

Abstract
Clinical tools to guide in the appropriate treatment selection in immunoglobulin light chain (AL) amyloidosis are not well developed. We evaluated the response and outcome for various regimens at first-line treatment (n=681) and first progression (n=240) stratified by the immunoparesis status at diagnosis. Immunoparesis was assessed by the average relative difference of the uninvolved immunoglobulins, classifying patients into a negative average relative difference (i.e. significant immunoparesis) or a positive average relative difference (no/modest immunoparesis). Treatment was categorized as autologous stem cell transplant and four non-transplant regimens (melphalan-based; bortezomib-based, immunomodulatory drug-based and dexamethasone alone). Patients with significant immunoparesis who underwent stem cell transplant had a significantly lower rate of very good partial response or better response (58%), progression-free survival (median 30 months) and overall survival (108 months), compared to those without significant immunoparesis (80%, 127 months, median not reached, respectively; P<0.001 for all comparisons). Among the non-transplant regimens, melphalan resulted in an unfavorable progression-free survival (11 vs. 27 months; P<0.001) and overall survival (30 vs. 74 months; P=0.001) in patients with significant immunoparesis compared to those without significant immunoparesis. In contrast, no significant difference in outcomes between the immunoparesis groups was seen for those treated with bortezomib or immunomodulatory drugs. At first progression, immunoparesis status did not impact response or survival of any regimen. Melphalan at first-line provided poorer outcomes for patients with significant immunoparesis, while bortezomib or immunomodulatory drugs were more likely to overcome the adverse prognosis associated with significant immunoparesis.
Introduction
AL amyloidosis is a disorder with considerable therapeutic challenges. The disease can produce profound organ dysfunction which may limit the intensity of the delivered treatment, and therefore treatment efficacy is impaired. Autologous stem cell transplantation (ASCT) is considered the treatment of choice, when applicable, given its superior long-term survival.1,2 However, for non-ASCT eligible patients, therapy selection based on baseline disease characteristics has not been well-developed. The lack of guidelines for treatment selection at diagnosis may lead to the selection of inappropriate treatment that would lead to irreversible organ damage and a shorter survival.
Recently, we have reported that significant immunoparesis is an adverse prognostic factor in newly diagnosed AL amyloidosis.3 We proposed that immunoparesis assessment can be carried out by two methods. First, qualitatively, considering the number of uninvolved immunoglobulins below their lower limit of normal (LLN) and second, quantitatively, in which the average relative difference (ARD) of the uninvolved immunoglobulins from their respective lower limits is assessed. It was found that patients with significant immunoparesis (represented by either a reduction of all the uninvolved immunoglobulins below the LLN or by a negative ARD value) had a higher rate of treatment failure and reduced progression-free survival (PFS) and overall survival (OS) compared to patients with no or only moderate immunoparesis. The quantitative assessment was, however, shown to have a better discriminate power for survival. In this study we explored the results of selected treatment regimens at induction and first progression and analyzed these outcomes based on the ARD status at diagnosis.
Methods
Six hundred and eighty-one patients (n=681) with systemic AL amyloidosis seen at the Mayo Clinic (Rochester, MN, USA) within 90 days of diagnosis between January 1, 2005 and August 31, 2015 were included. Data were extracted from a prospectively maintained database. The median follow-up of the surviving patients is 53 months (range 3–135 months). All patients gave written informed consent to have their medical records reviewed. The Mayo Clinic Foundation Institutional Review Board (IRB) approved this study.
Patients were excluded if they had prior treatment, amyloidosis associated with a lymphoproliferative disorder, an incidental positive bone marrow and/or fat aspirate without an amyloid-specific syndrome or a localized disease. Three hundred and seventeen patients who met inclusion criteria but lacked response evaluation data were not included in this study for the following reasons: no treatment/less than one cycle of treatment (n=58), lack of sufficient laboratory data for response evaluation, and/or early death rendering them inevaluable for response (n=259).
The diagnosis of AL amyloidosis was based on a tissue specimen positive for Congo red staining and with green birefringence under polarized light, followed by typing with immunohistochemistry, immunofluorescence, or mass spectrometry. All patients had immunoglobulin measurements before treatment. For immunoparesis assessment we used a quantitative measure, which utilizes the ARD of the uninvolved immunoglobulins. ARD was calculated as the mean value of the relative difference of the uninvolved immunoglobulins from their lower limit of normal (See the Online Supplementary File for a sample calculation). Based on this method, patients were stratified to those with a negative ARD value (i.e. significant immunoparesis) and those with a positive ARD value (i.e. no immunoparesis or modest immunoparesis only).
