

Abstract
Classical Hodgkin lymphoma is one of the most common lymphomas and shares clinical and genetic features with primary mediastinal B-cell lymphoma. In this retrospective study, we analyzed the recurrent hotspot mutation of the exportin 1 (XPO1, p.E571K) gene, previously identified in primary mediastinal B-cell lymphoma, in biopsies and plasma circulating cell-free DNA from patients with classical Hodgkin lymphoma using a highly sensitive digital PCR technique. A total of 94 patients were included in the present study. This widely expressed XPO1 E571K mutation is present in one quarter of classical Hodgkin lymphoma patients (24.2%). Mutated and wild-type classical Hodgkin lymphomas were similar regarding the main clinical features. Patients with a detectable XPO1 mutation at the end of treatment displayed a tendency toward shorter progression-free survival, as compared to patients with undetectable mutation in plasma cell-free DNA (2-year progression-free survival: 57.1%, 95% confidence interval: 30.1–100% versus 2-year progression-free survival: 90.5%, 95% confidence interval: 78.8–100%, respectively, P=0.0601). To conclude, the detection of the XPO1 E571K mutation in biopsy and plasma cell-free DNA by digital PCR may be used as a novel biomarker in classical Hodgkin lymphoma for both diagnosis and minimal residual disease, and pinpoints a crucial role of XPO1 in classical Hodgkin lymphoma pathogenesis. The detection of somatic mutation in the plasma cell-free DNA of patients represents a major technological advance in the context of liquid biopsies and noninvasive management of classical Hodgkin lymphoma.
Introduction
Hodgkin lymphoma (HL) is one of the most frequent lymphomas in the world, accounting for 10% of newly diagnosed lymphomas and 1% of all cancers, with an incidence of about 3 cases per 100,000 people per year in western countries.1 HL has been classified into classical HL (cHL), which accounts for 95% of all cases, and the rare nodular lymphocyte-predominant HL, which is considered to be a distinct entity.2 The majority of cHL cases are diagnosed in 20–30 year old patients, with a second smaller peak in adults older than 55 years of age.3 cHL is further subdivided into four subtypes: nodular sclerosis (60% of cases), mixed cellularity (30% of cases), lymphocyte-depleted and lymphocyte-rich HL.4 cHL patients have greatly benefited from multi-agent chemotherapy and improved radiation techniques, and 65–90% of patients can achieve disease-free survival after five years, depending on stage and clinical risk factors.5 Those with a rapid response to initial treatment have the best outcomes and may benefit from truncated, less-toxic treatment regimens.6 Nevertheless, approximately 20–25% of patients will ultimately experience either primary refractoriness to chemotherapy (within 3 months of doxorubicin-based chemotherapy), early disease relapse (within 12 months after the end of first-line treatment) or late disease relapse,7 underlying the need to understand the mechanisms involved and to identify predictive biomarkers.
The singularity of cHL is that tumor cells, designated as Hodgkin and Reed-Sternberg cells (HRS cells), usually account for only about 0.1–2% of cells in the tissue.8 The scarcity of HRS cells, embedded in an extensive inflammatory infiltrate, hampers their molecular analysis and thus the genomic landscape of cHL remains largely unknown. Constitutive activation of the NF-κB pathway in HRS cells has been demonstrated by targeted analyses using laser capture microdissection (LCM)9,10 or cell sorting by flow cytometry.11 Genomic gains of REL, encoding an NF-κB factor, are present in about 30% of cases,12,13 and β-2-microglobulin (B2M) is frequently mutated in cHL, which is strongly associated with the nodular sclerosis subtype and better overall survival.11
We recently detected an unexpected recurrent point mutation of XPO1 (exportin 1, also known as CRM1: chromosome region maintenance 1) located in exon 15 (c.1711G>A), leading to the Glu571Lys (p.E571K) missense substitution in relapsed/refractory (R/R) primary mediastinal large B-cell lymphoma (PMBL) patients included in the LYSA LNH03 trial program.14,15 PMBL is well known to share several genetic features with cHL, including mutations in SOCS1,16 STAT617 and PTPN.16 XPO1 is a member of the importin-β superfamily of nuclear export receptors (also termed karyopherins) that mediates the translocation of numerous RNAs and cellular regulatory proteins, including tumor suppressor proteins (TSPs) such as p53, BRCA1, Survivin, NPM, APC and FOXO. The exportin 1 hydrophobic groove binds to the leucine-rich nuclear export signal (NES) domain of its cargo proteins. Importantly, selective inhibitors of nuclear export (SINE), a new class of small molecule inhibitors, have been shown to effectively target XPO1 and retain TSPs in the nucleus, and are currently being evaluated in phase 1 and 2 clinical trials for various cancer types.18,19 This XPO1 E571K mutation has been detected in approximately 25% of PMBL cases but at a lower frequency (1/10) in sorted HRS cells.11 Because of the scarcity of HRS cells in biopsies of patients with cHL, finding recurrent mutations could be easier in the plasma cell-free DNA (cfDNA) of these patients, with potentially fewer heterogeneity issues than tumor tissue testing.20 Furthermore, the concept of “liquid biopsy” was recently highlighted in a series of diffuse large B-cell lymphoma (DLBCL) patients, for whom high-throughput sequencing of a panel of target genes was performed, with the successful detection of somatic variants both in the tumor and in the plasma.21 Tumor circulating cfDNA has also been detected in the plasma of cHL patients.22 Notably, Oki et al.23 successfully detected tumor-specific immunoglobulin gene segments in blood, indicating that the principle of liquid biopsy may also be relevant for cHL patients. We recently designed a highly sensitive and specific probe-based digital polymerase chain reaction (dPCR) assay for the detection of XPO1 E571K somatic mutations in plasma cfDNA24 that could be used as a tool to detect minimal residual disease (MRD).
