

Abstract
JUMP is a phase 3b expanded-access trial for patients without access to ruxolitinib outside of a clinical study; it is the largest clinical trial to date in patients with myelofibrosis who have been treated with ruxolitinib. Here, we present safety and efficacy findings from an analysis of 1144 patients with intermediate- or high-risk myelofibrosis, as well as a separate analysis of 163 patients with intermediate-1-risk myelofibrosis – a population of patients not included in the phase 3 COMFORT studies. Consistent with ruxolitinib’s mechanism of action, the most common hematologic adverse events were anemia and thrombocytopenia, but these led to treatment discontinuation in only a few cases. The most common non-hematologic adverse events were primarily grade 1/2 and included diarrhea, pyrexia, fatigue, and asthenia. The rates of infections were low and primarily grade 1/2, and no new or unexpected infections were observed. The majority of patients achieved a ≥50% reduction from baseline in palpable spleen length. Improvements in symptoms were rapid, with approximately half of all patients experiencing clinically significant improvements, as assessed by various quality-of-life questionnaires. The safety and efficacy profile in intermediate-1-risk patients was consistent with that in the overall JUMP population and with that previously reported in intermediate-2- and high-risk patients. Overall, ruxolitinib provided clinically meaningful reductions in spleen length and symptoms in patients with myelofibrosis, including those with intermediate-1-risk disease, with a safety and efficacy profile consistent with that observed in the phase 3 COMFORT studies. This trial was registered as NCT01493414 at ClinicalTrials.gov.
Introduction
Myelofibrosis (MF) is a chronic myeloproliferative neoplasm characterized by a dysregulated Janus kinase (JAK)/signal transducer and activator of transcription (STAT) pathway that can present as primary MF or evolve from polycythemia vera or essential thrombocythemia.1–4 Common aspects of the disease include bone marrow fibrosis, extramedullary hematopoiesis, anemia, splenomegaly, a debilitating symptom burden (e.g., fatigue, weight loss, night sweats, pruritus),4–7 and reduced survival.8 Patients are stratified as having low-, intermediate-1-, intermediate-2-, or high-risk MF by International Prognostic Scoring System (IPSS) criteria, with corresponding median survivals of 11, 8, 4, and 2 years.8
Ruxolitinib is a potent JAK1/JAK2 inhibitor that has demonstrated superiority over placebo9 and best available therapy10 in the pivotal phase 3 COMFORT (Controlled Myelofibrosis study with Oral JAK inhibitor Treatment) studies in patients with intermediate-2- or high-risk MF. Ruxolitinib treatment led to durable improvements in splenomegaly, symptoms, and quality-of-life (QOL) measures as well as improved survival.9–11 Based on these findings, ruxolitinib became the first JAK inhibitor approved for the treatment of MF. Despite the impact of these studies on the treatment landscape in higher-risk MF, not much is known about the efficacy and safety of ruxolitinib in patients with intermediate-1-risk MF – a group that also has significant constitutional symptoms, splenomegaly, and a reduced health-related QOL.8 Findings from ROBUST, a phase 2 study that evaluated ruxolitinib in patients with high-, intermediate-2-, and intermediate-1-risk MF, indicated that ruxolitinib treatment results in clinically meaningful reductions in spleen length and symptoms in most patients, including those classified as intermediate-1 risk.12
To allow for collection of additional safety and efficacy data for ruxolitinib and provide an access path to ruxolitinib for patients with MF, the JUMP (JAK Inhibitor RUxolitinib in Myelofibrosis Patients; NCT01493414) study was initiated. JUMP is a phase 3b expanded-access trial for patients in countries without access to ruxolitinib outside of a clinical study and includes those classified as intermediate-1 risk, a population that was not included in the COMFORT trials. Here, we present safety and efficacy findings from an analysis of 1144 patients with MF and 163 patients with intermediate-1-risk MF who started ruxolitinib treatment ≥1 year before the data cut-off.
Methods
Eligibility criteria
Eligible patients were aged ≥18 years with a diagnosis of primary or secondary MF by World Health Organization criteria3,13 and classified as IPSS high, intermediate-2, or intermediate-1 risk by the treating investigator.8 Patients with intermediate-1-risk MF were required to have a palpable spleen (≥5 cm from the costal margin). However, IPSS risk status was not recorded until implementation of protocol amendment 2, and most patients did not have IPSS risk status at the time of this data cut. Patients were required to have a baseline platelet count ≥50×109/L; those with a platelet count of 50×109/L to 100×109/L were included through amendments to the protocol.
Study design
JUMP is a single-arm, open-label phase 3b expanded-access global study. Patients received starting doses of ruxolitinib based on platelet counts at baseline: 5 mg twice daily (bid; 50×109/L to <100×109/L), 15 mg bid (100 to 200×109/L), or 20 mg bid (>200×109/L). Doses could be increased (up to 25 mg bid if platelet and neutrophil counts were adequate) in 5-mg bid increments (5 mg/day for patients starting ruxolitinib at 5 mg bid) when efficacy was insufficient. Dose decreases or interruptions were mandatory for safety reasons (e.g., declining platelet counts) and were made according to a protocol-specified dosing regimen (Online Supplementary Methods). Patients were followed up for 28 days after treatment discontinuation. The final analysis will be performed after all patients have completed 24 months of treatment, unless discontinuation criteria are met (Online Supplementary Methods).
Endpoints
The primary endpoint was assessment of ruxolitinib safety and tolerability by the frequency, duration, and severity of adverse events. Additional endpoints included the proportion of patients with ≥50% reduction in palpable spleen length, patient-reported outcomes [Functional Assessment of Cancer Therapy -Lymphoma (FACT-Lym) total score (TS) and Functional Assessment of Chronic Illness Therapy (FACIT) - Fatigue Scale], progression-free survival, survival without transformation to acute myeloid leukemia (AML-free survival), and overall survival (Online Supplementary Methods).