Treatment regimens at induction and first progression were grouped into autologous stem cell transplant (ASCT) and non-transplant regimens. The latter category includes the following regimen categories: melphalan-based regimen, bortezomib-based regimen (which includes 4 patients at first progression treated with other proteasome inhibitors) immunomodulatory drugs (IMiD)-based regimen (thalidomide, lenalidomide or pomalidomide), and dexamethasone alone. Eligibility criteria for ASCT at our center have been previously described.4
Two-hundred and ninety four patients (43% of the study population) progressed during follow-up. Of these, 51 patients (17%) progressed but were not treated and 3 additional patients had treatment of an unknown type. Therefore, 240 patients are evaluable for response and survival at first progression. Details of the specific regimens used at first-line of treatment and at first progression can be viewed in the Online Supplementary Material. For assignment of ARD group at first progression, we applied the baseline ARD groups at diagnosis, which represents an intrinsic feature of the disease not influenced by treatment.
The Pearson χ2 test and the Kruskal-Wallis test were used to ascertain differences between nominal and continuous variables, respectively. Evaluation of response, PFS and OS were performed in accordance with consensus criteria,5 the Kaplan-Meier method was used to measure survival analysis. Univariate Cox proportional regression analysis was used to examine the relationship between ARD groups, regimen types and survival outcome. P values less than 0.05 were considered significant. All statistical analyses were performed on JMP software (SAS, Cary, NC, USA).
Results
Baseline characteristics
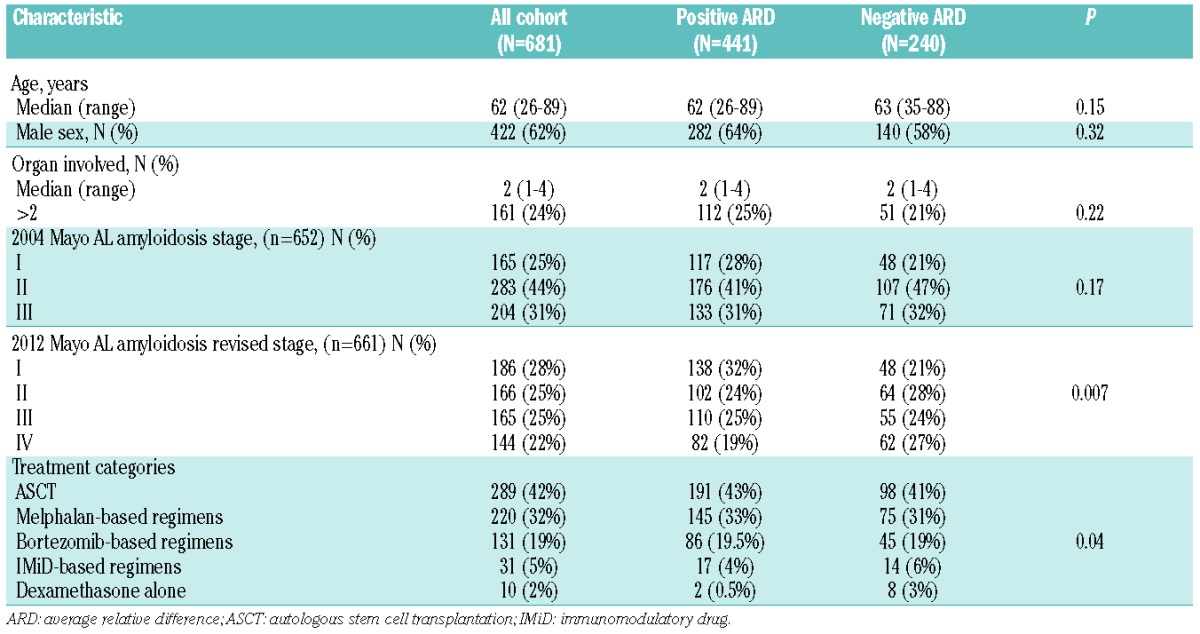
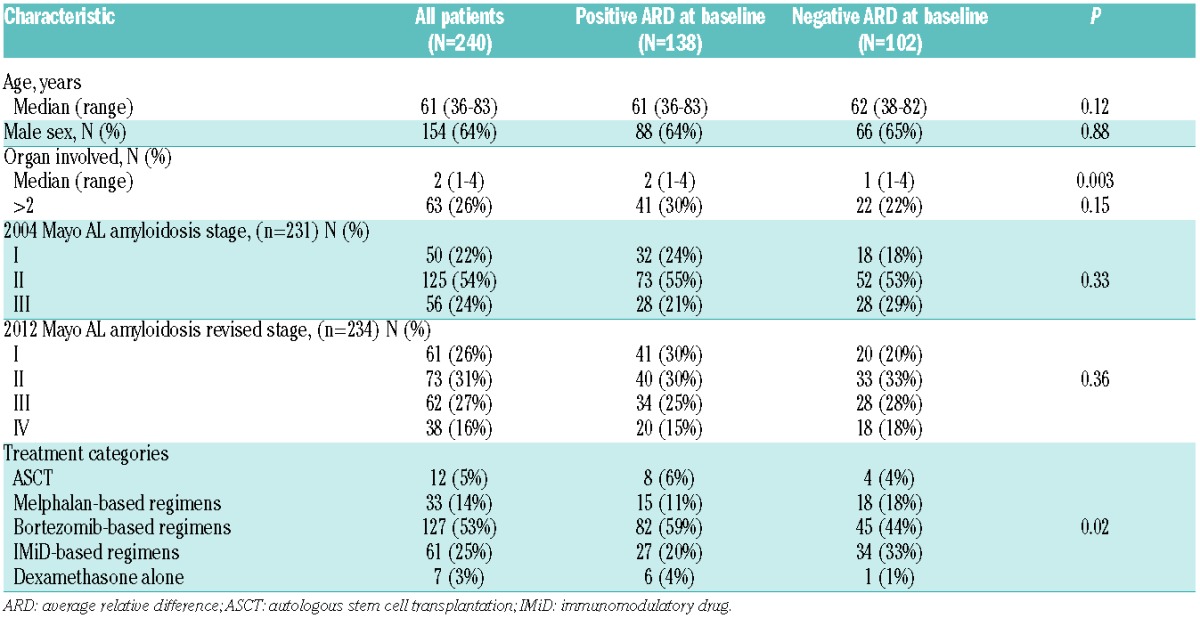
The median age of the 681 patients was 62 years [range 26–89]; 62% of patients were male. The median number of organs involved was 2 (range 1–4). The 2004 Mayo stage6 and the 2012 revised Mayo stage7 are listed in Table 1.
Table 1.
Baseline characteristics of all patients and by immunoparesis status at diagnosis.
First line treatment
