in the redox index of TKO neurons and astrocytes, indicating an increased respiration rate in these cells from 37.8 ± 3.9 in control (n = 37 cells) to 21.8 ± 3.1 in TKO (n = 49 cells) (p < 0.001; Fig. 2B). The total mitochondrial pool of NADH in TKO neurons and astrocytes was higher than in WT (132 ± 12% of control; p < 0.001; Fig. 2C). This effect might be due to an activation/upregulation of the TCA cycle (that leads to an increased mitochondrial NADH pool) to compensate for a higher rate of NADH consumption in the mitochondria (lower redox level in the same experiment).
α-Synuclein is a regulator of mitochondrial respiration
The lower ΔΨm and NADH redox state in cultured TKO cells suggest an altered mitochondrial respiration. Therefore, a more direct measurement of respiration was performed in WT and TKO brain mitochondria using a Clark-type oxygen electrode (Fig. 3). Brain mitochondria were isolated from WT and TKO mice and the basal rate of oxygen consumption was measured through the application of TCA cycle substrates (5 mm malate and 5 mm glutamate), which allowed the assessment of complex I activity and therefore mitochondrial respiration (V2). V2 respiration was significantly higher (by 26 ± 8.1%; n ≥ 36 measurements; 8 experimental days, p < 0.05; Fig. 3A,B) in TKO mitochondria compared with WT mitochondria. Importantly, the rate of V2 respiration was also significantly higher in AKO, BKO, GKO, and A/GKO mice (n ≥ 9 measurements; 3 experimental days; p < 0.05; Fig. 3B). This was repeated by application of alternate TCA cycle substrates, malate and sodium pyruvate (n = 7 measurements; Fig. 3C). These data are in agreement with the previous NADH experiment, which also suggested increased respiration in TKO.
Figure 3.
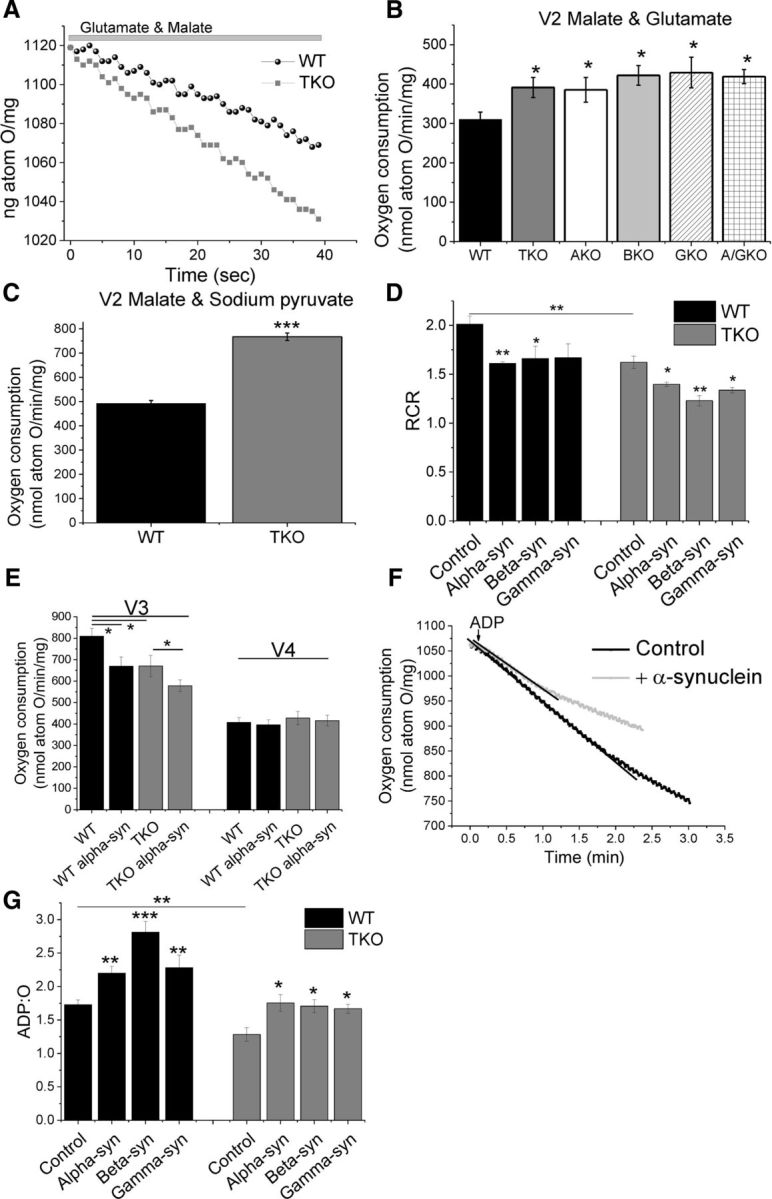
TKO mitochondria are uncoupled, respire faster, and have a lower ATP synthase efficiency. A, Representative traces of oxygen consumption in the presence of mitochondrial substrates (V2; 5 mm glutamate/malate). B, Quantification of basal respiration (V2) revealed a significantly faster respiration in synuclein-deficient mitochondria compared with WT mitochondria (n ≥ 3 experiments). C, Application of the alternate TCA substrates malate and sodium pyruvate mirrored the higher V2 observed in B (n = 3 experiments). D, Quantification of RCR showed that TKO mitochondria are uncoupled and that α-, β-, or γ-synuclein lowers the RCR. E, Quantification of V3 and V4 of WT and TKO mitochondria with and without α-synuclein. F, Representative traces of oxygen consumption (V3) in the absence and presence of monomeric α-synuclein. G, Quantification of ADP:O in WT and TKO mitochondria in the presence and absence of α-, β-, or γ-synuclein. *p < 0.05; **p < 0.01; ***p < 0.001.
