

Abstract
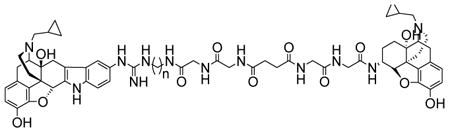
In an effort to develop antagonists for κ-μ opioid receptor heterodimers, a series of bivalent ligands 3 - 6 containing κ- and μ-antagonist pharmacophores were designed and synthesized. Evaluation of the series in HEK-293 cells revealed 4 (KMN-21) to selectively antagonize the activation of κ-μ heterodimers, suggesting possible bridging of receptors when the bivalent ligand spacer contains 21 atoms.
Graphical abstract
A series of bivalent ligands containing κ- and μ-antagonist pharmacophores were designed and reported as chemical tools for studying κ-μ heterodimers.
The opioid receptors belong to the superfamily of G-protein coupled receptors (GPCRs) and play important roles in pain perception and regulation. Three major types of opioid receptors including κ-, δ- and μ-opioid receptors have been well characterized and cloned.1 Numerous reports have demonstrated the dimerization/oligomerization of GPCRs,2 and the existence of dimers for opioid receptors in cultured cells.3-8 In addition, pharmacological evidence had suggested that multiple phenotypic κ-opioid receptor subtypes are related to heterodimeric opioid receptors. For example, κ-δ and κ-μ heterodimers have been linked to the κ2 subtype.7,9 Therefore it would be of value to develop chemical tools to assist in the pharmacological characterization of opioid receptor heterodimers.
In this regard, bivalent ligands have been developed as pharmacological tools to study the dimerization of opioid receptors.10-14 Recently, we had reported two selective bivalent ligands KDN-21 and KDAN-18, that have suggested the association of δ1 and κ2 phenotypes with κ-δ heterodimers.15-16 In view of the promising results from this bivalent ligand strategy and evidence showing the association of opioid receptors via homo- and hetero-dimerization,4-8 we have extended the bivalent strategy to design tools for κ-μ heterodimers.
Here we report on the synthesis and in vitro biological characterization of a series of bivalent ligands 3-6 containing a κ-opioid antagonist pharmacophore 5′-guanidinonaltrindole (5′-GNTI) (1)17 and a μ-opioid antagonist pharmacophore β-naltrexamine (β-NTX) (2)18 linked through a spacer of varying length. The design rationale of these bivalent ligands is based on our previous studies of κ-δ bivalent ligands15-16 and our desire to maintain a favorable hydrophilic and lipophilic balance coupled with flexibility. This included a spacer that contains (1) glycine units that maintain a favorable hydrophilic-lipophilic balance, (2) a succinyl unit that contributes to the flexibility for favorable interaction with heterodimers, and (3) an alkylamine moiety attached to pharmacophore 1 which permits variation of the spacer length by one atom increments (Figure 1).
Figure 1. Designed bivalent ligands.

The synthetic protocol for 3-6 is shown in Scheme 1 and Scheme 2. Briefly, coupling reaction between carboxylic acid 7 and naltrexamine 8, which were synthesized as reported15,18, gave intermediate 9, which on hydrolysis with TFA in dichloromethane afforded carboxylic acid intermediate 10 (Scheme 1). The reactions of monoprotected alkyldiamines 11-14 with KSCN followed by Cbz-protection yielded 15-18, which upon condensation with 19 in the presence of HgCl2 and Et3N followed by deprotection, provided 20-23 in good yield. Finally, standard amide coupling reactions of 10 with 20-23 followed by catalytic hydrogenation afforded the desired bivalent ligands 3-6 (Scheme 2). The chemical structures of bivalent ligands were characterized and confirmed by 1H-NMR and Fast-atom bombardment mass spectra (FABMS) (ESI). The purity of designed ligands was determined by reverse HPLC (Acetonitrile/H2O/TFA: 50/50/0.1 and MeOH/H2O/TFA: 30/70/0.1) to be >98%.
Scheme 1. The synthetic route for the carboxylic acid intermediate 10.

Scheme 2. The synthetic route for the designed bivalent ligands 3-6.