Induction treatment categories are listed in Table 1. A melphalan-based regimen was given to 32% of patients, bortezomib-based regimens to 19% of patients, an IMiD-based regimen to 5% of patients and dexamethasone alone for 2% of patients. Autologous stem cell transplant (ASCT) was performed in 32% of patients without prior induction, while an additional 10% of patients proceeded to ASCT following induction treatment making a total of 42% undergoing ASCT as a first-line treatment. There was no difference in treatment categories distribution, including ASCT, melphalan-based, bortezomib-based and IMiD-based regimens between immunoparesis groups (P=0.66), but not dexamethasone alone. Patients who underwent ASCT were younger (median age 59), with fewer organs involved (median one organ) and lower 2004 Mayo stage III (15%) and 2012 revised stages III–IV (25%), compared to patients treated with non-transplant regimens (median age 65; median involved organs 2; 2004 Mayo stage III 44%; 2012 revised stage III–IV 64%; all comparisons P<0.001). In patients who received non-transplant regimens, baseline characteristics were balanced between regimens in both immunoparesis groups (data not shown).
Hematological response to first-line treatment by immunoparesis status
The rate of very good partial response (VGPR) or better (≥VGPR) was 48% in patients with a negative ARD compared to 70% in patients with a positive ARD (P<0.001). While the rate of VGPR was similar between groups (25% and 26%, respectively), fewer patients in the negative ARD group reached a complete response (CR) compared to those with a positive ARD (23% vs. 44%). The rates of partial response and no response were 30% and 22% vs. 19% and 11%, respectively.
The rate of VGPR or better response by treatment categories can be seen in Figure 1. When comparing the ≥VGPR rate for each regimen between patients with a negative ARD to those with a positive ARD, the difference between groups was seen in those receiving ASCT (58% vs. 80%; P<0.001), a melphalan-based regimen (37% vs. 57%; P=0.006) and a bortezomib-based regimen (47% vs. 76%; P=0.001). However, no difference between groups was found in the IMiD-based regimen category (57% vs. 53%; P=0.81) or dexamethasone alone (25% vs. 50%; P=0.5)
Figure 1.