Several other states can be assessed to understand mitochondrial respiration. Application of ADP to the mitochondria allows the assessment of the V3 (ADP-dependent) state, representing the oxygen consumption during maximal ADP phosphorylation by the ATP synthase. Upon depletion of all ADP, mitochondrial oxygen consumption slows down, corresponding to state V4 (ADP-independent or absent phosphorylation). These two states allow the calculation of the respiratory control ratio (RCR). This V3/V4 ratio indicates the degree of coupling between the mitochondrial respiratory chain activity and oxidative phosphorylation (Chance and Williams, 1955).
The RCR was found to be significantly lower in TKO mitochondria (23 ± 1.8% decrease compared with WT; n ≥ 17 measurements; 8 independent experiments; Fig. 3D), indicating that these mitochondria are uncoupled. This is the result of significantly lower oxygen consumption in V3 in the TKO mitochondria (17 ± 6%), with no significant changes in V4 (Fig. 3E).
α-Synuclein affects ATP synthase efficacy
The oxygen electrode allows the analysis of ATP synthase efficiency, which is expressed in form of the ADP:O ratio. This ratio expresses how much oxygen is consumed per nmole of ADP to ATP conversion. The ADP:O ratio was significantly lower (25 ± 5%, n ≥ 17 measurements; 8 independent experiments, p < 0.01) in TKO compared with WT mitochondria, suggesting that oxidative phosphorylation is less efficient in TKO mice (Fig. 3G). To investigate whether α-synuclein has a direct effect on the ATP synthase efficiency, 100 nm α-synuclein was added and ADP:O assessed. Alpha-, β-, or γ-synuclein was added to the oxygen electrode chamber containing mitochondria and incubated for 60–90 s in the presence of TCA substrates. This was followed by the application of ADP, which allowed the ADP:O ratio to be calculated (Fig. 3F,G). In the presence of monomeric α-synuclein, a significant increase in ADP:O was observed in WT and TKO mitochondria (27 ± 6% and 36 ± 9%; respectively, n ≥ 6 measurements, 3 independent experiments; p < 0.001, p < 0.01; Fig. 3F). Importantly, β- and γ-synuclein also increase the ADP:O significantly (Fig. 3F). The RCR decreases upon α-, β-, or γ-synuclein addition. For example, the RCR decreases by 19 ± 0.2% in WT and 14 ± 0.4% in TKO mitochondria upon α-synuclein application (Fig. 3D). Further analysis of V3 and V4 revealed that application of α-synuclein reduces V3 (ADP dependent) by 18 ± 5.4% in WT and by 14 ± 4% in TKO mitochondria while not altering V4 (ADP independent) (Fig. 3E). These results suggest that synuclein deficiency or application of monomeric synuclein affects only ADP-dependent respiration (V3), which further suggests a monomeric synuclein effect on oxidative phosphorylation rather than respiration.
It should be noted that, in TKO mitochondria, only the higher concentration (100 nm) of α-synuclein provided a significant increase in the ADP:O ratio (36 ± 12.5%, n = 9 measurements; 3 independent experiments; p < 0.05; Fig. 4A). These results were mirrored in AKO mouse brain mitochondria, in which application of 100 nm α-synuclein increased the ADP:O by 31% (±3.7%, n = 8 measurements, 3 independent experiments; p < 0.01) compared with the AKO control background (Fig. 4B). Mutated α-synuclein A30P was included to serve as a control α-helical protein in the short incubation experiments. It has been shown that this mutation greatly impairs binding to phospholipids and, correspondingly, interaction with various phospholipid membranes (Jensen et al., 1998; Cole et al., 2002; Fortin et al., 2004). Importantly, A30P α-synuclein had no effect on ADP:O (Fig. 4C).
Figure 4.

ATP synthase efficiency can be rescued by application of monomeric synuclein. A, Quantification of the effects on ADP:O when 35 or 100 nm monomeric α-synuclein is applied to WT or TKO mitochondria (n = 3 experiments). B, Quantification of AKO ADP:O in the presence or absence of α-synuclein. C, Quantification of the effects on ADP:O when monomeric mutant A30P α-synuclein is applied to TKO mitochondria. D, Quantification of ADP:O in the presence of GDP (a UCP inhibitor) and/or α-synuclein. E, Representative traces of V2 activation by α-, β-, or γ-synuclein. E, Representative traces of oxygen consumption upon oligomycin, α-, β-, or γ-synuclein and FCCP application. G, Quantification of FCCP induced maximum respiration in the presence of α, β, or γ-synuclein. *p < 0.05; **p < 0.01; ***p < 0.001.
If the effect of synuclein on ADP:O is mediated through uncoupling proteins (UCPs), then a UCP inhibition would prevent the effect of synuclein. Inhibition of UCPs by GDP (500 μm, n = 3 experiments) did not change the ability of synuclein to increase ADP:O, suggesting that the effect of synuclein on ADP:O is emitted through an increased oxidative phosphorylation efficiency and not through a UCP-mediated process (Fig. 4D).
