

Abstract
Chromosome 22q11.2 deletion syndrome (22q11.2DS), a neurogenetic condition, is the most common microdeletion syndrome affecting 1 in 2,000–4,000 live births and involving haploinsufficiency of ∼50 genes resulting in a multisystem disorder. Phenotypic expression is highly variable and ranges from severe life-threatening conditions to only a few associated features. Most common medical problems include: congenital heart disease, in particular conotruncal anomalies; palatal abnormalities, most frequently velopharyngeal incompetence (VPI); immunodeficiency; hypocalcemia due to hypoparathyroidism; genitourinary anomalies; severe feeding/gastrointestinal differences; and subtle dysmorphic facial features. The neurocognitive profile is also highly variable, both between individuals and during the course of development. From infancy onward, motor delays (often with hypotonia) and speech/language deficits are commonly observed. During the preschool and primary school ages, learning difficulties are very common. The majority of patients with 22q11.2DS have an intellectual level that falls in the borderline range (IQ 70–84), and about one-third have mild to moderate intellectual disability. More severe levels of intellectual disability are uncommon in children and adolescents but are more frequent in adults. Individuals with 22q11.2DS are at an increased risk for developing several psychiatric disorders including attention deficit with hyperactivity disorder (ADHD), autism spectrum disorder (ASD), anxiety and mood disorders, and psychotic disorders and schizophrenia. In this review, we will focus on the developmental phenotypic transitions regarding cognitive development in 22q11.2DS from early preschool to adulthood, and on the changing behavioral/psychiatric phenotype across age, on a background of frequently complex medical conditions.
Keywords: chromosome 22, 22q11.2 deletion, Di George, developmental trajectories
Introduction
Chromosome 22q11.2 deletion syndrome (22q11.2DS), a neurogenetic condition, is the most common micro-deletion syndrome affecting 1 in 2,000–4,000 live births [Tezenas et al., 1996; Devriendt et al., 1998; Goodship et al., 1998; Oskarsdottir et al., 2004] and involving haploinsufficiency of ∼50 genes resulting in a multisystem disorder. Phenotypic expression is highly variable and ranges from severe life-threatening conditions to only a few associated features. Most common medical problems include: congenital heart defects, in particular conotruncal anomalies; palatal abnormalities, most frequently velopharyngeal incompetence (VPI); immunodeficiency; hypocalcemia due to hypoparathyroidism; genitourinary anomalies; severe feeding/gastrointestinal differences; and subtle dysmorphic facial features [McDonald-McGinn and Sullivan, 2011; Philip and Bassett, 2011]. The neurocognitive profile is also highly variable, both between individuals and during the course of development. From infancy onward, motor delays (often with hypotonia) and speech/language deficits are commonly observed. During the preschool and primary school ages, learning difficulties are very common. The majority of patients with 22q11.2DS have an intellectual level that falls in the borderline range (IQ 70–84), and about one-third have mild to moderate intellectual disability. More severe levels of intellectual disability are uncommon in children and adolescents, unless there is a secondary insult such as a hypoxic ischemic event during cardiac repair or a primary brain malformation such as polymicrogyria, but are more frequent in adults. Individuals with 22q11.2DS are at an increased risk for developing several psychiatric disorders including attention deficit with hyperactivity disorder (ADHD), autism spectrum disorder (ASD), anxiety and mood disorders, and psychotic disorders and schizophrenia. In this review, we will focus on the developmental phenotypic transitions regarding cognitive development in 22q11.2DS from early preschool to adulthood, and on the changing behavioral/psychiatric phenotype across age, on a background of frequently complex medical conditions.
Genetics And Presenting Phenotype
The association of thymic aplasia and congenital hypoparathyroidism was reported by Sedlakova [1955], Lobdell [1959], and by DiGeorge [1965]. Multiple etiologies have been identified over time including teratogens such as maternal diabetes [Sulik et al., 1986; Digilio et al., 1995] and maternal retinoic acid exposure [Coberly et al., 1996]; single gene disorders such as CHARGE syndrome due to mutations in CHD7 [Sanlaville et al., 2006; Jyonouchi et al., 2009]; and chromosome abnormalities involving 4q21.3-25ter [Fukushima et al., 1992], 10p13-14 [Elstner et al., 1984; Greenberg et al., 1988; Gottlieb et al., 1998], 11q23-ter [Grossfeld et al., 2004], and 22q11.2 [De la Chapelle et al., 1981; Kelley et al., 1982; Greenberg et al., 1984] deletions, all affecting neural crest cell migration, in particular the 3rd and 4th pharyngeal pouches, causing thymic hypoplasia leading to immunodeficiency, hypoplasia of the parathyroid glands leading to hypocalcemia, and structural abnormalities of the outflow tract of the heart leading to conotruncal cardiac anomalies [Graham, 2001].