Study oversight
The study was sponsored and designed by Novartis Pharmaceuticals Corporation, approved by the institutional review board at each participating institution, and conducted in accordance with applicable local regulations and the principles of the Declaration of Helsinki. All patients provided written informed consent. Data were analyzed and interpreted by the sponsor’s clinical and statistical teams in collaboration with authors not affiliated with the sponsor. A publication steering committee discussed the data and analyses and helped guide the publication plan for this study. The first author prepared the first draft of the manuscript, with assistance from a medical writer funded by Novartis Pharmaceuticals Corporation, and made the final decision to submit the manuscript for publication. All authors reviewed and amended the manuscript.
Statistical analysis
This analysis includes results from patients who started ruxolitinib treatment ≥1 year before the data cutoff. Only descriptive statistics are provided. Changes in spleen length were assessed in patients with baseline and post-baseline assessments. A 100% reduction from baseline in spleen length was defined as non-palpable. Survival assessments were performed at the end of the 28-day follow-up period. Progression-free and overall survival were estimated using the Kaplan-Meier method.
Results
Patients
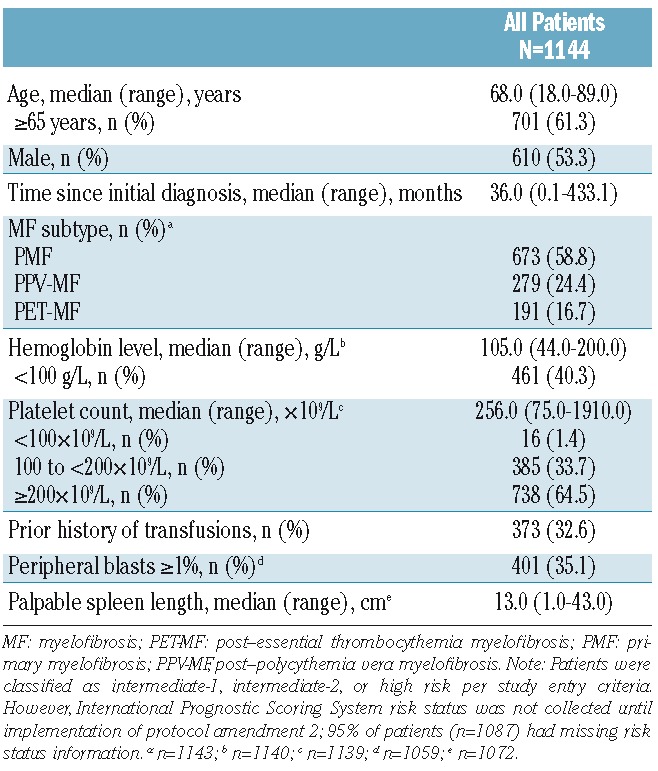
A total of 2233 patients were enrolled at more than 200 study sites in 26 countries (last patient first visit, 23 December 2014). This analysis includes results for 1144 patients who started ruxolitinib treatment ≥1 year before the data cutoff date (01 January 2014). Patients were from Europe (83.6%), South America (8.5%), North America (4.6%), and other regions (3.5%) (Online Supplementary Table S1). The baseline characteristics of the patients are shown in Table 1. The median age of the patients was 68 years (range, 18–89 years), with a median time since initial diagnosis of 36 months (range, 0.1–433.1 months). Overall, 53.3% of patients were male and 58.8% had primary MF.
Table 1.
Patients’ baseline characteristics.
Most patients completed treatment per protocol (i.e., had access to commercially available drug; 33.5%) or remained on treatment (35.5%; Online Supplementary Table S2). The most common reasons for treatment discontinuation included adverse events (13.8%), investigator-determined disease progression (7.1%), death (3.8%), consent withdrawal (3.8%), and physician’s decision (1.4%). Specific adverse events that led to discontinuation are shown in Online Supplementary Table S3.
The median exposure to ruxolitinib was 11.1 months. The mean total daily dose over time is shown in Figure 1. The median average daily dose was 37.2 mg for patients starting at 20 mg bid (63.6%), 23.4 mg for patients starting at 15 mg bid (32.7%), and 13.3 mg for patients starting at 5 mg bid (1.0%). The majority of patients (65.9%) had a dose modification, and 23.9% had a dose interruption.
Figure 1.
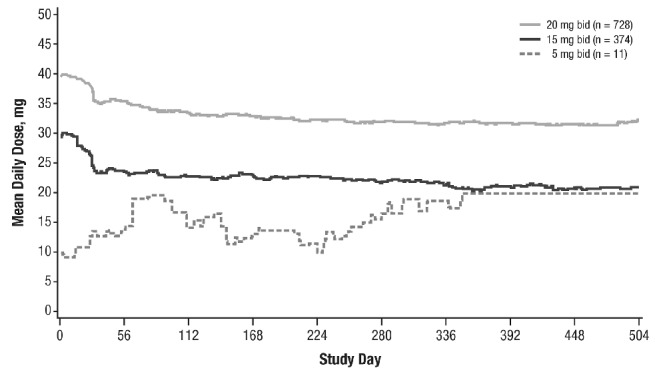
Mean total daily dosea,b of ruxolitinib. bid: twice daily; qd: once daily. aIncludes patients with complete drug administration dates (n=1141). b28 patients started at doses other than 5, 15, or 20 mg bid: 5 mg qd (n=1), 7.5 mg bid (n=1), 15 mg qd (n=6), 10 mg bid (n=16), 20 mg qd (n=4).