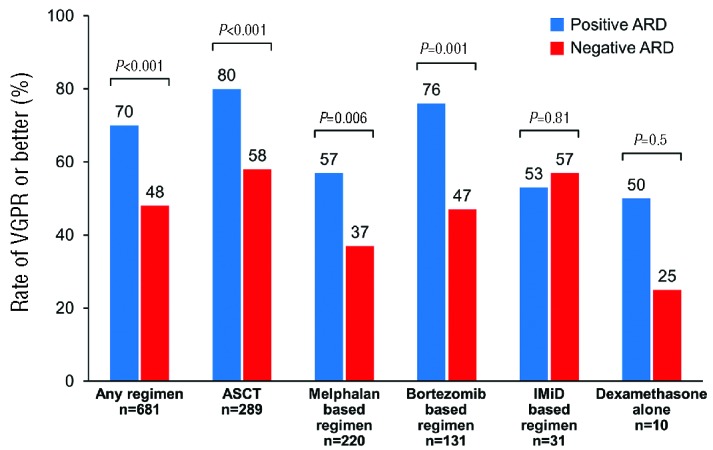
Rate of very good partial response (VGPR) or better by regimen type and immunoparesis status at first-line treatment. ASCT: autologous stem cell transplant; ARD: average relative difference; IMiD: immunomodulatory drug.
Organ response by immunoparesis status
Organ response, (i.e. response in at least one involved organ) was seen in 57% of patients, and was significantly lower in patients with a negative ARD compared to those with a positive ARD (48% vs. 62%, respectively; P<0.001) (Figure 2). No difference in the achievement of organ response between the two immunoparesis groups was seen in those treated with ASCT (66% vs. 75%; P=0.12), a bortezomib-based regimen (48% vs. 56%; P=0.41), an IMiD-based regimen (54% vs. 43%; P=0.56) and dexamethasone alone (14% vs. 0%; P=0.59). However, for patients treated with a melphalan-based regimen, those with a negative ARD were less likely to achieve an organ response compared to those with a positive ARD (28% vs. 52%; P<0.001).
Figure 2.
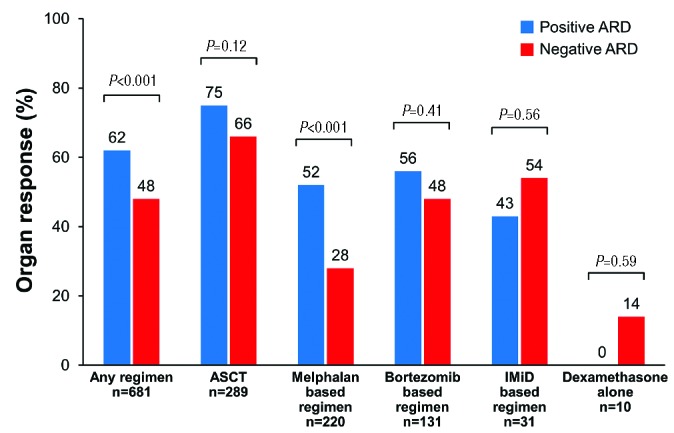
Rate of organ response by regimen type and immunoparesis status at first-line treatment. ASCT: autologous stem cell transplant; ARD: average relative difference; IMiD: immunomodulatory drug.
Effect of immunoparesis status on progression-free survival and overall survival by regimen type
Patients with a negative ARD, indicating significant immunoparesis, had a shorter PFS (median 16 months) compared to patients with a positive ARD [49 months, P<0.001; Hazard ratio (HR) 1.9, (95% confidence interval (CI) 1.5–2.3)]. The comparison of PFS between immunoparesis groups based on the given regimen demonstrated a significant difference in favor of the positive ARD group and was maintained in those undergoing ASCT [30 vs. 127 months, respectively, P<0.001; HR 2.3 (95% CI 1.6–3.2)] and for those treated with a melphalan-based regimen [11 vs. 27 months, P<0.001; HR 1.9 (95% CI 1.4–2.6)]. In contrast, no significant difference in PFS was noted between the negative and positive ARD groups in those treated with a bortezomib-based regimen [14 vs. 23 months, P=0.13; HR 1.4 (95% CI 0.9–2.3)], an IMiD-based regimen [20 vs. 13 months, P=0.84; HR 0.9 (95% CI 0.4–2)] or dexamethasone alone [5 vs. 6 months, P=0.66; HR 0.7 (95% CI 0.1–5)] (Figure 3).
Figure 3.