After synthesis, the antagonist activities of 3-6 were evaluated by measuring inhibition of Ca2+ release in HEK 293 cells that stably express κ- and μ-opioid receptors singly or together (Figure 2). A chimeric G protein was transiently transfected in this assay for Ca2+ release.19 The selective agonists used to evaluate the antagonism selectivity of the target compounds were U69,59320 (κ) and DAMGO (μ).21 While all of the bivalent ligands 3-6 (1 μM concentration) antagonized Ca2+ release in cells containing singly expressed receptors, only bivalent ligand 4 (KMN-21) significantly antagonized both U69593- and DAMGO-induced Ca2+ release in cells containing coexpressed κ- and μ-opioid receptors. These data suggest that bivalent ligand 4 with a spacer length of 21 atoms engages κ-μ opioid heterodimers more efficiently than homodimers. In this regard, it is noteworthy that the κ-δ bivalent ligand antagonist, KDN-21, also contains a 21-atom spacer, suggesting common bridging modes to κ-μ and κ-δ heterodimeric receptors.16 To further understand the interactions of bivalent ligand 4 with κ-μ heterodimers, additional studies would be required.
Figure 2.
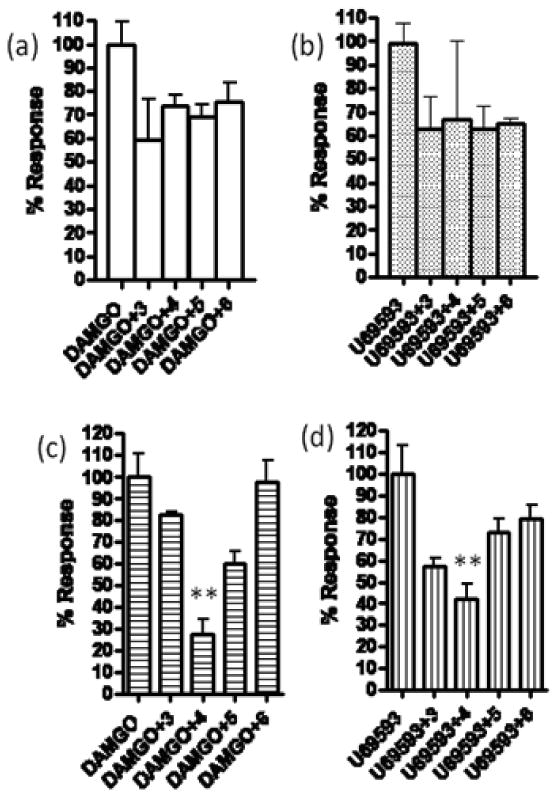
Compound 4 (KMN-21) is an antagonist in HEK-293 cells coexpressing μ- and κ-opioid receptors. Intracellular calcium release experiments were performed using appropriate agonist ligands and compounds 3-6 in HEK-293 cells stably expressing (a) mu, (b) kappa, (c& d) mu/kappa opioid receptors. Cells were transiently transfected with chimeric G-protein, Δ6-Gqi4-myr (200 ng for every 40,000 cells) and pre-incubated with antagonist compounds 3-6 (1 μM) and dye from calcium release kit (Molecular Devices). Intracellular calcium release was measured in a Flexstation apparatus (Molecular Devices) using DAMGO or U69593 (1 μM). Experiments were performed in triplicate (n=4) and significance was measured using ANOVA (** = p<0.01).
In summary, a series of bivalent ligands containing κ- and μ-opioid antagonist pharmacophores attached to variable length spacers were designed and synthesized as chemical tools to investigate κ-μ heterodimers. Biological evaluation in HEK 293 cells revealed that bivalent ligand 4 with 21 atoms in its spacer significantly antagonized Ca2+ release of activated κ- and μ-opioid receptors. Together with our previous results of different bivalent ligands,16-17 the results further exemplifies the power of bivalent ligands as pharmacological tools in investigating the dimerization of opioid receptors in particular and GPCRs in general.