Safety
Consistent with findings from the COMFORT studies,9,10 the most common hematologic adverse events were anemia (all grades, 56.3%; grade 3/4, 33.0%) and thrombocytopenia (all grades, 42.2%; grade 3/4, 12.5%; Table 2). However, only 2.6% (n=30) and 3.2% (n=37) of patients, respectively, discontinued treatment (Online Supplementary Table S3), indicating that these adverse events were manageable in most patients. Overall, 159 patients (13.9%) received erythropoiesis-stimulating agents as concomitant medication; preliminary results are summarized for these patients. Similar to what has been observed previously,9,10 mean hemoglobin levels decreased from baseline (108 g/L) during the first 8 to 12 weeks of the study but increased to near-baseline levels after week 12 (Figure 2A). Mean platelet counts decreased from baseline (319×109/L) during the first 4 weeks and then remained stable over time (Figure 2B). As expected, the rates of thrombocytopenia were higher in patients who had baseline platelet counts of 100×109/L to 200×109/L and received ruxolitinib at a starting dose of 15 mg bid (all grades, 58.8%; grade 3/4, 22.2%) than in patients who had baseline platelet counts >200×109/L and started treatment at 20 mg bid (all grades, 33.9%; grade 3/4, 7.1%). The rates of anemia were similar in both groups (all grades, 53.2% versus 57.9%; grade 3/4, 32.6% versus 33.1%, respectively).
Table 2.
Adverse events regardless of study drug relationship (in ≥5% of patients)a.
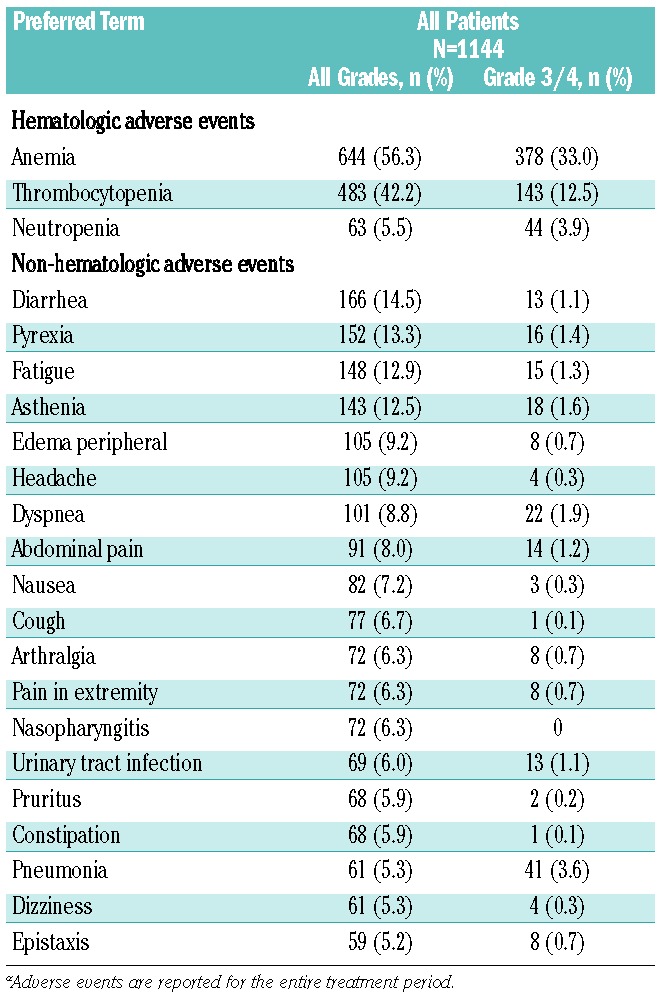
Figure 2.
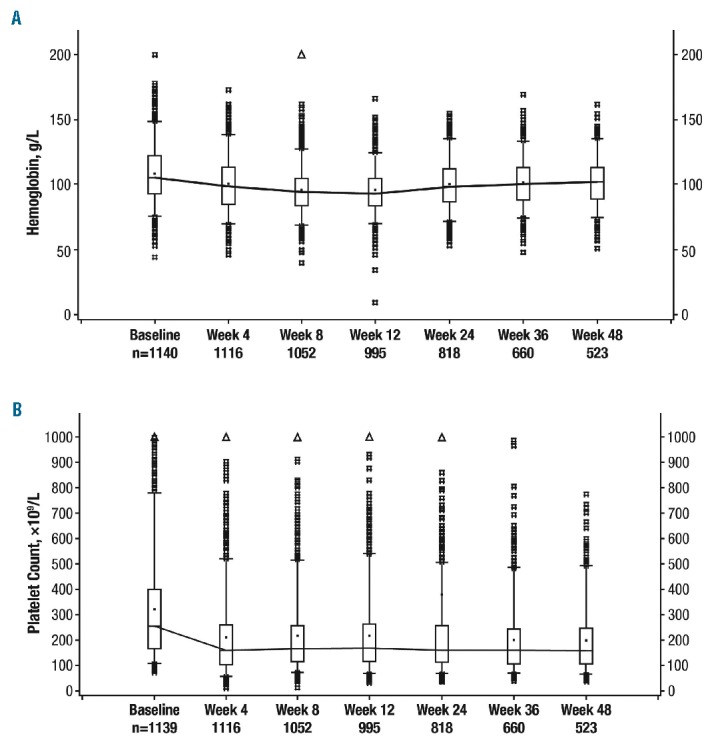
Hemoglobin levels and platelet counts over time. (A) Hemoglobin levels over time. Boxes represent the upper to lower quartiles and the bars represent the range; the line is connected through the mean values. (B) Platelet counts over time. Boxes represent the upper to lower quartiles and the bars represent the range; the line is connected through the mean values.
Serious adverse events were reported in 32.3% of patients (n=369). Serious adverse events occurring in >1% of patients included anemia (4.5%), pneumonia (3.9%), pyrexia (3.2%), cardiac failure (1.8%), dyspnea (1.7%), abdominal pain (1.6%), gastrointestinal hemorrhage (1.4%), thrombocytopenia (1.0%), cardiac atrial fibrillation (1.0%), and general physical health deterioration (1.0%). There were no reports of progressive multifocal leukoencephalopathy.
