

Abstract
Cardiovascular diseases (CVDs) have been the prime cause of mortality worldwide for decades. However, the underlying mechanism of their pathogenesis is not fully clear yet. It has been already established that reactive oxygen species (ROS) play a vital role in the progression of CVDs. ROS are chemically unstable reactive free radicals containing oxygen, normally produced by xanthine oxidase, nicotinamide adenine dinucleotide phosphate oxidase, lipoxygenases, or mitochondria or due to the uncoupling of nitric oxide synthase in vascular cells. When the equilibrium between production of free radicals and antioxidant capacity of human physiology gets altered due to several pathophysiological conditions, oxidative stress is induced, which in turn leads to tissue injury. This review focuses on pathways behind the production of ROS, its involvement in various intracellular signaling cascades leading to several cardiovascular disorders (endothelial dysfunction, ischemia-reperfusion, and atherosclerosis), methods for its detection, and therapeutic strategies for treatment of CVDs targeting the sources of ROS. The information generated by this review aims to provide updated insights into the understanding of the mechanisms behind cardiovascular complications mediated by ROS.
1. Chemical Characteristics of Reactive Oxygen Species (ROS)
Researchers have been continuously studying the potential role of oxidative damage in cardiovascular diseases (CVDs) for a few decades. In a simple term, the common risk factors for CVDs like diabetes mellitus, smoking, aging, hypercholesterolemia, and nitrate intolerance can further increase the possibility of the generation of ROS. Furthermore, these risk factors can trigger several pathways such as apoptosis of endothelial cells (EC), expression of adhesion molecules, activation of metalloproteinases, induction of proliferation and migration of smooth muscle cells, lipid peroxidation, and change in vasomotor functions, collectively leading to CVDs [1, 2]. ROS are chemically reactive molecules containing oxygen. Several ROS with unpaired electrons, for instance, superoxide anion (O2 ∙−), hydroxyl radical (OH∙−), and lipid radicals, are considered as free radicals. ROS, such as hydrogen peroxide (H2O2), peroxynitrite (ONOO−), and hypochlorous acid (HOCl), are not free radicals but possess an oxidizing effect resulting in oxidant stress. A chain reaction leads to the production of many reactive oxygen species from one ROS (Figure 1). For example, the reactions of radicals and fatty acids (polyunsaturated fatty acids, PUFAs) within the cytoplasmic membrane result in a fatty acid peroxyl radical which can attack the adjacent side chain of the fatty acid and commence production of other lipid radicals. Lipid radicals generated in this chain reaction get collected in the plasma membrane and may have an innumerable effect on cell function, including alteration in cell membrane permeability and dysfunction of membrane-bound receptors [1, 3].
Figure 1.
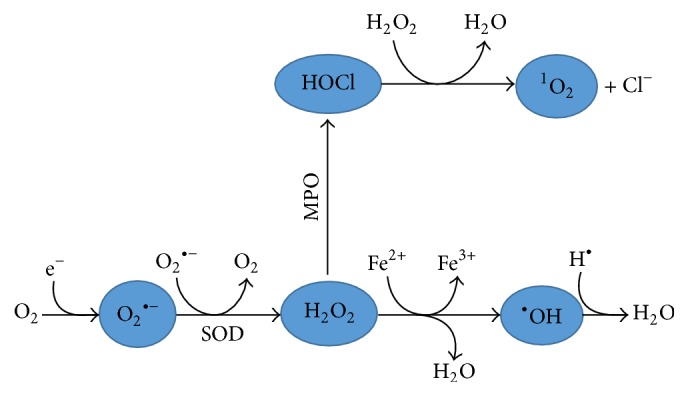
Production of ROS. The figure shows the pathway of ROS production in the human body with various enzymes involved. SOD: superoxide dismutase; MPO: myeloperoxidase.
2. Potential Sources of ROS for CVDs
In a physiological system, the imbalance between antioxidant defense mechanism and ROS production leads to oxidative stress and subsequent pathological conditions [4]. Most prominent ROS causing toxic insult to the human body are H2O2, O2 ∙−, ∙OH, and ONOO− [5]. In the blood vessel wall, each layer can produce ROS in pathological conditions [6]. Wattanapitayakul and Bauer reported that, within mitochondria, oxygen is usually utilized for energy production (in the form of ATP) and oxidative phosphorylation. During the mitochondrial electron transport (MET), harmful ROS are formed but they are balanced by antioxidant defense. However, in case of ischemia or hypoxia, MET is imbalanced, leading to ATP depletion, acidosis, mitochondrial depolarization, collection of noxious metabolites, intracellular Ca2+ overload, and cell death [7]. For example, approximately 1–3% of molecular oxygen is converted to unstable/reactive O2 ∙− in mitochondrial complexes I and III through a pathway involving oxidative phosphorylation [8]. In general, cardiac myocytes consume a high level of oxygen due to considerable higher number of mitochondria than other cells [9]. For this reason, cardiac myocytes also release ROS and cause oxidative stress to other cells [10]. But ROS do not have only a negative side, since production of ROS at physiological levels promotes cellular activities, controls the hormone level, maintains chemical balance, strengthens synaptic plasticity, and induces enzymes. Moreover, ROS also helps to fight against invading pathogens and induce an immune response against the pathogenic influence [5]. To a certain extent, ROS are neutralized by intracellular antioxidant enzymes such as glutathione peroxidase (GPx), superoxide dismutase (SOD), and catalase and consumption of other nonenzyme antioxidants like β-carotene, ascorbic acid, and tocopherols as a supplement [7]. In spite of being necessary to carry out cell signaling pathways, overproduction of ROS leads to injury of the cell membrane integrity causing altered permeability, change in proteins expression, and DNA damage [11]. For the majority of CVDs, the enzymatic sources of ROS include NAD(P)H oxidase, lipooxygenase, cyclooxygenase (COX), xanthine oxidase (XO), uncoupled nitric oxide synthases (NOS), cytochrome P450, and mitochondrial respiration [12–14] (Figure 2). The process of increased O2 ∙− generation, facilitated by XO enzyme, can be antagonized by a therapeutic approach with XO inhibitor, like allopurinol, to ameliorate cardiac conditions [15]. NADPH oxidase (Nox), commonly found on the cellular membrane, is stimulated during phagocytosis leading to increased ROS release [10]. In particular, the overexpression of Nox2 and Nox4 is linked to the remarkable oxidative stress observed during CVDs. A study done by Kuroda et al. showed that Nox4 knockout mice showed a low level of cardiac O2 − revealing that Nox4 is a potential source of superoxide in cardiac myocytes. Nox4 overexpression worsened the cardiac function and induced apoptosis and fibrosis in a mouse with response to pressure overload. Thus, Nox4 is a key contributor of oxidative stress in the mitochondrial redox systems leading to cardiac impairment during pressure overload. Therefore, the physiological role of Nox, translocating electrons throughout the membrane, can be deregulated in CVDs leading to cardiac dysfunction [16]. However, some pathways associated with ROS mediated CVDs are yet to be clarified. However, researchers are trying to reveal good, bad, and ugly roles of ROS in the physiological system. In contrast to the good face of ROS on signaling and immune response, at high concentrations, ROS can exhibit the deleterious effect on redox homeostasis leading to intracellular components damage as seen in neurodegenerative diseases, CVDs, and pulmonary disorders [5].
Figure 2.
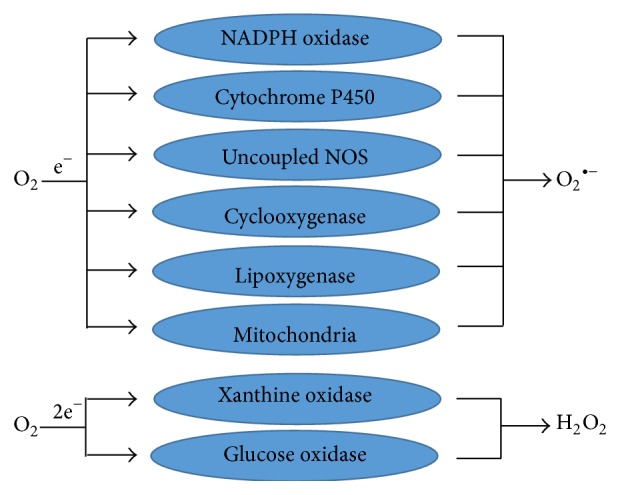
Sources of O2 ∙− and H2O2 in cells. The figure shows the enzymatic pathway of superoxide anion (O2 ∙−) and hydrogen peroxide (H2O2) generation in cells.
3. Oxidative Stress and Endothelial Dysfunction
Endothelial cells are lining the interior surface of blood and lymphatic vessels cells. Endothelial cells play an important role in homeostasis and immune and inflammatory reactions. EC regulates vascular tone by releasing various vasodilator factors such as nitric oxide (NO) endothelium derived hyperpolarizing factor, prostacyclin or vasoconstrictive factors such as thromboxane (TXA2), and endothelin-1 (ET-1). Endothelial dysfunction (ED) is a pathological state of the endothelium, which is a predictor of various CVDs, and is caused by imbalance between vasodilating and vasoconstricting substances [17]. ROS are considered as signaling molecules that contribute to ED in experimental and clinical atherosclerosis [3, 18]. NO is a potent vasodilator produced by the endothelium. Besides vasorelaxation, nitric oxide exerts various functions like antiplatelet, antithrombotic, and anti-inflammatory properties and permeability decreasing properties [19]. It is reported that ONOO− which is formed by the reaction of superoxide and free radical NO can oxidize tetrahydrobiopterin. If formed in a small amount, ONOO− exerts a similar physiological activity like NO. However, at a high concentration, it shows injurious activity by converting to harmful peroxynitrous acid and causing alteration of protein structure [20]. ED is associated with polymorphisms of various genes which include cytochrome P450, methylene tetrahydrofolate reductase, p22phox, angiotensin convertase enzyme, and glutathione-S-transferase [21]. Excess amount of ROS damages the endothelium, especially the terminal arteries leading to alteration of the intracellular reduction-oxidation homeostasis [22]. In a patient with diabetes mellitus, small vessel disease linked with mitochondrial disorders might also be due to oxidative stress. The result of diabetes mellitus in atherosclerosis is stimulated mainly by oxidative stress [23]. ROS may also activate mitogen activated protein kinase, which regulates the expression of monocyte chemoattractant protein 1 (MCP-1) and favors the chemotaxis of circulating monocyte to the site of atherosclerotic lesion. This demonstrates a potential link between arterial wall strain and atherosclerosis [24].