In the early 1980's, following the identification of patients with DiGeorge syndrome (DGS) and unbalanced translocations involving chromosome 22q11.2 [De la Chapelle et al., 1981; Kelley et al., 1982; Greenberg et al., 1984], visible cytogenetic deletions of 22q11.2 were identified in ∼25% of patients with DGS [Greenberg et al., 1988]. But the puzzle remained, what about the remaining 75% of patients with DGS? By the early 1990's, with the advent of fluorescent in situ hybridization (FISH) studies, this question was answered as the majority of patients with DGS were found to have a submicroscopic deletion of 22q11.2 [Scambler et al., 1991; Driscoll et al., 1992; Desmaze et al., 1993; Driscoll et al., 1993a]. Thereafter, the list of associated characteristics expanded rapidly to include palatal abnormalities, dysmorphic craniofacial features such as asymmetric crying facies, laryngotracheoesophageal abnormalities, speech, language and developmental delay [McDonald-McGinn et al., 1997; Ryan et al., 1997; McDonald-McGinn et al., 1999]. Concurrently leading to the recognition that patients with other previously described diagnoses actually had a chromosome 22q11.2 deletion, including the majority of patients with velocardiofacial syndrome (VCFS) [Driscoll et al., 1993b] and conotruncal anomaly face syndrome (CTAF) [Burn et al., 1993; Matsuoka et al., 1994], as well as a subset of patients with Opitz G/BBB syndrome [McDonald-McGinn et al., 1995; LaCassie and Arriaza, 1996; Fryburg et al., 1996] and Cayler cardiofacial syndrome [Gianotti et al., 1994]. This was understandable as each condition was originally described by clinicians concentrating on his/her own area of expertise, such as Endocrinology (DGS), Cardiology (CTAF) and Speech Pathology (VCFS) [McDonald-McGinn et al., 1997]. Currently the term DiGeorge “syndrome” continues to be utilized when the etiology is not due to a hemizygous 22q11.2 deletion but otherwise the diagnosis, with its broad phenotypic spectrum, has collectively become referred to by the cytogenetic abnormality, 22q11.2DS, as it is the single common denominator amongst all patients regardless of phenotypic presentation [Bassett et al., 2011; McDonald-McGinn et al., 2011].
Current detection methods still include FISH but Multiplex Ligation-dependent Probe Amplification (MLPA) [Sorensen et al., 2010] and single nucleotide polymorphism (SNP) arrays are now preferred as they both size the deletion and arrays do not require an elevated index of suspicion. Most deletions (90–94%) are de novo [McDonald-McGinn et al., 1997, 1999] as a result of non-homologous recombination due to the presence of low copy repeats (LCR 's) [Edelmann et al., 1999; Shaikh et al., 2000], also known as segmental duplications (SD's), which bracket the region leading to aberrant meiotic interchromosomal exchanges resulting in either a 22q11.2 deletion or duplication. Four discrete blocks are located in this region and each block is comprised of multiple repeats. These blocks are named LCR A–D, with A being the most proximal. They define the breakpoints and are used to describe the size of the resultant deletion/duplication. The typical 3 Mb (million base pair) deletion, involving haploinsufficiency of ∼50 genes, extends from LCR A–D, whereas a nested deletion is smaller extending from A–B, A–C, B–C, B–D or C–D [McDonald-McGinn et al., 2011]. FISH probes and the important developmental gene TBX1, thought to be responsible for many associated features such as congenital heart defects [Papangeli and Scambler, 2013; Jerome and Papaioannou, 2001], are located between A–B. In contrast, patients with nested B–D/C–D deletions would have been missed by FISH studies and do not include TBX1 (Fig. 1). Nonetheless, these patients have considerable overlapping features including conotruncal cardiac anomalies, palatal defects, and developmental differences, which may be explained by haploinsufficiency of other important developmental genes such as CRKL1 and SNAP29, a downstream effect of TBX1, or modifier genes on other chromosomes ([McDonald-McGinn et al., 2011].
Figure 1.