The most common non-hematologic adverse events (occurring in ≥5% of patients) were primarily grade 1/2 (Table 2) and included diarrhea, pyrexia, fatigue, and asthenia. Rates of non-hematologic grade 3/4 adverse events were low overall (<2%), except the rate of pneumonia (3.6%), which led to drug discontinuation in six patients (0.5%) (Online Supplementary Table S3). The rates of infections were also low; all-grade infections in ≥1% of patients included nasopharyngitis (6.3%), urinary tract infection (6.0%), pneumonia (5.3%), bronchitis (4.2%), herpes zoster (3.6%), influenza (3.0%), upper respiratory tract infection (2.9%), cystitis (2.5%), gastroenteritis (1.8%), respiratory tract infection (1.8%), and oral herpes (1.6%) (Online Supplementary Table S4). Other infections included tuberculosis in three patients (0.3%) and legionella pneumonia in one patient (0.1%); no hepatitis B reactivation was reported.
Efficacy
At weeks 24 and 48, 56.9% and 62.3% of evaluable patients achieved a ≥50% reduction from baseline in palpable spleen length, respectively (Figure 3A); additionally, 23.9% and 18.2% had 25% to <50% reductions at weeks 24 and 48, respectively. At each assessment, at least two-thirds of patients had a ≥25% reduction in palpable spleen length, and approximately 80% had a ≥25% reduction after week 4. Most patients (69.0%) experienced a ≥50% reduction in spleen length at any time by week 48 (Figure 3B), and 23.0% had spleens that became non-palpable. The proportion of patients who achieved a ≥50% reduction in spleen length at any time by week 48 was similar for patients with primary or secondary MF (66.9% versus 71.6%, respectively; see the Online Supplementary Results for additional details). At both weeks 24 and 48, ≥50% reductions from baseline in palpable spleen length were achieved by a higher proportion of patients starting treatment at 20 mg bid than by those starting at 15 mg bid (week 24, 61.8% versus 47.3%; week 48, 68.9% versus 48.7%). Likewise, the proportion of patients who achieved a ≥50% reduction from baseline in palpable spleen length at any time was higher in patients who started treatment at 20 mg bid (73.8% versus 60.1%).
Figure 3.
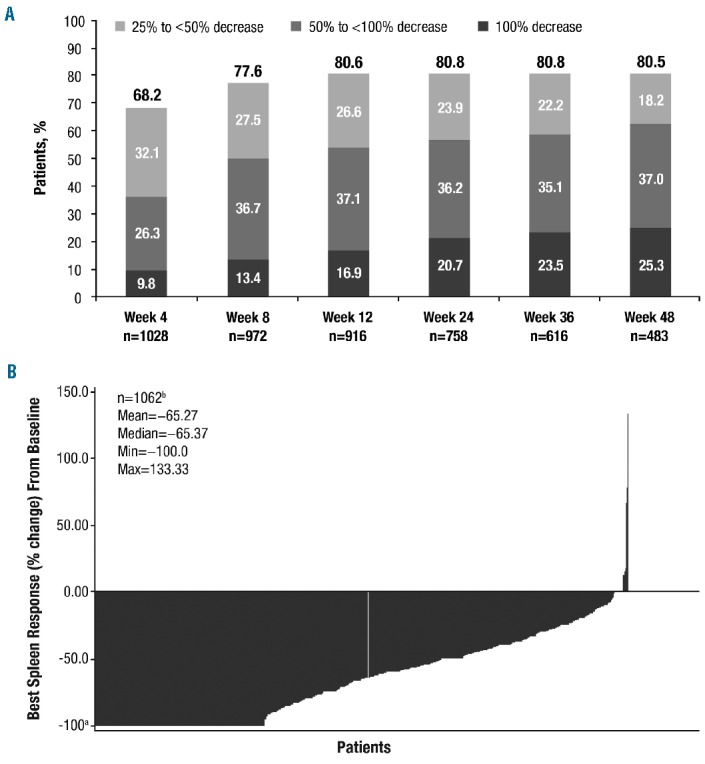
Spleen response. (A) Evaluable patients with a ≥25% decrease from baseline in palpable spleen length. (B) Best percent change from baseline in palpable spleen length at any time by week 48. Each bar represents data from an individual patient. Max: maximum; min: minimum. aNote: −100% change is defined as non-palpable. bOnly patients with spleen length assessments at baseline and at a post-baseline visit were included in this analysis.
The median time to the first ≥50% reduction in palpable spleen length was 5.1 weeks (range, 0.1–53.1 weeks). The Kaplan-Meier estimated probability of maintaining a spleen response for 24 weeks and 48 weeks was 93% (95% CI, 91%–95%) and 72% (95% CI, 54%–84%), respectively. The median duration of spleen response was not yet reached; 9.8% of patients had a loss of response (i.e., a return of spleen length to baseline size). Preliminary results suggest that patients who required concomitant erythropoiesis-stimulating agents on treatment also had clinically meaningful spleen responses: 70.1% of evaluable patients (101/144) who received erythropoiesis-stimulating agents achieved a ≥50% reduction from baseline in palpable spleen length at any time.
Clinically meaningful improvements in symptoms were seen as early as 4 weeks after the start of treatment and were maintained over time, as evaluated by the FACT-Lym TS (mean change from baseline was 11.0 at week 4 and 9.4 at week 48; Online Supplementary Figure S2A) and FACIT-Fatigue scale (mean change from baseline was 3.8 at week 4 and 3.0 at week 48; Online Supplementary Figure S2B). Approximately 44% to 46% and 46% to 52% of patients achieved a response (i.e., a minimally important difference at each time point) in the FACT-Lym TS (Figure 4A) and the FACIT-Fatigue scale (Figure 4B), respectively; symptom response rates were not affected by ruxolitinib starting dose. Response rates on the FACT-Lym TS and FACIT-Fatigue scales were similar when evaluated by MF subtype (Online Supplementary Figures S3 and S4). Similar improvements were observed in other QOL questionnaires, with the proportions of patients achieving a response ranging from 44% to 48% for the FACT-Lym subscale, 40% to 43% for the FACT-Lym Trial Outcome Index, and 39% to 42% for the FACT-General TS.
