

Abstract
Rationale
HFpEF patients experience elevated filling pressures and reduced ventricular compliance. The splicing factor RBM20 regulates the contour length of titin’s spring region and thereby determines the passive stiffness of cardiomyocytes. Inhibition of RBM20 leads to super compliant titin isoforms (N2BAsc) that reduce passive stiffness.
Objective
To determine the therapeutic potential of upregulating compliant titin isoforms in a HFpEF-like state in the mouse.
Methods and Results
Constitutive and inducible cardiomyocyte specific RBM20 inhibited mice were produced on a TtnΔIAjxn background to assess the effect of upregulating compliant titin at the cellular and organ levels. Genetic deletion of the I-band – A-band junction (IAjxn) in titin increases strain on the spring region and causes a HFpEF-like syndrome in the mouse without pharmacological or surgical intervention. The increased strain represents a mechanical analogue of deranged post-translational modification of titin that results in increased passive myocardial stiffness in HFpEF patients. Upon inhibition of RBM20 in TtnΔIAjxn mice, compliant titin isoforms were expressed and diastolic function was normalized, exercise performance was improved and pathologic hypertrophy was attenuated.
Conclusions
We report for the first time a benefit from upregulating compliant titin isoforms in a murine model with HFpEF-like symptoms. Constitutive and inducible RBM20 inhibition improves diastolic function resulting in greater tolerance to exercise. No effective therapies exists for treating this pervasive syndrome, therefore our data on RBM20 inhibition are clinically significant.
Keywords: Titin, RBM20, HFpEF, diastolic dysfunction, diastole, diastolic function, diastolic heart failure, compliance
Subject Terms: Contractile Function, Myocardial Biology, Physiology, Basic Science Research, Heart Failure
INTRODUCTION
Greater than half of all heart failure (HF) patients suffer from increased diastolic stiffness and impaired relaxation of the left ventricle while their ejection fraction (EF) is preserved (HFpEF)1. HFpEF is a complex syndrome that includes diastolic dysfunction, exercise intolerance, and concentric hypertrophic remodeling2, 3. No effective therapies exist for treating this pervasive syndrome due in part to the limited understanding of the underlying pathophysiology. HFpEF is likely due to a range of pathomechanisms that include dysfunction in sarcomeric proteins and slowing of the early phase of diastole that might be caused by changes in calcium handling/response proteins. Titin, the largest known protein and molecular spring in the heart, has emerged as a prime therapeutic target aimed at restoring compliance to the sarcomere and thereby improving diastolic function4. The titin gene (TTN) contains 364 exons that encode the third myofilament of the sarcomere that spans from Z-disk to M-band5. Titin’s I-band region acts as a molecular spring that is responsible for maintaining the structural integrity of the sarcomere6. Additionally, titin’s molecular spring is the chief contributor of passive stiffness in the heart, and functions as a mechano-sensor for stress and strain in the myocyte7. Alternative splicing produces two main cardiac isoforms; the smaller N2B isoform (~3.0 MDa) and the larger more compliant N2BA isoform (~3.3 MDa). Diastolic stiffness of the left ventricle is in large part dependent on the N2BA:N2B isoform ratio8. Stiffness can also be tuned through post-translational modification (PTM) of these primary isoforms, particularly through phosphorylation of titin’s spring region9–11.
PTM of titin’s spring region is deranged in HFpEF patients4, 12, which contributes to increased diastolic stiffness. Since protein kinase G (PKG) phosphorylation of titin’s N2B element enhances spring compliance, targeted treatment of HFpEF by enhancing PKG activity with the phosphodiesterase(PDE)5 inhibitor sildenafil was performed (summarized in the recent RELAX trial)13, however, exercise capacity was unimproved and clinical outcomes were similar to placebo13. Another potential therapeutic approach is upregulation of compliant titin isoforms. Alternative splicing of messenger RNA occurs primarily in transcripts encoding the spring region of titin and therefore regulates the elastic properties of titin through changes in contour length of the spring. Since the discovery of RBM20 as a titin splicing factor10, modification of titin size through RBM20 inhibition has become possible.
The current study evaluates whether manipulating post-transcriptional modification of titin isoforms through reduction of the titin splicing factor RBM20 has a beneficial effect on the diastolic function of mice with restrictive cardiomyopathy. A previously published mouse model14 deficient in titin’s IA junction (TtnΔIAjxn) places increased strain on the spring region of titin. The TtnΔIAjxn mouse is a choice tool for studying diastolic dysfunction as this model displays similar phenotypic characteristics found in the HFpEF patients, notably diastolic dysfunction, reduced exercise tolerance and concentric remodeling of the left ventricular chamber in response to stress. Furthermore, all known signaling elements in titin remain intact and therefore the pathology can be attributed solely to increases in titin-based strain. We aimed to determine if inhibition of RBM20 could recover function in this model back to a wild-type (WT) state. Our data establish that inhibiting titin splicing factor RBM20 in the TtnΔIAjxn mouse improves diastolic function, restores exercise tolerance, and attenuates afterload induced pathologic concentric remodeling.
METHODS
Generation of mice homozygous for TtnΔIAjxn and heterozygous for cardiac specific cRbm20ΔRRM
We bred two murine models the TtnΔIAjxn14 and the cardiac specific αMHC Cre cRbm20ΔRRM14 in order to compare effects of increased or decreased strain in titin, respectively. To study the effect of RBM20 inhibition in TtnΔIAjxn mice we crossed these two models to create hybrid TtnΔIAjxn mice with an intermediate expression level of WT RBM20, abbreviated ΔIAjxn/ΔRRM; details in Online Figure I. Additionally, we used a Tg(Myh6-cre/Esr1) mouse that has the Myh6 promoter (cardiac muscle αMHC) directing expression of a selective estrogen receptor modulator (SERM) inducible Cre recombinase (Mer-Cre-Mer, MCM) to cardiac myocytes. Mer-Cre-Mer ΔIAjxn/ΔRRM mice were injected intraperitoneal with raloxifene (40 mg/kg) or vehicle for 8 days to induce reduction of WT RBM20. After day 28 (from start of injections), mice were examined by echo and catheterization and then euthanized. Hearts were dissected, weighed and flash frozen at −80°C for further titin isoform analysis using standard procedures (see Online Methods). Western blot studies with an antibody to RBM20’s C-terminus11 revealed that the LV of the αMHC-Cre ΔIAjxn/ΔRRM expresses 40% of the total RBM20 as mutant protein; for the MCM ΔIAjxn/ΔRRM mice this value is 45% mutant RBM20 (see Fig. 3 B). Experiments were approved by the University of Arizona Institutional Animal Care and Use Committee and followed the U.S. National Institutes of Health Using
Figure 3. Inducible expression of super compliant titin isoforms recovers diastolic function in TtnΔIAjxn mice.