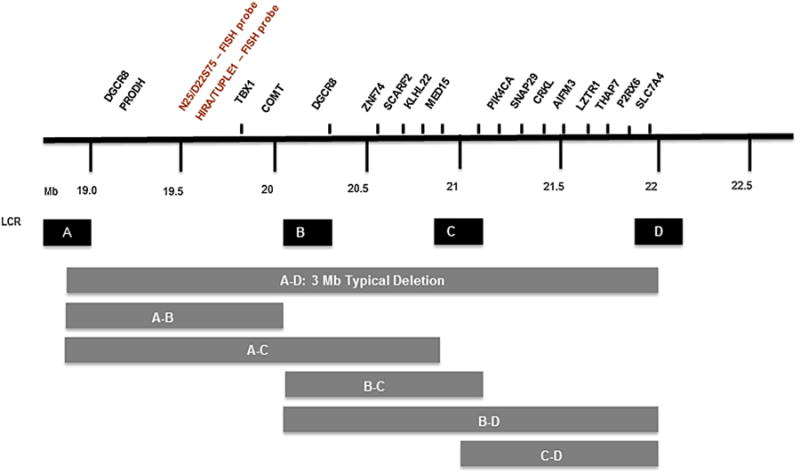
Low copy repeats, represented here as A, B, C, and D, bracket the 22q11.2 deletion and define the breakpoints with the standard ∼3Mb 22q11.2 deletion extending from A–D. Atypical nested deletions include A–B, A–C, B–C, B–D, and C–D. Notable genes within the deleted regions of chromosome 22q11.2 include PRODH, TBX1 and COMT within A–B and SNAP 29 and CRKL1 within C–D. Note that FISH probes D22S75 (N25) and HIRA (TUPLE1) are located within A–B and would be present in those patients with nested deletions excluding the A–B region.
Estimated prevalence figures vary from 1 in 2,000–4, 000 births [Tezenas et al., 1996; Devriendt et al., 1998 Goodship et al., 1998; Oskarsdottir et al., 2004] and are extrapolations of limited populations using FISH studies. However, based on the wide phenotypic variability and in light of data from a recent prenatal SNP array study [Costain et al., 2014], these figures may be underestimates and something that may be better defined following the introduction of non-invasive prenatal testing (NIPT) for microdeletions including 22q11.2DS [Saldivar et al., 2015]; the incidental identification of neonates with 22q11.2DS via newborn screening for severe combined immunodeficiency (SCID) [Chien et al., 2015]; and newborn screening studies for 22q11.2DS utilizing qPCR on Guthrie cards in selected populations [Tomita-Mitchell et al., 2010]. Moreover, the existing data do not take into account the rising prevalence due to increasing numbers of affected adults having their own affected children [McDonald-McGinn et al., 1999, 2001]. As a haploinsufficiency disorder, 50% of children born to affected adults will have the 22q11.2 deletion. Therefore, since survival with cardiac anomalies was low until the mid-1980's, the familial cases and thus the overall prevalence are expected to rise over time [McDonald-McGinn et al., 2011; Bassett et al., 2011].
Males and females are equally affected with 22q11.2DS and there is no evidence that the deletion is more frequently associated with any particular ethnic background, although non-Caucasians may be less likely to come to early diagnosis, perhaps related to more subtle dysmorphic features [McDonald-McGinn et al., 2005]. Unlike the early reports of DGS, mortality in childhood is low (∼5%) and is most often associated with complications of congenital heart defects [McDonald-McGinn et al., 2001]. However, premature death has been reported in adulthood [Bassett et al., 2009].
The presence and severity of associated features varies by age and the focus changes over time [McDonald-McGinn et al., 2011; Habel et al., 2014]. Likewise, the indications for testing vary by age (Fig. 2) and the diagnosis can be missed. This is especially true in adolescents, adults and non-Caucasians, in light of the broad inter and intrafamilial variability, even between identical twins [McDonald-McGinn, 2001; Goodship, 1995]. However, subtle dysmorphic features can often provide clues to the diagnosis and most frequently include: hooded eyelids with or without upslanting palpebral fissures and hyper-telorism; auricular anomalies in particular thick overfolded helices, attached lobes, protuberant or cupped ears, microtia, anotia, preauricular tags or pits; a prominent nasal root, bulbous nasal tip with hypoplastic alae nasi, and nasal dimple/crease/hemangioma; malar flatness; asymmetric crying facies, a small mouth especially in infancy, and micrognathia [McDonald-McGinn et al. 1997, 1999, 2001, 2011].
Figure 2.
Most common indications for 22q11.2 deletion testing varies by age.
Many patients are now being identified prenatally via fetal ultrasound/echocardiography, most often due to congenital heart disease. Other prenatal clues to the diagnosis include polyhydramnios, cleft palate, cleft lip/palate, polydactyly, clubfoot, renal anomalies, diaphragmatic hernia, spina bifida, and craniosynostosis [McDonald-McGinn, 2011].
In childhood, major phenotypic physical features involve: congenital heart defects, most often conotruncal cardiac anomalies, immunodeficiency including T-cell lymphopenia, IgA deficiency, delayed IgG production, humoral defects and autoimmune disease, and palatal anomalies, including VPI, overt cleft palate, cleft lip with or without cleft palate, and Pierre Robin, in three-quarters of patients; hypocalcemia/hypoparathyroidism in half of patients often presenting in the neonatal period or initially/as a recurrence during times of stress such as during puberty, illness or perioperatively; and genitourinary problems such as renal anomalies/agenesis, hydronephrosis, cryptorchidism, hypospadias, absent uterus, and gastrointestinal differences including gastroesophageal reflux disease, severe dysphagia, and constipation in one-third of patients [McDonald-McGinn et al., 2011].