All three synucleins were found to activate V2 (Fig. 4E). Preincubation of mitochondria with inhibitor of F0F1- ATP synthase oligomycin (2 μg/ml) completely blocked the effect of synucleins on V2 respiration, which suggests that activation of respiration by all synucleins is due to an effect on oxidative phosphorylation (Fig. 4F). Uncoupling of mitochondria with FCCP induces maximal respiration. Application of FCCP revealed that maximum respiration is not affected by synuclein, suggesting that synuclein does not affect the activity of mitochondrial ETC complexes (Fig. 4G).
Together, these data suggest that monomeric α-, β-, and γ-synuclein have a direct effect on oxidative phosphorylation and ATP synthase efficiency.
Lack of synuclein leads to lower ATP levels
The lower ATP synthase efficiency in TKO as shown by ADP:O should lead to decreased ATP levels in affected tissues. To determine whether an alteration in ATP synthase efficiency in TKO mitochondria was sufficient to reduce ATP levels in tissues, ATP levels in different brain regions of TKO and WT mice were assessed using a colorimetric ATP assay, as described in the “Materials and Methods” section. It was found that the ATP concentration in cortex and midbrain tissue of TKO mice was significantly lower (94.5 ± 0.4% and 90.1 ± 0.4%, respectively) than in those tissues of WT mice (p ≤ 0.05, n = 3; Fig. 5A).
Figure 5.
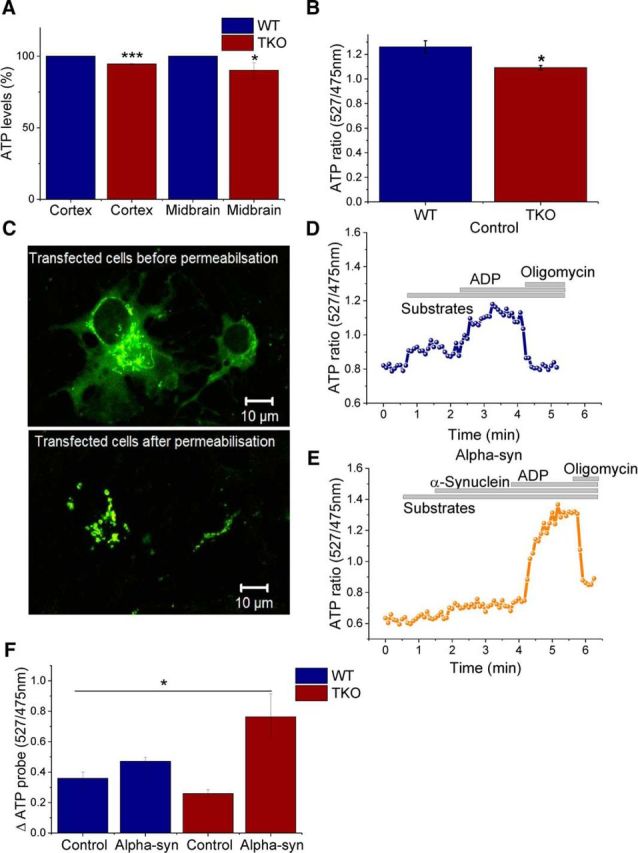
ATP levels in TKO brain tissue are significantly lower compared with WT. A, Quantification of ATP levels in WT and TKO cortex and midbrain tissue by colorimetric measurements (n = 3). B, Quantification of basal ATP levels in WT and TKO neuronal cocultures using a FRET-based mitochondrial ATP probe (n = 5). C, Representative images of cells transfected with the mitochondrial ATP probe before and after permeabilization. D, E, Representative trace of kinetic changes in mitochondrial ATP of WT cells in the presence of mitochondrial substrates (malate/glutamate), ADP, and oligomycin in the absence (D) or presence (E) of monomeric α-synuclein. F, ATP amplitude quantification upon ADP application in the presence or absence of monomeric α-synuclein in WT or TKO (n ≥ 4). *p < 0.05; ***p < 0.00. Scale bar, 10 μm.
Further evidence for the reduction of ATP level in TKO cells came from monitoring mitochondrial ATP. Primary neuronal/glial midbrain cultures were transfected with a FRET-based ATP probe (Imamura et al., 2009), allowing basal and kinetic changes in ATP to be monitored (Fig. 5C). The basal fluorescent signal reflects mitochondrial ATP levels and is in agreement with the data generated in Figure 5A because mitochondrial ATP levels were significantly reduced by 13 ± 1.3% (n = 5 experiments, p < 0.05) in TKO cells (Fig. 5B).
The transfected cells were then permeabilized in pseudo-intracellular solution (Fig. 5C) and the changes of ATP levels were assessed in the presence of mitochondrial substrates (5 mm malate and 5 mm glutamate), ADP, and oligomycin (2 μg/ml) with or without α-synuclein (Fig. 5D,E). The addition of ADP leads to an increase in the ATP level and therefore an increase in ATP fluorescence as the ATP synthase converts ADP to ATP. No significant differences were detected in the amplitude of ATP fluorescence in TKO compared with WT upon ADP addition (Fig. 5F). However, application of 100 nm α-synuclein monomers resulted in an increase in ATP fluorescence after ADP addition in both WT and TKO mitochondria (Fig. 5E,F).