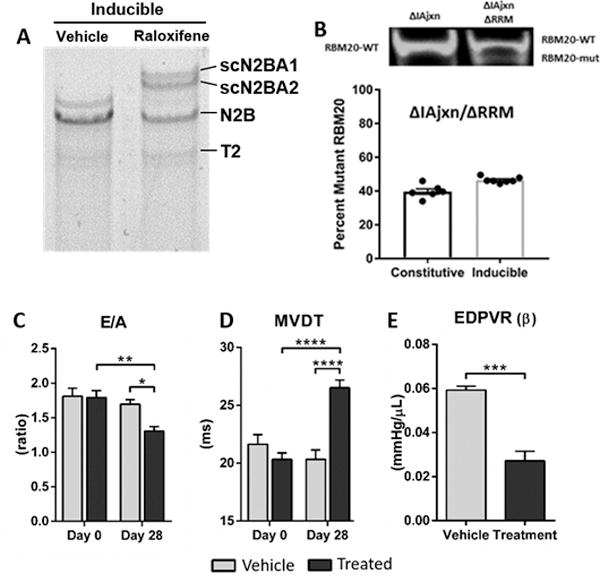
A) Representative image of 1% agarose gel for titin analysis of LV myocardium. Raloxifine results in expression of two large N2BA isoforms (scN2BA1 and 2). B) RBM20 expression in the constitutive and inducible ΔIAjxn/ΔRRM mouse. The top shows example Western blots and the bottom the ratio of mutant RBM20 to WT RBM20 in constitutive and inducible ΔIAjxn/ΔRRM LV samples. C) E/A ratio; D) MVDT: mitral valve deceleration time; E) EDPVR (β): end diastolic pressure volume relationship. Data shown as mean ± SEM. Age at day 0 = 100 ± 5 days; Age at day 28 = 128 ± 5 days for vehicle and treatment groups. Statistical significance calculated with two-way ANOVA and Bonferroni correction (D and E) or t-test (F), (* p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001 as indicated). (Vehicle: DMSO/Saline; Treatment: raloxifene (40 mg/kg), both administered IP daily for 8 days.)
Animals in Intramural Research guidelines for animal use
Protein expression analysis
Protein expression analysis was performed by using standard titin gel electrophoresis methods15.
Cell mechanics
Skinned cardiac myocyte mechanics were performed as previously reported16. (See online Methods for details.)
Hemodynamics
Anesthetized and conscious echocardiography was performed on mice and pressure volume relations were evaluated using cardiac catheterization. Cardiac function in diastole was assessed in mice using continuous flow two percent isoflurane inhaled anesthetic with a target heart rate of 450 ± 25 bpm. (See Online Methods for details.)
Exercise performance
Exercise tolerance was evaluated by measuring time and distance ran using a treadmill running test. (See Online Methods for details.)
TAC surgery
Male mice 3–4 months old were subjected to minimally invasive transverse aortic constriction (TAC)17 performed under Ketamine/Xylazine (120/12 mg/kg) anesthesia; details are explained in the Online Methods. Briefly, a 27-gauge needle was used for banding and mice were studied by echocardiography at 28 days and then euthanized. Hearts were immediately dissected, weighed and frozen for further analysis.
Myocyte cross section area
Cross-sectional area (CSA) of cardiomyocytes was performed as described18 with minor modifications. Hearts in diastole were removed from sham and TAC mice and the LV tissue was covered with OCT (Tissue-Tek) and frozen with liquid nitrogen–cooled isopentane and stored at −80°C. 8-μm thick sections were collected on VWR glass microscope slides and stored at −20°C overnight. The cross-sections were skinned and stained with anti-Laminin (Sigma) to demarcate cell borders and DAPI (Vector Laboratories) to stain nuclei. Images were collected on an Axio Imager M.1 microscope (Carl Zeiss) using an Axio Cam MRC (Carl Zeiss). CSAs were measured using the ImageJ program (National Institutes of Health) and their borders of stained cells that included nuclei were traced manually. The CSA of 100 myocytes from the myocardium of the left ventricle (6 mice per group) were collected from each sample using 4 randomly selected sections.
Statistics
Statistical analysis was performed in Graphpad Prism (GraphPad Software, Inc). Group significance was defined using one or two-way ANOVA followed by multiple testing correction, as appropriate. Results are shown as mean ± standard error of the mean. P<0.05 was considered significant.
RESULTS
Inhibition of Rbm20 upregulates super compliant titin isoforms (N2BAsc) and normalizes passive stiffness in cardiomyocytes of TtnΔIAjxn mice
TtnΔIAjxn mice are deficient in the IA junction of titin (see Figure 1 A for schematic) but have normal expression of the adult N2B and N2BA isoforms. However, both isoforms are reduced in size as revealed by their slight increase in mobility in gel electrophoresis (compare Fig. 1 B left and middle lanes). The larger and more compliant N2BA isoform (~3.3 MDa in WT and ~3.15 MDa in TtnΔIAjxn) comprises ~ 20 percent of total titin in both WT and TtnΔIAjxn mice (Fig. 1C, left and middle bar, Online Table I). It was recently shown that as a consequence of deleting the IA junction of titin, the attachment point of titin’s spring region is moved away from the Z-disk, resulting in greater titin strain in TtnΔIAjxn mice14.
Figure 1.
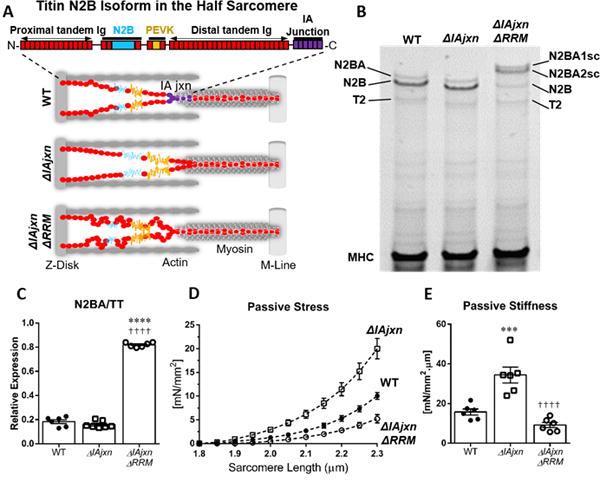
A) Half sarcomere models of the 3 genotypes used in this work: WT, ΔIAjxn and ΔIAjxn/ΔRRM. The domain composition of titin’s spring region (N2B isoform) is shown at the top. B) Representative image of 1% agarose gel for titin analysis of LV myocardium. WT myocardium expresses N2BA and N2B titin (T2 is a minor degradation product). The ΔIAjxn mouse expresses titin with a slightly higher mobility consistent with the deletion of the IA junction. Crossing the ΔIAjxn with the ΔRRM mouse (αMHC-Cre cRbm20ΔRRM+/−) results in expression of two very large N2BA isoforms that we refer to as super compliant (N2BAsc). C) Ratio of N2BA isoforms over total titin. D) Representative skinned cardiomyocyte with passive stress in cardiomyocytes. E) Passive stiffness in cardiomyocytes. Abbreviations: ΔIAjxn: TtnΔIAjxn; ΔRRM: αMHC-Cre cRbm20ΔRRM); N2BAsc: super compliant N2BA titin. Data shown as mean ± SEM. Statistical significance calculated by one-way ANOVA with Bonferroni correction. (*symbol versus WT; † symbol versus TtnΔIAjxn; 3 symbols: p<0.001; 4 symbols: p<0.0001).
cRbm20ΔRRM mice (αMHC-Cre) are deficient in titin splicing factor RBM20 and, as previously described11, they upregulate large N2BA titin isoforms that we refer to as super compliant titin (N2BAsc). To determine the therapeutic potential of RBM20 inhibition, we bred TtnΔIAjxn mice with αMHC-Cre cRbm20ΔRRM mice in order to produce TtnΔIAjxn mice that are heterozygous deficient in splicing factor RBM20 (see online Fig. I, left for more detail on the breeding scheme and Online Table II for tissue morphometry). TtnΔIAjxn mice constitutively inhibiting RBM20 express super compliant N2BA titin isoforms estimated ~3.35 and ~3.45 MDa (Fig. 1 B, right lane) encompassing ~80% of total titin in the murine left ventricle (Fig. 1 C, Online Table I). Thus, heterozygous inhibition of the titin splicing factor RBM20 is highly effective in upregulating compliant titin.