Figure 4.
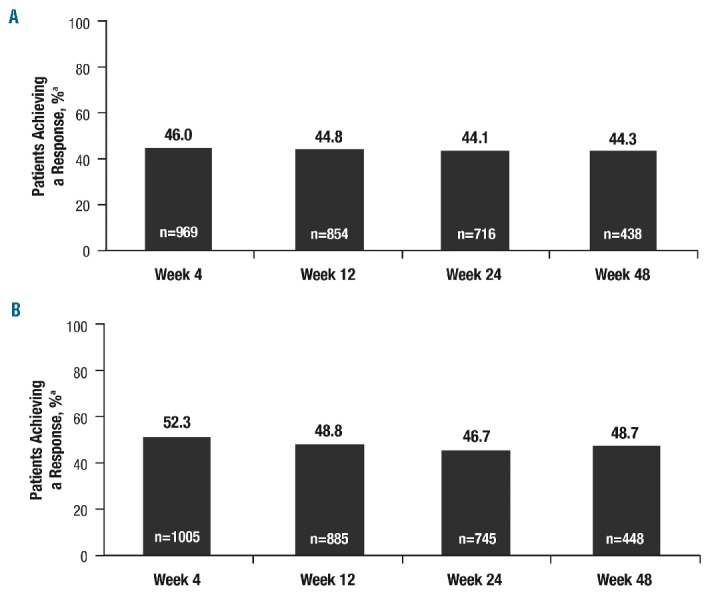
Quality of life responses. (A) Proportion of patients achieving a response in the FACT-Lymphoma total score. FACT: Functional Assessment of Cancer Therapy. aResponse was defined as the upper limit of the minimally important difference (FACT-Lym total score, 11.2 points). (B) Proportion of patients achieving a response on the FACIT-Fatigue Scale: FACIT: Functional Assessment of Chronic Illness Therapy. aResponse was defined as the upper limit of the minimally important difference (FACIT-Fatigue score, 3 points).
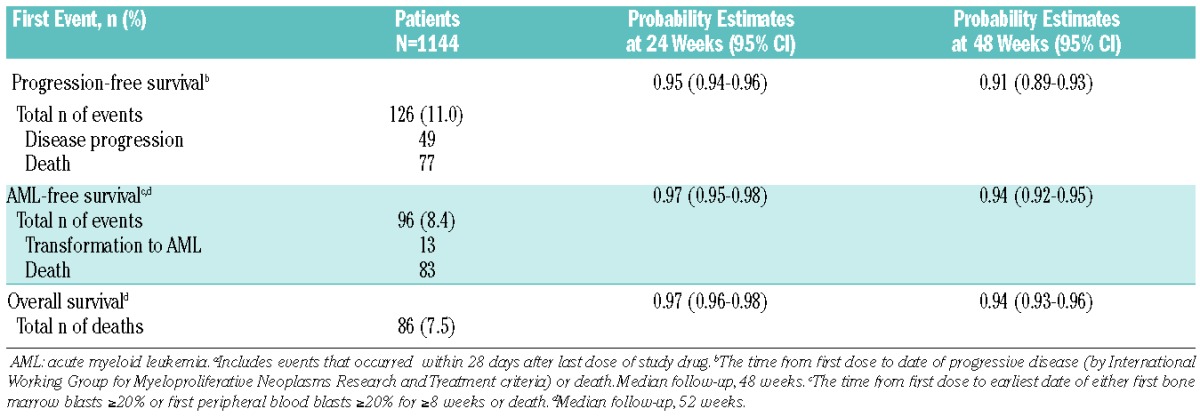
A total of 13 patients developed acute myeloid leukemia during the study or within 28 days following study discontinuation (Table 3). The estimated probability of AML-free survival at 48 weeks was 94% (95% CI, 92%–95%; Table 3). The estimated probability of progression-free survival at 48 weeks was 91% (95% CI, 89%–93%). The estimated probability of overall survival at 48 weeks was 94% (95% CI, 93%–96%) for the overall JUMP population, with 86 reported deaths (Table 3). Investigator-determined causes of death on study, regardless of causality, included myelofibrosis (n=11), cardiac failure (n=7), cardiac arrest (n=7), pneumonia (n=6), septic shock (n=5), multi-organ failure (n=4), transformation to acute myeloid leukemia (n=4), respiratory failure (n=3), sepsis (n=3), cardiorespiratory arrest (n=3), gastrointestinal hemorrhage (n=2), peritonitis (n=2), renal failure (n=1), and general health deterioration (n=2). All other causes were undetermined or reported for one patient each. The survival estimate for patients who received erythropoiesis-stimulating agents was similar to that for the overall population (99%; 95% CI, 96%–100%; deaths, n=10/159).
Table 3.