In this study, we investigated the prevalence and clinical relevance of XPO1 E571K in cHL cases and demonstrated that this recurrent mutation was detectable in both tumor and plasma, indicating that XPO1 mutations represent a new genetic biomarker, useful at the time of diagnosis or as a MRD marker.
Methods
Patients
We retrospectively considered adult patients treated for cHL at the Henri Becquerel Center (Rouen, France) between 2009 and 2015 using available frozen tumor DNA and formalin-fixed, paraffin-embedded (FFPE) samples. According to these inclusion criteria, 94 patients were included in the present analysis (FFPE samples: n=13; frozen tumor samples, n=81). Among these 94 patients, 50 were previously included in a biological monocentric prospective trial that aimed to assess the kinetics of cytokines (N° RCB 2009-A01117–50). These 50 patients (“cytokines” cohort) were included in this trial between 2010 and 2012 and had serial EDTA plasma samples, obtained from blood collection (Online Supplementary Methods) concomitant with the diagnostic biopsy and at the end of chemotherapy/radiotherapy treatment. All experiments were performed in accordance with the Declaration of Helsinki and the study was approved by our internal review board (N° 1601B). All patients gave informed consent for specimen collection, clinical data collection and biomarker analysis.
dPCR experiments
A chip-based digital PCR (dPCR) platform (QuantStudio3D® Digital PCR system; Thermo Fisher Scientific, Carlsbad, CA, USA) was used for mutation detection. Mutation analysis with dPCR was based on a 5′-exonuclease assay using TaqMan® MGB probes targeting the XPO1 E571K mutation. dPCR was performed on DNA from tumor tissue and circulating cfDNA from plasma specimens at diagnosis and at the end of treatment. The sequences of our designed Custom TaqMan® MGB probes and primers are listed in the Online Supplementary Table S1.
To consolidate the results obtained using this dPCR platform, we also analyzed all samples using a distinct droplet-based dPCR (ddPCRTM) platform (Qx200® droplet digital PCR system, Bio-rad laboratories, Hercules, CA, USA) using the same XPO1 dPCR assay, according to the manufacturer’s instructions. PCR cycling conditions and reagent compositions are described in the Online Supplementary Table S2. To assess the specificity of our dPCR assay with the ddPCR platform, allele burden was measured in twenty preamplified cfDNA samples from DLBCL control blood samples previously found negative for the mutation by next-generation sequencing (NGS). In this study, 0.1% was considered to be the relevant threshold25 to discriminate positive versus negative samples for XPO1 E571K by dPCR (Online Supplementary Table S3).
Ion torrent personal genome machine™ (PGM) sequencing
In the aim to validate results obtained by dPCR technologies, NGS experiments were performed using an Ion Torrent Personal Genome Machine™ (PGM, Thermo Fisher Scientific, Carlsbad, CA, USA). Libraries were prepared from 10 ng of genomic DNA with primers from our previously reported Lymphopanel21,26 targeting the XPO1 E571K nucleotide variant (Online Supplementary Methods).
Response evaluation and PET-CT
Responses after treatment were determined according to the International Workshop Group Response (Cheson) criteria.27 PET results were reported using the Deauville 5-point scale (Online Supplementary Methods).
Statistical analysis
The relationships between clinical and molecular parameters were assessed using non-parametric tests: Mann-Whitney U test, Kendall’s tau correlation coefficient or the associated test or Fisher’s exact test when appropriate. Results of the comparative tests were considered statistically significant if P<0.05. All statistical analyses were performed using R software v3.0.2.
Results
Mutations of XPO1 in cHL at the time of initial diagnosis
The main clinical features of the patients are summarized in Table 1. Patients were considered mutated if they had an XPO1 E571K mutation in their biopsy extracted DNA. The mutation was found at a frequency of 24.2% in biopsy extracted DNA by dPCR (Table 1). In this series of 94 cHL patients, the main clinical features at the time of diagnosis, including age, sex ratio, stage and mediastinal involvement were similar between mutated and wild-type cases (Table 1). The concordance of the XPO1 E571K mutation dPCR results within the 50 biopsy/plasma DNA pairs was highly significant (P=0.0179 Fisher’s exact test, Online Supplementary Table S4). PGM targeted sequencing analysis, carried out on the still available 48 DNA samples extracted from the “cytokines” cohort’s biopsies, confirmed dPCR results in 40/42 (NI=6) cHL (P<0.0001, Fisher’s exact test, Online Supplementary Table S5 and S6) and VAF were strongly correlated (Kendall’s tau coefficient=0.751, P=0.000193, Online Supplementary Figure S1). Digital PCR generates more copies (analysis points) of the sequence of interest than PGM (mean 16000 points by dPCR versus mean 4800 reads by PGM, data not shown), so the accuracy of the VAF might be lower by PGM than by digital PCR. The cases of discrepancies between PGM and digital PCR results involved FFPE DNA samples whose amplification by PGM is more difficult, with a lower sequencing depth. The XPO1 mutation was also investigated by dPCR in DNA from peripheral blood mononuclear cells (PBMC) of 4 mutated patients, and was not found in those samples (n=0/4, data not shown).
Table 1.
Comparison of XPO1 WT and MT clinical characteristics.