Previously we have shown that altering titin size and therefore strain on titin’s spring region directly affects passive stress in skinned left ventricular (LV) cardiomyocytes14. We tested whether inhibiting RBM20 in TtnΔIAjxn mice reduces the elevated diastolic stress in skinned myocytes to WT levels. Cardiomyocytes were skinned and isolated in relaxing solution and cells were attached at one end to a force transducer and at the other end to a servomotor and subjected to a stretch–hold–release protocol using standard procedures (see Online Methods for details). Passive stress was significantly increased in TtnΔIAjxn mice but was normalized in TtnΔIAjxn mice expressing super compliant titin (Fig. 1 D). Passive stiffness (slope of stress–sarcomere length (SL) relation within physiologic SL range) was similarly reduced to WT levels (Fig. 1 E).
Reduction of RBM20 expression restores diastolic function to TtnΔIAjxn mice
Results at the cellular level were further evaluated at the organ level to assess cardiac function in diastole (Fig. 2). Pulse wave Doppler in the 4-chamber apical view revealed significant differences in mitral valve deceleration time (MVDT) and E/A ratios of TtnΔIAjxn mice compared to WT control, indicating diastolic dysfunction (Fig. 2C and D and Table 1). The MVDT is significantly reduced in TtnΔIAjxn mice (21.9 ± 1.0 vs. 25.9 ± 0.7 ms in WT), a parameter inversely related to diastolic stiffness19. The MVDT was found to be at WT levels (26.5 ± 1.5 ms) in TtnΔIAjxn mice expressing N2BAsc titin, indicating restored LV chamber compliance. Additionally, a greater E/A ratio in TtnΔIAjxn mice (1.5 ± 0.1 vs. 1.3 ± 0.03 in WT) indicates restrictive LV filling20, and this ratio was improved in TtnΔIAjxn mice expressing super compliant titin (1.2 ± 0.03).
Figure 2. Diastolic function of Wildtype, ΔIAjxn and ΔIAjxn/ΔRRM mice.
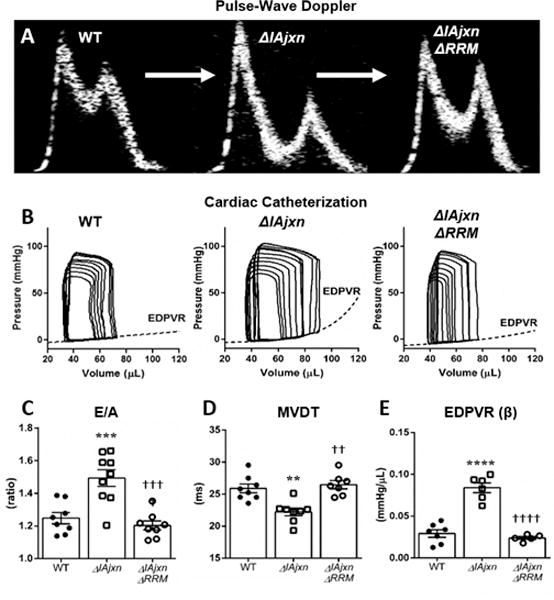
A) representative pulse-wave Doppler E and A waves of the 3 genotypes that were used; B) cardiac catheterization PV loops; C) E/A ratio; D) MVDT: mitral valve deceleration time; E) EDPVR: end-diastolic pressure volume relationship. Data shown as mean ± SEM. Statistical significance calculated with one-way ANOVA and Bonferroni correction. (*symbol versus WT; † symbol versus TtnΔIAjxn. (2 symbols: p<0.01; 3 symbols: p<0.001; 4 symbols: p<0.0001). * p<0.05 compared to WT; † p<0.05 compared to ΔIAjxn.)
Table 1.
Hemodynamics. Data shown as mean ± SEM
|
|
||||
|---|---|---|---|---|
| WT | TtnΔIAjxn | ΔIAjxn/ΔRRM | ||
|
|
||||
| Echocardiography | ||||
|
|
||||
| n | 8 | 8 | 8 | Conscious |
| HR (bpm) | 650.5 ± 10.3 | 650.5 ± 13.4 | 669.0 ± 9.3 | |
| FS (%) | 65.2 ± 1.8 | 57.5 ± 3.2 | 52.4 ± 1.7** | |
| LVID;d (mm) | 2.7 ± 0.1 | 2.8 ± 0.2 | 3.0 ± 0.1 | |
| LVID;s (mm) | 1.0 ± 0.1 | 1.2 ± 0.2 | 1.4 ± 0.1 | |
| LVPW;d (mm) | 1.1 ± 0.03 | 1.2 ± 0.1 | 1.1 ± 0.1 | |
| LVPW;s (mm) | 1.9 ± 0.1 | 1.8 ± 0.1 | 1.7 ± 0.1 | |
| LVDD/WT (ratio) | 2.5 ± 0.2 | 2.6 ± 0.3 | 2.7 ± 0.1 | |
|
|
||||
| Heart rate maintained at 450 ± 25 (bpm) | ||||
|
|
||||
| LA Diameter (mm) | 2.4 ± 0.2 | 2.7 ± 0.1 | 2.9 ± 0.1 | Anesthetized |
| MV Decel (ms) | 25.9 ± 0.7 | 21.9 ± 1.0** | 26.5 ± 1.5†† | |
| MV E (mm/s) | 770.5 ± 37.2 | 835.4 ± 22.7 | 780.7 ± 28.0 | |
| MV A (mm/s) | 621.6 ± 35.8 | 547.4 ± 14.2 | 650.9 ± 26.9† | |
| MV E/A | 1.3 ± 0.03 | 1.5 ± 0.04**** | 1.2 ± 0.03†††† | |
|
|
||||
| Cardiac Catheterization | ||||
|
|
||||
| n | 7 | 7 | 8 | |
| HR (BPM) | 431.3 ± 7.0 | 457.1 ± 23.2 | 492.5 ± 8.1 | |
| ESP (mmHg) | 94.4 ± 4.7 | 98.5 ± 7.0 | 93.5 ± 2.5 | |
| EDP (mmHg) | 2.0 ± 4.4 | 3.7 ± 1.0 | 1.4 ± 0.6 | |
| dPmax (mmHg/sec) | 6807.4 ± 431.4 | 8257.2 ± 667.2 | 9224.3 ± 261.0* | |
| dPmin (mmHg/sec) | −8116.3 ± 415.4 | −7073.2 ± 254.7 | −8894.5 ± 205.9††† | |
| ESV (uL) | 34.5 ± 7.6 | 33.9 ± 3.2 | 25.1 ± 3.0 | |
| EDV (uL) | 70.0 ± 7.6 | 73.6 ± 4.1 | 60.1 ± 3.6 | |
| SV (uL) | 35.5 ± 1.3 | 39.7 ± 3.4 | 37.1 ± 1.9 | |
| EF (%) | 54.0 ± 5.7 | 53.9 ± 3.5 | 62.2 ± 2.7 | |
| EA (mmHg/uL) | 2.7 ± 0.2 | 2.3 ± 0.2 | 2.4 ± 0.2 | |
| Tau Glantz (ms) | 12.2 ± 3.3 | 18.2 ± 4.5 | 9.8 ± 0.4 | |
| ESPVR | 4.3 ± 0.6 | 2.9 ± 0.5 | 3.1 ± 0.3 | |
| EDPVR | 0.03 ± 0.004 | 0.08 ± 0.01**** | 0.03 ± 0.004†††† | |
Abbreviations: ΔIAjxn: (TtnΔIAjxn); ΔRRM (αMHC-Cre cRbm20ΔRRM+/−). For additional abbreviations, see Non-standard Abbreviations and Acronyms list. Statistical significance calculated with one-way ANOVA with Bonferroni correction.