Less common but important associated physical features include: thyroid dysfunction, IUGR, failure to thrive, short stature/growth hormone deficiency; intestinal malrotation, Hirschsprung disease, imperforate anus, umbilical, and inguinal hernia; laryngeal web, esophageal atresia, tracheoesophageal fistula, choanal atresia, sensorineural, and conductive hearing loss, microtia/anotia; diaphragmatic hernia; cervical and thoracic vertebral anomalies, scoliosis, club foot, polydactyly, radial ray defects, craniosynostosis; microcephaly, asymmetric crying facies, unprovoked seizures, hypotonia, polymicrogyria, myleomeningocele; autoimmune disease including idiopathic thrombocytopenia, juvenile rheumatoid arthritis, immune mediated hypo and hyperthyroidism, neutropenia, vitiligo; malignancy including hepatoblastoma; scleracornea, microphthalmia/anophthalmia, ptosis, retinal coloboma, posterior embryotoxon, tortuous retinal vessels, hypertelorism; and enamel hypoplasia leading to chronic caries [McDonald-McGinn et al., 2011].
The considerable morbidity associated with 22q11.2DS in the face of extremely wide variability poses significant challenges for both individual and population based health care management. In light of this difficulty The International 22q11.2 Consortium developed practical guidelines for managing patients with 22q11.2DS that emphasizes the multi-system nature of the condition and includes recommendations for assessment by age and at diagnosis [Bassett et al., 2011].
Important additional genetic considerations include: reports of germline mosaicism affecting recurrence risk counseling for parents of children with de novo deletions [Sandrin-Garcia et al., 2002]; somatic mosaicism supporting parental 22q11.2DS testing in all cases [Halder et al., 2008; McDonald-McGinn et al., 2011]; phenocopies due to mutations in TBX1 only [Zweier et al., 2007]; the presence of co-occurring conditions such as 22q11.2DS and trisomy 8 mosaicism [McDonald-McGinn et al., 2005b] and 22q11.2DS and CHARGE syndrome due to a CHD7 mutation [McDonald-McGinn et al., 2011]; and the 22q11.2 deletion unmasking an autosomal recessive condition due to the presence of a mutation on the remaining allele such as with a GP1BB mutation leading to Bernard–Soulier syndrome [Budarf et al., 1995] or a SNAP29 mutation [McDonald-McGinn et al., 2013] leading to CEDNIK syndrome.
Cognitive Phenotype In 22Q11 DS Across Development
Developmental and educational concerns are frequently reported in 22q11.2DS. One of the first and most important questions parents and care-givers ask of children with 22q11.2DS is what the impact will be of the 22q11.2 deletion on the global cognitive development. During infancy and toddlerhood, cardiac defects, feeding difficulties, frequent infections, gross/fine motor difficulties and expressive language delays and speech problems dominate, but already from preschool age on learning difficulties and abnormal behavior become apparent. However, it is important to keep in mind that each infant/child/adolescent/adult with 22q11.2DS is unique, and that they may have concerns in a few or in many of the areas described. Both genes and environmental factors play essential roles in shaping brain growth and cognitive development throughout life.
What Do We Know about the Intellectual Abilities and Cognitive Profile in 22q11.2DS?
The majority of studies have focused on the intellectual abilities in children and adolescents. Less is known about the intellectual functioning of adults with 22q11 DS [Henry et al., 2002].
The level of intelligence in children and adolescents with 22q11 DS is highly variable and follows a normal distribution (similar to the IQ distribution in the general population), but is shifted about 30 IQ points to the left [De Smedt et al., 2007]. The average mean full scale IQ is in the mid-seventies (70–75) with about 55% having a borderline to normal intelligence (FSIQ > 70) and about 45% having a mild (to moderate) intellectual disability (ID) (FSIQ 55–70) and a minority experiencing moderate to severe intellectual disability [Swillen et al., 1997; Moss et al., 1999; Niklasson et al., 2009]. In addition, a significant number of individuals with 22q11DS show a discrepancy between verbal comprehension and perceptual reasoning abilities, favoring the verbal domain [Swillen et al., 1997; Moss et al., 1999; Shprintzen, 2008; De Smedt et al., 2007; Niklasson et al., 2009]. This VIQ > PIQ cognitive profile in children with 22q11DS seems to change with age: from adolescence on this VIQ > PIQ profile is less common [Campbell and Swillen, 2005].
