

Abstract
Lipoxygenase (LOX) genes are widely distributed in plants and play crucial roles in resistance to biotic and abiotic stress. Although they have been characterized in various plants, little is known about the evolution of legume LOX genes. In this study, we identified 122 full-length LOX genes in Arachis duranensis, Arachis ipaënsis, Cajanus cajan, Cicer arietinum, Glycine max, Lotus japonicus and Medicago truncatula. In total, 64 orthologous and 36 paralogous genes were identified. The full-length, polycystin-1, lipoxygenase, alpha-toxin (PLAT) and lipoxygenase domain sequences from orthologous and paralogous genes exhibited a signature of purifying selection. However, purifying selection influenced orthologues more than paralogues, indicating greater functional conservation of orthologues than paralogues. Neutrality and effective number of codons plot results showed that natural selection primarily shapes codon usage, except for C. arietinum, L. japonicas and M. truncatula LOX genes. GCG, ACG, UCG, CGG and CCG codons exhibited low relative synonymous codon usage (RSCU) values, while CCA, GGA, GCU, CUU and GUU had high RSCU values, indicating that the latter codons are strongly preferred. LOX expression patterns differed significantly between wild-type peanut and cultivated peanut infected with Aspergillus flavus, which could explain the divergent disease resistance of wild progenitor and cultivars.
Lipoxygenases (LOXs, EC1.13.11.12) are non-heme iron-containing enzymes that are widely distributed in plants and animals. LOX proteins catalyse the oxidation of polyunsaturated fatty acids into unsaturated fatty acid hydroperoxides1. LOX can induce synthesis of oxylipins, including acyclic and cyclic compounds2. Oxylipins can activate diverse pathways, including those associated with hydroperoxide lyase (HPL), peroxygenase (POX), allene oxide synthase (AOS), and divinyl ether synthase (DES)2,3. LOX proteins contain two major domains, a polycystin-1, lipoxygenase, alpha-toxin (PLAT) domain at the N-terminus and a lipoxygenase domain at the C-terminus4. The PLAT domain, which contains an eight-stranded antiparallel β-barrel, is found in a variety of membrane- or lipid-associated proteins5. This domain can bind to procolipase, which mediates membrane associations, such as polycystin-1 function5,6. The plant lipoxygenase domain contains about 38 amino acids with five conserved histidines, which are involved in iron binding7. LOX proteins can be classified into two types, 9-LOX and 13-LOX, based on their specificity of fatty acid oxygenation of linoleic acids (LA, 18:2) and linolenic acids (LeA, 18:3)8. Moreover, 13-LOX proteins can be further classified into two subgroups, type I 13-LOX and type II 13-LOX. Type I 13-LOX proteins harbour no transit peptides, exhibit high sequence similarity (>75%) between one another and are usually cytosolically localized9. Type II 13-LOX proteins are chloroplastic proteins carrying N-terminal transit peptides and showing moderate sequence similarity (>35%) to one another9.
To date, many LOX genes have been identified or cloned from plants including Arabidopsis thaliana10, Glycine max11, Medicago truncatula11, Pyrus bretschneideri12 and Vitis vinifera2 One review has reported that plant LOX genes are involved in responses to many pathways such as growth and developmental processes and resistance to abiotic stress2. Zhang, et al.13 showed that the expression levels of 18 Cucumis melo LOX genes are differentially regulated during melon development and ripening. In apple, MdLOX1a and MdLOX5e are involved in fruit aroma volatile production14. Yang, et al.15 found that 12 cucumber LOX genes are involved in response to abiotic stresses, including cold (4 °C), NaCl (200 mM) and KCl (200 mM). In Panax ginseng, PgLOX3 expression is increased under water deficit stress16. LOX can catalysed the initial step of the methyl jasmonate (JA) pathway, but little is known about its regulatory mechanism2,17. Chen, et al.4 found that 20 poplar LOX genes are regulated by methyl jasmonate (MeJA) treatment. LOX transcript levels in maize seeds were positively associated with resistance to fungi18,19. Podolyan, et al.3 reported that the expression of VvLOXC and VvLOXO increased under mechanical wounding and Botrytis cinerea infection in V. vinifera, while VvLOXA expression decreased in berries after pathogen infection. In cultivated peanut (Arachis hypogaea), transcriptome data showed at least 19 LOX genes were differentially expressed after Aspergillus flavus infection20,21.