The energy capacity of the cell is defined as the time between cessation of ATP production and the time of energetic collapse due to total ATP depletion and inability to maintain calcium homeostasis (Abramov et al., 2007; Yao et al., 2011). Live-cell imaging of the fluorescent probe MagFura-2 was used to assess the energy capacity of WT and TKO primary cocultured cells. Mg2+ is released from MgATP upon the hydrolysis of ATP, so the measurement of cellular free magnesium ([Mg2+]c) using the Mg2+-sensitive fluorescent probe MagFura-2 can be used as an indication of ATP consumption (Leyssens et al., 1996; Yao et al., 2011). Application of inhibitors of glycolysis and/or oxidative phosphorylation blocks ATP production in cells, which eventually leads to ATP depletion and subsequent Mg2+ release and an increase in Mag-Fura fluorescence (Fig. 6A).
Figure 6.
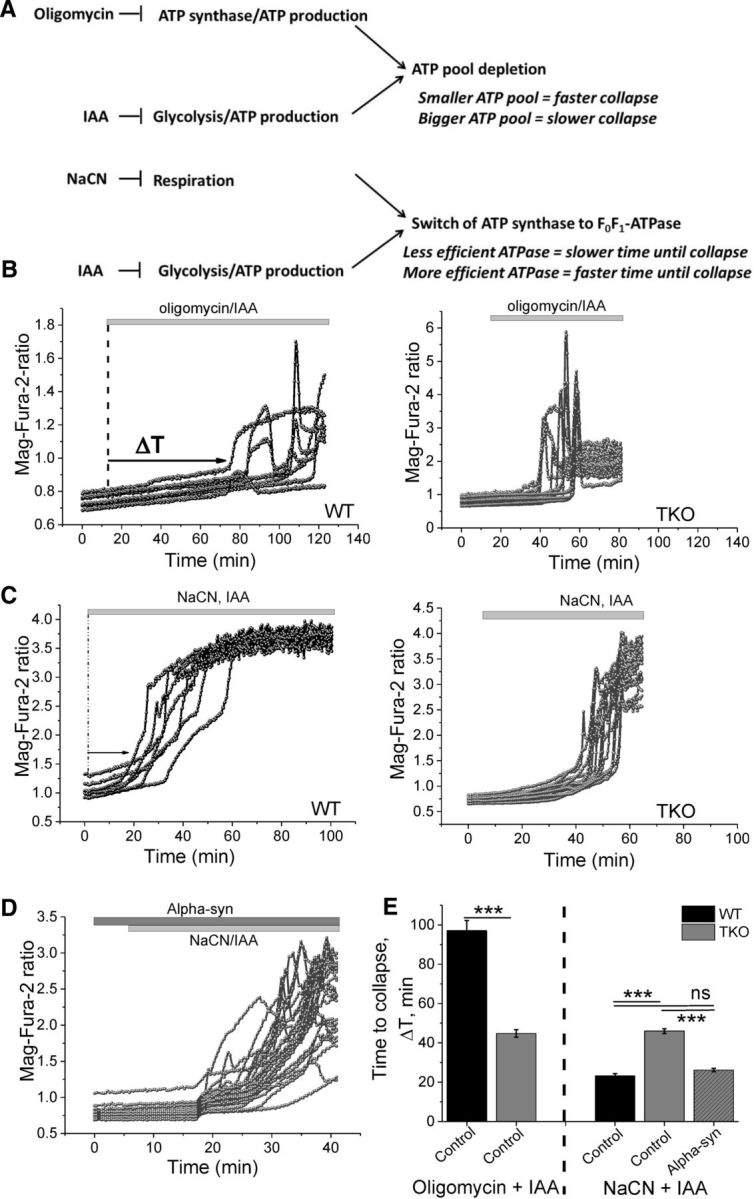
ATP synthase is more efficient in the presence of monomeric α-synuclein. A, Diagram displaying inhibitory effects of oligomycin, iodoacetic acid (IAA), and NaCN. B, Representative Mag-Fura traces of WT and TKO cocultures after treatment with oligomycin and IAA, which block oxidative phosphorylation and glycolysis. These inhibitors allow assessment of the total ATP pool. C, Representative Mag-Fura traces of WT and TKO cocultures after treatment with NaCN and IAA, which blocks mitochondrial respiration and glycolysis. D, Representative Mag-Fura traces of TKO cocultures after treatment with NaCN and IAA in the presence of 100 nm monomeric α-synuclein. E, Quantification of the time until collapse in cells exposed to oligomycin and IAA, NaCN and IAA, NaCN, or IAA in the presence of monomeric α-synuclein. n = 3 experiments; ***p < 0.001.
To estimate the overall ATP consumption in WT and TKO neuronal/glial cocultures, inhibitors of glycolysis (iodoacetic acid, 20 μm) and F1F0-ATP synthase (oligomycin, 2 μg/ml) were applied simultaneously. As shown in Figure 6B, complete inhibition of ATP production induced an earlier increase in Mag-Fura fluorescence, reflecting a faster energetic collapse in TKO cells (44.8 ± 1.9 min) compared with WT (97.1 ± 5.1 min; p < 0.001; n = 3 independent experiments; Fig. 6B,E), which can be explained by the previously described lower ATP levels in these cells.