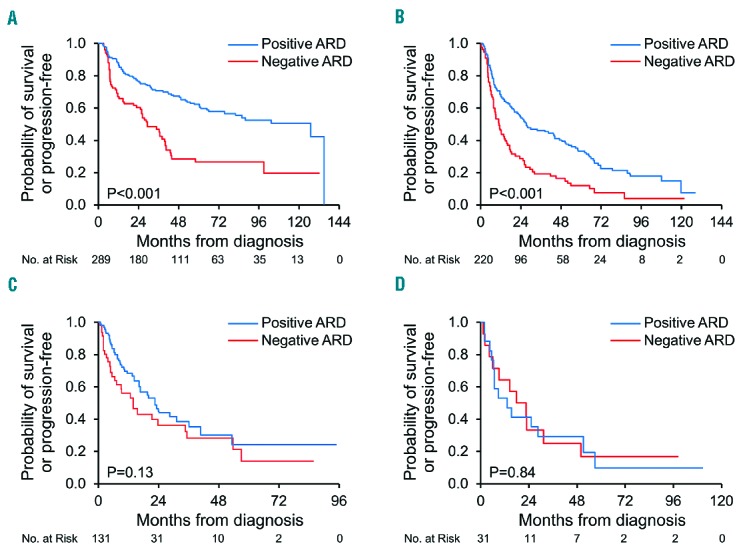
Progression-free survival by regimen type and immunoparesis status at first-line treatment. ARD: average relative difference A. Autologous stem cell transplantation (ASCT) B. Melphalan-based regimen C. Bortezomib-based regimen D. IMiD-based regimen.
Patients with a negative ARD had a shorter OS (median 66 months) compared to patients with a positive ARD [median 127 months, P<0.001; HR 1.6, (95% CI 1.3–2)]. In patients who underwent ASCT, inferior OS was seen in patients with a negative ARD (median 108 months) compared to patients with a positive ARD [median not reached, P=0.01; HR 1.9, (95% CI 1.1–3)]. Similarly, patients with a negative ARD treated with a melphalan-based regimen had an inferior OS compared to patients with a positive ARD treated with the same regimen type [30 vs. 74 months, P=0.001; HR 1.7, (95% CI 1.2–2.5)]. However, no significant difference in OS between immunoparesis groups was observed for a bortezomib-based regimen [57 vs. 41 months, P=0.61; HR 1.2, (95% CI 0.6–2.0)], an IMiD-based regimen [32 vs. 16 months, P=0.4; HR 0.7, (95% CI 0.3–1.6)] or dexamethasone alone [20 months vs. median not reached, P=0.57; HR 1.8, (95% CI 0.3–34)] (Figure 4). Of note, patients with a positive ARD treated with an IMiD-based regimen had a significantly shorter OS compared to other non-transplant regimens in this group (P=0.01).
Figure 4.
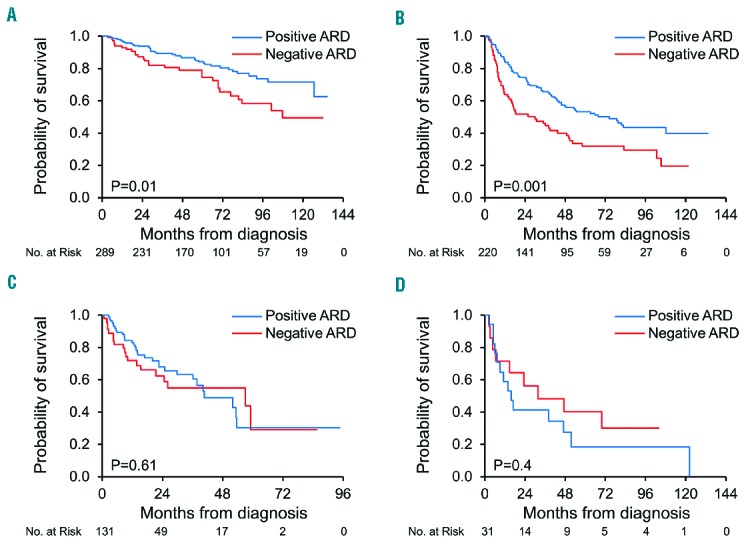
Overall survival by regimen type and immunoparesis status at first-line treatment. ARD: average relative difference. A. Autologous stem cell transplantation (ASCT) B. Melphalan-based regimen C. Bortezomib-based regimen D. IMiD-based regimen.
Treatment at first progression
The progression rate in patients with a negative ARD was 53% compared to 39% in patients with a positive ARD (P<0.001). The median time to first progression was 14 months, shorter in patients with a negative ARD (12 months) compared to patients with a positive ARD (17 months; P=0.01). The characteristics of patients at first progression are listed in Table 2. The most common regimen used at first progression was bortezomib-based (53%), followed by IMiD-based (25%), melphalan-based (14%), ASCT (5%) and dexamethasone alone (3%). A different treatment distribution was noted at first progression by baseline immunoparesis status. Patients with a baseline negative ARD were more likely to receive a melphalan–based (18%) or an IMiD-based regimen (33%) compared to patients with a positive ARD (11% and 20%, respectively). In contrast, patients with a positive ARD were more likely to receive a bortezomib-based regimen (59%) compared to those with a negative ARD (44%). ASCT was used at a similar rate between groups (4% in the negative ARD groups and 6% in the positive ARD group) (P for all comparisons=0.02). Moreover, patients receiving a melphalan-based regimen at first progression were less likely to receive ASCT at first-line (30%) compared to other non-transplant regimens at first progression (49%; P=0.04), with no difference between ARD groups.