Recently, the genome sequences of Arachis duranensis, Arachis ipaënsis, Cajanus cajan, Cicer arietinum, Glycine max, Lotus japonicus and Medicago truncatula have been completed and released22,23,24,25,26,27,28. These species belong to the Papilionoideae subfamily, and their phylogenetic relationships are shown in Fig. 1. The availability of whole genome sequences enables the comparative analysis of LOX genes in these legume species. Previous studies have identified 143 unique LOX genes from four model and seventeen crop plants, including monocotyledonous and dicotyledonous plants, which can be classified into two subfamilies based on a predicted chloroplast-targeting peptide29. However, these sequences were collected from public databases such as NCBI and UniProt in previous study29. In this study, we identified LOX genes from the seven aforementioned leguminous genomes using a bioinformatics approach. Both the overall phylogenetic relationships and codon usage patterns were analyzed. The expression patterns of some LOX genes in A. duranensis and its orthologous genes in cultivated peanut (A. hypogeae L.) were determined after A. flavus infection using quantitative real-time PCR (qRT-PCR). This provides helpful insights into the evolution of LOX genes and their function in A. flavus resistance.
Figure 1. Phylogenetic tree of seven legumes.
The topology of phylogenetic tree is revised based on references 22 and 23.
Results and discussion
Identification and classification of LOX genes in seven legumes
We identified LOX genes in A. duranensis (AdLOX), A. ipaënsis (AiLOX), C. arietinum (CaLOX), C. cajan (CcLOX), G. max (GmLOX), L. japonicus (LjLOX) and M. truncatula (MtLOX) using a bioinformatics approach. From these genome sequences, we retained 122 full-length LOX gene sequences for phylogenetic analysis, including 14 AdLOX, 13 AiLOX, 10 CaLOX, 16 CcLOX, 36 GmLOX, 5 LjLOX and 28 MtLOX sequences (Table 1 and Table S1). A total of 19 GmLOX and 15 MtLOX sequences have been released in a previous study that used four G. max and two M. truncatula regions11. However, in this study, we identified 34 GmLOX and 28 MtLOX sequences to identify additional LOX sequences. The number of LOX genes identified was significantly positively correlated with the number of whole-genome duplications (WGDs, r = 0.81, P < 0.05), but had no correlation with genome size (r = 0.09, P > 0.05), indicating WGD events directly affect the number of LOX genes. Fox example, G. max (1100 Mb, 3 WGD) contains 36 LOX genes, and A. ipaënsis (1560 Mb, 1 WGD) contains 13 LOX genes, while M. truncatula (375 Mb, 3 WGD) contains 28 LOX genes (Table 1). Shin, et al.11 found that GmLOX and MtLOX genes have a common ancestor, but GmLOX genes expanded through an ancient polyploidy event prior to taxon divergence, followed by a soybean-specific duplication. Further, there is positive but not significant correlation (r = 0.92, P > 0.05) between the number of LOX genes and genome size among species exhibiting an equal number of WGD events. One WGD event each was detected in A. duranensis (1250 Mb), A. ipaënsis (1560 Mb), C. arietinum (738 Mb) and L. japonicas (315 Mb) (Table 1). Accordingly, the numbers of LOX genes are 14 (AdLOX), 13 (AiLOX), 10 (CaLOX) and 5 (LjLOX), respectively (Table 1).
Table 1. The number and type of LOX genes in seven legumes.
| Name | 9-LOX | Type I 13-LOX | Type II 13-LOX | Total | Genome size | Whole-genome duplication |
|---|---|---|---|---|---|---|
| AdLOX | 2 | 7 | 5 | 14 | 1250a | 1a |
| AiLOX | 1 | 7 | 5 | 13 | 1250a | 1a |
| CaLOX | 2 | 1 | 7 | 10 | 738b | 1b |
| CcLOX | 3 | 4 | 9 | 16 | 833c | 0c |
| GmLOX | 5 | 17 | 14 | 36 | 1100d | 3d |
| LjLOX | 0 | 3 | 2 | 5 | 315e | 1e |
| MtLOX | 3 | 18 | 7 | 28 | 375f | 3f |
A phylogenetic tree was generated using amino acid sequences, and it shows three clear clades, including 9-LOX, type I 13-LOX and type II 13-LOX (Fig. 2). Compared to 13-LOX, relative fewer 9-LOX amino acid sequences were identified. In 9-LOX, only one paralogous gene pair (Glyma.03G091000.1 and Glyma.16G082600.1) was identified (Fig. 2, green colour), suggesting few duplication events occurred in 9-LOX. Most gene families can be classified into several groups. Fox example, WRKY proteins can be classified into three groups, and group II can be further classified into five subgroups based on the numbers and types of domains30,31,32. Nucleotide-binding site (NBS)-Leucine-rich repeat (LRR) proteins can be further classified into TIR-NBS-LRR and CNL-NBS-LRR based on the presence of toll/mammalian interleukin-1 receptor (TIR) or coiled-coil (CC) domains at N-terminuses33,34,35. The origin of these proteins is always a key research focus35,36. Little is known about the origin of LOX proteins. Typically, researchers have classified LOX proteins according to fatty acid oxygenation specificity8. Based on a phylogenetic tree generated in this study, the divergence of 9-LOX occurred between the divergence of type I 13-LOX and type II 13-LOX (Fig. 2). Furthermore, 9-LOX sequences were clustered into two groups (Fig. 2, shown in green and orange), indicating that 9-LOX sequences have independent origins.