The ATP synthase can be reversed to the F1F0-ATPase by mitochondrial respiration inhibitors, which results in a maximal ATP consumption rate (Campanella et al., 2008). To force the cells into the F1F0-ATPase mode, iodoacetic acid (20 μm) and an inhibitor of the respiratory chain (NaCN, 1 mm) were applied (Fig. 6A). Under these conditions, the rate in [Mg2+]c as a function of ATP consumption and time to cell collapse was faster in WT cells (23.2 ± 1.2 min) compared with TKO (46 ± 0 min; Fig. 6C,E). These observations suggest that F1F0-ATPase activity is lower in TKO than in WT cells. Interestingly, preincubation of TKO cocultures with 100 nm α-synuclein led to a significant reduction in the time until energy collapse (26.2 ± 0.9 min; p < 0.001), matching the observations in WT cultures (23.2 ± 1.2 min; Fig. 6D,E, 3 independent experiments, p = NS). These results suggest that exogenous α-synuclein is able to rescue ATPase activity in TKO cells back to control levels.
These data suggest that the ATPase activity is reduced in α-synuclein-deficient cells and that α-synuclein can increase ATPase activity. Therefore, α-synuclein is also able to modulate the activity of complex V in ATPase mode in addition to the previous data showing its ability to modulate the activity and efficiency of ATP synthase.
α-Synuclein increases ATP synthase activity
The obtained data suggest that α-synuclein acts on the ATP synthase and its activity. To verify whether α-synuclein regulates ATP synthase activity, brain mitochondria were isolated and the F0F1-ATPase immunocaptured in the wells of a microplate, as described in the “Materials and Methods” section. This assay allows measurements of α-synuclein effects on ATP synthase activity in the absence of any other mitochondrial proteins. Monomeric α-synuclein was added to the wells immediately before absorbance was measured at 340 nm. The addition of α-synuclein led to a significant increase in ATP synthase activity (36 ± 5.8%) compared with control wells (100 ± 8%; Fig. 7A; n = 4 experiments; p < 0.05). This assay provided evidence that α-synuclein is able to act directly on the ATP synthase independent of any other mitochondrial proteins.
Figure 7.

Monomeric α-synuclein binds and improves ATP synthase efficiency. A, Quantification of ATP synthase activity in the presence and absence of 100 nm monomeric α-synuclein. Bi, Representative images of PLA showing α-synuclein and ATP synthase subunit α interaction in SH-SY5Y and rat neuronal cocultures. The nucleus is stained in blue (DAPI). Note that the α-synuclein antibody does not detect endogenous rat α-synuclein. Bii, Representative image of a technical control in which one primary antibody was omitted. C, Western blot showing an interaction between α-synuclein and the ATP synthase α-subunit (ATP5A) in the eluent that was exposed to exogenous α-synuclein. The membrane was reprobed with the antibody specific for α-synuclein to visualize the bait used for the co-IP. D, Schematic diagram illustrating the effects of α-synuclein deficiency and rescue within the mitochondria. *p < 0.05; n = 4 experiments. Scale bar, 10 μm.
Direct interaction of monomeric α-synuclein and ATP synthase subunit α
To provide further evidence that α-synuclein has a direct effect on ATP synthase, a PLA, which detects in situ protein interaction, was performed. Antibodies against ATP synthase subunit α (ATP5A) and α-synuclein were used in this assay. Human neuroblastoma cells (SH-SY5Y) and rat neuronal cocultures were used to provide evidence for a protein interaction across two species. It should be noted that the most robust commercially available monoclonal antibodies against ATP synthase subunit α are raised in mice, which prevents the use of TKO mouse primary neuronal cultures. SH-SY5Y cells and rat primary cocultures were preexposed to exogenous α-synuclein for 1 h at 37°C. An interaction of exogenously applied α-synuclein and ATP synthase subunit α is represented by fluorescence emission at 624 nm in form of distinct puncta (Fig. 7B). The PLA signal provided evidence that that α-synuclein not only penetrates cells, but also localizes to the mitochondria in a close proximity (<40 nm) to the ATP synthase subunit α (Fig. 7B).
To provide further evidence for a direct interaction between the exogenous monomeric α-synuclein and ATP synthase subunit α, rat brain mitochondria were exposed to exogenous human monomeric α-synuclein for 15 min and lysates were subjected to immunoprecipitation with an antibody specific for α-synuclein. The immunoprecipitates were then probed with an ATP synthase subunit α antibody (ATP5A). The membrane was then stripped and reprobed with an α-synuclein antibody to visualize its presence. As shown by PLA, an interaction between monomeric α-synuclein and ATP5A was observed (Fig. 7C).
These results strongly suggest that exogenously applied α-synuclein interacts directly with ATP synthase.
Discussion
A growing body of evidence suggests that pathological modifications of the structure, expression, and/or intracellular localization of misfolded aggregated α-synuclein cause mitochondrial dysfunction(s), which becomes a crucial event in the pathogenesis of PD and other synucleinopathies. In contrast, information about the physiological effects of unfolded monomeric α-synuclein in mitochondria is scarce. This current study assessed the physiological role(s) of this protein in brain mitochondria by examining the effects of recombinant monomeric α-synuclein at low concentrations on cells and mitochondria obtained from WT and TKO mice lacking all three members of the synuclein family, in which no functional compensation by endogenous synucleins can occur.