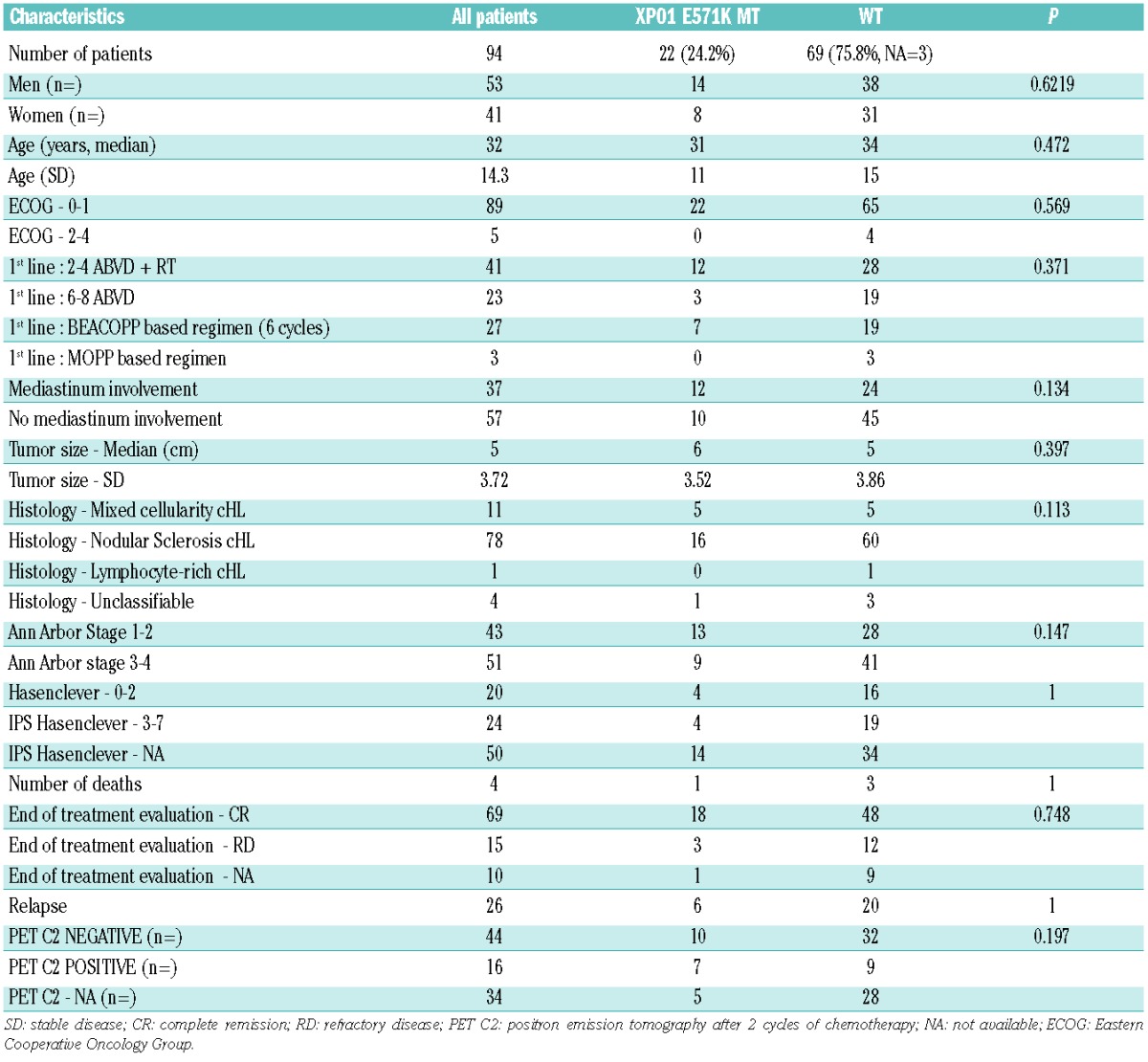
The E571K XPO1 mutation VAF in biopsy extracted DNA ranged from 0.13% to 4.78% (median 0.435%). The VAF in plasmatic cfDNA ranged from 0.13% to 14.74% (median 0.24%, Online Supplementary Table S7). The correlation between cfDNA VAF and tumor DNA VAF was low but significant (Kendall’s tau=0.462, P=0.0387, Figure 1). This highlights the fact that, although HRS cells in the tumor are very sparse, the mutation frequencies seem to be correctly represented in the plasma.
Figure 1.
Correlation between XPO1 E571K VAF in biopsies and cfDNA.
There was a trend toward higher cfDNA concentration (ng/mL of plasma) in advanced stage disease compared to localized disease (P=0.0894, Online Supplementary Figure S2) but no correlation between cfDNA concentration and XPO1 mutation VAF (P=0.735, Online Supplementary Figure S3). XPO1 mutations were found in both mixed cellularity and nodular sclerosis subtypes, indicating that the mutation is not a feature of a specific histological subtype. No XPO1 mutations were detected in the only lymphocyte-rich cHL case of the cohort (Table 1).
Of note, 5 patients displayed the XPO1 mutation exclusively in the tumor DNA biopsy (Online Supplementary Table S4); 3 of these 5 patients had a localized stage II disease with a small tumor size (4–6 cm diameter), whereas the remaining 2 patients had advanced stage disease (1 stage III, 1 stage IV). Furthermore, 8 patients had an XPO1 mutation detectable only in the cfDNA. These results illustrate that considering only tumor biopsy DNA may lead to an underestimation of the mutation prevalence, and suggest that joint analysis of cfDNA and biopsy extracted DNA is more suitable to detect XPO1 mutations in cHL.
Cell lines analysis
We also analyzed 6 HL-derived cell lines by dPCR and PGM: KM-H2 (DSMZ: ACC 8), HDLM-2 (ACC 17), L-540 (ACC 72), L-428 (ACC 197), L-1236 (ACC 530), and U-H01 (ACC 626), and we found the XPO1 E571K mutation in the L-1236 and U-H01 cell lines, with VAF of 18.5% and 85%, respectively (Online Supplementary Figure S4). The karyotype of the L-1236 and U-H01 cell lines revealed amplification of the 2p15 - 2p16 region where XPO1 is located (chr2:61703069–61767418:2p15 region). Our data are in favor of duplication of the mutated XPO1 allele in the U-H01 cell line and of the wild-type allele in the L-1236 cell line. We did not find any other XPO1 variants within exons 15 to 18.