Figure 2. Phylogenetic tree of LOX amino acid sequences in seven legumes.

The phylogenetic tree was constructed using MEGA 6.0. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model and branch support estimates are based on 1,000 bootstrap replicates.
LOX homologous genes in seven legumes
In this study, 63 orthologous and 36 paralogous genes were identified (Fig. 3). To determine if selection pressures were uniform among orthologues and paralogues in full-length LOX genes as well as PLAT and lipoxygenase domain sequences, we calculated the nonsynonymous/synonymous (Ka/Ks) values of these three types of sequence. All estimated Ka/Ks values were less than 1 (Fig. S1), suggesting each of these sequences underwent purifying selection. However, Ka/Ks values of paralogous genes exceeded those of orthologous genes among full-length LOX genes as well as PLAT and lipoxygenase domain sequences (t-test, P < 0.05, Fig. 4). The results suggested constrained purifying selection influenced orthologues more than paralogues, indicating that the biological function of orthologues is more conserved than that of paralogues. In addition, purifying selection on the lipoxygenase domain has exceeded that on the PLAT domain among orthologous and paralogous genes, indicating the biological function of the lipoxygenase domain is more conserved than that of PLAT domains.
Figure 3. Chromosomal location and homologous gene relationships among LOX genes from A. duranensis, A. ipaënsis, G. max and M. truncatula.

The chromosomal location information of LOX genes was obtained from the source websites for each sequence. The map was generated using Circos v0.69.
Figure 4. Ka/Ks values calculated using full-length, PLAT and lipoxygenase domains from homologous genes.
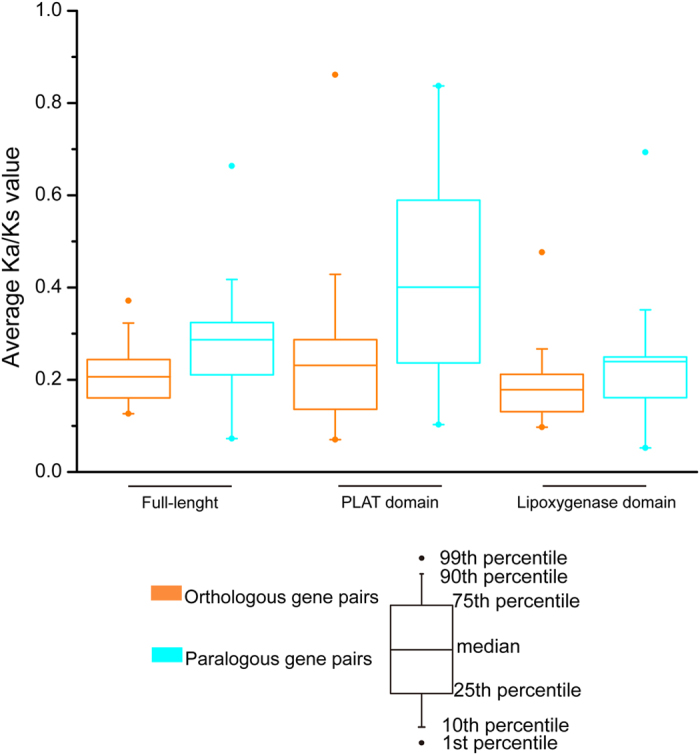
PAL2NAL was used to convert amino acid sequences into the corresponding nucleotide sequences. PAML 4.0 was used to calculate the nonsynonymous/synonymous substitution (Ka/Ks) ratio. Ka/Ks values of 1, >1 and <1 indicated neutral, positive, and purifying selection, respectively.
CcLOX, CaLOX and LjLOX genes were excluded from the chromosomal location analysis because most of these genes lacked localization information. GmLOX genes were distributed across 11 of 20 chromosomes, MtLOX genes were detected in 7 of 8 chromosomes and AdLOX and AiLOX genes were identified on 6 of 10 chromosomes, respectively (Fig. 3, Table S1). Many paralogous genes were among GmLOX and MtLOX sequences, while no paralogous genes were found among AdLOX and AiLOX sequences (Fig. 3). However, most orthologous genes were identified among AdLOX and AiLOX sequences in corresponding chromosomes (Fig. 3), suggesting that the biological functions of orthologous genes among AdLOX and AiLOX genes were more conserved than those of GmLOX and MtLOX genes.