Within the 22q11DS child/adolescent population there is a wide variability in intelligence: some patients function within the limits of borderline-normal intelligence while others function in the range of moderate-severe intellectual disability (ID). Current findings indicate that the genetic architecture of ID is complex, consistent with other neurodevelopmental disorders, with an important role for rare variants with large effects [Najmabadi et al., 2011]. In the 22q11DS literature, several factors have been put forward to explain this wide variability in IQ: origin of the deletion (patients with a familial deletion have been associated with lower IQ scores as compared to de novo deletions) [Swillen et al., 1997; De smedt et al., 2007], genetic variation within the 22q11.2 region [Gothelf et al., 2005; Raux et al., 2007], and environmental factors such as socioeconomic status [Shashi et al., 2010] and parental IQ and siblings IQ [Olszewski et al., 2014]. Several other possible factors that contribute to this variability in IQ have not been systematically studied yet and therefore should be the focus of future research, for example: the size of the deletion, genes within the region (COMT, PRODH, TBX1, CRKL1, etc.), the remainder of the genome/genetic background, personality and temperament, and risk and protective factors in the environment such as the impact of therapy/remediation/anticipatory guidance, quality of life, coping strategies in the family, availability of social network support and resources.
What Do We Know about the Developmental Trajectory of Intelligence in 22q11 DS?
In Figure 3 the developmental cognitive trajectories from preschool to adolescence are presented. Several cross-sectional studies have found a negative correlation between age and IQ scores in 22q11DS, particularly a decline in VIQ, suggesting that at least some of these individuals show a gradual decline in cognitive development as they grow into adulthood [Gothelf et al., 2005; Green at al., 2009]. Recent longitudinal studies [Duijff et al., 2012; Duijff et al., 2013] have shown that already from primary school age on several divergent cognitive trajectories become apparent: a) a relative stable IQ-trajectory: a number of children showed adequate progress in their performance to keep up with the gradual increase of the level of cognitive requirements with age; b) a decrease in IQ score or a growing into deficit trajectory due to insufficient cognitive development leading to an increasing discrepancy with age-required norms, and c) an absolute decline in cognitive abilities as manifested by lower subtest raw scores for at least two subtests in a subgroup of children. The authors stated that “It could be speculated that the observed decline (in a subgroup) is likely to be the first symptom of schizophrenic disorder in 22q11DS. Interestingly, the absolute decline does not occur in all subjects, but rather in a subgroup, parallel to the observation that only a subgroup of people with 22q11DS will develop schizophrenia. However, in order to answer this question a continued follow-up into adulthood is required”. A recent collaborative study by the international 22Q11 Brain Behavior Consortium (22q11 DS IBBC, an international group of more than 100 scientists from 22 sites) on the (longitudinal) cognitive development of 829 patients ages 8–24 years, showed that on average children with 22q11DS show a cognitive decline of 7 FSIQ-points or 9 VIQ-points [Vorstman et al., 2015]. In the subgroup that developed psychotic symptoms, this decline was significantly steeper. Based on VIQ trajectories, those who subsequently developed a psychotic disorder and those who did not, could be distinguished already from age 11 onwards. In accordance to what is observed in general population regarding the early precursors of psychosis, a decline in verbal IQ precedes the onset of psychosis in 22q11 DS.
Figure 3.

Developmental cognitive trajectories in 22q11 DS.
What Are the Implications of This Variability in IQ and These Divergent Cognitive Trajectories?
Depending on their overall cognitive capacities (borderline intelligence vs. intellectual disability), children and adolescents with 22q11.2DS will follow either normal school with additional learning and educational support (starting from an individualized educational plan (IEP), or they will need special education with IEPs that are adapted to the individual needs of the child/adolescent. Secondly, given the changing cognitive phenotype with age and the possible cognitive decline with age, the cognitive abilities of children and adolescents should be followed-up and re-evaluated at a regular basis. Additionally, since in an important subgroup of children and adolescents a decline in IQ occurs, a continuous adaptation of the expectations and the learning environment will be necessary in order to have a good balance between the capacities of the child/adolescent and the demands of the environment. In this way, anticipatory guidance can be implemented at home and in school, and unnecessary stress can be prevented.
From a research perspective, given the early cognitive decline in 22q11.2DS, this microdeletion syndrome/CNV is an interesting model to investigate possible genes, genetic mechanisms and central neurotransmitter systems in the 22q11.2 region that contribute to cognitive deterioration. A nice illustration of this approach is a recent study by Evers et al. [2014]. Their results suggest that a subgroup of adults with 22q11DS may be affected by a neurodegenerative process affecting at least three neurotransmitter systems (serotonin, dopamine and norepinephrine), but the precise mechanism in the cognitive deterioration as seen in 22q11DS, has to be elucidated.