Response rate and survival analysis
The complete remission rate at the end of treatment (85.7% versus 80%, P=0.537) was similar in MT and WT patients. There was no statistically different rate of positivity of interim PET-CT after two cycles (C2) of chemotherapy (PET-2) in MT patients as compared with WT patients (41.2% versus 21.95% P=0.197). Concerning survival analysis, with a median follow-up of 34.5 months, overall survival (OS) and progression-free survival (PFS) were similar between MT and WT patients with a 2-year OS and PFS probability of 95% [CI 95%: 85.9–100%] and 82.2% [CI 95%:67.8–99.7%], respectively, in MT patients, and of 98.1% [CI 95%: 94.5–100%] and 64.8% [CI 95%: 52.9–79.5%], respectively, in WT patients (P=0.637 and 0.248 for OS and PFS, respectively, Online Supplementary Figure S5). Of note, PET-2-positive patients had a shorter 2y-PFS, as compared to PET-2-negative patients (2y-PFS=62.5%, CI 95%:42.8–91.4% versus 2y-PFS=86.1%, CI 95 %:75.5–98.2%, respectively, P=0.00159, Online Supplementary Figure S6).
cfDNA as a biomarker for MRD
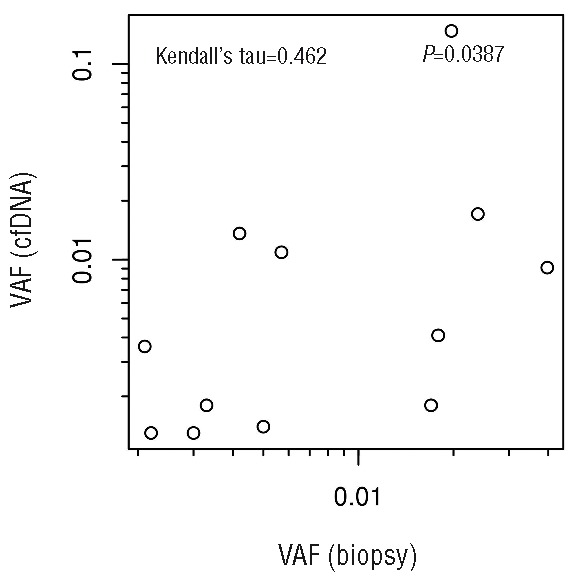
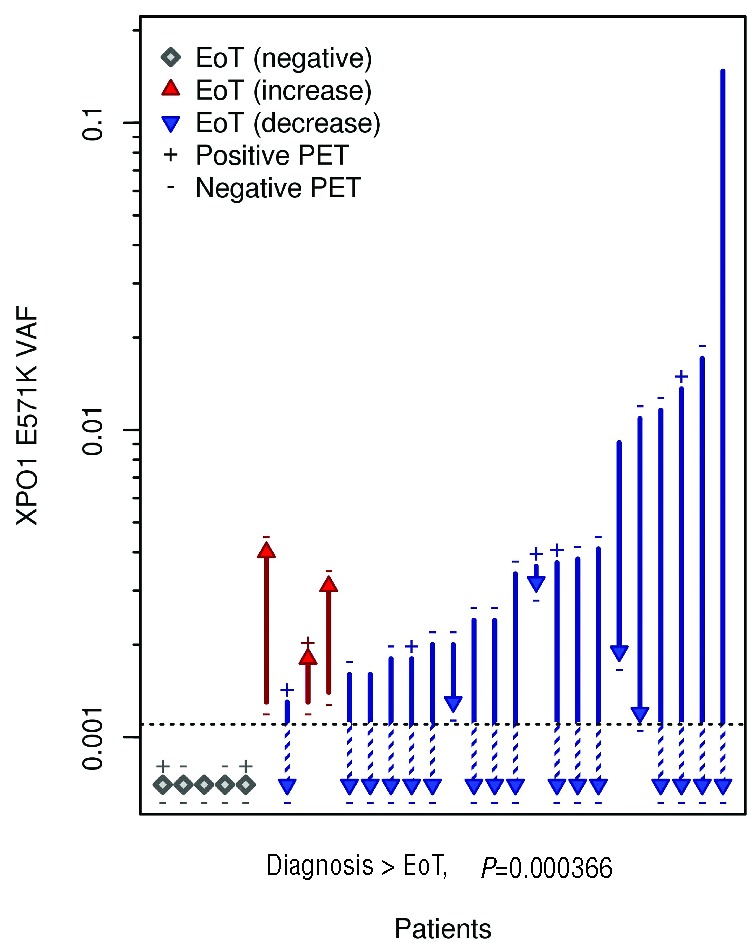
At the end of treatment plasma cfDNA was analyzed for 28 patients; the remaining 66 patients had either no available plasma sample at the end of treatment (n=46), or no XPO1 mutation at diagnosis in either the plasma or the biopsy (n=20). Figure 2 schematically shows the variation of the XPO1 E571K mutation VAF in the plasma cfDNA of the 28 evaluable patients between diagnosis and completion of treatment, with PET-CT at C2 and the end of treatment. For the majority of patients (n = 20), the cfDNA VAF decreased between diagnosis and the completion of treatment. The mutation became undetectable (using the threshold of 0.1%) in plasma for 16 patients. Five patients had plasma cfDNA with undetectable XPO1 E571K mutation at diagnosis and at the end of treatment (“negative” patients, Figure 2), but were mutated in biopsy extracted DNA. Patients with a detectable XPO1 mutation at the end of treatment displayed a trend toward shorter 2y-PFS, as compared to patients with undetectable mutation in plasma cell-free DNA (2y-PFS=57.1%, CI 95%:30.1–100% versus 2y-PFS=90.5%, CI 95%:78.8–100%, respectively, P=0.0601, Figure 3). Furthermore, patients with increasing cfDNA VAF at the end of treatment (n=3/28) seem to display shorter OS and PFS, but the sample size is too low to conclude this with certainty (2y-PFS=33.3%, CI 95%:6.7–100% versus 2y-PFS=90%, CI 95%:77.8–100%, respectively, P=0.0439, data not shown). Of note, in 2/3 cases, PET was negative at the time of the detection of an MRD increase. In the full cohort of 94 patients, 26 patients experienced relapse during follow-up. Of the 7 patients with a positive plasma at the end of treatment, 4 (57%) relapsed during follow-up (Figure 3), which suggests that the assessment of plasma might be more sensitive than PET because only 1 patient had a positive PET at the end of treatment.