A. duranensis and A. ipaënsis diverged 2.9–3.5 million years ago (MYA)37, and the cultivated peanut was domesticated 3500–4500 years ago24. Full genome sequences revealed most orthologous genes appeared in the A. duranensis and A. ipaënsis genomes24. However, it is difficult to distinguish homologous genes from A. duranensis and A. ipaënsis in the cultivated peanut genome. In this study, we found that most AiLOX genes were longer than their homologous AdLOX genes (Fig. S2). The difference between homologous AdLOX and AiLOX genes is attributed to intron length differences, especially the first intron. This result can be helpful for the future analysis and assembly of the cultivated peanut genome.
Codon usage bias of LOX genes in seven legumes
The average GC content was lower than the AT content in the seven legume species (Table 2). We found that the GC content at the three codon positions was, in decreasing order, GC1 (GC at the first codon position) > GC3 (GC at the third codon position) > GC2 (GC at the second codon position) in AdLOX, AiLOX, CcLOX, GmLOX and LjLOX. However, in CaLOX and MtLOX, GC content was as follows: GC1 > GC2 > GC3 (Table 2). In a neutrality plot, if the correlation between GC12 (average of GC1 and GC2) and GC3 is significant and the slope of the regression is close to 1, mutation pressure is the main force shaping codon usage38. When natural selection is the dominant factor, the slope of the regression is close to 038. In this study, GC12 and GC3 from AdLOX (r = 0.76393, P < 0.05 and slope = 0.1786), AiLOX (r = 0.77378, P < 0.05 and slope = 0.21234), CcLOX (r = 0.82218, P < 0.05 and slope = 0.16592) and GmLOX (r = 0.49242, P < 0.05 and slope = 0.1393) showed a significant positive correlation, and the slope was close to 0 (Fig. S3). These results suggested that codon usage was primarily shaped by natural selection. GC12 and GC3 from CaLOX, LjLOX and MtLOX showed a positive correlation but it was not significant (Fig. S3). These results indicated that different evolutionary pressures shaped variation in these legumes. If codons are constrained by mutation pressure (i.e. nucleotide composition), the gene would be on the curve in an effective number of codons (ENC) plot39. Genes from seven legumes were all below the curve line, suggesting that other evolutionary pressures (i.e. natural selection) likely influence codon usage (Fig. S4). Nevertheless, if GC3s values (GC content at synonymous codons) are narrow, natural selection is involved in codon usage patterns40. In the seven legumes, GC3s values were narrowly distributed (AdLOX: 0.325–0.434, AiLOX: 0.314–0.424, CaLOX: 0.267–0.339, CcLOX: 0.303–0.557, GmLOX: 0.312–0.522, LjLOX: 0.385–0.449, and MtLOX: 0.276–0.362, Fig. S4). Neutrality and ENC plots showed that natural selection is main force shaping codon usage, while the codon usage of CaLOX, LjLOX and MtLOX may involve other processes.
Table 2. GC content of LOX genes in seven legumes.
| Gene | GC1 content | GC2 content | GC3 content | Average GC content |
|---|---|---|---|---|
| AdLOX | 50.08 | 38.11 | 40.28 | 42.82 |
| AiLOX | 49.95 | 38.26 | 39.78 | 42.66 |
| CaLOX | 48.44 | 37.53 | 31.26 | 39.08 |
| CcLOX | 51.15 | 38.45 | 40.69 | 43.43 |
| GmLOX | 50.03 | 38.48 | 40.13 | 43.88 |
| LjLOX | 50.81 | 39.71 | 42.67 | 44.4 |
| MtLOX | 49.55 | 37.89 | 34.61 | 40.68 |
Relative synonymous codon usage (RSCU) is the observed frequency of a codon divided by the expected frequency. RSCU < 1 indicates less-used codons, and RSCU > 1 indicates that the codons are used more frequently than expected41. LOX genes in the seven legumes showed a strong preference for AT-ending codons based on the above criterion (RSCU > 1, Fig. 5 and Table S2). The higher AT contents at the third position than GC contents may explain these results (Table 2). Based on a heat map, RSCU values can be classified into two groups (Fig. 5). The low RSCU group included GCG, ACG, UCG, CGG and CCG. The RSCU value (~2.5) of AGA was the highest and CCA, GGA, GCU, CUU and GUU had relatively high RSCU values (~1.5); these were included in the high RSCU group. These results indicated GCG, ACG, UCG, CGG and CCG are not preferentially used codons and CCA, GGA, GCU, CUU and GUU are strongly preferred. Seven legume LOX genes could be classified into two groups based on RSCU values. AdLOX, AiLOX, CcLOX, GmLOX and LjLOX clustered into one group, and CaLOX and MtLOX were included in another group. These results are consistent with the GC content results. The GC contents at the three codon positions in CaLOX and MtLOX were similar and the five additional LOX genes had nearly equal GC contents at the three positions, suggesting GC content at the three positions may influence RSCU values.