4. ROS in Ischemia-Reperfusion (I/R) Damage
Several researches support the fact that ROS are involved in ischemic occlusion leading to cardiac damage [39, 40]. Apart from carrying out the function of cellular O2 storage and supply by oxymyoglobin (Mb), it also acts as a potent preventive source against I/R injury [40]. During the outset of I/R injury, O2 ∙− release has been observed in an isolated rat heart. Zhu and Zuo speculate that generation of O2 ∙− is linked with Mb because of the lower myocardium oxygen tension. Results revealed that the rise of fluorescence in the ischemic heart was terminated by a SOD mimic, carbon monoxide (CO), or by Mb gene knockout. Likewise, O2 ∙− was not formed in intracellular EC but rather from the myocytes, which are considered a potential source of Mb. This suggests that Mb is an important factor responsible for production of O2 ∙− during ischemia [40]. An enzyme responsive to stress, named sirtuin-6 (SIRT6), displays cardiac protection from I/R injury as revealed in partial SIRT6 knockout mice as well as in vitro cultured cardiomyocyte. SIRT6 is a deacetylase and mono-ADP ribosyltransferase enzyme responsive to oxidative stress and can protect the cell against oxidative stress. This protective activity was achieved by initiating the expression of catalase and manganese SOD antioxidant-encoding gene resulting in reduced cellular oxidative stress [41]. Restoration of the coronary artery blood flow reverse to the ischemic myocardium can have a detrimental effect on the microvascular function, causing arrhythmias [42]. In the endothelium, the rise of ROS release and the opening of mitochondrial permeability transition (MPT) pore play an important role in the protection from I/R damage [43]. However, Kim and Lemasters showed that mitochondrial ROS, accompanied by normalization of pH, stimulate initiation of MPT pore followed by death of myocytes after reperfusion. However, Ca2+ overloading does not promote onset of MPT pore [44]. Research demonstrated that the reason behind the protective effect was the involvement of ROS and potent vasodilator NO in regulating downstream pathways by stimulating adenosine triphosphate sensitive potassium channel in mitochondria [45]. After open cardiac surgery, the I/R injury can influence postsurgical consequences because of the lipid peroxidation mediated by ROS [46]. In comparison to the heart obtained from juvenile rat, the cardiac dysfunction due to I/R was dramatically concealed in the heart obtained from congenital heart disease (CHD) model rats. Moreover, the ratio of n-3/n-6 PUFA was remarkably raised in I/R phase in CHD rats, whereas it was not observed in juvenile rats suggesting that the rise in n-3/n-6 ratio could result in the upregulation of cell defense system against oxidation via n-3 PUFA oxidation product 4-hydroxy-2-hexenal causing higher tolerance to I/R damage [46].
5. ROS and Atherosclerosis
Excess production of ROS plays an important role in inflammation, disturbed blood flow/abnormal shear stress, and arterial wall remodeling. ROS causes remodeling through proliferation of smooth muscle cell and increased inflammation [39]. Repeated continuous exposure to nonstreamline shear stress of arterial regions generates O2 induced by endothelial Nox resulting in adhesion of monocytes [47]. The upregulation of adhesion molecules including P-selectin, VCAM-1, and E-selectin causes further inflammation by adhesion of white blood cells. Development of inflammatory response increases ROS production by phagocytosis, which is important in the early stage of atherosclerosis [48, 49]. The Nox family of superoxide producing proteins is an important source of ROS in signal transduction. Nox are found to be expressed in phagocytic cells, EC, smooth muscle cells, and fibroblasts. Experiments conducted on arteries from human volunteers with coronary artery disease and animal experimental model with hypertension, diabetes, or atherosclerosis demonstrated that Nox1, Nox2, and Nox5 stimulate endothelial dysfunction, inflammation, and programmed cell death; however, isoform Nox4 protects the vascular system by increasing bioavailability of nitric oxide and stoppage of cell death pathways [50]. Some research presents the controversial role of Nox4 displaying either protective or a deleterious role of Nox4.
Nox4 are found abundantly in kidney, vascular cells, and osteoclasts [16]. Angiotensin II type 1 receptor activation and hypertension are linked to increased expression of Nox1 and Nox4 that could lead to vascular damage during chronic hypertension [51]. MCP-1 is essential for the formation of endothelial cell tumors (hemangioendotheliomas) which is redox sensitive. It was found that only the Nox4 isoform was present in endothelial cell tumors cells whereas knockdown of Nox4 gene remarkably decreased the expression of MCP-1 as well as hemangioendothelioma formation. This was due to the fact that, in hemangioendothelioma cells, Nox4 delivers H2O2 to the nuclear compartment causing oxidative alteration of DNA [52]. Inflammation mediates all stages of atherosclerosis and ROS sources might include infiltrated monocytes/macrophages, dysfunctional EC, and smooth muscle cells that migrated from tunica media to tunica intima layers of the wall of an artery. ROS oxidized-LDL is available in the arterial wall and macrophages scavenge it resulting in the formation of foam cells. This is one of the important steps in the progression and development of atherosclerosis [53]. Also, the calcium-dependent zinc containing endopeptidase, matrix metalloproteinase, secreted from EC, foam cells, and vascular smooth cells, is activated during oxidative stress in part due to inflammation and nonlaminar shear stress, resulting in the ruptures of thrombosis [54].
6. Oxidative Stress and Mitochondria
Mitochondria play an important role in cellular signaling pathways, particularly in the modulation of calcium stores within the cell, generation of ROS, respiration, and biogenesis. So, changes in mitochondrial function lead to development of human diseases [55, 56]. Mitochondrial DNA (mtDNA) damage is linked to the atherosclerotic lesions in apolipoprotein E (apoE) knockout mice and also introduces atherogenesis in young apoE knockout mice [57]. Raised levels of mtDNA damage have been seen in the vascular tissue of CVD patients [58]. Mitochondrial dysfunction is due to decreased manganese SOD, increased damage of mtDNA, and increased atherosclerosis in apoE knockout mice [59]. There is excessive mitochondrial damage in atherosclerosis model. Oxidized-LDL stimulates mitochondrial complex I activity which depends on the induction of oxidative stress [60, 61]. Composed of 46 subunits, human mitochondrial complex I is the key enzyme responsible for oxidative phosphorylation. Dysfunction of the mitochondrial oxidative phosphorylation in a physiological system is responsible for occurrence of CVDs in humans, and mitochondrial diseases are linked to mitochondrial respiratory-chain pathologies and mutations of mitochondrial DNA. Studies reported that various stress induced in the cells causes structural and functional disturbance of mitochondria [62, 63]. Dysfunction of mitochondria provokes a signaling pathway for cell death resulting in organ failure and diseases. Mitochondria based pathological conditions including obesity, cancer, stroke, diabetes, neurodegenerative diseases, heart failure, and aging, however, are caused by intrusion of mitochondrial Ca2+, ATP, or ROS metabolism [61, 63]. Myocardial ischemia-reperfusion injury leads to mitochondrial Ca2+ overload and consequent generation of ROS and opening of the mitochondrial permeability transition pore [60–62], resulting in apoptosis. Compounds which can reduce mitochondrial Ca2+ overload, decrease mitochondrial ROS collection, and prevent mitochondrial energy generation are all potential sources of therapies for preventing disease. Mitochondria produce oxidative stress which plays an important role in mediating programmed cell death (apoptosis) and damage to mtDNA and leads to human aging, cancer, and CVDs. Oxidative damage of the mitochondrial membrane results in depolarization of membrane and uncoupled oxidative phosphorylation and altered cellular respiration. Altered mitochondrial respiratory chain can hinder the pivotal role of providing the energy to the cell as ATP, leading to various disease progression [60–64].
7. Methods for Detection of ROS in CVDs
Since ROS are highly unstable and very reactive, researchers always face the problem of precisely monitoring them in biological systems. One way to find out the possibility of ROS in CVDs subjects involves exploring experimental proof of oxidative reactions. Fluorescent probes and electron spin resonance probes tools for detection of ROS are limited in animal and human experiment due to technical problems [65]. The following list of direct methods can show at least in part indirect evidence of ROS effect in CVDs (Table 1).
Table 1.
Direct methods for detection of ROS in CVDs.
| Methods | ROS detected | Applications/mechanism | Reference |
|---|---|---|---|
| Fluorescent protein-based redox probes | Cytoplasmic and mitochondrial H2O2 | Used to detect redox status and ROS by introducing adenoviruses or plasmids inside cells. Afterwards, cells form chimeric proteins efficient to detect alteration in the redox status or ROS. | [25, 26] |
|
| |||
| Dihydroethidium (DHE) and mitochondrion-targeted probe mitoSOX | Cellular and mitochondrial O2 ∙− | Can detect mitochondrial O2 ∙− by adding a triphenylphosphonium group for promoting its collection in the mitochondria. Similar to DHE, mitoSOX reacts with O2 ∙− to give 2-hydroxy-mito-ethidium (2-OH-Mito-E+) so as to be identified and measured using HPLC. | [27–29] |
|
| |||
| Cyclic hydroxylamine spin probes | Total cellular and mitochondrial O2 ∙− | Allows measurement of O2 ∙− in tissue, in in vitro cells, and in vivo. | [30–32] |
|
| |||
| Boronate-based fluorescent probes | H2O2 and ONOO∙− | As probes have a fluorophore which is secured by boronate, when subjected to H2O2, the boronate encounters a nucleophilic attack, followed by its displacement from the fluorophore, thus causing emission of light. | [33, 34] |
|
| |||
| Immunospin trapping | Free radical adduct formation in the mitochondria, cells, and tissue samples | 5,5-Dimethyl-1-pyrroline-N-oxide reacts with protein radicals to form epitopes which can be particularly characterized immunologically. | [35, 36] |
|
| |||
| In vivo using X- and L-band ESR spectroscopy | Short-lived free radicals in whole living animals | Detection is done in vivo by infusion of cyclic hydroxylamines or nitrone spin traps, followed by ex vivo study of the tissue or blood using X-band (9 GHz) electron spin resonance spectroscopy. | [37, 38] |
7.1. Biomarkers of ROS
7.1.1. Lipid Peroxidation Mediated by ROS
LDL collects in the blood vessels walls and lipid species undergo oxidation in the presence of several ROS [66]. It is reported that oxidative modification of LDL plays an important role in the atherosclerosis process [67]. Macrophages take up oxidized-LDL through scavenger receptor pathways resulting in cholesterol ester-rich foam cells and EC dysfunction, in part, by role of lectin-like oxidized-LDL receptor-1 [68, 69]. In atherosclerotic plaques, the availability of oxidized-LDL has been observed by using immunohistochemical staining for modified primary apolipoprotein B-100, the protein moiety in LDL [38]. Elevated levels of autoantibodies against oxidized-LDL or malondialdehyde-modified LDL particles are linked to atherosclerosis and coronary artery diseases (CADs) including acute coronary syndrome. In healthy people, circulating levels of oxidized-LDL can be identified by techniques such as ultrasound while in diseased subjects it is commonly seen in clinical case of CAD [70–72].