p<0.05;
p<0.01;
p<0.001;
p<0.0001.
(*symbol versus WT; † symbol versus TtnΔIAjxn. (1 symbol: p<0.05; 2 symbols: p<0.01; 3 symbols: p<0.001; 4 symbols: p<0.0001).)
Diastolic dysfunction was further demonstrated in TtnΔIAjxn mice using high fidelity pressure-volume admittance catheters (Fig. 2 B and E and Table 1). Occlusion of the inferior vena cava allowed us to determine the steepness of the end-diastolic pressure-volume relationship (EDPVR), β, which is a load-independent parameter. The EDPVR (β) is the coefficient of stiffness in the LV and was found to be (0.03 ± 0.004 mmHg/μL) in WT mice. TtnΔIAjxn mice had an elevated EDPVR (β) of (0.08 ± 0.01 mmHg/μL) that was normalized in TtnΔIAjxn mice expressing N2BAsc titin (0.02 ± 0.002 mmHg/μL). Therefore, consistent with our skinned cell experiments, compliance in the LV chamber was recovered in TtnΔIAjxn mice deficient in splicing factor RBM20.
We further examined diastolic recovery of TtnΔIAjxn mice with inducible (α-MHC-MCM) cRbm20ΔRRM mice that express N2BAsc on a TtnΔIAjxn background (see breeding scheme in Fig. S1, right for further details). This evaluation is important because it allows us to test whether diastolic dysfunction, once it has occurred, is recoverable therapeutically by upregulating compliant titin. Inducible mice were echoed at day 0 and then injected intraperitoneal for 8 days with either vehicle or selective estrogen receptor modulator (SERM) raloxifene (see Methods for additional details, and see Online Table III for raloxifene control experiments). After 8 days of raloxifene injections, greater than 60% N2BAsc titin expression occurred at 28 days (Fig. 3 A and Online Figure II). To evaluate the expression levels of mutant and wildtype RBM20, we used an antibody raised against the C-terminus of RBM20 that labels a single band in wildtype control mice and a doublet in heterozygous RBM20ΔRRM mice (Fig. 3 B, top). The top band reflects the level of wildtype RBM20 and the bottom band the level of mutant RBM20 (i.e. missing the RNA Recognition Motif (RRM)). We used this antibody on LV samples of constitutive (α-MHC ΔIAjxn/ΔRRM) and inducible (α-MHC-MCM ΔIAjxn/ΔRRM) models. Since the mice were heterozygous for the ΔRRM allele, mutant RBM20 is expected to make up 50% of total protein. However, there is slightly less than 50% mutant RBM20 (Fig. 3 B, bottom) indicating upregulation of wildtype RBM20 in the heterozygous mice. Furthermore, the samples from inducible ΔIAjxn/ΔRRM mice have a higher level of mutant RBM20 (45% of total) than the constitutive mice (40%), suggesting that the degree of upregulation of wildtype RBM20 is higher in the inducible mice. These results combined with the protein and functional data suggest that inactivating ~40–45% of total RBM20 results in robust upregulation of super compliant titin.
Inducible (α-MHC-MCM ΔIAjxn/ΔRRM) mice were echoed at day 28 to assess diastolic function and then evaluated with a terminal cardiac catheterization procedure for studying the effect of expressing N2BAsc titin. Similar to our constitutive model (α-MHC ΔIAjxn/ΔRRM), diastolic function was improved as shown by pulse-wave Doppler revealing a greater duration of the mitral valve deceleration time and a significant reduction in the E/A ratio (Fig. 3 C and D). Moreover, pressure-volume analysis showed significant improvement of the EDPVR (β) upon induction of N2BAsc expression (Fig. 3 E). Systolic function was unaltered between vehicle and treatment groups (Online Table IV). Additionally, WT controls undergoing the same protocol of raloxifene treatment were evaluated by echo and systolic function was unaffected (see Online Table III). Thus, acutely reducing strain in titin has a beneficial effect on the diastolic function of the stiff left ventricle.
Assessment of associated HFpEF symptoms
Similar to HFpEF patients21, 22, TtnΔIAjxn mice display exercise intolerance14. We tested whether upregulating compliant titin has a beneficial effect on exercise tolerance by submitting mice to incremental speed running on a treadmill. Mice with compliant titin ran longer and for a greater distance in both the constitutive (Fig. 4 A and B) and the inducible N2BAsc expressing models (Fig. 4 C and D). Overall these findings suggest that increasing titin compliance in the heart has a beneficial effect on exercise performance in the TtnΔIAjxn murine model.
Figure 4. Mice with upregulated compliant titin display enhanced exercise capacity in a treadmill test.
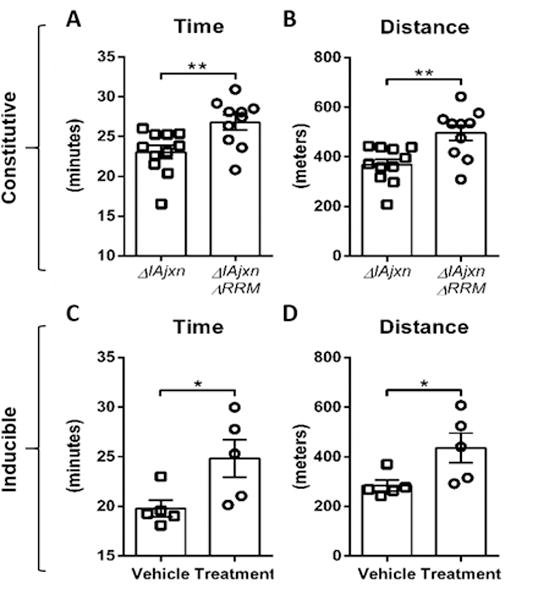
A–B) Time and distance of TtnΔIAjxn mice and TtnΔIAjxn mice constitutively expressing N2BAsc; C–D) Time and distance of TtnΔIAjxn mice and TtnΔIAjxn mice with inducible expression of N2BAsc. Data shown as mean ± SEM. (Constitutive: αMHC-Cre cRbm20ΔRRM+/−; inducible: αMHC–MerCreMer cRbm20ΔRRM+/−). (Vehicle: DMSO; Treatment: raloxifene, both administered IP daily for 8 days with exercise testing 28 days after last injection) Note that the IAjxn is also deleted from the skeletal muscles of these mice and that therefore future control experiments are needed on cardiac-specific TtnIAjxn deletion mice.