Table 2.
Characteristics of patients at first progression by immunoparesis status at diagnosis.
Response at first progression
The rate of VGPR or better at first progression did not differ between the immunoparesis groups. Fifty-seven percent of patients with a baseline negative ARD achieved ≥VGPR compared to 56% in patients with a positive baseline ARD (P=0.97). No difference was seen in the depth of response between groups (CR 19% vs. 15%; VGPR 23% vs. 28%, respectively; P=0.32). Organ response also did not differ between negative and positive ARD groups (42% vs. 40%; P=0.73).
The comparison of the rate of ≥VGPR by regimens between patients with a negative ARD to those with a positive ARD can be viewed in Figure 5. ASCT at first progression was utilized in 12 patients, and yielded the highest VGPR or better response rate (100% in patients with a negative ARD and 86% in patients with a positive ARD; P=0.46). As for the non-transplant regimens, no difference in the rate of ≥VGPR between the negative and positive ARD groups was noted for any regimen (melphalan-based regimen 29% vs. 43%, P=0.43; bortezomib-based 74% vs. 60%, P=0.14; IMiD-based 50% vs. 50%, P=1.0; dexamethasone alone 0% vs. 33%, P=0.39).
Figure 5.
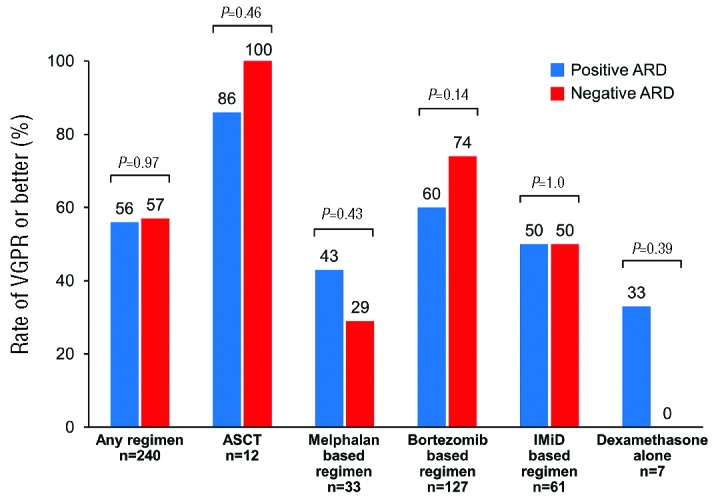
Rate of ≥VGPR or better by regimen type and immunoparesis status at first progression. ASCT: autologous stem cell transplant; ARD: average relative difference; IMiD: immunomodulatory drug.
Survival from first progression
PFS from first progression was comparable between those with a negative ARD and those with a positive ARD (median 17 vs. 20 months, respectively, P=0.95; HR 1 95% CI 0.7–1.4) (Figure 6A). No significant difference in PFS was seen between the negative and positive ARD groups for any regimen at first progression [ASCT median not reached in both, P=0.23; HR not estimable); melphalan-based median 8 months vs. 13 months, P=0.36; HR 1.5 (95% CI 0.6–3.5); bortezomib-based 27 vs. 22 months, P=0.64; HR 0.9 (95% CI 0.5–1.5); IMiD-based 17 months both, P=0.59; HR 0.8 (95% CI 0.4–1.7); dexamethasone alone 3 vs. 7 months, P=0.17; HR 5.5 (95% CI 0.2–138)].
Figure 6.
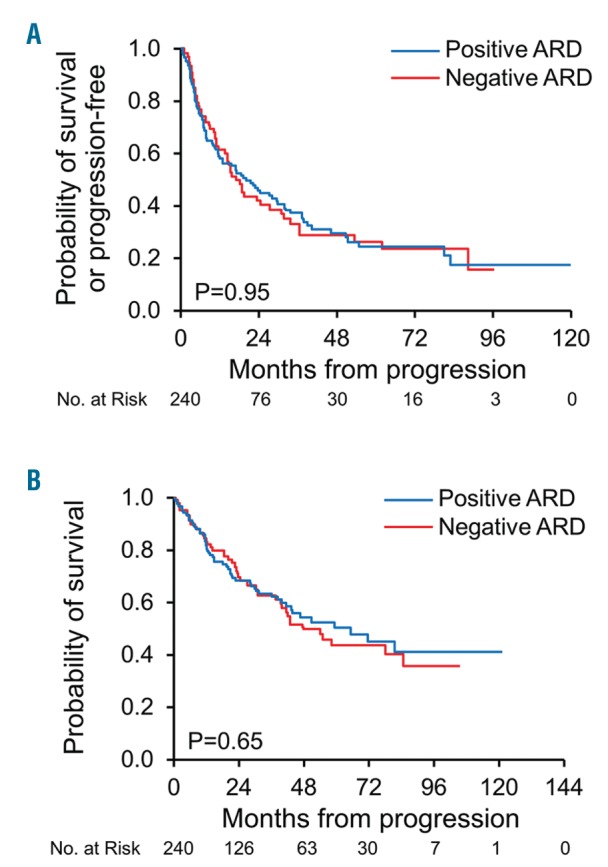
Survival curves at first progression by baseline immunoparesis status, ARD: average relative difference. A. Progression-free survival from progression. B. Overall survival from progression.