7.1.2. MPO
High levels of the enzyme myeloperoxidase which produces hypochlorous acid (HOCl) are found in human atheroma and are an important predictor of CAD, as well as in patients with unstable angina and mitochondrial infarction (MI). Increased numbers of myeloperoxidase-expressing macrophages are found in eroded or ruptured plaques [73]. Also, MPO and hypochlorite-modified proteins are colocalized in atherosclerotic lesions. Also, in human study research, strong inverse relation occurs between MPO serum concentrations and brachial artery flow-mediated dilation, which is another clinical marker of atherosclerosis [73, 74]. So, lowering MPO levels could lower occurrence of CVDs. Human studies have demonstrated that humans with total or near-total deficiency of MPO have a lower chance of developing CADs [73]. Reduced expression of MPO by its gene promoter polymorphism showed reduced CAD manifestations but increased MPO expression by MPO gene promoter polymorphism demonstrated raised CAD [75]. Since the level of plasma MPO is sensitive to heparin dosing [76], neutrophil activation [77], and the procedure of collection [78], there is a need for development of an appropriate method for its sampling.
7.1.3. Plasma F2-Isoprostanes
F2-isoprostanes are considered as the best biomarkers of oxidative stress status and lipid peroxidation in an in vivo model. F2-isoprostanes are found in an esterified form in normal biological tissues and are available in free form in biological fluids, demonstrating “physiological” levels of oxidative stress. The F2-isoprostanes might be produced from membrane phospholipids or circulating LDL [79, 80]. The generation of F2-isoprostanes can be via the action of several cell types like monocytes which are involved in atherosclerosis and the oxidized products have been restricted to a particular area within foam cells and atherosclerotic plaques individual specimens [81]. Various human studies have demonstrated a link between CAD and isoprostane levels [82]. Raised levels of isoprostanes in urine are an independent risk factor of CAD and are found to be increased in patients having unstable angina. Raised levels of isoprostanes are important markers of ischemic tissue injury, chronic heart failure, congestive heart failure, and cardiac remodeling [79, 82, 83]. Therefore, F2-isoprostanes could be used for the prediction of cardiovascular events.
8. Molecular Role of ROS in Muscle Contraction
During skeletal muscle contraction, ROS are generated which can affect muscle adaptation and function. Zuo et al. studied whether ROS are generated in the process of muscle contraction in isolated single skeletal muscle fibers (using Xenopus laevis muscle), as well as whether these ROS generated by contraction have an impact on fatigue development. To detect the ROS generation, myofibers were loaded with fluorescent probe (dihydrofluorescein-diacetate) which reacts with ROS to form fluorescein. Fluorescein signal was raised remarkably in both the first (42 ± 14%) and the third periods (39 ± 10%) of maximal tetanic contraction. However, with the treatment of reference antioxidant compound, ebselen, there was no rise of fluorescein during the second contractile period suggesting that ROS generation is high during contractile activity and antioxidant treatment can halt ROS production without any effect on myofiber contractility [84]. In spite of the various pathways of ROS generation, the study of key pathways of their production is still undergoing. In particular, ROS generation in response to exercise, hypoxia, and heat in the diaphragmatic skeletal muscle (a key muscle during respiration) is a topic of interest [85, 86]. During the state of heat stress, O2 ∙− is generated by skeletal muscle which can be quantified by cytochrome c reduction as it is correlated with arachidonic acid metabolism. The blockage of enzyme phospholipase A2 using manoalide remarkably reduced O2 ∙− release. However, neither the blockage of COX with nonselective COX inhibitor indomethacin nor the blockage of CYP P-450 contingent monooxygenase with SKF-525A reduces O2 ∙− generation. In contrast, lipoxygenase blockage with common inhibitors cinnamyl-3,4-dihydroxy-α-cyanocinnamate and 5,8,11,14-eicosatetraynoic acid drastically halted the signal. Moreover, O2 ∙− generation was notably reduced by diethylcarbamazine (5-LOX inhibitor) suggesting that metabolism of arachidonic acid involving LOX is a key mediator of generation of extracellular O2 ∙− in skeletal muscle [86]. The role of ROS in myocardial I/R injury has been widely studied [87]. Vanden Hoek et al. proposed the generation of a high amount of ROS in case of ischemia before reperfusion by an in vitro experiment in isolated cardiomyocyte during simulated I/R. The fluorescent probes 2′,7′-dichlorofluorescein and dihydroethidium (DHE) were significantly oxidized during ischemia, revealing ROS production. After an hour of ischemia, reperfusion leads to further generation of OH− and H2O2. In contrast, treatment of antioxidant compounds (1,10-phenanthroline and 2-mercaptopropionyl glycine) during ischemia injury halted oxidant production, raised the viability of cardiomyocytes, and opposed contraction following ischemia. The ROS production in response to residual O2 as in case of ischemia causes cellular injury observed in the reperfusion stage [88]. In a similar study on cardiomyocytes model of ischemia performed by Becker et al., an inhibitor of mitochondrial site III (myxothiazol) reduced oxidation. However, the inhibitor of mitochondrial site IV (cyanide) along with NOS inhibitor (nitro-L-arginine methyl ester), XO inhibitor (allopurinol), and Nox inhibitor (apocynin) showed no effect, suggesting that excessive O2 ∙− production is observed in ischemia prior to reperfusion through ubisemiquinone area of the MET chain [89]. Another study suggests that sublethal H2O2 production in isolated cardiomyocytes during the period of simulated ischemia modulates cell death later in reperfusion step, mainly due to the burst of reperfusion oxidant [90].
9. Therapeutic Strategy Targeting ROS Sources in CVD
9.1. Antioxidant
Antioxidants are a prime choice to fight against ROS. Therefore, it is crucial to formulate the strategies to halt abnormal ROS production inside the human body as well as improve innate antioxidant protection capacity. A cross-sectional research performed by Lane et al. reported that dietary supplements of vitamins E, C, and A help to lower occurrence of peripheral arterial disease [91]. Regular consumption of diet rich in vegetables and fruits (as a source of antioxidant vitamins) lowers the prevalence of CVDs, and, globally, it is recommended for enough daily intake of vegetables and fruit [92, 93]. These antioxidants vitamins A, C, and E, CoQ10, lycopene, and quercetin have been studied to explore their therapeutic and/or preventative effects on ventricular remodeling, atherosclerosis, heart failure, myocardial infarction, and ischemia-reperfusion heart injury [94–97]. Various herbal plants such as Nelumbo nucifera [98], Juglans regia [99], and Rumex nepalensis [100] are a rich source of compounds exhibiting remarkable antioxidant activity along with cardioprotective activity. Among the antioxidants, the most commonly used vitamin C and vitamin E as a cardioprotective supplement are discussed in the following section also including the information about their failure to revert the CVDs in some study.
9.1.1. Vitamin C
It is well known that vitamin C helps in regulation of blood pressure. A considerable study favors the notion that vitamin C restores high blood pressure related baroreflex dysfunction [101–103]. Some study revealed that oxidative stress decreases the baroreflex sensitivity leading to the constant hypertensive state. As shown in a hypertensive rat model by Botelho-Ono et al., treatment of vitamin C (150 mg/kg, IV) remarkably lowered heart rate, with improvement in baroreflex sensitivity compared to the untreated hypertensive group. Also, treatment of NADPH oxidase inhibitor apocynin (30 μg/kg, intravenous) maintained baroreflex sensitivity revealing that ROS generated via NADPH oxidase pathway plays a key role in the modification of baroreflex sensitivity in hypertension, whereas treatment of antioxidants (vitamin C) restored this change [102]. Similarly, Nishi et al. [101] also reported that chronic administration of vitamin C at a dose of 150 mg/kg/day drastically lowers the mean arterial pressure (MAP) in a hypertensive rat's model as compared to vitamin C untreated rat. Furthermore, the study also revealed increased expression of angiotensin II type 1 (AT-1) receptor in vitamin C untreated hypertensive rat, with downregulation of AT-1 in the vitamin C-treated group. Likewise, in a human clinical trial, Bruno et al. showed that IV infusion of vitamin C (3 g, over 5 min) significantly reduces both sympathetic nerve activity and blood pressure in essential hypertension patients (n = 32) but not in normotensive patients (n = 20). This study highlights the notion that the decrease in heart rate and sympathetic nerve activity leading to reduced blood pressure after application with vitamin C was because of makeover of baroreflex function [103]. Endothelial dysfunction, a cause of CVDs, is corrected in a human study (n = 93) by treatment with vitamin C (2 g) alone or in combination with vitamin E (600 mg) as shown by Uzun et al. Results showed enhanced vasodilation following an endothelium dependent pathway in the radial artery of a subject with coronary artery disease receiving vitamin C and/or vitamin E [104]. Vitamin C promotes synthesis and deposition of collagen (type IV) in the basement membrane of EC, induces endothelial proliferation, scavenges radicals to prevent EC apoptosis, and increases endothelial NO production. However, there is variation in the beneficial effect of synthetic vitamin C and natural (food-derived) vitamin C on CVDs. One reason may be that, along with dietary vitamin C (from fruits and vegetables), we also consume other phytochemicals which may potentiate its availability to systemic circulation for action. This hypothesis is supported by a study done by Agarwal et al. on a human cohort. The results revealed that although vitamin C supplement did not help to decrease the progression of carotid artery intima-media thickness (IMT) in CVDs like atherosclerosis, dietary vitamin C did [105]. In contrast to the beneficial action of vitamin C to alleviate CVD symptoms, there is some controversy because of the results of the clinical trial which oppose this fact. For instance, a clinical trial done by Ward et al. showed that although monotherapy of vitamin C (for 6 weeks) decreases the systolic blood pressure in hypertensive subjects, combination therapy of vitamin C and grapes seed polyphenol increased it. Moreover, the endothelium independent and dependent vasorelaxation as well as oxidative stress marker were not significantly different from vitamin C/grape seed polyphenol therapy as compared to a hypertensive individual without therapy. This finding recommends that hypertensive individuals on vitamin C and polyphenol supplements therapy should take the necessary precaution [106].