In addition to exercise intolerance, concentric remodeling in the heart is a common clinical manifestation in HFpEF patients23. We therefore tested the responsiveness of mice with differing strain in titin to afterload stress. Mice were subjected to minimally invasive transverse aortic constriction (TAC) surgery, a procedure widely used to elicit afterload-dependent hypertrophic remodeling in the left ventricle. Compared to WT mice, TtnΔIAjxn mice displayed an exaggerated hypertrophic response to TAC (p = 0.0001), whereas, the LV hypertrophy response in TtnΔIAjxn mice expressing N2BAsc titin was attenuated compared with TtnΔIAjxn mice (Fig. 5 A). The degree of remodeling these mice experienced in the left ventricle was determined by measuring the left ventricular end diastolic diameter (LVDD) and the wall thickness (WTH) in diastole. These parameters were obtained using M-mode echocardiography of the left ventricle in the short axis view. By calculating WTH/LVDD we can assess the degree to which the left ventricle is concentrically remodeled in response to TAC. Robust concentric remodeling after 4 weeks of pressure overload was observed in WT and TtnΔIAjxn mice with the largest values in TtnΔIAjxn mice, however, remodeling was lessened significantly in TtnΔIAjxn mice expressing N2BAsc titin (Fig. 5 B, see also Online Figure III with cross-sectional area analysis). In addition to reduced pathologic remodeling, TtnΔIAjxn mice with upregulated compliant titin also exhibited better diastolic function as revealed by Doppler imaging (Fig. 5 C–D). This data suggests that varying the length of titin isoforms alters the responsiveness to pressure overload hypertrophy in the heart and that increasing titin compliance is advantageous for preventing diastolic dysfunction associated with afterload induced concentric remodeling of the heart.
Figure 5.
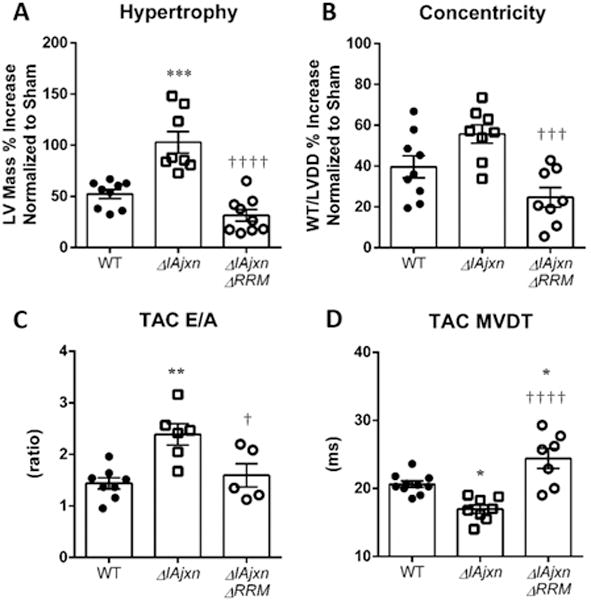
TtnΔIAjxn mice have an exaggerated response to TAC that is normalized in ΔIAjxn/ΔRRM mice. A) Hypertrophy; B) Concentricity: WTH: wall thickness in diastole; LVDD: left ventricular diameter in diastole. C) E/A ratio. D) MVDT: mitral valve deceleration time. Data shown as mean ± SEM. Statistical significance calculated by one or two-way ANOVA with Bonferroni correction: * p<0.05; ** p<0.01; *** p<0.001; **** p<0.0001; (versus WT ****, versus TtnΔIAjxn ††††). (αMHC-Cre cRbm20ΔRRM+/− mice on aΔIAjxn background were used.)
DISCUSSION
We report for the first time a rescue of HFpEF-like symptoms in the mouse through inhibition of the titin splicing factor RBM20. The TtnΔIAjxn murine model acts as a mechanical analogue of the titin-based increase in passive myocardial stiffness found in HFpEF patients4. Increased passive stiffness is present in the adult TtnΔIAjxn mouse, in the absence of pharmacological or surgical intervention. By using the TtnΔIAjxn mouse as a model of HFpEF we demonstrate that inhibiting RBM20 restores diastolic function, improves exercise tolerance and attenuates afterload induced pathologic remodeling of the left ventricle. Due to our still limited understanding of HFpEF pathophysiology, there are no specific therapies to treat this widespread disease, therefore our findings on RBM20 inhibition are clinically significant.
Cardiac titin’s I-band region is a ~1.0 MDa spring composed of three distinct elements: the tandemly arranged immunoglobulin-like (Ig) element, the cardiac specific N2B element, and the proline, glutamate, valine, and lysine rich (PEVK) element24. During diastole, stretch occurs in titin’s spring region and this provides elasticity to the sarcomere25. Alternative splicing of titin occurs predominantly in the spring region and is responsible for the variances found in the two main adult cardiac isoforms of titin, N2B and N2BA26. The more compliant N2BA isoform has a longer extensible spring region owing to the longer PEVK and Ig segments. The N2B isoform is the dominant isoform in both mice and men, with human LV expressing N2B and N2BA isoforms at a ratio of approximately 60:4027,4. Both isoforms extend the same distance for a given sarcomere length (near Z-disk to beginning of A-band), however, the force to stretch titin is greater in the N2B isoform attributable to its shorter contour length8. By reducing the level of functional RBM20 protein, additional exon incorporation into titin’s spring elements minimizes passive stiffness in a manner inversely related to the size of titin11. The TtnΔIAjxn murine model has 18 exons deleted from the mouse Ttn gene that encode 3 Ig domains and 11 FnIII domains that constitute a 153 kDa polypeptide fragment in titin known as the I-band – A-band junction. This model is unique from previously published titin I-band truncation models (N2B28, PEVK29 and Ig16) in that the entire spring region of titin remains intact, including all spring element phospho-sites. Removal of titin’s IA junction, results in anchoring of the C-terminal attachment of titin’s distal Ig spring element further away (~ 70 nm) from the Z-disk, resulting in enhanced strain in titin’s spring region and consequently an increase in force per titin molecule14. This increased force mimics the pathologic conditions of oxidative stress and deranged phosphorylation evident in HFpEF patients4, 30. Interestingly, the TtnΔIAjxn mouse recapitulates many characterisitcs of HFpEF, e.g., diastolic dysfunction, exercise intolerance and hypertrophy. Herein, we studied whether reducing functional RBM20 expression ameliorates these symptoms.