Figure 2.
Plasmatic cfDNA XPO1 E571K mutation VAF at diagnosis and after treatment completion (after 6–8 cycles of chemotherapy for advanced stage disease, or after 3–4 cycles of chemotherapy and radiotherapy for localized stage disease), with interim and the end of treatment (EoT) PET-CT results. The interim PET at C2 is represented at the base of the arrow and the end of treatment PET is represented at the tip of the arrow. The arrows show the variations of the XPO1 mutation VAF in the plasma cfDNA between diagnosis and the end of treatment. The dotted line represents the detection limit (0.001).
Figure 3.
OS and PFS according to the XPO1 E571K cfDNA results at the end of treatment (EoT). Negative: patients with undetectable XPO1 mutation, Positive: patients with detectable XPO1 E571K mutation in cfDNA at EoT.
Finally, 1 patient (patient N°42) had an XPO1 E571K mutation at diagnosis in both biopsy and plasma samples as well as in cfDNA at the end of treatment (VAF 4.04%, 0.91% and 0.19%, respectively, Online Supplementary Figure S7). The mutation was also detected in plasma cfDNA 3 months after the end of treatment (VAF 0.12%) and in the relapse biopsy 10 months after the end of first-line ABVD chemotherapy (VAF relapse 2%, Figure 4). Unfortunately, we were unable to extract plasma cfDNA at relapse for this patient.
Figure 4.
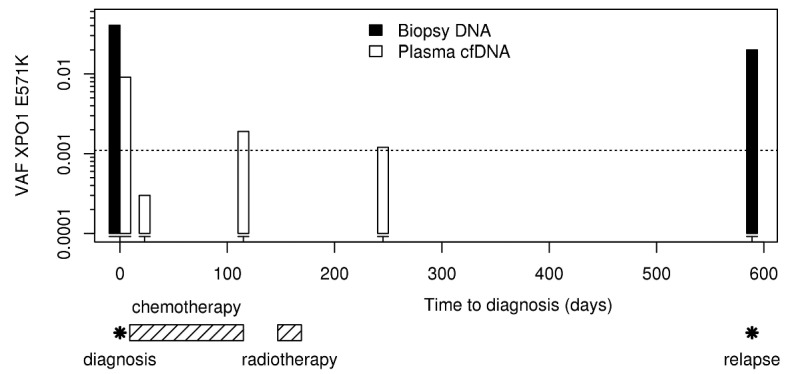
Monitoring XPO1 mutations in biopsy and cfDNA of a refractory cHL patient (Patient N° 42) throughout her clinical history. The dotted line represents the detection limit (0.001).
Discussion
Herein we describe for the first time the existence of a widely present XPO1 E571K somatic mutation in a large cohort of patients with cHL, as identified by dPCR and confirmed by NGS experiments. This observation is novel and could add new information on driver events and tumorigenesis in this disease. In total, 24.2% of our patients with cHL harbored the XPO1 E571K mutation. It is remarkable that 29% of all XPO1 mutations were found only in the plasma but not in the tumor because of the well-known tumor cell sparsity in HL. This indicates that cfDNA assessment might play an important role to define somatic mutations in this disease at the time of diagnosis. The detection of this mutation in the plasma cfDNA of patients represents a major technological progress in the onset of liquid biopsies in Hodgkin lymphoma.
There is a pathological overlap between PMBL and, specifically, cHL of the nodular sclerosis subtype; whether both might derive from thymic B cells is currently an area of discussion.28 The fact that the XPO1 E571K mutation is so enriched in both of these subtypes reinforces the hypothesis of a common origin and a strong oncogenic role of this gene. Missense substitutions targeting XPO1 have also previously been reported at a low frequency (<5%) in chronic lymphocytic leukemia (CLL) and esophageal squamous cell carcinoma (ESCC), suggesting that these XPO1 mutations may also play a role in several oncogenic processes.29–31 Importantly, alternative XPO1 variants, including those located at the hotspot,14,15,32,33 were not assessed by our dPCR approaches and we may have underestimated the rate of XPO1 mutations in cHL. Nevertheless, we did not detect those alternative XPO1 variants by NGS on cHL biopsies. To date, the role of XPO1 and the impact of the highly selected E571K mutation in the pathogenesis of cHL remain totally unknown. Several proteins known to play a major role in cHL oncogenesis, including STAT1/STAT6, FOXO1 or CIITA are also identified as cargo proteins.34–36 Whether the XPO1 mutations may interfere with the nucleus/cytoplasm shuttle of these proteins remains to be determined.