It was found that AKO and TKO neurons and astrocytes have a significant lower ΔΨm. Further, TKO mitochondria respire faster, as established by oxygen electrode measurements and NADH redox index calculations (complex I activity). These lower ΔΨm and faster respiration were not due to a diminished mitochondrial substrate supply because the NADH pool was higher in TKO cells.
Oxygen electrode measurements confirmed a faster V2 in TKO brain mitochondria. These data are consistent with the lower RCR values in TKO mitochondria, indicating an uncoupling between oxidative phosphorylation and respiration. In agreement with the previous sets of data, the APD:O ratio was found to be significantly lower in TKO mitochondria, indicating that oxidative phosphorylation/ATP synthase activity is decreased in these cells. The finding of a decreased oxidative phosphorylation/ATP synthase activity was further supported by a lower ATP pool in TKO cells.
To establish whether the observed cellular effects were caused by a lack of α-synuclein and were not an indirect consequence or downstream effect of synuclein deficiency, exogenous monomeric α-synuclein was applied. Previous studies have shown that α-synuclein can pass membranes and associate with mitochondria (Cremades et al., 2012; Guardia-Laguarta et al., 2014; Robotta et al., 2014). This current study used low physiological doses of monomeric α-synuclein (Sierks et al., 2011) to establish whether the mitochondrial dysfunction induced by synuclein deficiency could be reversed. Here, it was shown by PLA that recombinant, exogenously applied α-synuclein is able to penetrate cells (human and rat origin) and localize to the mitochondria, which is in agreement with previous studies. Furthermore, the PLA and co-IP provided strong evidence of a direct interaction between monomeric α-synuclein and the ATP synthase subunit α.
Application of recombinant α-synuclein led to an increase in ATP levels through improving ATP synthase efficiency (as shown by the ADP:O ratio). This was specific to its WT form because A30P synuclein did not result in an improved ATP synthase efficiency. Application of exogenous α-synuclein was able to rescue the mitochondrial phenotype of the TKO model significantly. Complete restoration to WT levels was not always achieved and this may be attributed to concentration-dependent and/or time-dependent effects of exogenously added protein in this experimental paradigm. Nevertheless, we were also able to demonstrate that, when ATP synthase is acting as an ATPase, the application of α-synuclein was able to increase the activity of the ATPase. We have provided evidence that the effect of α-synuclein on ATP efficiency is due to a direct effect of α-synuclein on the ATP synthase itself.
One of the possible explanations for the increased capacity in oxidative phosphorylation may be an α-synuclein-mediated effect on mitochondrial UCPs. However, synuclein works through a UCP-independent mechanism for two reasons: (1) UCP inhibition by GDP does not alter the effect of α-synuclein on ADP:O and (2) if synuclein improves mitochondrial coupling, then it should increase the RCR upon application of synuclein, but the RCR decreases.
Increased ATP synthase efficiency may be particularly important for the proper function of remote cellular compartments such as presynaptic terminals, which require a high energy output from a limited number of available mitochondria. High levels of α-synuclein in presynaptic terminals, together with its ability to regulate ATP synthase activity and mitochondrial ATP production positively, may be crucial to maintaining the bioenergetic needs of neuronal synapses. This may represent a mechanism for monomeric α-synuclein involvement in synaptic function. It is feasible that this mechanism may become vital for functional compensation when the efficiency of other intraneuronal processes start declining and might, at least in part, explain why synuclein-deficient mice develop an age-dependent synaptic pathology (Al-Wandi et al., 2010; Greten-Harrison et al., 2010; Anwar et al., 2011). Interestingly, α-synuclein has been implicated in other synaptic processes such as synaptic vesicle exocytosis and endocytosis (Burré et al., 2010; Vargas et al., 2014), again, not as a functionally critical component, but rather, as a factor increasing their efficiency.
Many PD models have been linked to oxidative stress, mitochondrial dysfunction, and synaptic deficits (Parihar et al., 2008; Gandhi et al., 2009; Gandhi and Abramov, 2012; Angelova et al., 2016). The observed pathology may be due to a toxic gain of function of the aggregated α-synuclein protein or perhaps a loss of its physiological role.
Our study provided clear evidence that α-synuclein not only interacts with the ATP synthase, but also acts as a direct regulator of ATP synthase efficiency. This allows the hypothesis that α-synuclein regulates ATP synthase activity and ATP levels, but in times of stress or PD mutations, may lead to neuronal cell toxicity.
Footnotes
This work was supported by the National Institute for Health Research Biomedical Research Centre (A.Y.A.), the Wellcome Trust Programme (Grant 075615/Z/04/z to V.L.B.), and the Russian Scientific Fund (Grant 14-14-01138 to N.N.N.). S.G. is a Wellcome Trust Intermediate Clinical Research Fellow. We thank Hiromi Imamura (Japan Science and Technology Agency) for the AT 1.03 construct.
The authors declare no competing financial interests.
This is an Open Access article distributed under the terms of the Creative Commons Attribution License Creative Commons Attribution 4.0 International, which permits unrestricted use, distribution and reproduction in any medium provided that the original work is properly attributed.