Evaluation of TtnΔIAjxn mice with and without N2BAsc expression was performed at the cell and organ levels. The increased stiffness of cardiac myocytes from TtnΔIAjxn mice (Fig. 1 E) are in agreement with previous reports showing that altering the size of titin directly affects passive stiffness11, 14. That this causes increased LV chamber stiffness is supported by multiple chamber-level measurements. The greater ratio of the early mitral inflow (E) to late atrial kick (A) revealed by pulse-wave Doppler imaging is indicative of increased diastolic stiffness (Fig. 2 C). Moreover, LV chamber stiffening abbreviates the time of deceleration of the early mitral inflow31 and the reduced deceleration time in TtnΔIAjxn mice (Fig. 2 D) also support restrictive filling. In accordance with echocardiography, cathererization of the left ventricle revealed a steeper slope of the EDPVR demonstrating diastolic stiffness in the LV of TtnΔIAjxn mice. Remarkably, inhibiting RBM20 both chronically and acutely in TtnΔIAjxn mice restored these parameters back to WT levels (Figs. 2 and 3). Assessing inducible expression of compliant titin is imperative since HFpEF patients typically develop symptoms later in adult life, therefore, an acute therapy to restore diastolic function in the elderly HFpEF patient is necessary. Upon inducing compliant titin isoform expression in TtnΔIAjxn mice, diastolic dysfunction was corrected and these mice preformed better in exercise tolerance studies (as further discussed below). Thus, our hemodynamic studies in mice with inhibited RBM20 suggest that this intervention is beneficial for restoring compliance to the restrictive left ventricle.
The advantage of RBM20 inhibition was further evidenced by exercise testing. Exercise is considered a beneficial therapy in heart failure patients32, but how titin compliance relates to exercise capacity is not well understood. Since our models are cardiac specific for RBM20 reduction, we could assess exercise performance as it directly relates to increasing the strain in cardiac titin. A limitation to note is that the TtnΔIAjxn mouse deletes the IA junction in both skeletal and cardiac muscles (it is a global KO) and that future control studies are necessary to test exercise tolerance in a cardiac-specific TtnΔIAjxn mouse. When TtnΔIAjxn mice were challenged with treadmill running, mice with upregulated compliant titin (raloxifine group) performed significantly better those that did not (vehicle control). Exercise intolerance leading to fatigue and shortness of breath has a major impact on the quality of life in HFpEF patients, therefore our finding of improved exercise performance through upregulating titin compliance is encouraging.
Titin has been hypothesized to function as biomechancial sensor and our present studies support that titin mechanosensation contributes to increased afterload induced LV hypertrophy. We constricted the transverse aorta of mice to induce acute pressure overload stress to the heart. Compared to WT controls, TtnΔIAjxn mice exhibited a 50% greater increase in LV mass characterized by robust concentric remodeling (Fig. 5). In contrast, RBM20 inhibited TtnΔIAjxn mice (with reduced titin strain) had the least amount of LV mass incease in respone to TAC (Fig. 5 A). Hypertrophy signaling in the myocyte is extremely complex33 and mechanotransduction remains incompletely understood. We explored a previously proposed titin-based signal transduction mechanisms that involved the titin binding proteins FHL1 and FHL2. Four-and-a half LIM domain (FHL) proteins are expressed in striated muscle where they facilitate diverse protein-protein interactions involved in signal transduction34. Previous studies have shown localization of FHL1 and FHL2 at the N2B element in titin35, 36. Futhermore, regulation of extracellular signal-regulated kinase 2 (ERK2) has been reported by both FHL1 and FHL2 in cardiomyocytes37, 38. It has also been reported that in response to pressure oveload, FHL1 null mice display an attenuated hypertrophy response35 whereas the TAC response of FHL2 null mice does not significantly differ from WT34. Our data are in agreement with these findings as FHL1 is significanlty upregulated and FHL2 protein levels remain unaltered in response to TAC (see Online Figure IV). Thus, FHL1 might be involved in the increased hypertrophy response in the TtnΔIAjxn model and its normalization when N2BAsc is expressed. Further studies are necessary to investigate titin as a sensor for mechanotransduction including the roles of FHL1 and FHL2.
HFpEF is a complex disease with multiple etiologies that induce restrictive filling in the left ventricle of which titin-based stiffening is likely to be only one of several underlying causes. Our present work revealed that increasing the compliance of titin’s spring region has a beneficial effect on diastolic function of the heart in a titin specific HFpEF mouse model. HFpEF patients suffer from diastolic dysfunction that leads to a myriad of symptoms including exercise intolerance and reduced quality of life39. Innovative therapies are urgently needed to address diastolic stiffness and our study reveals novel insights into the pathophysiology of HFpEF and the central role played by titin. Lengthening the spring region of titin through alternative splicing reduces passive myocardial stiffness and improves a HFpEF-like phenotype in the TtnΔIAjxn mouse. Diastolic function was recovered both chronically and acutely resulting in an enhanced exercise performance compared to littermate controls. Furthermore, afterload induced pathologic remodeling was attenuated in mice expressing N2BAsc titin. Future efforts should be focused on testing RBM20 inhibition in larger animal models to determine how much inhibition is required to achieve benefit in diastolic function without causing side-effects, especially when inhibition of RBM20 is long-term. In summary, our work supports that ameliorating diastolic dysfunction and pathological chamber remodeling might be achievable through therapeutically targeting titin.
Supplementary Material
Novelty and Significance.
What Is Known?
Tuning mechanisms in the elastic protein titin that alter titin’s compliance are deranged in heart failure patients with preserved ejection fraction (HFpEF).
The titin-based murine model TtnΔIAjxn has increased diastolic stiffness and HFpEF-like symptoms (diastolic dysfunction, exercise intolerance).
RBM20, a splicing factor of titin, regulates the compliance of titin in a dose-dependent manner.
What New Information Does This Article Contribute?
Reduction of RBM20 by 40–45% in TtnΔIAjxn mice is sufficient to greatly increase titin compliance.
Reduction of RBM20 in TtnΔIAjxn mice recovers diastolic function, and improves exercise tolerance and pressure-overload induced ventricular remodeling.
Increasing titin’s compliance improves HFpEF-like symptoms in the TtnΔIAjxn mouse model. HFpEF affects greater than half of all heart failure patients, yet no effective therapies are available. This study reveals that inhibiting RBM20 increases titin compliance and is a potential therapeutic HFpEF target.
Acknowledgments
We are grateful to our current and former lab members (and in particular Mr Xiangdang Liu for genotyping) who contributed to this work and to the University of Arizona Phenotyping Core Facility.
SOURCES OF FUNDING
This work was supported by training grant T32HL007249 and Van Denburgh ARCS award to (M.B.), National Institutes of Health grants HL062881 and HL118524, and Foundation Leducq (TNE-13CVD04) (H.L.).
Nonstandard Abbreviations and Acronyms
- E/A
LV pulse wave Doppler E-wave: A-wave ratio
- Ea
arterial elastance
- Eccentricity
LVDD/WTH
- EDP
end‐diastolic pressure
- EDPVR
end diastolic pressure volume relationship
- EDV
end‐diastolic volume
- ESP
end‐systolic pressure
- ESPVR
end‐systolic PV relationship
- ESV
end systolic volume
- FHL1
four and a Half LIM domains 1
- FHL2
four and a Half LIM domains 2
- FS
fractional shortening
- HF
heart failure
- HFpEF
heart failure with preserved ejection fraction
- IAjnx
I-band – A-band junction
- Ig
immunoglobulin-like
- LA
left atrium PV
- LV
left ventricle
- LVIDd
left ventricular internal diastolic diameter
- LVIDs
left ventricular internal systolic diameter
- LVPWd
left ventricular posterior wall thickness in diastole
- LVPWs
left ventricular posterior wall thickness in systole.