Genomic analyses of HL have been hampered until recently by the scarcity of HRS cells that are tremendously thinned out (to 1%) by a very large number of robust, nonmalignant infiltrating inflammatory cells that include macrophages, reactive lymphocytes, plasma cells and fibroblasts.4 Reactive infiltrating cells, especially T cells, usually make up at least 99% of the cells in the tumor mass,37 which may explain the low XPO1 E571K VAF observed in our series of cHL patients. We cannot affirm that the mutation in XPO1 is indeed present in HRS cells and not in some bystander cells, but the presence of the mutation in two Hodgkin cell lines and its absence in PBMC are in favor of the presence of this XPO1 mutation in HRS tumor cells specifically. The XPO1 E571K mutation in the L-1236 cell line was first detected by whole-exome sequencing (WES).38
In this particular disease, highly sensitive techniques like dPCR and targeted NGS are essential to highlight low frequency mutations. High-throughput techniques such as low-coverage whole genome sequencing,39 sequencing of circulating cfDNA22 or targeted exome sequencing of isolated HRS cells11 have recently helped to investigate genetic lesions underlying cHL, but sample sizes were very low. WES-based experiments of sorted Reed-Sternberg (RS) cells11,40,41 recently identified new point mutations in cHL cases, including several mutations already reported in PMBL, such as CIITA, SOCS1, STAT6 and B2M mutations, as well as one case harboring the E571K XPO1 mutation, but this study was restricted to 10 cases,11 with an insufficient median depth of sequencing (48X), which is potentially limiting for the discovery of genomic alterations.
A recent NGS study on circulating cfDNA of 9 nodular sclerosis HL cases showed genomic imbalances in HRS cells that can be identified at diagnosis with rapid normalization of circulating cfDNA profiles upon therapy initiation, suggesting a potential role for circulating cfDNA profiling in early response monitoring.22 Although cHL is curable even in advanced stages, up to 20% of patients will not be cured or achieve remission with initial therapy,7 and overall more than 25% of cHL patients will ultimately relapse, warranting the identification of a specific and trackable biomarker. In the present study, we performed highly sensitive analysis of a large cohort of 94 cHL patients and showed a recurrent XPO1 E571 somatic point mutation that could be considered as a new biomarker in approximately one quarter of cHL patients. Nevertheless, the applicability as a widely used biomarker would be low since three quarters of the patients do not have a XPO1 E571K mutation and of the positive 25%, an additional 26% are negative for XPO1 mutations in the plasma.
In line with this aspect we have identified a trend toward unfavorable prognostic impact in terms of PFS in patients with detectable XPO1 E571K mutation in plasma cfDNA at the end of treatment, which could prove to be statistically significant in a larger cohort. We observed that 57% of the patients who ultimately relapsed were positive for XPO1 mutations in the plasma after the end of therapy. In fact, XPO1 assessment seems to be more sensitive than PET-CT since only 1 patient had a positive PET at the end of treatment.
These results suggest that the clearance of the XPO1 mutation in plasma cfDNA may represent a new prognostic marker for mutated patients. A study in a larger prospective cohort is warranted and already planned to draw definitive conclusions. In accordance with this observation, it has been reported that pretreatment levels of plasma Epstein-Barr virus (EBV) DNA, as determined by quantitative real-time polymerase chain reaction (QRT-PCR), are associated with inferior outcomes among a large cohort of patients with previously untreated, advanced-stage Hodgkin lymphoma.42 Another interesting potential biomarker is the combination of serum CD163 and tumor-specific TARC proteins that predicted disease response in a small cohort of 47 patients with HL.43 These results still require validation, and the usefulness of monitoring the MRD by XPO1 somatic mutation detection in cfDNA, compared with other potential biomarkers, has to be assessed.
One of the limitations of our study, in addition to its retrospective nature with possible selection bias, is the absence of plasma available at the time of diagnosis for 44 patients which potentially led to underestimating the prevalence of the mutation in these patients, for whom only biopsies were analyzed. The few discrepancies between the detection of the mutation in the plasma and the tumor can be explained partly by the poor quality of some of our biopsies with tumor cell scarcity, potentially rendering the mutation detection in biopsy extracted DNA impossible, and also by the absence of tumor DNA release in plasma by certain tumors and the short half-life (10–15 minutes) of circulating DNA in plasma.44 Although additional XPO1 mutations were found in the plasma compared to the tumor, the sensitivity of the test remains low, with 26% of XPO1 mutations missed in the plasma at diagnosis. Another possibility for the lack of detection of this mutation in our cHL cases is the potential subclonality of the XPO1 E571K mutation. This point warrants further research. In addition, it has been previously described that plasma cfDNA concentrations do not uniformly correlate with disease stage in solid tumors and biological mechanisms that underlined tumor cfDNA amount release are incompletely understood.45,46 The results presented here are limited to patients with histologically confirmed cHL and may not be generalizable to HIV-associated HL, nodular lymphocyte-predominant HL or HL developing in the post-transplant setting. Finally, selective inhibitors of nuclear export (SINE), have been shown to effectively target XPO1 by retaining TSPs in the nucleus.47 Given the frequency of XPO1 E571 mutations in cHL, suggesting a crucial role of XPO1 in cHL pathophysiology, further investigation concerning the impact of XPO1 E571 mutations in response to SINE is currently being investigated.48
To conclude, we identify herein a widely spread XPO1 E571K mutation that is present in about one quarter of cHL patients. The presence of the XPO1 E571K mutation in plasma cfDNA may serve as a novel biomarker in cHL. These data have to be confirmed in a large dedicated prospective study, and it remains to be established whether this mutation adds new relevant value as compared to PET-scans and whether it can be targeted by SINE compounds for the treatment of cHL.