9.1.2. Vitamin E
Various studies have been carried out to examine the beneficial effect of vitamin E on CVDs. Serbinova et al. compared the effect of palm oil vitamin E with tocopherol alone and found that palm oil vitamin E was comparatively more successful in safeguarding the cardiac ischemia-reperfusion damage in the isolated heart [107]. A recent meta-analysis study done by Ashor et al. [108] to observe the potency of vitamins on atrial stiffness in adults disclosed that antioxidant vitamins possess a beneficial effect by reducing arterial stiffness. Moreover, the efficacy was dependent on the duration of treatment and dose supplemented. Those subjects having reduced levels of vitamin E in the blood attained an improved pharmacological effect from this intervention. In another double-blind, placebo-controlled study performed by Stephens et al. [109] on a coronary disease patient, it was found that 400 or 800 IU per day dose of vitamin E notably decreased the incidence of nonfatal myocardial infarction in study subjects.
9.2. Failure of Antioxidant Vitamin Therapy to Revert the CVDs
Although considerable research supports the fact that antioxidants possess therapeutic benefit to fight against disease progression, however, clinical trials are unsuccessful in showing the benefit [110]. Hasty et al. found that vitamin E supplementation for 12 weeks was not successful in alleviating the oxidative damage in western-type diet fed low density lipoprotein receptor knockout (LDLR−/−) mice model of obesity/hyperlipidemia. Although diet was enough to drastically increase the plasma lipid profile like free fatty acid, triglyceride, and total cholesterol (a marker of atherogenesis) in LDLR−/− obese mice with respect to lean mice, there was no beneficial effect observed after supplementation of vitamin E. Furthermore, there was no reduction in the urinary isoprostanes (a biomarker of oxidative stress) levels suggesting that vitamin E does not account for the cardioprotective effect [111]. A human clinical trial done in 730 volunteers (either sex, ≥65 years) for more than 20 years demonstrated that lower vitamin C supplementation was associated with high mortality rate by stroke in elderly people. However, there was no remarkable link between vitamin C diet status and CHD [112]. Similarly, another human clinical trial done in a larger population (6996 men and 2545 women) also showed that vitamin E had no beneficial effect on cardiovascular outcomes in patients who are more prone to cardiovascular events even after treatment for a long period of 4.5 years [113].
9.3. Pharmacological Agent
Various pharmacology agents such as statins and angiotensin-converting enzyme (ACE) inhibitors show pleiotropic effects to halt the oxidative stress. Particularly in the myocardium, oxidative stress and cell signaling proteins like Rac, Rho, and Ras are responsible for the cardiac hypertrophic response [114]. A recent in vivo investigation revealed that phagocyte-type Nox could be a potential source of ROS in the myocardium [115]. Nox-dependent ROS production seems to be linked with cardiac hypertrophy mediated by pressure overload [116] and angiotensin II infusion [117]. Even though the principal effect of statin therapy in CVDs is mainly vascular, in vivo studies recommend that there are also protective effects on the myocardium. Since Rac1 is essential for Nox function and cardiac hypertrophy resulted to some extent by oxidative stress, probably the statins could reduce cardiac hypertrophy by antioxidant pathway. Particularly, statins were successful in blocking angiotensin II-mediated oxidative injury in a rat model of cardiac hypertrophy [118]. Also, this activity of statins was seen in a clinical trial done in a cardiac hypertrophy subject that presented with hypercholesterolemia [119]. In a patient with heart failure, Nox mediated ROS generation raised in the left ventricular myocardium and associated with the rise in Rac1 GTPase activity, whereas statin therapy was able to reduce Rac1 activity in the heart [120].
9.3.1. Statins
Statins category medicines are not a direct scavenger of ROS; however, they act in an indirect way by hindering the 3-hydroxy-3-methyl-glutaryl-coenzyme A (HMG CoA) reductase pathway involved in cholesterol synthesis. Metabolism with HMG CoA reductase leads to the production of intermediate pyrophosphates, considered as a crucial point for O2 ∙− formation via Nox. The therapeutic effect of statins to lower the occurrence of CVDs is achieved by their capacity to promote endothelial nitric oxide synthase (eNOS) expression as well as antioxidant nature [118, 121–123]. Factors causing endothelial damage involve ROS such as oxidized-LDL and hypoxia can reduce the expression of eNOS whereas statins group medicine can reverse the eNOS downregulation, highlighting their ability to ameliorate the vessel NO bioavailability and atherosclerotic plaque stability [124–126]. Furthermore, statins also block tumor necrosis factor-α (TNF-α) mediated downregulation of eNOS [127]. Stimulation of angiotensin II hormone will further activate Rac1 and ADP-ribosylation factor 6 (ARF6), a controller of NADPH oxidase function. ARF6 is crucial for ROS production because, in the knocked down situation of this GTPase, angiotensin II cannot stimulate O2 ∙− (superoxide anion) generation. Likewise, ARF6 also regulates NADPH oxidase 1 (Nox1) expression [128]. A study done by Copaja et al. showed that induction of cardiac myofibroblasts and fibroblasts apoptosis by simvastatin followed a cholesterol synthesis independent pathway but was dependent on Rho GTPases protein isoprenylation. On comparison, cardiac myofibroblasts were less sensitive to apoptosis induction than cardiac fibroblasts by simvastatin. Therefore, it is likely that simvastatin could circumvent harmful cardiac remodeling followed by some fibrotic restoration of the injured tissues [129]. Taken together, the abovementioned beneficial role of statins in prevention of coronary heart disease can promote the development of statin therapy against ROS mediated CVDs.
9.3.2. ACE Inhibitors
ACE is an enzyme that metabolizes angiotensin I to angiotensin II. ACE inhibitors were designed to treat hypertension. During arterial wall remodeling, increased angiotensin II activity leads to thickening of the tunica media and narrowing of the vessel diameter, a key feature of atherosclerosis [130, 131]. In particular, increased angiotensin II level is associated with the proportional release of vascular O2 ∙− [132]. In a rabbit model of hypercholesterolemia, it was observed that O2 ∙− bioavailability was decreased in thoracic aorta due to the antioxidative activity of NO [133]. In contrast, NO concentration can decrease after reaction with O2 ∙−, thereby worsening the atheroma plague formation [134]. Although eNOS and neuronal NOS control normal metabolic functions, upregulation of inducible NOS leads to enhanced production of NO displaying deleterious inflammatory responses by forming peroxynitrite by NO and superoxide [135]. Circulating angiotensin II level can be enhanced by angiotensin II type I receptor blocker (ARB) leading to stimulation of angiotensin II type II receptors followed by vasodilation due to NO production [136, 137]. A clinical trial in hypertensive patients undergoing candesartan (a type of ARB) therapy displayed remarkable diminishing of carotid artery intima-media thickness, mainly because of augmented NO production as well as decreased oxidative stress [136]. Likewise, cotreatment of ACE and ARB inhibitor showed a synergistic inhibitory action against oxidative stress in a balloon-injured rat carotid artery [138].
10. Conclusions
The exact mechanism of CVD is complex and is not yet fully understood. ROS plays an important role in the progression and development of CVD. There is a link between ROS and the pathophysiology of CVD. We have developed a greater understanding of production of ROS, detection of ROS, and therapeutic strategy to prevent production of ROS and cardiovascular disease. However, more works to improve the detection and treatment of the ROS mediated dysfunction are necessary in the upcoming days.
Competing Interests
The authors declare that they have no competing interests.
Authors' Contributions
All the authors contributed equally to this review.