- MHC
myosin heavy chain
- MV
DT mitral valve deceleration time
- PEVK
proline, glutamate, valine, and lysine
- PKG
protein kinase G
- PTM
posttranslational modification
- RBM20
RNA binding motif 20
- RRM
RNA recognition motif
- SERM
selective estrogen receptor modulator
- SL
sarcomere length
- TAC
transverse aortic constriction
- WTH
wall thickness
- αBC
alpha-B crystallin
Footnotes
DISCLOSURES
None.
References
- 1.Writing Committee M. Yancy CW, Jessup M, Bozkurt B, Butler J, Casey DE, Jr, Drazner MH, Fonarow GC, Geraci SA, Horwich T, Januzzi JL, Johnson MR, Kasper EK, Levy WC, Masoudi FA, McBride PE, McMurray JJ, Mitchell JE, Peterson PN, Riegel B, Sam F, Stevenson LW, Tang WH, Tsai EJ, Wilkoff BL, American College of Cardiology Foundation/American Heart Association Task Force on Practice G 2013 accf/aha guideline for the management of heart failure: A report of the american college of cardiology foundation/american heart association task force on practice guidelines. Circulation. 2013;128:e240–327. doi: 10.1161/CIR.0b013e31829e8776. [DOI] [PubMed] [Google Scholar]
- 2.Butler J, Fonarow GC, Zile MR, Lam CS, Roessig L, Schelbert EB, Shah SJ, Ahmed A, Bonow RO, Cleland JG, Cody RJ, Chioncel O, Collins SP, Dunnmon P, Filippatos G, Lefkowitz MP, Marti CN, McMurray JJ, Misselwitz F, Nodari S, O’Connor C, Pfeffer MA, Pieske B, Pitt B, Rosano G, Sabbah HN, Senni M, Solomon SD, Stockbridge N, Teerlink JR, Georgiopoulou VV, Gheorghiade M. Developing therapies for heart failure with preserved ejection fraction: Current state and future directions. JACC Heart Fail. 2014;2:97–112. doi: 10.1016/j.jchf.2013.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Haq MA, Wong C, Mutha V, Anavekar N, Lim K, Barlis P, Hare DL. Therapeutic interventions for heart failure with preserved ejection fraction: A summary of current evidence. World J Cardiol. 2014;6:67–76. doi: 10.4330/wjc.v6.i2.67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, Redfield MM, Bull DA, Granzier HL, LeWinter MM. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: Contributions of collagen and titin. Circulation. 2015;131:1247–1259. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bang ML, Centner T, Fornoff F, Geach AJ, Gotthardt M, McNabb M, Witt CC, Labeit D, Gregorio CC, Granzier H, Labeit S. The complete gene sequence of titin, expression of an unusual approximately 700-kda titin isoform, and its interaction with obscurin identify a novel z-line to i-band linking system. Circ Res. 2001;89:1065–1072. doi: 10.1161/hh2301.100981. [DOI] [PubMed] [Google Scholar]
- 6.Labeit S, Kolmerer B. Titins: Giant proteins in charge of muscle ultrastructure and elasticity. Science. 1995;270:293–296. doi: 10.1126/science.270.5234.293. [DOI] [PubMed] [Google Scholar]
- 7.Granzier HL, Irving TC. Passive tension in cardiac muscle: Contribution of collagen, titin, microtubules, and intermediate filaments. Biophys J. 1995;68:1027–1044. doi: 10.1016/S0006-3495(95)80278-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Trombitas K, Redkar A, Centner T, Wu Y, Labeit S, Granzier H. Extensibility of isoforms of cardiac titin: Variation in contour length of molecular subsegments provides a basis for cellular passive stiffness diversity. Biophys J. 2000;79:3226–3234. doi: 10.1016/S0006-3495(00)76555-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hidalgo C, Granzier H. Tuning the molecular giant titin through phosphorylation: Role in health and disease. Trends Cardiovasc Med. 2013;23:165–171. doi: 10.1016/j.tcm.2012.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Guo W, Schafer S, Greaser ML, Radke MH, Liss M, Govindarajan T, Maatz H, Schulz H, Li S, Parrish AM, Dauksaite V, Vakeel P, Klaassen S, Gerull B, Thierfelder L, Regitz-Zagrosek V, Hacker TA, Saupe KW, Dec GW, Ellinor PT, MacRae CA, Spallek B, Fischer R, Perrot A, Ozcelik C, Saar K, Hubner N, Gotthardt M. Rbm20, a gene for hereditary cardiomyopathy, regulates titin splicing. Nature medicine. 2012;18:766–773. doi: 10.1038/nm.2693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Methawasin M, Hutchinson KR, Lee EJ, Smith JE, 3rd, Saripalli C, Hidalgo CG, Ottenheijm CA, Granzier H. Experimentally increasing titin compliance in a novel mouse model attenuates the frank-starling mechanism but has a beneficial effect on diastole. Circulation. 2014;129:1924–1936. doi: 10.1161/CIRCULATIONAHA.113.005610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.van Heerebeek L, Hamdani N, Falcao-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, Verheugt FW, Niessen HW, Paulus WJ. Low myocardial protein kinase g activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–839. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- 13.Redfield MM, Chen HH, Borlaug BA, et al. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: A randomized clinical trial. JAMA. 2013;309:1268–1277. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Granzier HL, Hutchinson KR, Tonino P, Methawasin M, Li FW, Slater RE, Bull MM, Saripalli C, Pappas CT, Gregorio CC, Smith JE., 3rd Deleting titin’s i-band/a-band junction reveals critical roles for titin in biomechanical sensing and cardiac function. Proc Natl Acad Sci U S A. 2014;111:14589–14594. doi: 10.1073/pnas.1411493111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chung CS, Hutchinson KR, Methawasin M, Saripalli C, Smith JE, 3rd, Hidalgo CG, Luo X, Labeit S, Guo C, Granzier HL. Shortening of the elastic tandem immunoglobulin segment of titin leads to diastolic dysfunction. Circulation. 2013;128:19–28. doi: 10.1161/CIRCULATIONAHA.112.001268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.King NM, Methawasin M, Nedrud J, Harrell N, Chung CS, Helmes M, Granzier H. Mouse intact cardiac myocyte mechanics: Cross-bridge and titin-based stress in unactivated cells. J Gen Physiol. 2011;137:81–91. doi: 10.1085/jgp.201010499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hu P, Zhang D, Swenson L, Chakrabarti G, Abel ED, Litwin SE. Minimally invasive aortic banding in mice: Effects of altered cardiomyocyte insulin signaling during pressure overload. American Journal of Physiology - Heart and Circulatory Physiology. 2003;285:H1261–H1269. doi: 10.1152/ajpheart.00108.2003. [DOI] [PubMed] [Google Scholar]