Behavioral-Psychiatric Phenotype In 22Q11 DS Across Age
Children with intellectual disabilities (ID) show a greater tendency to engage in problem behavior than individuals without ID [Dykens, 2000; Dekker et al., 2002; Emerson, 2003; De Ruiter et al., 2007] and there is evidence in the literature that the prevalence of problem behavior among individuals with certain genetic syndromes conditions associated with ID is higher than in individuals with idiopathic ID [Bodfish and Lewis, 2002; Arron et al., 2011; Powis and Oliver, 2014]. This is also the case for 22q11 DS.
What Do We Know from the Behavioral-psychiatric Phenotype in 22q11 DS?
Cross-sectional studies using parental and teachers reports have shown that children with 22q11DS showed significantly more internalizing behavior than peers [Swillen et al., 1999; Briegel et al., 2008], children with speech-language delays [Swillen et al., 2001] and children with clefts [Jansen et al., 2007]. Specifically, children with 22q11DS are described as showing more withdrawn behavior, social interaction problems with peers, attention problems, and anxiety problems. This is consistent with several clinical studies in 22q11DS in which high rates of attention deficit disorder with hyperactivity (ADHD) [Niklasson et al., 2001; Gothelf et al., 2005], autism spectrum disorders [Niklasson et al., 2001, 2009; Fine et al., 2005; Antshel et al., 2006; Vorstman et al., 2006] and anxiety and affective disorders [Antshel et al., 2006; Jolin et al., 2009] are reported. There has been some controversy regarding the diagnosis of ASD in 22q11 DS; recent studies have shown that individuals with 22q11 DS and ASD had significant problems in social interaction and communication while stereotyped behaviors were not reported, suggesting a unique type of ASD [Kates et al., 2007; Bruining et al., 2010].
The increased rate of most psychiatric disorders in children with 22q11 DS is similar to that in children with other developmental disabilities [Feinstein et al., 2002; Baker and Skuse, 2005]. However, by late adolescence and early adulthood, this picture seems to change as up to one-third of the patients with 22q11DS develop psychotic disorders mostly resembling schizophrenia and schizoaffective disorder [Murphy et al., 1999; Bassett et al., 2003; Gothelf et al., 2005]. Psychotic disorders are far more common in adolescents and young adults with 22q11 DS than in matched IQ subjects without the syndrome. In addition, many adults with 22q11DS suffer from depression or generalized anxiety disorder [Fung et al., 2010]. The largest study of lifetime psychiatric diagnoses to date in 22q11DS (n = 1402; ages 6–68 years) combined data from 22q11DS cohorts from 22 sites in the world [Schneider et al., 2014 and 22q11 DS IBBC] and found remarkably similar prevalence and developmental trends across countries. Attention-deficit/hyperactivity disorder, autism spectrum disorder and anxiety disorders were the most common diagnoses during childhood, whereas rates of psychosis and mood disorders increased dramatically during adolescence and young adulthood. Anxiety disorders were especially prevalent among children and adolescents. Generalized anxiety disorder, specific phobia, and social phobia were the most frequent anxiety disorders during childhood and adolescence. Whereas the rate of specific phobia is similar to that in individuals with intellectual disability, social phobia and generalized anxiety disorder appear to be overrepresented in 22q11.2 deletion syndrome [Dekker and Koot, 2003]. This finding, combined with the high rate of autism spectrum disorders, indicates that difficulties in the social domain may represent key characteristics of the syndrome [Baker and Vorstman, 2012]. Sex differences in the rates of several psychiatric disorders were comparable to those reported in the general population. Specifically, disruptive disorders and ADHD were more frequent in males than females, as documented in the general population [Maughan et al., 2004; Merikangas et al., 2010]. There was a predominance of females among those diagnosed with anxiety and mood disorders as adults. Numerous studies have observed that sex differences in the rate of internalizing disorders emerge around puberty [Hayword and Sanborn, 2002], suggesting the possible impact of hormonal changes in the development of affective symptoms in females. In contrast, the male predominance of autism spectrum disorders and to a lesser extent psychotic disorders in the general population was not observed in 22q11.2 deletion syndrome. This may be related to the strong genetic contribution to the pathogenesis of social deficits and psychosis in 22q11.2 deletion syndrome and deserves investigation in future studies [Schneider et al., 2014].
What Are the Implications of This Changing Behavioral-psychiatric Phenotype?
Given the increased risk for developing behavioral problems and psychiatric disorders, early psychological/psychiatric follow-up and intervention is warranted in individuals with 22q11 DS. It will be key to find a balance between follow-up and intervention, and to monitor in a flexible way the changing and increasing environmental demands with age. In school-age children with 22q11.2 deletion syndrome, special emphasis is needed with regard to the diagnosis and management of attentional deficits, as these can interfere with learning and academic achievement. The presence of attentional deficits should be screened systematically and at regular intervals. The emergence of social deficits during adolescence can represent a major source of disability in individuals with 22q11.2 deletion syndrome. Interventions focusing on appropriate adaptation of social demands and on socio-cognitive remediation programs and cognitive-behavioral therapy to improve social skills are recommended. As part of anticipatory care, individuals with 22q11.2 deletion syndrome should be screened for anxiety and mood disorders throughout their lifetimes. Given the markedly elevated risk for schizophrenia spectrum disorders, individuals with 22q11.2 deletion syndrome should be closely monitored for prodromal and fullblown symptoms of psychosis [Schneider et al., 2014]. Clinically significant psychotic symptoms should be treated in accordance with the most recent recommendations for effective management. Antipsychotic medications are mainstays of treatment for schizophrenia spectrum disorders, as are psycho-education, rehabilitation, and active stress reduction strategies.