References
- 1.Vogiatzi G., Tousoulis D., Stefanadis C. The role of oxidative stress in atherosclerosis. Hellenic Journal of Cardiology. 2009;50(5):402–409. [PubMed] [Google Scholar]
- 2.Panth N., Park S.-H., Kim H. J., Kim D.-H., Oak M.-H. Protective effect of Salicornia europaea extracts on high salt intake-induced vascular dysfunction and hypertension. International Journal of Molecular Sciences. 2016;17(7, article 1176) doi: 10.3390/ijms17071176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bayir H. Reactive oxygen species. Critical Care Medicine. 2005;33(12):S498–S501. doi: 10.1097/01.CCM.0000186787.64500.12. [DOI] [PubMed] [Google Scholar]
- 4.Heitzer T., Schlinzig T., Krohn K., Meinertz T., Münzel T. Endothelial dysfunction, oxidative stress, and risk of cardiovascular events in patients with coronary artery disease. Circulation. 2001;104(22):2673–2678. doi: 10.1161/hc4601.099485. [DOI] [PubMed] [Google Scholar]
- 5.Zuo L., Zhou L. T., Pannell B. K., Ziegler A. C., Best T. M. Biological and physiological role of reactive oxygen species—the good, the bad and the ugly. Acta Physiologica. 2015;214(3):329–348. doi: 10.1111/apha.12515. [DOI] [PubMed] [Google Scholar]
- 6.Reid M. B. Redox modulation of skeletal muscle contraction: what we know and what we don't. Journal of Applied Physiology. 2001;90(2):724–731. doi: 10.1063/1.1381002. [DOI] [PubMed] [Google Scholar]
- 7.Wattanapitayakul S. K., Bauer J. A. Oxidative pathways in cardiovascular disease: roles, mechanisms, and therapeutic implications. Pharmacology and Therapeutics. 2001;89(2):187–206. doi: 10.1016/s0163-7258(00)00114-5. [DOI] [PubMed] [Google Scholar]
- 8.Murphy M. P. How mitochondria produce reactive oxygen species. Biochemical Journal. 2009;417(1):1–13. doi: 10.1042/bj20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorov D. B., Juhaszova M., Sollott S. J. Mitochondrial reactive oxygen species (ROS) and ROS-induced ROS release. Physiological Reviews. 2014;94(3):909–950. doi: 10.1152/physrev.00026.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handy D. E., Loscalzo J. Redox regulation of mitochondrial function. Antioxidants & Redox Signaling. 2012;16(11):1323–1367. doi: 10.1089/ars.2011.4123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Taverne Y. J. H. J., Bogers A. J. J. C., Duncker D. J., Merkus D. Reactive oxygen species and the cardiovascular system. Oxidative Medicine and Cellular Longevity. 2013;2013:15. doi: 10.1155/2013/862423.862423 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Paravicini T. M., Touyz R. M. NADPH oxidases, reactive oxygen species, and hypertension: clinical implications and therapeutic possibilities. Diabetes Care. 2008;31(supplement 2):S170–S180. doi: 10.2337/dc08-s247. [DOI] [PubMed] [Google Scholar]
- 13.Coyle C. H., Martinez L. J., Coleman M. C., Spitz D. R., Weintraub N. L., Kader K. N. Mechanisms of H2O2-induced oxidative stress in endothelial cells. Free Radical Biology and Medicine. 2006;40(12):2206–2213. doi: 10.1016/j.freeradbiomed.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 14.Scherz-Shouval R., Elazar Z. ROS, mitochondria and the regulation of autophagy. Trends in Cell Biology. 2007;17(9):422–427. doi: 10.1016/j.tcb.2007.07.009. [DOI] [PubMed] [Google Scholar]
- 15.Bedard K., Krause K.-H. The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology. Physiological Reviews. 2007;87(1):245–313. doi: 10.1152/physrev.00044.2005. [DOI] [PubMed] [Google Scholar]
- 16.Kuroda J., Ago T., Matsushima S., Zhai P., Schneider M. D., Sadoshima J. NADPH oxidase 4 (Nox4) is a major source of oxidative stress in the failing heart. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(35):15565–15570. doi: 10.1073/pnas.1002178107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zuo L., Pasniciuc S., Wright V. P., Merola A. J., Clanton T. L. Sources for superoxide release: lessons from blockade of electron transport, NADPH oxidase, and anion channels in diaphragm. Antioxidants and Redox Signaling. 2003;5(5):667–675. doi: 10.1089/152308603770310347. [DOI] [PubMed] [Google Scholar]
- 18.Taniyama Y., Griendling K. K. Reactive oxygen species in the vasculature: molecular and cellular mechanisms. Hypertension. 2003;42(6):1075–1081. doi: 10.1161/01.hyp.0000100443.09293.4f. [DOI] [PubMed] [Google Scholar]
- 19.Napoli C., Ignarro L. J. Nitric oxide and atherosclerosis. Nitric Oxide. 2001;5(2):88–97. doi: 10.1006/niox.2001.0337. [DOI] [PubMed] [Google Scholar]
- 20.Verhaar M. C., Westerweel P. E., Van Zonneveld A. J., Rabelink T. J. Free radical production by dysfunctional eNOS. Heart. 2004;90(5):494–495. doi: 10.1136/hrt.2003.029405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jones L. C., Hingorani A. D. Genetic regulation of endothelial function. Heart. 2005;91(10):1275–1277. doi: 10.1136/hrt.2005.061325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Paz Scribano M., del Carmen Baez M., Florencia B., et al. Effects of atorvastatin on oxidative stress biomarkers and mitochondrial morphofunctionality in hyperfibrinogenemia-induced atherogenesis. Advances in Medicine. 2014;2014:6. doi: 10.1155/2014/947258.947258 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lipinski B. Pathophysiology of oxidative stress in diabetes mellitus. Journal of Diabetes and Its Complications. 2001;15(4):203–210. doi: 10.1016/s1056-8727(01)00143-x. [DOI] [PubMed] [Google Scholar]
- 24.Chen X.-L., Zhang Q., Zhao R., Medford R. M. Superoxide, H2O2, and iron are required for TNF-α-induced MCP-1 gene expression in endothelial cells: Role of Rac1 and NADPH oxidase. American Journal of Physiology—Heart and Circulatory Physiology. 2004;286(3):H1001–H1007. doi: 10.1152/ajpheart.00716.2003. [DOI] [PubMed] [Google Scholar]
- 25.Malinouski M., Zhou Y., Belousov V. V., Hatfield D. L., Gladyshev V. N. Hydrogen peroxide probes directed to different cellular compartments. PLoS ONE. 2011;6(1) doi: 10.1371/journal.pone.0014564.e14564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Belousov V. V., Fradkov A. F., Lukyanov K. A., et al. Genetically encoded fluorescent indicator for intracellular hydrogen peroxide. Nature Methods. 2006;3(4):281–286. doi: 10.1038/nmeth866. [DOI] [PubMed] [Google Scholar]
- 27.Dikalov S., Griendling K. K., Harrison D. G. Measurement of reactive oxygen species in cardiovascular studies. Hypertension. 2007;49(4):717–727. doi: 10.1161/01.hyp.0000258594.87211.6b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zielonka J., Srinivasan S., Hardy M., et al. Cytochrome c-mediated oxidation of hydroethidine and mito-hydroethidine in mitochondria: identification of homo- and heterodimers. Free Radical Biology and Medicine. 2008;44(5):835–846. doi: 10.1016/j.freeradbiomed.2007.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dikalova A. E., Bikineyeva A. T., Budzyn K., et al. Therapeutic targeting of mitochondrial superoxide in hypertension. Circulation Research. 2010;107(1):106–116. doi: 10.1161/CIRCRESAHA.109.214601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dikalov S., Fink B., Skatchkov M., Bassenge E. Comparison of glyceryl trinitrate-induced with pentaerythrityl tetranitrate-induced in vivo formation of superoxide radicals: effect of vitamin C. Free Radical Biology and Medicine. 1999;27(1-2):170–176. doi: 10.1016/s0891-5849(99)00066-0. [DOI] [PubMed] [Google Scholar]
- 31.Dikalov S., Skatchkov M., Fink B., Bassenge E. Quantification of superoxide radicals and peroxynitrite in vascular cells using oxidation of sterically hindered hydroxylamines and electron spin resonance. Nitric Oxide. 1997;1(5):423–431. doi: 10.1006/niox.1997.0139. [DOI] [PubMed] [Google Scholar]
- 32.Dikalov S. I., Dikalova A. E., Mason R. P. Noninvasive diagnostic tool for inflammation-induced oxidative stress using electron spin resonance spectroscopy and an extracellular cyclic hydroxylamine. Archives of Biochemistry and Biophysics. 2002;402(2):218–226. doi: 10.1016/S0003-9861(02)00064-4. [DOI] [PubMed] [Google Scholar]
- 33.Chang M. C. Y., Pralle A., Isacoff E. Y., Chang C. J. A selective, cell-permeable optical probe for hydrogen peroxide in living cells. Journal of the American Chemical Society. 2004;126(47):15392–15393. doi: 10.1021/ja0441716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miller E. W., Albers A. E., Pralle A., Isacoff E. Y., Chang C. J. Boronate-based fluorescent probes for imaging cellular hydrogen peroxide. Journal of the American Chemical Society. 2005;127(47):16652–16659. doi: 10.1021/ja054474f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gomez-Mejiba S. E., Zhai Z., Akram H., et al. Immuno-spin trapping of protein and DNA radicals: ‘tagging’ free radicals to locate and understand the redox process. Free Radical Biology and Medicine. 2009;46(7):853–865. doi: 10.1016/j.freeradbiomed.2008.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Mason R. P. Using anti-5,5-dimethyl-1-pyrroline N-oxide (anti-DMPO) to detect protein radicals in time and space with immuno-spin trapping. Free Radical Biology and Medicine. 2004;36(10):1214–1223. doi: 10.1016/j.freeradbiomed.2004.02.077. [DOI] [PubMed] [Google Scholar]