References
- Abramov AY, Canevari L, Duchen MR. Beta-amyloid peptides induce mitochondrial dysfunction and oxidative stress in astrocytes and death of neurons through activation of NADPH oxidase. J Neurosci. 2004;24:565–575. doi: 10.1523/JNEUROSCI.4042-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci. 2007;27:1129–1138. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Al-Wandi A, Ninkina N, Millership S, Williamson SJ, Jones PA, Buchman VL. Absence of alpha-synuclein affects dopamine metabolism and synaptic markers in the striatum of aging mice. Neurobiol Aging. 2010;31:796–804. doi: 10.1016/j.neurobiolaging.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelova PR, Ludtmann MH, Horrocks MH, Negoda A, Cremades N, Klenerman D, Dobson CM, Wood NW, Pavlov EV, Gandhi S, Abramov AY. Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J Cell Sci. 2016;129:1792–1801. doi: 10.1242/jcs.180737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anwar S, Peters O, Millership S, Ninkina N, Doig N, Connor-Robson N, Threlfell S, Kooner G, Deacon RM, Bannerman DM, Bolam JP, Chandra SS, Cragg SJ, Wade-Martins R, Buchman VL. Functional alterations to the nigrostriatal system in mice lacking all three members of the synuclein family. J Neurosci. 2011;31:7264–7274. doi: 10.1523/JNEUROSCI.6194-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartolomé F, Abramov AY. Measurement of mitochondrial NADH and FAD autofluorescence in live cells. Methods Mol Biol. 2015;1264:263–270. doi: 10.1007/978-1-4939-2257-4_23. [DOI] [PubMed] [Google Scholar]
- Burré J, Sharma M, Tsetsenis T, Buchman V, Etherton MR, Südhof TC. Alpha-synuclein promotes SNARE-complex assembly in vivo and in vitro. Science. 2010;329:1663–1667. doi: 10.1126/science.1195227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campanella M, Casswell E, Chong S, Farah Z, Wieckowski MR, Abramov AY, Tinker A, Duchen MR. Regulation of mitochondrial structure and function by the F1Fo-ATPase inhibitor protein, IF1. Cell Metab. 2008;8:13–25. doi: 10.1016/j.cmet.2008.06.001. [DOI] [PubMed] [Google Scholar]
- Chance B, Williams GR. A simple and rapid assay of oxidative phosphorylation. Nature. 1955;175:1120–1121. doi: 10.1038/1751120a0. [DOI] [PubMed] [Google Scholar]
- Chandra S, Fornai F, Kwon HB, Yazdani U, Atasoy D, Liu X, Hammer RE, Battaglia G, German DC, Castillo PE, Südhof TC. Double-knockout mice for alpha- and beta-synucleins: effect on synaptic functions. Proc Natl Acad Sci U S A. 2004;101:14966–14971. doi: 10.1073/pnas.0406283101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole NB, Murphy DD, Grider T, Rueter S, Brasaemle D, Nussbaum RL. Lipid droplet binding and oligomerization properties of the Parkinson's disease protein alpha-synuclein. J Biol Chem. 2002;277:6344–6352. doi: 10.1074/jbc.M108414200. [DOI] [PubMed] [Google Scholar]
- Cremades N, Cohen SI, Deas E, Abramov AY, Chen AY, Orte A, Sandal M, Clarke RW, Dunne P, Aprile FA, Bertoncini CW, Wood NW, Knowles TP, Dobson CM, Klenerman D. Direct observation of the interconversion of normal and toxic forms of alpha-synuclein. Cell. 2012;149:1048–1059. doi: 10.1016/j.cell.2012.03.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deas E, Cremades N, Angelova PR, Ludtmann MH, Yao Z, Chen S, Horrocks MH, Banushi B, Little D, Devine MJ, Gissen P, Klenerman D, Dobson CM, Wood NW, Gandhi S, Abramov AY. Alpha-synuclein oligomers interact with metal ions to induce oxidative stress and neuronal death in Parkinson's disease. Antioxid Redox Signal. 2016;24:376–391. doi: 10.1089/ars.2015.6343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortin DL, Troyer MD, Nakamura K, Kubo S, Anthony MD, Edwards RH. Lipid rafts mediate the synaptic localization of alpha-synuclein. J Neurosci. 2004;24:6715–6723. doi: 10.1523/JNEUROSCI.1594-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Abramov AY. Mechanism of oxidative stress in neurodegeneration. Oxid Med Cell Longev. 2012;2012:428010. doi: 10.1155/2012/428010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gandhi S, Wood-Kaczmar A, Yao Z, Plun-Favreau H, Deas E, Klupsch K, Downward J, Latchman DS, Tabrizi SJ, Wood NW, Duchen MR, Abramov AY. PINK1-associated Parkinson's disease is caused by neuronal vulnerability to calcium-induced cell death. Mol Cell. 2009;33:627–638. doi: 10.1016/j.molcel.2009.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten-Harrison B, Polydoro M, Morimoto-Tomita M, Diao L, Williams AM, Nie EH, Makani S, Tian N, Castillo PE, Buchman VL, Chandra SS. alphabetagamma-Synuclein triple knockout mice reveal age-dependent neuronal dysfunction. Proc Natl Acad Sci U S A. 2010;107:19573–19578. doi: 10.1073/pnas.1005005107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths EJ, Rutter GA. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim Biophys Acta. 2009;1787:1324–1333. doi: 10.1016/j.bbabio.2009.01.019. [DOI] [PubMed] [Google Scholar]