OS from first progression was similar between groups [median 42 months in those with a negative ARD compared to 47 months in those with a positive ARD, P=0.65; HR 1.1 (95%CI 0.8–1.5)] (Figure 6B). No significant difference in OS was seen between the negative and positive ARD groups for any given regimen at first progression [ASCT median not reached in both groups, P=0.18; HR not estimable; melphalan-based median 31 months vs. 40 months, P=0.9; HR 0.9 (95%CI 0.4–2.5); bortezomib-based median not reached vs. 72 months, P=0.42; HR 0.8 (95%CI 0.4–1.4); IMiD-based 54 months vs. 68 months, P=0.95; HR 1 (95%CI 0.5–2.2); dexamethasone alone 5 vs. 37 months, P=0.17; HR 5.5 (95%CI 0.2–138)].
Discussion
This study provides data on the differential response to various chemotherapeutic regimens in AL amyloidosis at first-line and first progression stratified by the immunoparesis status at diagnosis. For newly diagnosed patients, ASCT provided the best outcomes within each immunoparesis group, but with a greater benefit seen in patients without significant immunoparesis (i.e. positive ARD) compared to those with significant immunoparesis (i.e. negative ARD). For non-transplant first-line regimens, a comparison between immunoparesis groups shows that while the advantage for those with a positive ARD was seen with a melphalan-based regimen, this advantage disappeared with bortezomib or IMiD-based regimens. The response and outcome at first progression were comparable between immunoparesis groups, suggesting neutralization of the adverse prognostic effect of immunosuppression at progression. However, when the data was analyzed by regimen, melphalan-based and dexamethasone alone generally produced poorer response and survival compared to other regimens, possibly reflecting selection bias.
ASCT provided an improved survival rate in each immunoparesis group. The advantage of ASCT was seen as a higher rate of ≥VGPR as well as a longer PFS and OS. ASCT is, however, applicable only in a fraction of patients, which carry a more favorable prognosis, as reflected by younger age, a median of one involved organ and lower risk-stratified stage. In this study, ASCT (with or without prior induction) was performed in over 40% of patients, and reflects a referral bias. It is important to note that although patients with significant immunoparesis were shown to have more cardiac involvement, less renal involvement and a higher tumor burden (dFLC, bone marrow plasma cell percentage),3 they were as likely to proceed to ASCT as those without significant immunoparesis (41% vs. 43%, respectively). Even with ASCT, patients with a negative ARD had a lower ≥VGPR rate, PFS and OS compared to patients with a positive ARD, although organ response was achieved at a similar rate (66% vs. 75%, respectively). It appears that ASCT is the treatment of choice regardless of immunoparesis status, but response and response duration in patients with significant immunoparesis are lower than in those without significant immunoparesis.
Exploration of the treatment options in the non-transplant regimens reveals that patients with significant immunoparesis had a poorer response to melphalan-based regimens. This was reflected by a low rate of ≥VGPR (which represents the therapeutic endpoint in AL amyloidosis),7 as well as significantly lower organ response rate. In comparison, patients lacking significant immunoparesis treated with similar regimens had higher hematological (57%) and organ response (53%) rates. Moreover, a PFS and OS advantage in favor of patients without significant immunoparesis was seen in those treated with high-dose melphalan or low-intensity melphalan, but not for bortezomib or IMiDs. This finding suggests that in ASCT ineligible patients, melphalan has a greater impact in those without significant immunoparesis, while those with significant immunoparesis are less likely to benefit from melphalan. The reason for this is unclear, but might reflect different disease biology based on immunoparesis status. Melphalan, an alkylating agent is, unlike bortezomib and IMiDs, genotoxic.8 As such, it has the potential to impact DNA integrity and accelerate progression of a genomically unstable plasma cell clone. At progression, melphalan therapy produced the poorest results, which also supports this hypothesis. However, this clearly needs further investigation.