Acknowledgments
We thank Mike Powers for capable technical assistance. This research is supported by grant DA01533 from National Institute on Drug Abuse.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References and Notes
- 1.Dhawan BN, Cesselin F, Raghubir R, Reisine T, Bradley PB, Portoghese PS. Pharmacol Rev. 1996;48:567. [PubMed] [Google Scholar]
- 2.Angers S, Salahpur A, Bouvier M. Annu Rev Pharmacol Toxicol. 2002;42:409. doi: 10.1146/annurev.pharmtox.42.091701.082314. [DOI] [PubMed] [Google Scholar]
- 3.Levac BA, O'Dowd BF, George SR. Curr Opin Pharmacol. 2002;2:76. doi: 10.1016/s1471-4892(02)00124-8. [DOI] [PubMed] [Google Scholar]
- 4.Rios CD, Jordan BA, Gomes I, Devi LA. Pharmacol Ther. 2001;92:71. doi: 10.1016/s0163-7258(01)00160-7. [DOI] [PubMed] [Google Scholar]
- 5.Portoghese PS, Lunzer MM. Eur J Pharmacol. 2003;467:233. doi: 10.1016/s0014-2999(03)01599-1. [DOI] [PubMed] [Google Scholar]
- 6.Gomes I, Jordan BA, Trapaidze N, Nagy V, Devi LA. J Neurosci. 2000;20:RC110. doi: 10.1523/JNEUROSCI.20-22-j0007.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jordan BA, Devi LA. Nature. 1999;399:697. doi: 10.1038/21441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wang D, Sun X, Bohn LM, Sadee W. Molecular Pharmacology. 2005;67:2173. doi: 10.1124/mol.104.010272. [DOI] [PubMed] [Google Scholar]
- 9.Simonin F, Slowe S, Becker JA, Matthes HW, Filliol D, Chluba J, Kitchen I, Kieffer BL. Eur J Pharmacol. 2001;414:189. doi: 10.1016/s0014-2999(01)00822-6. [DOI] [PubMed] [Google Scholar]
- 10.Portoghese PS, Ronsisvalle G, Larson DL, Yim CB, Sayre LM, Takemori AE. Life Sci. 1982;31:1283. doi: 10.1016/0024-3205(82)90362-9. [DOI] [PubMed] [Google Scholar]
- 11.Erez M, Takemori AE, Portoghese PS. J Med Chem. 1982;25:847. doi: 10.1021/jm00349a016. [DOI] [PubMed] [Google Scholar]
- 12.Portoghese PS, Larson DL, Yim CB, Sayre LM, Ronsisvalle G, Lipkowski AW, Takemori AE, Rice KC, Tam SW. J Med Chem. 1985;28:1140. doi: 10.1021/jm00147a002. [DOI] [PubMed] [Google Scholar]
- 13.Portoghese PS, Larson DL, Sayre LM, Yim CB, Ronsisvalle G, Tam SW, Takemori AE. J Med Chem. 1986;29:1855. doi: 10.1021/jm00160a010. [DOI] [PubMed] [Google Scholar]
- 14.Portoghese PS, Ronsisvalle G, Larson DL, Takemori AE. J Med Chem. 1986;29:1650. doi: 10.1021/jm00159a014. [DOI] [PubMed] [Google Scholar]
- 15.Bhushan RG, Sharma SK, Xie ZH, Daniels DJ, Portoghese PS. J Med Chem. 2004;47:2969. doi: 10.1021/jm0342358. [DOI] [PubMed] [Google Scholar]
- 16.Daniels DJ, Kulkarni A, Xie ZH, Bhushan RG, Portoghese PS. J Med Chem. 2005;48:1713. doi: 10.1021/jm034234f. [DOI] [PubMed] [Google Scholar]
- 17.Jones RM, Portoghese PS. Eur J Pharmacol. 2000;396:49. doi: 10.1016/s0014-2999(00)00208-9. [DOI] [PubMed] [Google Scholar]
- 18.Sayre LM, Portoghese PS. J Org Chem. 1980;45:3366. [Google Scholar]
- 19.Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL. Proc Natl Acad Sci USA. 2005;102:9050. doi: 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lahti RA, Miekelson MM, Mccall JM, Von Voightlander PF. Eur J Pharmacol. 1985;109:281. doi: 10.1016/0014-2999(85)90431-5. [DOI] [PubMed] [Google Scholar]
- 21.Handa BK, Lane AC, Lord JA, Morgan BA, Rance MJ, Smith CF. Eur J Pharmacol. 1981;70:531. doi: 10.1016/0014-2999(81)90364-2. [DOI] [PubMed] [Google Scholar]