- 37.Jiang J., Liu K. J., Shi X., Swartz H. M. Detection of short-lived free radicals by low-frequency electron paramagnetic resonance spin trapping in whole living animals. Archives of Biochemistry and Biophysics. 1995;319(2):570–573. doi: 10.1006/abbi.1995.1332. [DOI] [PubMed] [Google Scholar]
- 38.Sano T., Umeda F., Hashimoto T., Nawata H., Utsumi H. Oxidative stress measurement by in vivo electron spin resonance spectroscopy in rats with streptozotocin-induced diabetes. Diabetologia. 1998;41(11):1355–1360. doi: 10.1007/s001250051076. [DOI] [PubMed] [Google Scholar]
- 39.He F., Zuo L. Redox roles of reactive oxygen species in cardiovascular diseases. International Journal of Molecular Sciences. 2015;16(12):27770–27780. doi: 10.3390/ijms161126059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhu X., Zuo L. Characterization of oxygen radical formation mechanism at early cardiac ischemia. Cell Death & Disease. 2013;4(9, article e787):7. doi: 10.1038/cddis.2013.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang X.-X., Wang X.-L., Tong M.-M., et al. SIRT6 protects cardiomyocytes against ischemia/reperfusion injury by augmenting FoxO3α-dependent antioxidant defense mechanisms. Basic Research in Cardiology. 2016;111(2):1–19. doi: 10.1007/s00395-016-0531-z. [DOI] [PubMed] [Google Scholar]
- 42.Halliwell B., Whiteman M. Measuring reactive species and oxidative damage in vivo and in cell culture: how should you do it and what do the results mean? British Journal of Pharmacology. 2004;142(2):231–255. doi: 10.1038/sj.bjp.0705776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Adam-Vizi V., Chinopoulos C. Bioenergetics and the formation of mitochondrial reactive oxygen species. Trends in Pharmacological Sciences. 2006;27(12):639–645. doi: 10.1016/j.tips.2006.10.005. [DOI] [PubMed] [Google Scholar]
- 44.Kim J.-S., Jin Y., Lemasters J. J. Reactive oxygen species, but not Ca2+ overloading, trigger pH- and mitochondrial permeability transition-dependent death of adult rat myocytes after ischemia-reperfusion. American Journal of Physiology—Heart and Circulatory Physiology. 2006;290(5):H2024–H2034. doi: 10.1152/ajpheart.00683.2005. [DOI] [PubMed] [Google Scholar]
- 45.Rigoulet M., Yoboue E. D., Devin A. Mitochondrial ROS generation and its regulation: mechanisms involved in H2O2 signaling. Antioxidants & Redox Signaling. 2011;14(3):459–468. doi: 10.1089/ars.2010.3363. [DOI] [PubMed] [Google Scholar]
- 46.Asada D., Itoi T., Nakamura A., Hamaoka K. Tolerance to ischemia reperfusion injury in a congenital heart disease model. Pediatrics International. 2016 doi: 10.1111/ped.13022. [DOI] [PubMed] [Google Scholar]
- 47.Huang X., Zhang J., Liu J., et al. C-reactive protein promotes adhesion of monocytes to endothelial cells via NADPH oxidase-mediated oxidative stress. Journal of Cellular Biochemistry. 2012;113(3):857–867. doi: 10.1002/jcb.23415. [DOI] [PubMed] [Google Scholar]
- 48.Lee H.-H., Paudel K. R., Kim D.-W. Terminalia chebula fructus inhibits migration and proliferation of vascular smooth muscle cells and production of inflammatory mediators in RAW 264.7. Evidence-Based Complementary and Alternative Medicine. 2015;2015:10. doi: 10.1155/2015/502182.502182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Paudel K. R., Panth N., Kim D.-W. Circulating endothelial microparticles: a key hallmark of atherosclerosis progression. Scientifica. 2016;2016:9. doi: 10.1155/2016/8514056.8514056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Touyz R. M., Briones A. M., Sedeek M., Burger D., Montezano A. C. NOX isoforms and reactive oxygen species in vascular health. Molecular Interventions. 2011;11(1):27–35. doi: 10.1124/mi.11.1.5. [DOI] [PubMed] [Google Scholar]
- 51.Akasaki T., Ohya Y., Kuroda J., et al. Increased expression of gp91phox homologues of NAD(P)H oxidase in the aortic media during chronic hypertension: Involvement of the renin-angiotensin system. Hypertension Research. 2006;29(10):813–820. doi: 10.1291/hypres.29.813. [DOI] [PubMed] [Google Scholar]
- 52.Gordillo G., Fang H., Park H., Roy S. Nox-4-dependent nuclear H2O2 drives DNA oxidation resulting in 8-OHdG as urinary biomarker and hemangioendothelioma formation. Antioxidants and Redox Signaling. 2010;12(8):933–943. doi: 10.1089/ars.2009.2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Harrison D., Griendling K. K., Landmesser U., Hornig B., Drexler H. Role of oxidative stress in atherosclerosis. The American Journal of Cardiology. 2003;91(3):7–11. doi: 10.1016/s0002-9149(02)03144-2. [DOI] [PubMed] [Google Scholar]
- 54.Harrison D. G. Cellular and molecular mechanisms of endothelial cell dysfunction. The Journal of Clinical Investigation. 1997;100(9):2153–2157. doi: 10.1172/jci119751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.White M. Y., Edwards A. V. G., Cordwell S. J., Van Eyk J. E. Mitochondria: a mirror into cellular dysfunction in heart disease. Proteomics: Clinical Applications. 2008;2(6):845–861. doi: 10.1002/prca.200780135. [DOI] [PubMed] [Google Scholar]
- 56.Sharma L. K., Lu J., Bai Y. Mitochondrial respiratory complex I: structure, function and implication in human diseases. Current Medicinal Chemistry. 2009;16(10):1266–1277. doi: 10.2174/092986709787846578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu E., Calvert P. A., Mercer J. R., et al. Mitochondrial DNA damage can promote atherosclerosis independently of reactive oxygen species through effects on smooth muscle cells and monocytes and correlates with higher-risk plaques in humans. Circulation. 2013;128(7):702–712. doi: 10.1161/CIRCULATIONAHA.113.002271. [DOI] [PubMed] [Google Scholar]
- 58.Fetterman J. L., Holbrook M., Westbrook D. G., et al. Mitochondrial DNA damage and vascular function in patients with diabetes mellitus and atherosclerotic cardiovascular disease. Cardiovascular Diabetology. 2016;15(53):1–7. doi: 10.1186/s12933-016-0372-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fleming I., Michaelis U. R., Bredenkötter D., et al. Endothelium-derived hyperpolarizing factor synthase (cytochrome P450 2C9) is a functionally significant source of reactive oxygen species in coronary arteries. Circulation Research. 2001;88(1):44–51. doi: 10.1161/01.res.88.1.44. [DOI] [PubMed] [Google Scholar]
- 60.Madamanchi N. R., Runge M. S. Mitochondrial dysfunction in atherosclerosis. Circulation Research. 2007;100(4):460–473. doi: 10.1161/01.res.0000258450.44413.96. [DOI] [PubMed] [Google Scholar]
- 61.Ballinger S. W., Patterson C., Knight-Lozano C. A., et al. Mitochondrial integrity and function in atherogenesis. Circulation. 2002;106(5):544–549. doi: 10.1161/01.CIR.0000023921.93743.89. [DOI] [PubMed] [Google Scholar]
- 62.DiMauro S., Schon E. A. Mitochondrial respiratory-chain diseases. The New England Journal of Medicine. 2003;348(26):2656–2668. doi: 10.1056/nejmra022567. [DOI] [PubMed] [Google Scholar]
- 63.Brookes P. S., Yoon Y., Robotham J. L., Anders M. W., Sheu S.-S. Calcium, ATP, and ROS: a mitochondrial love-hate triangle. American Journal of Physiology—Cell Physiology. 2004;287(4):C817–C833. doi: 10.1152/ajpcell.00139.2004. [DOI] [PubMed] [Google Scholar]
- 64.Di Lisa F., Bernardi P. Mitochondrial function and myocardial aging. A critical analysis of the role of permeability transition. Cardiovascular Research. 2005;66(2):222–232. doi: 10.1016/j.cardiores.2005.02.009. [DOI] [PubMed] [Google Scholar]
- 65.Utsumi H., Yasukawa K., Soeda T., et al. Noninvasive mapping of reactive oxygen species by in vivo electron spin resonance spectroscopy in indomethacin-induced gastric ulcers in rats. Journal of Pharmacology and Experimental Therapeutics. 2006;317(1):228–235. doi: 10.1124/jpet.105.095166. [DOI] [PubMed] [Google Scholar]
- 66.Lee H.-H., Paudel K. R., Jeong J., et al. Antiatherogenic effect of Camellia japonica fruit extract in high fat diet-fed rats. Evidence-Based Complementary and Alternative Medicine. 2016;2016:8. doi: 10.1155/2016/9679867.9679867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Paudel K. R., Lee U.-W., Kim D.-W. Chungtaejeon, a Korean fermented tea, prevents the risk of atherosclerosis in rats fed a high-fat atherogenic diet. Journal of Integrative Medicine. 2016;14(2):134–142. doi: 10.1016/s2095-4964(16)60249-2. [DOI] [PubMed] [Google Scholar]
- 68.Mitra S., Goyal T., Mehta J. L. Oxidized LDL, LOX-1 and atherosclerosis. Cardiovascular Drugs and Therapy. 2011;25(5):419–429. doi: 10.1007/s10557-011-6341-5. [DOI] [PubMed] [Google Scholar]
- 69.Paudel K. R., Karki R., Kim D.-W. Cepharanthine inhibits in vitro VSMC proliferation and migration and vascular inflammatory responses mediated by RAW264.7. Toxicology in Vitro. 2016;34:16–25. doi: 10.1016/j.tiv.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 70.Jun M. Y., Karki R., Paudel K. R., Sharma B. R., Adhikari D., Kim D.-W. Alkaloid rich fraction from Nelumbo nucifera targets VSMC proliferation and migration to suppress restenosis in balloon-injured rat carotid artery. Atherosclerosis. 2016;248:179–189. doi: 10.1016/j.atherosclerosis.2016.03.020. [DOI] [PubMed] [Google Scholar]
- 71.Ehara S., Ueda M., Naruko T., et al. Elevated levels of oxidized low density lipoprotein show a positive relationship with the severity of acute coronary syndromes. Circulation. 2001;103(15):1955–1960. doi: 10.1161/01.CIR.103.15.1955. [DOI] [PubMed] [Google Scholar]