Progression-free, leukemia-free, and overall survival estimates.a
Retreatment after ruxolitinib interruption
Overall, 207 of the 1144 patients had a therapy interruption of ≥7 days before restarting treatment. Compared with the overall patient population, this subgroup had a higher proportion of patients with primary MF (68.1% versus 58.8%) and hemoglobin level <100 g/L (50.7% versus 40.3%) and a lower median platelet count at baseline (199.5×109/L versus 256×109/L). Interruptions lasted from 7 to 14 days in 41.1% of patients, 15 to 21 days in 26.6% of patients, and >21 days in 32.4% of patients. Treatment interruptions were mostly due to adverse events (92.3%). Adverse events occurring before treatment interruption were primarily grade 3/4 anemia (34.8%) or thrombocytopenia (33.8%); grade 3/4 adverse events occurring in >2% of patients included decreased platelet count (8.2%), pneumonia (4.4%), neutropenia (4.4%), leukopenia (3.4%), abdominal pain (2.9%), and increased gamma-glutamyltransferase (2.4%). Most patients (67.1%) had only one interruption. There were no reports of a withdrawal effect following treatment interruption, and, although exposure to ruxolitinib was more than three times longer after restarting treatment, the rates of adverse events before interruption and after restarting treatment were similar overall. The median duration of exposure was 1.9 months from baseline to interruption and 6.5 months after restarting treatment. The mean daily dose was 30.5 mg prior to treatment interruption and 19.4 mg between restarting treatment and the end of the follow-up.
Despite treatment interruption, 68.2% of patients (133/195) experienced a ≥50% reduction in palpable spleen length at any time. This was achieved by 77 patients (58%) before ruxolitinib interruption and 56 patients (42%) after restarting ruxolitinib. From spleen length at restarting treatment, 24% of patients achieved a further 50% reduction, 9% experienced a spleen length increase ≥50%, and 67% remained stable in between. Clinically meaningful improvements in symptoms, as assessed by the FACT-Lym TS, were observed as early as week 4 (mean change from baseline, 9.6), with a trend toward improvement at week 48 (mean change, 6.1).
Patients with intermediate-1-risk myelofibrosis
JUMP included patients classified as intermediate-1-risk, a group of patients not included in the COMFORT studies. Overall, 163 intermediate-1-risk patients who started treatment ≥1 year before data cutoff were identified; because risk status was not initially collected on study, a second, later data cutoff date (01 July 2014) was used. The median age in this cohort of patients was 62 years (range, 25–79 years), with a median time since diagnosis of 17.9 months (range, 0.2–276 months); the median palpable spleen length at baseline was 12 cm (range, 4–45 cm; Online Supplementary Table S5). Overall, 21.5% and 8.0% of patients had hemoglobin <10 g/dL and platelet counts <100×109/L at baseline, respectively.
In general, ruxolitinib was well tolerated by intermediate-1-risk patients. At the time of data cutoff, most patients had completed the study per protocol (4.3%) or remained on treatment (76.1%). Reasons for treatment discontinuation included adverse events (11%), withdrawal of consent (2.5%), disease progression (2.5%), investigator’s decision (1.8%), physician’s decision (1.8%), death (1.2%), and protocol deviation (0.6%). The majority of patients received starting doses of 20 mg bid (65.0%) or 15 mg bid (23.9%), with a median exposure of 14.4 months (range, 0.1–19.1 months) and a median daily dose of 30.0 mg/day (range, 6.3–49.4 mg/day). Overall, 64% of patients had dose modifications and 27% had a dose interruption (23% due to adverse events).
In this cohort of patients, ruxolitinib demonstrated an adverse event profile consistent with that previously reported in intermediate-2- and high-risk patients with MF. The most common hematologic adverse events were anemia (all grades, 54.0%; grade 3/4, 24.5%), thrombocytopenia (all grades, 40.5%; grade 3/4, 11.0%; Online Supplementary Table S6). Anemia and thrombocytopenia led to treatment discontinuation in one patient (0.6%) and three patients (1.8%), respectively. Similar to what was observed in the overall JUMP population, mean hemoglobin levels decreased from baseline (118.5 g/L) during the first 8 to 12 weeks and increased to near-baseline levels after week 24. Mean platelet counts decreased from baseline (309.4×109/L) during the first 4 weeks but then remained stable over time. Rates of non-hematologic grade 3/4 adverse events were low overall (<2%), with the exception of asthenia (2.5%; Online Supplementary Table S6). Adverse events leading to treatment discontinuation were reported for 11% of patients (n=18). These occurred in one patient each, with the exception of pyrexia (n=2; 1.2%). Rates of infections were also low: all-grade infections in ≥5% of patients included herpes zoster (8.0%) and bronchitis (6.1%). Grade 3/4 infections occurring in more than two patients included pneumonia and sepsis (both 1.8%). Hepatitis B was reported in one patient (0.6%; grade 3/4) and led to treatment discontinuation. No tuberculosis was reported in this cohort.
Serious adverse events were reported for 22.1% of patients. Serious adverse events occurring in >1% of patients included esophageal variceal hemorrhage and sepsis (both 1.8%) and bronchopneumonia, cardiac failure, pyrexia, pneumonia, acute renal failure, respiratory failure, syncope, and urinary tract infection (1.2% each). There were no reports of progressive multifocal leukoencephalopathy in this cohort of patients. Overall, there were three deaths among intermediate-1-risk patients. Primary causes of death included cardiac failure, sepsis, and undetermined cause (n=1 each).
Similar to patients with higher-risk MF, the majority of intermediate-1-risk patients in JUMP experienced clinically meaningful reductions in spleen size and improvements in disease-related symptoms. At weeks 24 and 48, 63.8% and 60.5% of patients achieved a ≥50% reduction from baseline in palpable spleen length, respectively. Additionally, 19.6% and 21.0% of patients had 25% to <50% reductions at weeks 24 and 48, respectively (Online Supplementary Figure S5A). At each assessment, ≥75% of patients had a ≥25% reduction from baseline in palpable spleen length. By week 72, 77.6% of patients had achieved a ≥50% reduction, including 21% (n=34) with complete resolution of splenomegaly (Online Supplementary Figure S5B). The median time to a ≥50% reduction in palpable spleen length was 4.7 weeks (range, 3.1–60.1 weeks), and the estimated probability of maintaining a response was 91% (95% CI, 83%–95%) at 48 weeks and 88% (95% CI, 79%–94%) at 60 weeks. Overall, 9.6% of patients had a loss of response. In contrast to what was observed in higher-risk patients, the proportion of patients who achieved ≥50% reductions from baseline in palpable spleen length at week 24 was similar between those starting with a ruxolitinib dose of 20 mg bid and those starting with 15 mg bid (65.2% versus 62.9%, respectively). At week 48, the proportion of patients who achieved a spleen response was slightly higher in those who started treatment at 20 mg bid (66.2% versus 55.9%). However, the proportion of patients who achieved a ≥50% reduction from baseline in palpable spleen length at any time was higher in patients who started treatment at 20 mg bid (81.9% versus 68.4%).