While IMiDs produced a relatively good response rate and survival in patients with significant immunoparesis, they were associated with a reduced OS in patients without significant immunoparesis. IMiDs in AL amyloidosis are generally not well tolerated and produce modest benefit,9,10 and therefore are not considered as first-line treatment for most patients. Based on the data presented, the selection of an IMiD as first-line should be discouraged in those without significant immunoparesis but can be considered for those with significant immunoparesis. The number of patients treated with IMiDs in this study was small, so this conclusion must be taken with caution and should be confirmed by other studies. When dexamethasone alone was utilized, the results were generally poor.
Patients at first progression were still able to achieve ≥VGPR in over 50% of patients in both groups, but had a shorter duration of response with a second-line of therapy. PFS and OS were comparable between patients with or without significant immunoparesis, independent of the salvage regimen. However, patients treated with melphalan-based regimens had poorer response, PFS and OS in immunoparesis groups. This may reflect a bias, as patients treated with low-dose melphalan at first progression were less likely to have been ASCT-eligible at diagnosis and carry a poorer prognosis.
In conclusion, first-line ASCT provides the best response and survival in patients with AL amyloidosis, irrespective of immunoparesis status. Better results for ASCT, however, were seen in those without significant immunoparesis. For non-transplant regimens, bortezomib and IMiDs were more likely to overcome the poorer prognosis associated with significant immunoparesis, while low-dose melphalan was associated with the least benefit for patients with significant immunoparesis. These findings should be assessed in prospective studies.
Footnotes
Check the online version for the most updated information on this article, online supplements, and information on authorship & disclosures: www.haematologica.org/content/101/9/1102
References
- 1.D’Souza A, Dispenzieri A, Wirk B, et al. Improved Outcomes After Autologous Hematopoietic Cell Transplantation for Light Chain Amyloidosis: A Center for International Blood and Marrow Transplant Research Study. J Clin Oncol. 2015; 33(32):3741–3749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Cordes S, Dispenzieri A, Lacy MQ, et al. Ten-year survival after autologous stem cell transplantation for immunoglobulin light chain amyloidosis. Cancer. 2012; 118(24):6105–6109. [DOI] [PubMed] [Google Scholar]
- 3.Muchtar E, Dispenzieri A, Kumar SK, et al. Immunoparesis in newly diagnosed AL amyloidosis is a marker for response and survival. Leukemia. 2016. [Epub ahead of print] 10.1038/leu.2016.140 [DOI] [PubMed] [Google Scholar]
- 4.Dispenzieri A, Buadi F, Kumar SK, et al. Treatment of Immunoglobulin Light Chain Amyloidosis: Mayo Stratification of Myeloma and Risk-Adapted Therapy (mSMART) Consensus Statement. Mayo Clin Proc. 2015;90(8):1054–1081. [DOI] [PubMed] [Google Scholar]
- 5.Comenzo RL, Reece D, Palladini G, et al. Consensus guidelines for the conduct and reporting of clinical trials in systemic light-chain amyloidosis. Leukemia. 2012; 26(11):2317–2325. [DOI] [PubMed] [Google Scholar]
- 6.Dispenzieri A, Gertz MA, Kyle RA, et al. Prognostication of survival using cardiac troponins and N-terminal pro-brain natriuretic peptide in patients with primary systemic amyloidosis undergoing peripheral blood stem cell transplantation. Blood. 2004;104(6):1881–1887. [DOI] [PubMed] [Google Scholar]
- 7.Kumar S, Dispenzieri A, Lacy MQ, et al. Revised prognostic staging system for light chain amyloidosis incorporating cardiac biomarkers and serum free light chain measurements. J Clin Oncol. 2012; 30(9):989–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ranaldi R, Palma S, Tanzarella C, Lascialfari A, Cinelli S, Pacchierotti F. Effect of p53 haploinsufficiency on melphalan-induced genotoxic effects in mouse bone marrow and peripheral blood. Mutat Res. 2007; 615(1–2):57–65. [DOI] [PubMed] [Google Scholar]
- 9.Dispenzieri A, Lacy MQ, Rajkumar SV, et al. Poor tolerance to high doses of thalidomide in patients with primary systemic amyloidosis. Amyloid. 2003;10(4):257–261. [DOI] [PubMed] [Google Scholar]
- 10.Sanchorawala V, Wright DG, Rosenzweig M, et al. Lenalidomide and dexamethasone in the treatment of AL amyloidosis: results of a phase 2 trial. Blood. 2007;109(2):492–496. [DOI] [PubMed] [Google Scholar]