- 72.Meisinger C., Baumert J., Khuseyinova N., Loewel H., Koenig W. Plasma oxidized low-density lipoprotein, a strong predictor for acute coronary heart disease events in apparently healthy, middle-aged men from the general population. Circulation. 2005;112(5):651–657. doi: 10.1161/CIRCULATIONAHA.104.529297. [DOI] [PubMed] [Google Scholar]
- 73.Baldus S., Heeschen C., Meinertz T., et al. Myeloperoxidase serum levels predict risk in patients with acute coronary syndromes. Circulation. 2003;108(12):1440–1445. doi: 10.1161/01.cir.0000090690.67322.51. [DOI] [PubMed] [Google Scholar]
- 74.Apple F. S., Pearce L. A., Chung A., Ler R., Murakami M. M. Multiple biomarker use for detection of adverse events in patients presenting with symptoms suggestive of acute coronary syndrome. Clinical Chemistry. 2007;53(5):874–881. doi: 10.1373/clinchem.2006.080192. [DOI] [PubMed] [Google Scholar]
- 75.Kutter D., Devaquet P., Vanderstocken G., Paulus J. M., Marchal V., Gothot A. Consequences of total and subtotal myeloperoxidase deficiency: risk or benefit? Acta Haematologica. 2000;104(1):10–15. doi: 10.1159/000041062. [DOI] [PubMed] [Google Scholar]
- 76.Léculier C., Couprie N., Adeleine P., Leitienne P., Francina A., Richard M. The effects of high molecular weight- and low molecular weight-heparins on superoxide ion production and degranulation by human polymorphonuclear leukocytes. Thrombosis Research. 1993;69(6):519–531. doi: 10.1016/0049-3848(93)90056-T. [DOI] [PubMed] [Google Scholar]
- 77.de Gaetano G., Cerletti C., Evangelista V. Recent advances in platelet-polymorphonuclear leukocyte interaction. Haemostasis. 1999;29(1):41–49. doi: 10.1159/000022459. [DOI] [PubMed] [Google Scholar]
- 78.Shih J., Datwyler S. A., Hsu S. C., et al. Effect of collection tube type and preanalytical handling on myeloperoxidase concentrations. Clinical Chemistry. 2008;54(6):1076–1079. doi: 10.1373/clinchem.2007.101568. [DOI] [PubMed] [Google Scholar]
- 79.Milne G. L., Musiek E. S., Morrow J. D. F2-isoprostanes as markers of oxidative stress in vivo: an overview. Biomarkers. 2005;10:10–23. doi: 10.1080/13547500500216546. [DOI] [PubMed] [Google Scholar]
- 80.Shao B., Heinecke J. W. HDL, lipid peroxidation, and atherosclerosis. Journal of Lipid Research. 2009;50(4):599–601. doi: 10.1194/jlr.E900001-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Patrignani P., Santini G., Panara M. R., et al. Induction of prostaglandin endoperoxide synthase-2 in human monocytes associated with cyclo-oxygenase-dependent F2-isoprostane formation. British Journal of Pharmacology. 1996;118(5):1285–1293. doi: 10.1111/j.1476-5381.1996.tb15535.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Vassalle C., Botto N., Andreassi M. G., Berti S., Biagini A. Evidence for enhanced 8-isoprostane plasma levels, as index of oxidative stress in vivo, in patients with coronary artery disease. Coronary Artery Disease. 2003;14(3):213–218. doi: 10.1097/00019501-200305000-00004. [DOI] [PubMed] [Google Scholar]
- 83.Halliwell B., Lee C. Y. J. Using isoprostanes as biomarkers of oxidative stress: some rarely considered issues. Antioxidants & Redox Signaling. 2010;13(2):145–156. doi: 10.1089/ars.2009.2934. [DOI] [PubMed] [Google Scholar]
- 84.Zuo L., Nogueira L., Hogan M. C. Reactive oxygen species formation during tetanic contractions in single isolated Xenopus myofibers . Journal of Applied Physiology. 2011;111(3):898–904. doi: 10.1152/japplphysiol.00398.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Zuo L., Best T. M., Roberts W. J., Diaz P. T., Wagner P. D. Characterization of reactive oxygen species in diaphragm. Acta Physiologica. 2015;213(3):700–710. doi: 10.1111/apha.12410. [DOI] [PubMed] [Google Scholar]
- 86.Zuo L., Christofi F. L., Wright V. P., Bao S., Clanton T. L. Lipoxygenase-dependent superoxide release in skeletal muscle. Journal of Applied Physiology. 2004;97(2):661–668. doi: 10.1152/japplphysiol.00096.2004. [DOI] [PubMed] [Google Scholar]
- 87.Zhou T., Chuang C.-C., Zuo L. Molecular characterization of reactive oxygen species in myocardial ischemia-reperfusion injury. BioMed Research International. 2015;2015:9. doi: 10.1155/2015/864946.864946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Vanden Hoek T. L., Li C., Shao Z., Schumacker P. T., Becker L. B. Significant levels of oxidants are generated by isolated cardiomyocytes during ischemia prior to reperfusion. Journal of Molecular and Cellular Cardiology. 1997;29(9):2571–2583. doi: 10.1006/jmcc.1997.0497. [DOI] [PubMed] [Google Scholar]
- 89.Becker L. B., Vanden Hoek T. L., Shao Z.-H., Li C.-Q., Schumacker P. T. Generation of superoxide in cardiomyocytes during ischemia before reperfusion. American Journal of Physiology—Heart and Circulatory Physiology. 1999;277(6):H2240–H2246. doi: 10.1152/ajpheart.1999.277.6.H2240. [DOI] [PubMed] [Google Scholar]
- 90.Robin E., Guzy R. D., Loor G., et al. Oxidant stress during simulated ischemia primes cardiomyocytes for cell death during reperfusion. The Journal of Biological Chemistry. 2007;282(26):19133–19143. doi: 10.1074/jbc.m701917200. [DOI] [PubMed] [Google Scholar]
- 91.Lane J. S., Magno C. P., Lane K. T., Chan T., Hoyt D. B., Greenfield S. Nutrition impacts the prevalence of peripheral arterial disease in the United States. Journal of Vascular Surgery. 2008;48(4):897–904. doi: 10.1016/j.jvs.2008.05.014. [DOI] [PubMed] [Google Scholar]
- 92.World Cancer Research Fund/American Institute for Cancer Research. Food, Nutrition and the Prevention of Cancer: A Global Perspective. Washington, DC, USA: American Institute for Cancer Research; 1997. [Google Scholar]
- 93.WHO. Report of the joint WHO/FAO expert consultation. Diet, nutrition and the prevention of chronic diseases. WHO Technical Report Series. 2003;(916 (TRS 916)) [PubMed]
- 94.Rapola J. M., Virtamo J., Ripatti S., et al. Randomised trial of α-tocopherol and β-carotene supplements on incidence of major coronary events in men with previous myocardial infarction. The Lancet. 1997;349(9067):1715–1720. doi: 10.1016/s0140-6736(97)01234-8. [DOI] [PubMed] [Google Scholar]
- 95.Singh R. B., Wander G. S., Rastogi A., et al. Randomized, double-blind placebo-controlled trial of coenzyme Q10 in patients with acute myocardial infarction. Cardiovascular Drugs and Therapy. 1998;12(4):347–353. doi: 10.1023/A:1007764616025. [DOI] [PubMed] [Google Scholar]
- 96.Das S., Otani H., Maulik N., Das D. K. Lycopene, tomatoes, and coronary heart disease. Free Radical Research. 2005;39(4):449–455. doi: 10.1080/10715760500053685. [DOI] [PubMed] [Google Scholar]
- 97.Su J.-F., Guo C.-J., Wei J.-Y., Yang J.-J., Jiang Y.-G., Li Y.-F. Protection against hepatic ischemia-reperfusion injury in rats by oral pretreatment with quercetin. Biomedical and Environmental Sciences. 2003;16(1):1–8. [PubMed] [Google Scholar]
- 98.Paudel K. R., Panth N. Phytochemical profile and biological activity of Nelumbo nucifera . Evidence-based Complementary and Alternative Medicine. 2015;2015:16. doi: 10.1155/2015/789124.789124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Panth N., Paudel K. R., Karki R. Phytochemical profile and biological activity of juglans regia. Journal of Integrative Medicine. 2016;14 doi: 10.1016/S2095-4964(16)60274-1. [DOI] [PubMed] [Google Scholar]
- 100.Devkota S. R., Paudel K. R., Sharma K., et al. Investigation of antioxidant and anti-inflammatory activity of roots of Rumex nepalensis. World Journal of Pharmacy and Pharmaceutical Sciences. 2015;4(3):582–594. [Google Scholar]
- 101.Nishi E. E., Ribeiro Campos R., Toledo Bergamaschi C., de Almeida V. R., Ribeiro D. A. Vitamin C prevents DNA damage induced by renovascular hypertension in multiple organs of Wistar rats. Human & Experimental Toxicology. 2010;29(7):593–599. doi: 10.1177/0960327109358267. [DOI] [PubMed] [Google Scholar]
- 102.Botelho-Ono M. S., Pina H. V., Sousa K. H. F., Nunes F. C., Medeiros I. A., Braga V. A. Acute superoxide scavenging restores depressed baroreflex sensitivity in renovascular hypertensive rats. Autonomic Neuroscience: Basic and Clinical. 2011;159(1-2):38–44. doi: 10.1016/j.autneu.2010.07.025. [DOI] [PubMed] [Google Scholar]
- 103.Bruno R. M., Daghini E., Ghiadoni L., et al. Effect of acute administration of vitamin C on muscle sympathetic activity, cardiac sympathovagal balance, and baroreflex sensitivity in hypertensive patients. American Journal of Clinical Nutrition. 2012;96(2):302–308. doi: 10.3945/ajcn.112.035022. [DOI] [PubMed] [Google Scholar]
- 104.Uzun A., Yener U., Cicek O. F., et al. Does vitamin C or its combination with vitamin e improve radial artery endothelium-dependent vasodilatation in patients awaiting coronary artery bypass surgery? Cardiovascular Journal of Africa. 2013;24(7):255–259. doi: 10.5830/CVJA-2013-046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Agarwal M., Mehta P. K., Dwyer J. H., et al. Differing relations to early atherosclerosis between vitamin C from supplements vs. food in the Los Angeles atherosclerosis study: a prospective cohort study. The Open Cardiovascular Medicine Journal. 2012;6(1):113–121. doi: 10.2174/1874192401206010113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ward N. C., Hodgson J. M., Croft K. D., Burke V., Beilin L. J., Puddey I. B. The combination of vitamin C and grape-seed polyphenols increases blood pressure: a randomized, double-blind, placebo-controlled trial. Journal of Hypertension. 2005;23(2):427–434. doi: 10.1097/00004872-200502000-00026. [DOI] [PubMed] [Google Scholar]