Clinically meaningful improvements in symptoms were seen as early as 4 weeks after the start of treatment, as evaluated per the FACT-Lym TS and FACIT-Fatigue scale (Online Supplementary Figure S6). Approximately 30% to 40% and 34% to 47% of patients achieved a response at each time point in the FACT-Lym TS and on the FACIT-Fatigue scale, respectively (Online Supplementary Figure S6). Improvements were also observed on other FACT-Lym scales, with the proportion of patients achieving a response ranging from 30% to 43% for the FACT-Lym subscale, 23% to 31% for the FACT-Lym Trial Outcome Index, and 29% to 36% for the FACT-General TS.
Discussion
The JUMP study is the most extensive study in MF and includes the largest cohort of patients treated with ruxolitinib reported to date. Compared with the COMFORT studies, the JUMP trial seems to have a slightly older population with a longer evolution of MF prior to study entry and a greater proportion of patients with primary MF. Overall, the tolerability of ruxolitinib in JUMP was similar to that observed in the COMFORT studies.9,10 Consistent with the mechanism of action of ruxolitinib, the most common adverse events were anemia and thrombocytopenia; however, these adverse events were manageable and led to treatment discontinuation in only 2.6% and 3.2% of patients, respectively. Although findings are preliminary, concomitant administration of erythropoiesis-stimulating agents was well tolerated and did not have a negative impact on the efficacy of ruxolitinib, as was seen in COMFORT-II.14 The rates of infections in JUMP were low and the infections were primarily grade 1/2; no new or unexpected infections were observed. Additionally, no cases of progressive multifocal leukoencephalopathy were reported. Herpes zoster (3.6%) and influenza (3.0%) were the most common viral infections. No hepatitis B reactivation was reported in the 1144-patient cohort, and one case was reported in the intermediate-1-risk group of patients. Prophylaxis for herpes zoster or other infections should be considered on a case-by-case basis and may vary by region and local risk. Of the 86 deaths in the overall survival analysis, 83 were recorded as deaths in the leukemia-free survival analysis; 13 patients had transformation to acute myeloid leukemia. However, the limited follow-up for survival in this study should be taken into consideration. As observed in other studies with ruxolitinib,9,10,12 the majority of patients in JUMP experienced clinically meaningful reductions in spleen size and improvements in disease-related symptoms.
Ruxolitinib was also generally safe and well tolerated and provided meaningful reductions in splenomegaly and symptoms in patients who restarted ruxolitinib after treatment interruption. Consistent with ruxolitinib’s mechanism of action, hematologic adverse events were the primary cause of treatment interruptions. In the 207 patients who restarted treatment after therapy interruption, ruxolitinib provided reductions in palpable spleen length and symptoms prior to and after treatment interruption. After restarting treatment, patients were able to stay on ruxolitinib at a median dose of ≈10 mg bid, and most patients did not require another interruption. Rates of adverse events did not increase after restarting treatment. Additionally, the discontinuation rate after restarting treatment was comparable to that observed in the overall study population. Of note, there was no evidence of a withdrawal effect after discontinuation of ruxolitinib treatment in this cohort of patients.
JUMP also includes patients with intermediate-1-risk MF, a risk group that was not included in the COMFORT studies. Importantly, the IPSS was used to determine prognostic score in this study because at the time the protocol was released in January 2011, the Dynamic IPSS had only recently been published (October 2010). As mentioned previously, the adverse event profile of ruxolitinib in intermediate-1-risk patients was consistent with that previously reported in higher-risk patients.9,10 Rates of non-hematologic adverse events were similar in both groups of patients, although those with higher-risk MF reported higher rates of fatigue (12.9% versus 5.5%). Herpes zoster reactivation was observed in both groups, with intermediate-1–risk patients reporting higher rates of reactivation (8.0% versus 3.6%). Patients with intermediate-1-risk MF achieved clinically meaningful reductions in spleen size and symptom improvement consistent with those seen in intermediate-2- and high-risk patients enrolled in this study. At week 24, slightly more patients with intermediate-1-risk MF had achieved a ≥50% reduction from baseline in palpable spleen length than had patients in the overall population (63.8% versus 56.9%, respectively); the rates were similar at week 48 (60.5% versus 62.3%). Additionally, a similar proportion of patients in each cohort achieved a ≥50% reduction from baseline in palpable spleen length at any time (overall population, 69%; intermediate-1-risk patients, 77.6%). The median time to a spleen response was also similar (4.7 versus 5.1 weeks). Likewise, the proportion of patients who achieved clinically meaningful improvements in symptoms (≈30% to 40%) was within the expected range and consistent with that seen in the overall JUMP population (≈45% to 50%). These findings support those observed in the UK ROBUST study12 as well as those from real-world clinical evidence of ruxolitinib use in patients with lower-risk MF20 and indicate that ruxolitinib is an effective treatment for patients with intermediate-1-risk disease.
Overall, findings from the JUMP study confirm the efficacy and safety of ruxolitinib in the treatment of patients with MF. Ruxolitinib provided clinically meaningful reductions in spleen size and symptoms, including for those patients with intermediate-1-risk disease, with a safety and efficacy profile consistent with that observed in the phase 3 COMFORT studies. Furthermore, JUMP is a global study conducted in a setting that resembles routine clinical practice. Findings from this study will help guide clinicians in the management of their patients with MF and may help shape the current treatment paradigm, ultimately maximizing the benefits that patients can derive from treatment.