Figure 5. Hierarchal clustering of RSCU values for each codon in seven legumes.

Relative synonymous codon usage (RSCU) was calculated using codonW. RSCU < 1 indicated the codons are less used, and RSCU > 1 indicated that the codons are used more frequently than expected.
Expression of LOX genes in wild and cultivated peanuts after A. flavus infection
Previous studies indicated that wild peanut is more resistant to diseases than cultivated peanut, and transferring resistance genes from wild peanut to cultivars can improve disease resistance in cultivated peanut42. Recently, sequencing results indicated that the genome size of A. hypogaea (~2.8 Gb) is similar to the sum of the A. duranensis (~1.25 Gb) and A. ipaënsis genomes (~1.56 Gb), indicating that most genes from the two wild peanuts are probably present in cultivated peanut24. Here, we hypothesize that LOX gene activities explain the higher resistance in wild peanut than cultivated peanut.
A. flavus produces carcinogenic mycotoxins known as aflatoxins, which are toxic to animals, including humans. Aflatoxin contamination is an important factor limiting peanut production. According to a previous study, LOX gene expression is related to the response to A. flavus infection20. In this study, expression patterns of five LOX genes were compared between A. duranensis and A. hypogaea using qRT-PCR after A. flavus infection. The expression of AE16G (type I 13-LOX) steadily increased upon A. flavus infection. After 7 days of infection, the RNA level was more than 10 times higher than the level observed on the first day after infection in A. duranensis (Fig. 6). However, the expression of this gene did not change significantly in A. hypogaea after infection (Fig. 6). Similar expression patterns were found in C88Z1 (type II 13-LOX) and KZX2M (9-LOX, partial sequence) (Fig. 6). Increased expression of C3RV0 (9-LOX) was observed in both A. duranensis and A. hypogaea during the first 5 days of infection. On the seventh day after infection, the expression of C3RV0 continued to increase in A. duranensis, but decreased drastically in A. hypogaea (Fig. 6). KXZ9V (type I 13-LOX, partial sequence) showed a different expression pattern than that of the other four LOX genes upon A. flavus infection (Fig. 6). Lower expression levels were observed in A. duranensis than in A. hypogaea. Expression reached the highest level on the third day after infection, and then decreased beginning on the fifth day (Fig. 6). It is noteworthy that the expression levels in A. duranensis continuously increased after A. flavus infection, but not in A. hypogeae (Fig. 6).
Figure 6. Expression of LOX genes in A. duranensis and A. hypogaea after A. flavus infection.

The Y-axis indicates the relative expression level and the X-axis (1, 3, 5, and 7 d) indicates the number of days after A. flavus infection. The standard errors are shown using vertical lines.
Orthologous genes in polyploids and their parents have at least three possible fates, including expression-level dominance, transgressive segregation and homeolog expression bias43. The results of this study showed that total gene expression in cultivated peanut is lower or higher than that in diploid parents, consistent with the transgressive segregation model. Many processes, such as DNA methylation and cis-regulation, may explain this phenomenon according to previous reviews43,44. DNA methylation on cytosines at CG, CHG and CHH sites is an important epigenetic factor influencing transcriptomic changes44. In Arabidopsis, DNA methylation levels differ between F1 hybrids and parent plants (A. thaliana Ler and C24); 18–26% of GC sites exhibit DNA methylation in parents compared with 36–37% in hybrids45. A total of 77 genes sensitive to methylome remodelling are transcriptionally repressed in F1 hybrids45. Lee and Chen46 demonstrated that DNA methylation regulation is involved in the expression of orthologous genes in Arabidopsis suecica and its diploid parents A. thaliana and Cardaminopsis arenosa. Liu, et al.47 found the expression level of Gossypium barbadense WRKY1 is higher than that in Gossypium hirsutum WRKY1 after Verticillium dahliae infection, and the GhWRKY1 promoter lacks an ethylene-responsive element compared to the GBWRKY1 promoter, indicating promoter differences probably resulted in differences in expression patterns. Understanding the expression regulation of disease-resistance genes in tetraploid peanut may facilitate the development of an efficient method to improve disease resistance in cultivated peanut.