- 18.Buck D, Smith JE, 3rd, Chung CS, Ono Y, Sorimachi H, Labeit S, Granzier HL. Removal of immunoglobulin-like domains from titin’s spring segment alters titin splicing in mouse skeletal muscle and causes myopathy. J Gen Physiol. 2014;143:215–230. doi: 10.1085/jgp.201311129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ohno M, Cheng CP, Little WC. Mechanism of altered patterns of left ventricular filling during the development of congestive heart failure. Circulation. 1994;89:2241–2250. doi: 10.1161/01.cir.89.5.2241. [DOI] [PubMed] [Google Scholar]
- 20.DeMaria AN, Wisenbaugh TW, Smith MD, Harrison MR, Berk MR. Doppler echocardiographic evaluation of diastolic dysfunction. Circulation. 1991;84:I288–295. [PubMed] [Google Scholar]
- 21.Owan TE, Hodge DO, Herges RM, Jacobsen SJ, Roger VL, Redfield MM. Trends in prevalence and outcome of heart failure with preserved ejection fraction. N Engl J Med. 2006;355:251–259. doi: 10.1056/NEJMoa052256. [DOI] [PubMed] [Google Scholar]
- 22.Melenovsky V, Borlaug BA, Rosen B, Hay I, Ferruci L, Morell CH, Lakatta EG, Najjar SS, Kass DA. Cardiovascular features of heart failure with preserved ejection fraction versus nonfailing hypertensive left ventricular hypertrophy in the urban baltimore community: The role of atrial remodeling/dysfunction. J Am Coll Cardiol. 2007;49:198–207. doi: 10.1016/j.jacc.2006.08.050. [DOI] [PubMed] [Google Scholar]
- 23.Zile MR, Gottdiener JS, Hetzel SJ, McMurray JJ, Komajda M, McKelvie R, Baicu CF, Massie BM, Carson PE. Prevalence and significance of alterations in cardiac structure and function in patients with heart failure and a preserved ejection fraction. Circulation. 2011;124:2491–2501. doi: 10.1161/CIRCULATIONAHA.110.011031. [DOI] [PubMed] [Google Scholar]
- 24.Granzier H, Labeit S. Cardiac titin: An adjustable multi-functional spring. J Physiol. 2002;541:335–342. doi: 10.1113/jphysiol.2001.014381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Granzier H, Helmes M, Trombitas K. Nonuniform elasticity of titin in cardiac myocytes: A study using immunoelectron microscopy and cellular mechanics. Biophys J. 1996;70:430–442. doi: 10.1016/S0006-3495(96)79586-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Trombitas K, Wu Y, Labeit D, Labeit S, Granzier H. Cardiac titin isoforms are coexpressed in the half-sarcomere and extend independently. Am J Physiol Heart Circ Physiol. 2001;281:H1793–1799. doi: 10.1152/ajpheart.2001.281.4.H1793. [DOI] [PubMed] [Google Scholar]
- 27.Neagoe C, Kulke M, del Monte F, Gwathmey JK, de Tombe PP, Hajjar RJ, Linke WA. Titin isoform switch in ischemic human heart disease. Circulation. 2002;106:1333–1341. doi: 10.1161/01.cir.0000029803.93022.93. [DOI] [PubMed] [Google Scholar]
- 28.Radke MH, Peng J, Wu Y, McNabb M, Nelson OL, Granzier H, Gotthardt M. Targeted deletion of titin n2b region leads to diastolic dysfunction and cardiac atrophy. Proc Natl Acad Sci U S A. 2007;104:3444–3449. doi: 10.1073/pnas.0608543104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Granzier HL, Radke MH, Peng J, Westermann D, Nelson OL, Rost K, King NM, Yu Q, Tschope C, McNabb M, Larson DF, Labeit S, Gotthardt M. Truncation of titin’s elastic pevk region leads to cardiomyopathy with diastolic dysfunction. Circ Res. 2009;105:557–564. doi: 10.1161/CIRCRESAHA.109.200964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Szelenyi Z, Fazakas A, Szenasi G, Kiss M, Tegze N, Fekete BC, Nagy E, Bodo I, Nagy B, Molvarec A, Patocs A, Pepo L, Prohaszka Z, Vereckei A. Inflammation and oxidative stress caused by nitric oxide synthase uncoupling might lead to left ventricular diastolic and systolic dysfunction in patients with hypertension. J Geriatr Cardiol. 2015;12:1–10. doi: 10.11909/j.issn.1671-5411.2015.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Vlahovic A, Popovic AD. evaluation of left ventricular diastolic function using doppler echocardiography. Medicinski pregled. 1999;52:13–18. [PubMed] [Google Scholar]
- 32.Vromen T, Kraal JJ, Kuiper J, Spee RF, Peek N, Kemps HM. The influence of training characteristics on the effect of aerobic exercise training in patients with chronic heart failure: A meta-regression analysis. International journal of cardiology. 2016;208:120–127. doi: 10.1016/j.ijcard.2016.01.207. [DOI] [PubMed] [Google Scholar]
- 33.van Berlo JH, Maillet M, Molkentin JD. Signaling effectors underlying pathologic growth and remodeling of the heart. J Clin Invest. 2013;123:37–45. doi: 10.1172/JCI62839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chu PH, Ruiz-Lozano P, Zhou Q, Cai C, Chen J. Expression patterns of fhl/slim family members suggest important functional roles in skeletal muscle and cardiovascular system. Mechanisms of development. 2000;95:259–265. doi: 10.1016/s0925-4773(00)00341-5. [DOI] [PubMed] [Google Scholar]
- 35.Sheikh F, Raskin A, Chu PH, Lange S, Domenighetti AA, Zheng M, Liang X, Zhang T, Yajima T, Gu Y, Dalton ND, Mahata SK, Dorn GW, 2nd, Brown JH, Peterson KL, Omens JH, McCulloch AD, Chen J. An fhl1-containing complex within the cardiomyocyte sarcomere mediates hypertrophic biomechanical stress responses in mice. J Clin Invest. 2008;118:3870–3880. doi: 10.1172/JCI34472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lange S, Auerbach D, McLoughlin P, Perriard E, Schafer BW, Perriard JC, Ehler E. Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by dral/fhl-2. J Cell Sci. 2002;115:4925–4936. doi: 10.1242/jcs.00181. [DOI] [PubMed] [Google Scholar]
- 37.Raskin A, Lange S, Banares K, Lyon RC, Zieseniss A, Lee LK, Yamazaki KG, Granzier HL, Gregorio CC, McCulloch AD, Omens JH, Sheikh F. A novel mechanism involving four-and-a-half lim domain protein-1 and extracellular signal-regulated kinase-2 regulates titin phosphorylation and mechanics. J Biol Chem. 2012;287:29273–29284. doi: 10.1074/jbc.M112.372839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Purcell NH, Darwis D, Bueno OF, Muller JM, Schule R, Molkentin JD. Extracellular signal-regulated kinase 2 interacts with and is negatively regulated by the lim-only protein fhl2 in cardiomyocytes. Molecular and Cellular Biology. 2004;24:1081–1095. doi: 10.1128/MCB.24.3.1081-1095.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gladden JD, Linke WA, Redfield MM. Heart failure with preserved ejection fraction. Pflugers Arch. 2014;466:1037–1053. doi: 10.1007/s00424-014-1480-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.