Acknowledgments
We thank Catherine Bouard and Raj Vadde, employees of Novartis, for their substantial contributions to the JUMP study. Editorial assistance was provided by Karen Chinchilla, PhD, and was funded by Novartis.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/9/1065
References
- 1.Mesa RA, Verstovsek S, Cervantes F, et al. Primary myelofibrosis (PMF), post polycythemia vera myelofibrosis (post-PV MF), post essential thrombocythemia myelofibrosis (post-ET MF), blast phase PMF (PMF-BP): consensus on terminology by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT). Leuk Res. 2007;31(6):737–740. [DOI] [PubMed] [Google Scholar]
- 2.Swerdlow SH, Campo E, Harrison NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. Vol 2 4th ed. Geneva, Switzerland: World Health Organization; 2008. [Google Scholar]
- 3.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. [DOI] [PubMed] [Google Scholar]
- 4.Tefferi A. Primary myelofibrosis: 2013 update on diagnosis, risk-stratification, and management. Am J Hematol. 2013;88(2): 141–150. [DOI] [PubMed] [Google Scholar]
- 5.Mesa RA, Niblack J, Wadleigh M, et al. The burden of fatigue and quality of life in myeloproliferative disorders (MPDs): an international Internet-based survey of 1179 MPD patients. Cancer. 2007;109(1):68–76. [DOI] [PubMed] [Google Scholar]
- 6.Abdel-Wahab OI, Levine RL. Primary myelofibrosis: update on definition, pathogenesis, and treatment. Annu Rev Med. 2009;60:233–245. [DOI] [PubMed] [Google Scholar]
- 7.Mesa RA, Schwager S, Radia D, et al. The Myelofibrosis Symptom Assessment Form (MFSAF): an evidence-based brief inventory to measure quality of life and symptomatic response to treatment in myelofibrosis. Leuk Res. 2009;33(9):1199–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cervantes F, Dupriez B, Pereira A, et al. New prognostic scoring system for primary myelofibrosis based on a study of the International Working Group for Myelofibrosis Research and Treatment. Blood. 2009;113(13):2895–2901. [DOI] [PubMed] [Google Scholar]
- 9.Verstovsek S, Mesa RA, Gotlib J, et al. A double-blind, placebo-controlled trial of ruxolitinib for myelofibrosis. N Engl J Med. 2012;366(9):799–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison C, Kiladjian JJ, Al-Ali HK, et al. JAK inhibition with ruxolitinib versus best available therapy for myelofibrosis. N Engl J Med. 2012;366(9):787–798. [DOI] [PubMed] [Google Scholar]
- 11.Cervantes F, Kiladjian JJ, Niederwieser D, et al. Long-term efficacy, safety, and survival findings from COMFORT-II, a phase 3 study comparing ruxolitinib with best available therapy for the treatment of myelofibrosis. Blood. 2012;120(21):801. [DOI] [PubMed] [Google Scholar]
- 12.Mead AJ, Milojkovic D, Knapper S, et al. Response to ruxolitinib in patients with intermediate-1-, intermediate-2-, and high-risk myelofibrosis: results of the UK ROBUST trial. Br J Haematol. 2015; 170(1):29–39. [DOI] [PubMed] [Google Scholar]
- 13.Barosi G, Mesa RA, Thiele J, et al. Proposed criteria for the diagnosis of post-polycythemia vera and post-essential thrombocythemia myelofibrosis: a consensus statement from the International Working Group for Myelofibrosis Research and Treatment. Leukemia. 2008;22(2):437–438. [DOI] [PubMed] [Google Scholar]
- 14.McMullin MF, Harrison CN, Niederwieser D, et al. The use of erythropoiesis-stimulating agents with ruxolitinib in patients with myelofibrosis in COMFORT-II: an open-label, phase 3 study assessing efficacy and safety of ruxolitinib versus best available therapy in the treatment of myelofibrosis. Exp Hematol Oncol. 2015;4:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tefferi A, Barosi G, Mesa RA, et al. International Working Group (IWG) consensus criteria for treatment response in myelofibrosis with myeloid metaplasia, for the IWG for Myelofibrosis Research and Treatment (IWG-MRT). Blood. 2006;108(5): 1497–1503. [DOI] [PubMed] [Google Scholar]
- 16.Cella DF, Tulsky DS, Gray G, et al. The Functional Assessment of Cancer Therapy scale: development and validation of the general measure. J Clin Oncol. 1993;11(3): 570–579. [DOI] [PubMed] [Google Scholar]
- 17.Webster K, Cella D, Yost K. The Functional Assessment of Chronic Illness Therapy (FACIT) Measurement System: properties, applications, and interpretation. Health Qual Life Outcomes. 2003;1:79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Carter GC, Liepa AM, Zimmermann AH, Morschhauser F. Validation of the Functional Assessment of Cancer Therapy-Lymphoma (FACT-LYM) in patients with relapsed/refractory mantle cell lymphoma. Blood 2008;112(11):2376 [Google Scholar]
- 19.Cella D, Eton DT, Lai JS, Peterman AH, Merkel DE. Combining anchor and distribution-based methods to derive minimal clinically important differences on the Functional Assessment of Cancer Therapy (FACT) anemia and fatigue scales. J Pain Symptom Manage. 2002;24(6):547–561. [DOI] [PubMed] [Google Scholar]
- 20.Davis KL, Cote I, Kaye JA, et al. Real-world assessment of clinical outcomes in patients with lower-risk myelofibrosis receiving treatment with ruxolitinib. Adv Hematol. 2015;2015;848473. [DOI] [PMC free article] [PubMed] [Google Scholar]