Acknowledgments
The authors would like to thank all the patients who provided samples for the study.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/9/1094
References
- 1.Diehl V, Thomas RK, Re D. Part II: Hodgkin’s lymphoma–diagnosis and treatment. Lancet Oncol. 2004;5(1):19–26. [DOI] [PubMed] [Google Scholar]
- 2.Campo E, Swerdlow SH, Harris NL, et al. The 2008 WHO classification of lymphoid neoplasms and beyond: evolving concepts and practical applications. Blood. 2011; 117(19):5019–032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2015. CA Cancer J Clin. 2015;65(1):5–29. [DOI] [PubMed] [Google Scholar]
- 4.Swerdlow SH, Campo E, Harris NL, et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues, Fourth Edition. Lyon: IARC 2008. [Google Scholar]
- 5.Hasenclever D, Diehl V. A prognostic score for advanced Hodgkin’s disease. International Prognostic Factors Project on Advanced Hodgkin’s Disease. N Engl J Med. 1998;339(21):1506–1514. [DOI] [PubMed] [Google Scholar]
- 6.Hutchings M, Loft A, Hansen M, et al. FDG-PET after two cycles of chemotherapy predicts treatment failure and progression-free survival in Hodgkin lymphoma. Blood. 2006;107(1):52–59. [DOI] [PubMed] [Google Scholar]
- 7.Canellos GP, Rosenberg SA, Friedberg JW, Lister TA, Devita VT. Treatment of Hodgkin lymphoma: a 50-year perspective. J Clin Oncol. 2014;32(3):163–168. [DOI] [PubMed] [Google Scholar]
- 8.Schmitz R, Stanelle J, Hansmann M-L, Küppers R. Pathogenesis of classical and lymphocyte-predominant Hodgkin lymphoma. Annu Rev Pathol. 2009;4:151–714. [DOI] [PubMed] [Google Scholar]
- 9.Slovak ML, Bedell V, Hsu Y-H, et al. Molecular karyotypes of Hodgkin and Reed-Sternberg cells at disease onset reveal distinct copy number alterations in chemosensitive versus refractory Hodgkin lymphoma. Clin Cancer Res. 2011; 17(10):3443–3454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schumacher MA, Schmitz R, Brune V, et al. Mutations in the genes coding for the NF- B regulating factors I B and A20 are uncommon in nodular lymphocyte-predominant Hodgkin’s lymphoma. Haematologica. 2010;95(1):153–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Reichel J, Chadburn A, Rubinstein PG, et al. Flow sorting and exome sequencing reveal the oncogenome of primary Hodgkin and Reed-Sternberg cells. Blood. 2015; 125(7):1061–1072. [DOI] [PubMed] [Google Scholar]
- 12.Martín-Subero JI, Gesk S, Harder L, et al. Recurrent involvement of the REL and BCL11A loci in classical Hodgkin lymphoma. Blood. 2002;99(4):1474–1477. [DOI] [PubMed] [Google Scholar]
- 13.Joos S, Menz CK, Wrobel G, et al. Classical Hodgkin lymphoma is characterized by recurrent copy number gains of the short arm of chromosome 2. Blood. 2002; 99(4):1381–1387. [DOI] [PubMed] [Google Scholar]
- 14.Mareschal S, Dubois S, Viailly P-J, et al. Whole exome sequencing of relapsed/refractory patients expands the repertoire of somatic mutations in diffuse large B-cell lymphoma. Genes Chromosomes Cancer. 2015; 55(3):251–267. [DOI] [PubMed] [Google Scholar]
- 15.Dubois S, Viailly PJ, Mareschal S, et al. Next Generation Sequencing in Diffuse Large B Cell Lymphoma Highlights Molecular Divergence and Therapeutic Opportunities: a LYSA Study. Clin Cancer Res. 2016. January 27 [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 16.Steidl C, Gascoyne RD. The molecular pathogenesis of primary mediastinal large B-cell lymphoma. Blood. 2011; 118(10):2659–2669. [DOI] [PubMed] [Google Scholar]
- 17.Ritz O, Guiter C, Castellano F, et al. Recurrent mutations of the STAT6 DNA binding domain in primary mediastinal B-cell lymphoma. Blood. 2009; 114(6):1236–1242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Etchin J, Sanda T, Mansour MR, et al. KPT-330 inhibitor of CRM1 (XPO1)-mediated nuclear export has selective anti-leukaemic activity in preclinical models of T-cell acute lymphoblastic leukaemia and acute myeloid leukaemia. Br J Haematol. 2013; 161(1):117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012; 120(23): 4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Diaz LA, Bardelli A. Liquid biopsies: genotyping circulating tumor DNA. J Clin Oncol. 2014;32(6):579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bohers E, Viailly PJ, Dubois S, et al. Somatic mutations of cell-free circulating DNA detected by Next Generation Sequencing reflect the genetic changes in both Germinal Center B-Cell like and Activated B-Cell like Diffuse Large B-Cell Lymphoma tumors at the time of diagnosis. Haematologica. 2015;100(7):280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vandenberghe P, Wlodarska I, Tousseyn T, et al. Non-invasive detection of genomic imbalances in Hodgkin/Reed-Sternberg cells in early and advanced stage Hodgkin’s lymphoma by sequencing of circulating cell-free DNA: a technical proof-of-principle study. Lancet Haematol. 2015; 2(2):e55–65. [DOI] [PubMed] [Google Scholar]
- 23.Oki Y, Neelapu SS, Fanale M, et al. Detection of classical Hodgkin lymphoma specific sequence in peripheral blood using a next-generation sequencing approach. Br J Haematol. 2015;169(5):689–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Camus V, Sarafan-Vasseur N, Bohers E, et al. Digital PCR for quantification of recurrent and potentially actionable somatic mutations in circulating free DNA from patients with diffuse large B-cell lymphoma. Leuk Lymphoma. 2016;1–9. [DOI] [PubMed] [Google Scholar]