- 107.Serbinova E., Khwaja S., Catudioc J., et al. Palm oil vitamin E protects against ischemia/reperfusion injury in the isolated perfused langendorff heart. Nutrition Research. 1992;12(supplement 1):S203–S215. doi: 10.1016/s0271-5317(05)80464-6. [DOI] [PubMed] [Google Scholar]
- 108.Ashor A. W., Siervo M., Lara J., Oggioni C., Mathers J. C. Antioxidant vitamin supplementation reduces arterial stiffness in adults: a systematic review and meta-analysis of randomized controlled trials. Journal of Nutrition. 2014;144(10):1594–1602. doi: 10.3945/jn.114.195826. [DOI] [PubMed] [Google Scholar]
- 109.Stephens N. G., Parsons A., Schofield P. M., et al. Randomised controlled trial of vitamin E in patients with coronary disease: cambridge Heart Antioxidant Study (CHAOS) The Lancet. 1996;347(9004):781–786. doi: 10.1016/s0140-6736(96)90866-1. [DOI] [PubMed] [Google Scholar]
- 110.Goszcz K., Deakin S. J., Duthie G. G., Stewart D., Leslie S. J., Megson I. L. Antioxidants in cardiovascular therapy: panacea or false hope? Frontiers in Cardiovascular Medicine. 2015;2, article 29:1–22. doi: 10.3389/fcvm.2015.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hasty A. H., Gruen M. L., Terry E. S., et al. Effects of vitamin E on oxidative stress and atherosclerosis in an obese hyperlipidemic mouse model. The Journal of Nutritional Biochemistry. 2007;18(2):127–133. doi: 10.1016/j.jnutbio.2006.03.012. [DOI] [PubMed] [Google Scholar]
- 112.Gale C. R., Martyn C. N., Winter P. D., Cooper C. Vitamin C and risk of death from stroke and coronary heart disease in cohort of elderly people. The British Medical Journal. 1995;310(6994):1563–1566. doi: 10.1136/bmj.310.6994.1563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Yusuf S., Dagenais G., Pogue J., Bosch J., Sleight P. Vitamin E supplementation and cardiovascular events in high-risk patients. The Heart Outcomes Prevention Evaluation Study Investigators. The New England Journal of Medicine. 2000;342(3):154–160. doi: 10.1056/NEJM200001203420302. [DOI] [PubMed] [Google Scholar]
- 114.Thorburn J., Xu S., Thorburn A. MAP kinase- and Rho-dependent signals interact to regulate gene expression but not actin morphology in cardiac muscle cells. The EMBO Journal. 1997;16(8):1888–1900. doi: 10.1093/emboj/16.8.1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Bendall J. K., Cave A. C., Heymes C., Gall N., Shah A. M. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hyper-trophy in mice. Circulation. 2002;105:293–296. doi: 10.1161/hc0302.103712. [DOI] [PubMed] [Google Scholar]
- 116.Li J.-M., Gall N. P., Grieve D. J., Chen M., Shah A. M. Activation of NADPH oxidase during progression of cardiac hypertrophy to failure. Hypertension. 2002;40(4):477–484. doi: 10.1161/01.HYP.0000032031.30374.32. [DOI] [PubMed] [Google Scholar]
- 117.Nakagami H., Takemoto M., Liao J. K. NADPH oxidase-derived superoxide anion mediates angiotensin II-induced cardiac hypertrophy. Journal of Molecular and Cellular Cardiology. 2003;35(7):851–859. doi: 10.1016/s0022-2828(03)00145-7. [DOI] [PubMed] [Google Scholar]
- 118.Takemoto M., Node K., Nakagami H., et al. Statins as antioxidant therapy for preventing cardiac myocyte hypertrophy. The Journal of Clinical Investigation. 2001;108(10):1429–1437. doi: 10.1172/jci200113350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Lee T. M., Chou T. F., Tsai C. H. Association of pravastatin and left ventricular mass in hypercholesterolemic patients: role of 8-iso-prostaglandin f2alpha formation. Journal of Cardiovascular Pharmacology. 2002;40(6):868–874. doi: 10.1097/00005344-200212000-00007. [DOI] [PubMed] [Google Scholar]
- 120.Maack C., Kartes T., Kilter H., et al. Oxygen free radical release in human failing myocardium is associated with increased activity of rac1-GTPase and represents a target for statin treatment. Circulation. 2003;108(13):1567–1574. doi: 10.1161/01.cir.0000091084.46500.bb. [DOI] [PubMed] [Google Scholar]
- 121.Cholesterol Treatment Trialists' (CTT) Collaborators. Efficacy and safety of cholesterol-lowering treatment: prospective meta-analysis of data from 90,056 participants in 14 randomised trials of statins. The Lancet. 2005;366(9493):1267–1278. doi: 10.1016/S0140-6736(05)67394-1. [DOI] [PubMed] [Google Scholar]
- 122.Shishehbor M. H., Brennan M.-L., Aviles R. J., et al. Statins promote potent systemic antioxidant effects through specific inflammatory pathways. Circulation. 2003;108(4):426–431. doi: 10.1161/01.CIR.0000080895.05158.8B. [DOI] [PubMed] [Google Scholar]
- 123.Node K., Fujita M., Kitakaze M., Hori M., Liao J. K. Short-term statin therapy improves cardiac function and symptoms in patients with idiopathic dilated cardiomyopathy. Circulation. 2003;108(7):839–843. doi: 10.1161/01.cir.0000084539.58092.de. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mcquillan L. P., Leung G. K., Marsden P. A., Kostyk S. K., Kourembanas S. Hypoxia inhibits expression of enos via transcriptional and posttranscriptional mechanisms. American Journal of Physiology—Heart and Circulatory Physiology. 1994;267(5):1921–1927. doi: 10.1152/ajpheart.1994.267.5.H1921. [DOI] [PubMed] [Google Scholar]
- 125.Endres M., Laufs U., Liao J. K., Moskowitz M. A. Targeting eNOS for stroke protection. Trends in Neurosciences. 2004;27(5):283–289. doi: 10.1016/j.tins.2004.03.009. [DOI] [PubMed] [Google Scholar]
- 126.Chavakis E., Dernbach E., Hermann C., Mondorf U. F., Zeiher A. M., Dimmeler S. Oxidized LDL inhibits vascular endothelial growth factor—induced endothelial cell migration by an inhibitory effect on the Akt/endothelial nitric oxide synthase pathway. Circulation. 2001;103(16):2102–2107. doi: 10.1161/01.cir.103.16.2102. [DOI] [PubMed] [Google Scholar]
- 127.Jantzen F., Konemann S., Wolff B., et al. Isoprenoid depletion by statins antagonizes cytokine-induced down-regulation of endothelial nitric oxide expression and increases no synthase activity in human umbilical vein EC. Journal of Physiology and Pharmacology. 2007;58(3):503–514. [PubMed] [Google Scholar]
- 128.Bourmoum M., Charles R., Claing A. The GTPase ARF6 controls ROS production to mediate angiotensin II-induced vascular smooth muscle cell proliferation. PLoS ONE. 2016;11(1) doi: 10.1371/journal.pone.0148097.e0148097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Copaja M., Venegas D., Aránguiz P., et al. Simvastatin induces apoptosis by a Rho-dependent mechanism in cultured cardiac fibroblasts and myofibroblasts. Toxicology and Applied Pharmacology. 2011;255(1):57–64. doi: 10.1016/j.taap.2011.05.016. [DOI] [PubMed] [Google Scholar]
- 130.Schmidt-Ott K. M., Kagiyama S., Phillips M. I. The multiple actions of angiotensin II in atherosclerosis. Regulatory Peptides. 2000;93(1–3):65–77. doi: 10.1016/s0167-0115(00)00178-6. [DOI] [PubMed] [Google Scholar]
- 131.Pushpakumar S., Kundu S., Pryor T., et al. Angiotensin-II induced hypertension and renovascular remodelling in tissue inhibitor of metalloproteinase 2 knockout mice. Journal of Hypertension. 2013;31(11):2270–2281. doi: 10.1097/hjh.0b013e3283649b33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Laursen J. B., Rajagopalan S., Galis Z., Tarpey M., Freeman B. A., Harrison D. G. Role of superoxide in angiotensin II—induced but not catecholamine-induced hypertension. Circulation. 1997;95(3):588–593. doi: 10.1161/01.cir.95.3.588. [DOI] [PubMed] [Google Scholar]
- 133.Müller S., König I., Meyer W., Kojda G. Inhibition of vascular oxidative stress in hypercholesterolemia by eccentric isosorbide mononitrate. Journal of the American College of Cardiology. 2004;44(3):624–631. doi: 10.1016/j.jacc.2004.02.062. [DOI] [PubMed] [Google Scholar]
- 134.Ignarro L. J., Napoli C. Novel features of nitric oxide, endothelial nitric oxide synthase, and atherosclerosis. Current Diabetes Reports. 2005;5(1):17–23. doi: 10.1007/s11892-005-0062-8. [DOI] [PubMed] [Google Scholar]
- 135.Zuo L., Koozechian M. S., Chen L. L. Characterization of reactive nitrogen species in allergic asthma. Annals of Allergy, Asthma & Immunology. 2014;112(1):18–22. doi: 10.1016/j.anai.2013.10.007. [DOI] [PubMed] [Google Scholar]
- 136.Cosentino F., Savoia C., De Paolis P., et al. Angiotensin II type 2 receptors contribute to vascular responses in spontaneously hypertensive rats treated with angiotensin II type 1 receptor antagonists. American Journal of Hypertension. 2005;18(4):493–499. doi: 10.1016/j.amjhyper.2004.11.007. [DOI] [PubMed] [Google Scholar]
- 137.Ono H., Minatoguchi S., Watanabe K., et al. Candesartan decreases carotid intima-media thickness by enhancing nitric oxide and decreasing oxidative stress in patients with hypertension. Hypertension Research. 2008;31(2):271–279. doi: 10.1291/hypres.31.271. [DOI] [PubMed] [Google Scholar]
- 138.Yagi S., Morita T., Katayama S. Combined treatment with an AT1 receptor blocker and angiotensin converting enzyme inhibitor has an additive effect on inhibiting neointima formation via improvement of nitric oxide production and suppression of oxidative stress. Hypertension Research. 2004;27(2):129–135. doi: 10.1291/hypres.27.129. [DOI] [PubMed] [Google Scholar]