Conclusion
In this study, we identified LOX genes in seven legumes. The full-length, PLAT and lipoxygenase domain sequences from orthologous and paralogous genes exhibited signatures of purifying selection. Constrained purifying selection influenced orthologous genes more than paralogous genes. Natural selection was the driving force shaping codon usage, while CaLOX, LjLOX and MtLOX genes may have been influenced by other processes. Legume LOX genes preferentially used CCA, GGA, GCU, CUU and GUU. The expression pattern of LOX genes differed significantly between wild-type and cultivated peanuts.
Materials and Methods
LOX sequences in seven legume genomes
We downloaded the genome sequences of A. duranensis (http://peanutbase.org/), A. ipaënsis (http://peanutbase.org/), C. cajan (http://gigadb.org/dataset/100028), C. arietinum (http://nipgr.res.in/CGAP/home.php), G. max (https://phytozome.jgi.doe. gov/pz/portal.html), L. japonicus (http://www.kazusa. or.jp/lotus/) and M. truncatula (http://jcvi.org/medicago/display.php?pageName = General§ion = Download). To obtain LOX coding sequences, we downloaded the HMM profile of the lipoxygenase domain (PF00305) from pfam database (http://pfam.janelia.org). Each LOX gene was extracted using the HMMER program48. To verify the reliability of the results, all amino acid sequences were checked in the pfam database. If PLAT (PF01477) and lipoxygenase domains were both present, the sequence was consider a LOX gene4.
Phylogenetic analysis
Multiple sequence alignment and phylogenetic tree construction were carried out using MAFFT 7.049 and MEGA 6.050, respectively. The phylogenetic tree was estimated using maximum likelihood with the Jones-Taylor-Thornton (JTT) model based on 1,000 bootstrap replicates. If genes from different species clustered in the phylogenetic tree with a bootstrap value of greater than 70, these genes were considered orthologous. Similarly, if genes from a single species clustered in a phylogenetic tree with a bootstrap value of greater than 70, these genes were considered paralogous. The PAL2NAL program51 was used for the conversion of amino acid sequences into the corresponding nucleotide sequences. PAML 4.052 was used to calculate the nonsynonymous/synonymous substitution (Ka/Ks) ratio. Generally, Ka/Ks values of 1, >1 and <1 indicate neutral, positive, and purifying selection, respectively.
The chromosomal locations of LOX genes in legume plant genomes were obtained from the source website for each sequence. The map was generated using the Circos v0.69 program53.
GC content at synonymous codons (GC3s), effective number of codons (ENC) and relative synonymous codon usage (RSCU) were calculated using the codonW program (http://codonw.sourceforge.net). GC1, GC2 and GC3 values were calculated using an in-house Perl script.
Gene expression analysis
The A. flavus inoculation method was described by Zhang, et al.20 Briefly, mature peanut seeds were surface-sterilized and placed on humid filter paper at 28 °C for 3 days. The germinated peanut seeds were used for inoculation by immersion in a suspension of ~3 × 107 spores/ml of A. flavus. Seeds immersed in sterile distilled water were used as controls. Seeds were placed in Petri dishes at 28 °C, and were harvested at 1, 3, 5 and 7 days after treatment.
qRT-PCR primers were designed based on the A. duranensis genome sequence using the Beacon Designer 8.0 program. The primers can distinguish genes from the A subgenome and B subgenome in cultivated peanut (Luhua14). The primers are shown in Table S3.
The hexadecyltrimethylammonium bromide (CTAB) method was used to extract total RNA. qRT-PCR was carried out using Fast Start Universal SYBR Green Master (ROX) and a 7500 Real-time PCR machine (ABI).
Additional Information
How to cite this article: Song, H. et al. Identification of lipoxygenase (LOX) genes from legumes and their responses in wild type and cultivated peanut upon Aspergillus flavus infection. Sci. Rep. 6, 35245; doi: 10.1038/srep35245 (2016).
Supplementary Material
Acknowledgments
This study was supported by National High Tech Project, NSFC Project (2011BAD35B04, 2013AA102602, 31500217), Young Talents Training Program of Shandong Academy of Agricultural Sciences and Shandong Province Germplasm Innovation and Utilization Project.
Footnotes
Author Contributions H.S. conceived and designed research, analyzed data and wrote the manuscript. P.W. analyzed data and participated in the discussion of the results. C.L. and S.H. contributed to the RNA extraction and real-time PCR. J.L.B. participated in the discussion of the results. W.X. and X. Z. contributed to the evaluation and discussion of the results and manuscript revision.