- 25.Armbruster DA, Pry T. Limit of Blank, Limit of Detection and Limit of Quantitation. Clin Biochem Rev. 2008; 29(Suppl 1):S49–52. [PMC free article] [PubMed] [Google Scholar]
- 26.Dubois S, Mareschal S, Picquenot J-M, et al. Immunohistochemical and genomic profiles of diffuse large B-cell lymphomas: Implications for targeted EZH2 inhibitor therapy? Oncotarget. 2015;6(18):16712–16724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheson BD, Pfistner B, Juweid ME, et al. Revised response criteria for malignant lymphoma. J Clin Oncol. 2007; 25(5):579–586. [DOI] [PubMed] [Google Scholar]
- 28.Traverse-Glehen A, Pittaluga S, Gaulard P, et al. Mediastinal gray zone lymphoma: the missing link between classic Hodgkin’s lymphoma and mediastinal large B-cell lymphoma. Am J Surg Pathol. 2005; 29(11): 1411–21. [DOI] [PubMed] [Google Scholar]
- 29.Lin D-C, Hao J-J, Nagata Y, et al. Genomic and molecular characterization of esophageal squamous cell carcinoma. Nat Genet. 2014;46(5):467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jeromin S, Weissmann S, Haferlach C, et al. SF3B1 mutations correlated to cytogenetics and mutations in NOTCH1, FBXW7, MYD88, XPO1 and TP53 in 1160 untreated CLL patients. Leukemia. 2014;28(1):108–117. [DOI] [PubMed] [Google Scholar]
- 31.Bond J, Bergon A, Durand A, et al. Cryptic XPO1-MLLT10 translocation is associated with HOXA locus deregulation in T-ALL. Blood. 2014;124(19):3023–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guieze R, Robbe P, Clifford R, et al. Presence of multiple recurrent mutations confers poor trial outcome of relapsed/refractory CLL. Blood. 2015;126(18):2110–2117. [DOI] [PubMed] [Google Scholar]
- 33.Puente XS, Pinyol M, Quesada V, et al. Whole-genome sequencing identifies recurrent mutations in chronic lymphocytic leukaemia. Nature. 2011;475(7354):101–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chiu E, Gold T, Fettig V, LeVasseur MT, Cressman DE. Identification of a Nuclear Export Sequence in the MHC CIITA. J Immunol. 2015;194(12):6102–6111. [DOI] [PubMed] [Google Scholar]
- 35.Xie L, Ushmorov A, Leithauser F, et al. FOXO1 is a tumor suppressor in classical Hodgkin lymphoma. Blood. 2012;119(15): 3503–3511. [DOI] [PubMed] [Google Scholar]
- 36.Hao Y, Chapuy B, Monti S, et al. Selective JAK2 inhibition specifically decreases Hodgkin lymphoma and mediastinal large B-cell lymphoma growth in vitro and in vivo. Clin Cancer Res. 2014; 20(10):2674–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Küppers R, Engert A, Hansmann M-L. Hodgkin lymphoma. J Clin Invest. 2012; 122(10):3439–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Liu Y, Abdul Razak FR, Terpstra M, et al. The mutational landscape of Hodgkin lymphoma cell lines determined by whole-exome sequencing. Leukemia. 2014;28(11): 2248–2251. [DOI] [PubMed] [Google Scholar]
- 39.Salipante SJ, Adey A, Thomas A, et al. Recurrent somatic loss of TNFRSF14 in classical Hodgkin lymphoma. Genes Chromosomes Cancer. 2015; 55(3):278–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Weniger MA, Melzner I, Menz CK, et al. Mutations of the tumor suppressor gene SOCS-1 in classical Hodgkin lymphoma are frequent and associated with nuclear phospho-STAT5 accumulation. Oncogene. 2006;25(18):2679–2684. [DOI] [PubMed] [Google Scholar]
- 41.Ritz O, Rommel K, Dorsch K, et al. STAT6-mediated BCL6 repression in primary mediastinal B-cell lymphoma (PMBL). Oncotarget. 2013;4(7):1093–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kanakry JA, Li H, Gellert LL, et al. Plasma Epstein-Barr virus DNA predicts outcome in advanced Hodgkin lymphoma: correlative analysis from a large North American cooperative group trial. Blood. 2013;121(18): 3547–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jones K, Vari F, Keane C, et al. Serum CD163 and TARC as disease response biomarkers in classical Hodgkin lymphoma. Clin Cancer Res. 2013;19(3):731–742. [DOI] [PubMed] [Google Scholar]
- 44.Elshimali Y, Khaddour H, Sarkissyan M, Wu Y, Vadgama J. The Clinical Utilization of Circulating Cell Free DNA (CCFDNA) in Blood of Cancer Patients. Int J Mol Sci. 2013;14(9):18925–18958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Beau-Faller M, Gaub MP, Schneider A, et al. Plasma DNA microsatellite panel as sensitive and tumor-specific marker in lung cancer patients. Int J Cancer J Int Cancer. 2003; 105(3):361–370. [DOI] [PubMed] [Google Scholar]
- 46.Szpechcinski A, Chorostowska-Wynimko J, Struniawski R, et al. Cell-free DNA levels in plasma of patients with non-small-cell lung cancer and inflammatory lung disease. Br J Cancer. 2015;113(3):476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lapalombella R, Sun Q, Williams K, et al. Selective inhibitors of nuclear export show that CRM1/XPO1 is a target in chronic lymphocytic leukemia. Blood. 2012;120(23): 4621–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jardin F, Pujals A, Pelletier L, et al. Recurrent Mutations of the Exportin 1 Gene (XPO1) in Primary Mediastinal B-Cell Lymphoma: a LYSA Study. Blood. 2015;126(23):129.26160186 [Google Scholar]