From a research perspective, more large scale prospective studies that focus on the interaction between genesclinical/somatic phenotype-cognitive/psychiatric phenotype and environmental factors will be needed to understand the complex interplay of cognitive, mood and psychotic symptoms that are present in 22q11DS and that contribute to the variable neuropsychiatric phenotypes of 22q11 DS [Jonas et al., 2014].
Summary And Conclusions
Persons with 22q11DS present with a distinctive but dynamic and developing cognitive, behavioral and psychiatric phenotype due to the interaction between genes – environment.
This profile is often colored by a complex associated medical phenotype which frequently results in multiple hospitalizations beginning at an early age.
Children with a parent who also has 22q11.2DS are at greater risk for a poorer long term outcome.
Educators/teachers and psychologists/child psychiatrists are important members of the multidisciplinary team of professionals who provide services to a child/adolescent and adult with 22q11.2 DS.
Information on the cognitive trajectory of children/adolescents/adults with 22q11DS has recently become available but more longitudinal studies are important as a means of elucidating the cognitive changes in individuals with the syndrome throughout the lifespan.
More large scale prospective studies that focus on the interaction between genes-clinical/somatic phenotype-cognitive/psychiatric phenotype and environmental factors will be needed to understand the complex interplay of cognitive, mood and psychotic symptoms that are present in 22q11DS and that contribute to the variable neuropsychiatric phenotypes of 22q11 DS.
There is an urgent need for studies with focus on risk and protective factors in the environment that contribute to the variable medical/neurocognitive/neuropsychiatric phenotypes in patients with 22q11DS.
Contributor Information
Prof. Ann Swillen, Department of Human Genetics, KU Leuven and at the Department of Rehabilitation Sciences, KU Leuven (University of Leuven, Belgium). Trained as an educational psychologist, she is also affiliated to the Center of Human Genetics Leuven (University Hospital Gasthuisberg), an international center of excellence in the field of clinical and molecular genetics. She can be reached by email at.
Prof. Donna M. McDonald-McGinn, Email: mcginn@email.chop.edu, 22q and You Center; Clinical Genetics Center; Section of Genetic Counseling at The Children's Hospital of Philadelphia. In addition, Pediatrics at the Perelman School of Medicine of The University of Pennsylvania. She can be reached by email at.
References
- Antshel K, Fremont W, Roizen N, Shprintzen R, Higgins AM, Dhamoon A, Kates WR. ADHD, major depressive disorder, and simple phobias are prevalent psychiatric conditions in youth with velocardiofacial syndrome. J Am Acad Child Adolesc Psychiatry. 2006;45:596–603. doi: 10.1097/01.chi.0000205703.25453.5a. [DOI] [PubMed] [Google Scholar]
- Antshel K, Aneja A, Strunge L, Peebles J, Fremont WP, Stallone K, Abdulsabur N, Higgins AM, Shprintzen RJ, Kates WR. Autistic spectrum disorders in velo-cardio facial syndrome (22q11.2 deletion) J Autism Dev Disord. 2007;37:1776–1786. doi: 10.1007/s10803-006-0308-6. [DOI] [PubMed] [Google Scholar]
- Arron K, Oliver C, Moss J, Berg K, Burbidge C. The prevalence and phenomenology of self-injurious and aggressive behaviour in genetic syndromes. J Intellect Disabil Res. 2011;55:109–120. doi: 10.1111/j.1365-2788.2010.01337.x. [DOI] [PubMed] [Google Scholar]
- Baker K, Skuse D. Adolescents and young adults with 22q11 deletion syndrome: Psychopathology in an at-risk group. Br J Psychiatry. 2005;186:115–120. doi: 10.1192/bjp.186.2.115. [DOI] [PubMed] [Google Scholar]
- Baker K, Vorstman J. Is there a core neuropsychiatric phenotype in 22q11.2 deletion syndrome? Curr Opin Neurol. 2012;25:131–137. doi: 10.1097/WCO.0b013e328352dd58. [DOI] [PubMed] [Google Scholar]