References
- Siedow J. N. Plant lipoxygenase: structure and function. Annu Rev Plant Biol 42, 145–188 (1991). [Google Scholar]
- Porta H. & Rocha-Sosa M. Plant lipoxygenases. Physiological and molecular features. Plant Physiol 130, 15–21 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podolyan A., White J., Jordan B. & Winefield C. Identification of the lipoxygenase gene family from Vitis vinifera and biochemical characterisation of two 13-lipoxygenases expressed in grape berries of Sauvignon Blanc. Funct Plant Biol 37, 767–784 (2010). [Google Scholar]
- Chen Z. et al. Thelipoxygenase gene family in Poplar: identification, classification, and expression in response to MeJA treatment. PLoS ONE 10, e0125526 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A. & Sandford R. The PLAT domain: a new piece in the PKD1 puzzle. Current Biology 9, R588–R590 (1999). [DOI] [PubMed] [Google Scholar]
- Minor W., Tomchick D. R., Phan P., Cymborowski M. & Holman T. R. Structural and functional characterization of second-coordination sphere mutants of soybean lipoxygenase-1. Biochemistry 40, 7509–7517 (2001). [DOI] [PubMed] [Google Scholar]
- Steczko J., Donoho G. P., Clemens J. C., Dixon J. E. & Axelrod B. Conserved histidine residues in soybean lipoxygenase: functional consequences of their replacement. Biochemistry 31, 4053–4057 (1992). [DOI] [PubMed] [Google Scholar]
- Feussner I., Kühn H. & Wasternack C. Lipoxygenase-dependent degradation of storage lipids. Trends Plant Sci 6, 268–273 (2001). [DOI] [PubMed] [Google Scholar]
- Feussner I. & Wasternack C. The lipoxygenase pathway. Annu Rev Plant Biol 53, 275–297 (2002). [DOI] [PubMed] [Google Scholar]
- Bannenberg G., Martínez M., Hamberg M. & Castresana C. Diversity of the enzymatic activity in the lipoxygenase gene family of Arabidopsis thaliana. Lipids 44, 85–95 (2009). [DOI] [PubMed] [Google Scholar]
- Shin J. H. et al. The lipoxygenase gene family: a genomic fossil of shared polyploidy between Glycine max and Medicago truncatula. BMC Plant Biol 8, 133 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M. et al. Characterization of the lipoxygenase (LOX) gene family in the Chinese white pear (Pyrus bretschneideri) and comparison with other members of the Rosaceae. BMC Genomics 15, 444 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. et al. The phylogeny and expression profiles of the lipoxygenase (LOX) family genes in the melo (Cucumis melo L.) genome. Sci Hortic 170, 94–102 (2014). [Google Scholar]
- Vogt J., Schiller D., Ulrich D., Schwab W. & Dunemann F. Identification of lipoxgenase (LOX) genes putatively involved in fruit flavour formation in apple (Malus × domestica). Tree Genet Genomes 9, 1493–1511 (2013). [Google Scholar]
- Yang X. Y., Jiang W. J. & Yu H. J. The expression profiling of the lipoxygenase (LOX) family genes during fruit development, abiotic stress and hormonal treatments in cucumber (Cucumis sativus L.). Int J Mol Sci 13, 2481–2500 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bae K. S. et al. Molecular characterization of lipoxygenase genes and their expression analysis against biotic and abiotic stresses in Panax ginseng. Eur J Plant Pathol 10.1007/s10658-10015-10847-10659 (2016). [Google Scholar]
- Howe G. A. & Jander G. Plant immunity to insect herbivores. Annu Rev Plant Biol 59, 41–66 (2008). [DOI] [PubMed] [Google Scholar]
- Wilson R. A., Gardner H. W. & Keller N. P. Cultivar-dependent expression of a maize lipoxygenase responsive to seed infesting fungi. Mol Plant Microbe Interact 14, 980–987 (2001). [DOI] [PubMed] [Google Scholar]
- Gao X. et al. Inactivation of the lipoxygenase ZmLOX3 increases susceptibility of maize to Aspergillus spp. Mol Plant Microbe Interact 22, 222–231 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H. et al. Peanut resistance gene expression in response to Aspergillus flavus infection during seed germination. J Phytopathol 163, 212–221 (2015). [Google Scholar]
- Wang H. et al. Comparative transcript profiling of resistant and susceptible peanut post-harvest seeds in response to aflatoxin production by Aspergillus flavus. BMC Plant Biol 16, 54 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X. et al. Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc Natl Acad Sci USA 113, 6785–6790 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varshney R. K. et al. Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat Biotechnol 31, 240–246 (2013). [DOI] [PubMed] [Google Scholar]
- Bertioli D. J. et al. The genome sequences of Arachis duranensis and Arachis ipaensis, the diploid ancestors of cultivated peanut. Nat Genet 48, 438–446 (2016). [DOI] [PubMed] [Google Scholar]
- Sato S. et al. Genome structure of the legume, Lotus japonicus. DNA Res 15, 227–239 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young N. D. et al. The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmutz J. et al. Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183 (2010). [DOI] [PubMed] [Google Scholar]