- Guardia-Laguarta C, Area-Gomez E, Rüb C, Liu Y, Magrané J, Becker D, Voos W, Schon EA, Przedborski S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J Neurosci. 2014;34:249–259. doi: 10.1523/JNEUROSCI.2507-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hettiarachchi NT, Parker A, Dallas ML, Pennington K, Hung CC, Pearson HA, Boyle JP, Robinson P, Peers C. alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J Neurochem. 2009;111:1192–1201. doi: 10.1111/j.1471-4159.2009.06411.x. [DOI] [PubMed] [Google Scholar]
- Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova-Kostova AT, Abramov AY. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open. 2013;2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imamura H, Nhat KP, Togawa H, Saito K, Iino R, Kato-Yamada Y, Nagai T, Noji H. Visualization of ATP levels inside single living cells with fluorescence resonance energy transfer-based genetically encoded indicators. Proc Natl Acad Sci U S A. 2009;106:15651–15656. doi: 10.1073/pnas.0904764106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jakes R, Spillantini MG, Goedert M. Identification of two distinct synucleins from human brain. FEBS Lett. 1994;345:27–32. doi: 10.1016/0014-5793(94)00395-5. [DOI] [PubMed] [Google Scholar]
- Jensen PH, Nielsen MS, Jakes R, Dotti CG, Goedert M. Binding of alpha-synuclein to brain vesicles is abolished by familial Parkinson's disease mutation. J Biol Chem. 1998;273:26292–26294. doi: 10.1074/jbc.273.41.26292. [DOI] [PubMed] [Google Scholar]
- Leyssens A, Nowicky AV, Patterson L, Crompton M, Duchen MR. The relationship between mitochondrial state, ATP hydrolysis, [Mg2+]i and [Ca2+]i studied in isolated rat cardiomyocytes. J Physiol. 1996;496:111–128. doi: 10.1113/jphysiol.1996.sp021669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luth ES, Stavrovskaya IG, Bartels T, Kristal BS, Selkoe DJ. Soluble, prefibrillar alpha-synuclein oligomers promote complex I-dependent, Ca2+-induced mitochondrial dysfunction. J Biol Chem. 2014;289:21490–21507. doi: 10.1074/jbc.M113.545749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ninkina N, Peters OM, Connor-Robson N, Lytkina O, Sharfeddin E, Buchman VL. Contrasting effects of alpha-synuclein and gamma-synuclein on the phenotype of cysteine string protein alpha (CSPalpha) null mutant mice suggest distinct function of these proteins in neuronal synapses. J Biol Chem. 2012;287:44471–44477. doi: 10.1074/jbc.M112.422402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parihar MS, Parihar A, Fujita M, Hashimoto M, Ghafourifar P. Mitochondrial association of alpha-synuclein causes oxidative stress. Cell Mol Life Sci. 2008;65:1272–1284. doi: 10.1007/s00018-008-7589-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plun-Favreau H, Burchell VS, Holmström KM, Yao Z, Deas E, Cain K, Fedele V, Moisoi N, Campannella M, Miguel Martins L, Wood NW, Gourine AV, Abramov AY. HtrA2 deficiency causes mitochondrial uncoupling through the F1F0-ATP synthase and consequent ATP depletion. Cell Daet Dis. 2012;28:e335. doi: 10.1038/cddis.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson DC, Schmidt O, Ninkina N, Jones PA, Sharkey J, Buchman VL. Developmental loss and resistance to MPTP toxicity of dopaminergic neurones in substantia nigra pars compacta of gamma-synuclein, alpha-synuclein and double alpha/gamma-synuclein null mutant mice. J Neurochem. 2004;89:1126–1136. doi: 10.1111/j.1471-4159.2004.02378.x. [DOI] [PubMed] [Google Scholar]
- Robotta M, Gerding HR, Vogel A, Hauser K, Schildknecht S, Karreman C, Leist M, Subramaniam V, Drescher M. Alpha-synuclein binds to the inner membrane of mitochondria in an alpha-helical conformation. Chembiochem. 2014;15:2499–2502. doi: 10.1002/cbic.201402281. [DOI] [PubMed] [Google Scholar]
- Sierks MR, Chatterjee G, McGraw C, Kasturirangan S, Schulz P, Prasad S. CSF levels of oligomeric alpha-synuclein and beta-amyloid as biomarkers for neurodegenerative disease. Integr Biol (Camb) 2011;3:1188–1196. doi: 10.1039/c1ib00018g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A. 1998;95:6469–6473. doi: 10.1073/pnas.95.11.6469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vargas KJ, Makani S, Davis T, Westphal CH, Castillo PE, Chandra SS. Synucleins regulate the kinetics of synaptic vesicle endocytosis. J Neurosci. 2014;34:9364–9376. doi: 10.1523/JNEUROSCI.4787-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venda LL, Cragg SJ, Buchman VL, Wade-Martins R. alpha-Synuclein and dopamine at the crossroads of Parkinson's disease. Trends Neurosci. 2010;33:559–568. doi: 10.1016/j.tins.2010.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao Z, Gandhi S, Burchell VS, Plun-Favreau H, Wood NW, Abramov AY. Cell metabolism affects selective vulnerability in PINK1-associated Parkinson's disease. J Cell Sci. 2011;124:4194–4202. doi: 10.1242/jcs.088260. [DOI] [PMC free article] [PubMed] [Google Scholar]