- Varshney R. K. et al. Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat Biotechnol 30, 83–89 (2012). [DOI] [PubMed] [Google Scholar]
- Feng B. et al. Molecular analysis of lipoxygenase (LOX) genes in common wheat and phylogenetic investigation of LOX proteins from model and crop plants. J Cereal Sci 52, 387–394 (2010). [Google Scholar]
- Eulgem T., Rushton P., Robatzek S. & Somssich I. The WRKY superfamily of plant transcription factors. Trends Plant Sci 5, 199–206 (2000). [DOI] [PubMed] [Google Scholar]
- Song H. et al. Global analysis of WRKY genes and their response to dehydration and salt stress in soybean. Front Plant Sci 7, 9 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H. et al. Genome-wide identification and characterization of WRKY gene family in peanut. Front Plant Sci 7, 534 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyers B. C., Kozik A., Griego A., Kuang H. H. & Michelmore R. W. Genome-wide analysis of NBS-LRR-encoding genes in Arabidopsis. Plant Cell 15, 809–834 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H. & Nan Z. Genome-wide analysis of nucleotide-binding site disease resistance genes in Medicago truncatula. Chin Sci Bull 59, 1129–1138 (2014). [Google Scholar]
- Yue J. X., Meyers B. C., Chen J. Q., Tian D. & Yang S. Tracing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes. New Phytol 193, 1049–1063 (2012). [DOI] [PubMed] [Google Scholar]
- Rinerson C. I., Rabara R. C., Tripathi Q. J., Shen P. J. & Rushton P. J. The evolution of WRKY transcription factors. BMC Plant Biol 15, 66 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moretzsohn M. C. et al. A study of the relationships of cultivated peanut (Arachis hypogaea) and its most closely related wild species using intron sequences and microsatellite markers. Ann Bot 11, 113–126 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sueoka N. Directional mutation pressure and neutral molecular evolution. Proc Natl Acad Sci USA 85, 2653–2657 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W. J. et al. Comparative analysis of codon usage patterns among mitochondrion, chloroplast and nuclear genes in Triticum aestivum L. J Integr Plant Biol 49, 246–254 (2007). [Google Scholar]
- Kawabe A. & Miyashita N. T. Patterns of codon usage bias in three dicot and four monocot plant species. Genes Genet Syst 78, 343–352 (2003). [DOI] [PubMed] [Google Scholar]
- Sharp P. M., Tuohy T. M. & Mosurski K. R. Codon usage in yeast: cluster analysis clearly differentiates highly and lowly expressed genes. Nucleic Acids Res 14, 5125–5143 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varman P. V., Ganesan K. N. & Mothilal A. Wild germplasm: potential source for resistance breeding in groundnut Journal of Ecobiology 12, 223–228 (2000). [Google Scholar]
- Yoo M. J., Liu X., Pires C., Soltis P. S. & Soltis D. E. Nonadditive gene expression in polyploids. Annu Rev Genet 48, 485–517 (2014). [DOI] [PubMed] [Google Scholar]
- Yao H., Dogra Gray A., Auger D. L. & Birchler J. A. Genomic dosage effects on heterosis in triploid maize. Proc Natl Acad Sci USA 10, 2665–2669 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen H. et al. Genome-wide analyisis of DNA methylation and gene expression changes in two Arabidopsis ecotypes and their reciprocal hybrids. Plant Cell 24, 875–892 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H. S. & Chen Z. J. Protein-coding genes are epigenetically regulated in Arabidopsis polyploids. Proceeding of the National Academy of Sciences, USA 98, 6753–6758 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J. et al. Molecular characters and different expression of WRKY1 gene from Gossypium barbadense L. and Gossypium hirsutum L. Biotechnology & Biotechnological Equipment (2016). [Google Scholar]
- Finn R. D., Clements J. & Eddy S. R. HMMER web server: interactive sequence similarity searching. Nucleic Acids Res 39, W29–W37 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh K. & Standley D. M. MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Bio Evol 30, 772–780 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamura K., Stecher G., Peterson D., Filipski A. & Kumar S. MEGA6: Molecular evolutionary genetics analysis version 6.0. Mol Bio Evol 30, 2725–2729 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suyama M., Torrents D. & Bork P. PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res 34, 609–612 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. PAML 4: phylogenetic analysis by maximum likelihood. Mol Bio Evol 24, 1586–1591 (2007). [DOI] [PubMed] [Google Scholar]
- Krzywinski M. et al. Circos: an information aesthetic for comparative genomics. Genome Res 19, 1639–1645 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.