- Bassett AS, Caluseriu O, Weksberg R, Young DA, Chow EW. Catechol-O-methyl transferase and expression of schizophrenia in 73 adults with 22q11 deletion syndrome. Biol Psychiatry. 2007;61:1135–1140. doi: 10.1016/j.biopsych.2006.07.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow E, AbdelMalik P, Gheorghiu M, Husted J, Weksberg R. The schizophrenia phenotype in 22q11 deletion syndrome. Am J Psychiatry. 2003;160:1580–1586. doi: 10.1176/appi.ajp.160.9.1580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, Chow EW, Husted J, Hodgkinson KA, Oechslin E, Harris L, Silversides C. Premature death in adults with 22q11.2 deletion syndrome. J Med Genet. 2009;46:324–330. doi: 10.1136/jmg.2008.063800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bassett AS, McDonald-McGinn DM, Devriendt K, Digilio MC, Goldenberg P, Habel A, Marino B, Oskarsdottir S, Philip N, Sullivan K, Swillen A, Vorstman J. International 22q11.2 deletion syndrome consortium. Practical guidelines for managing patients with 22q11.2 deletion syndrome. J Pediatr. 2011;159:332–339. doi: 10.1016/j.jpeds.2011.02.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bearden CE, Jawad A, Lynch D, Sokol S, Kanes S, McDonald-McGinn D, Saitta SC, Harris SE, Moss E, Wang PP, Zackai E, Emanuel BS, Simon TJ. Effects of a functional COMT polymorphism on prefrontal cognitive function in patients with 22q11.2 deletion syndrome. Am J Psychiatry. 2004;161:1700–1702. doi: 10.1176/appi.ajp.161.9.1700. [DOI] [PubMed] [Google Scholar]
- Briegel W, Schneider M, Schwab KO. 22q11. 2 deletion syndrome: Behaviour problems of children and adolescents and parental stress. Child Care Health Dev. 2008;34:795–800. doi: 10.1111/j.1365-2214.2008.00850.x. [DOI] [PubMed] [Google Scholar]
- Bruining H, de SL, Swaab H, de Jonge M, Kas M, van Engeland H, Vorstman J. Dissecting the clinical heterogeneity of autism spectrum disorders through defined genotypes. PLoS One. 2010;5:e10887. doi: 10.1371/journal.pone.0010887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budarf ML, Konkle BA, Ludlow LB, Michaud D, Li M, Yamashiro DJ, McDonald-McGinn D, Zackai EH, Driscoll DA. Identification of a patient with Bernard-Soulier syndrome and a deletion in the DiGeorge/velo-cardio-facial chromosomal region in 22q11. 2. Hum Mol Genet. 1995;4:763–766. doi: 10.1093/hmg/4.4.763. [DOI] [PubMed] [Google Scholar]
- Burn J, Takao A, Wilson D, Cross I, Momma K, Wadey R, Scambler P, Goodship J. Conotruncal anomaly face syndrome is associated with a deletion within chromosome 22q11. J Med Genet. 1993;30:822–824. doi: 10.1136/jmg.30.10.822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell L, Swillen A. The cognitive spectrum in velo-cardio-facial syndrome. In: Murphy KC, Scambler PJ, editors. Velo-Cardio-Facial Syndrome. Cambridge: Cambridge University Press; 2005. pp. 147–164. [Google Scholar]
- Chien YH, Chiang SC, Chang KL, Yu HH, Lee WI, Tsai LP, Hsu LW, Hu MH, Hwi WL. Incidence of severe combined immunodeficiency through newborn screening in a Chinese population. J Formos Med Assoc. 2015;114:12–16. doi: 10.1016/j.jfma.2012.10.020. [DOI] [PubMed] [Google Scholar]
- Coberly S, Lammer E, Alashari M. Retinoic acid embryopathy: Case report and review of literature. Pediatr Pathol Lab Med. 1996;16:823–3610. [PubMed] [Google Scholar]
- Costain G, McDonald-McGinn DM, Bassett AS. Prenatal genetic testing with chromosomal microarray analysis identifies major risk variants for schizophrenia and other late-onset disorders. Am J Psychiatry. 2014;170:1498. doi: 10.1176/appi.ajp.2013.13070880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Debbané M, Glaser B, David MK, Feinstein C, Eliez S. Psychotic symptoms in children and adolescents with 22q11. 2 deletion syndrome: Neuropsychological and behavioral implications. Schizophr Res. 2006;84:187–193. doi: 10.1016/j.schres.2006.01.019. [DOI] [PubMed] [Google Scholar]
- Dekker MC, Koot HM, van der Ende J, Verhulst FC. Emotional and behavioral problems in children and adolescents with and without intellectual disability. J Child Psychol Psychiatry. 2002;43:1087–1098. doi: 10.1111/1469-7610.00235. [DOI] [PubMed] [Google Scholar]
- Dekker MC, Koot HM. DSM-IV disorders in children with borderline to moderate intellectual disability. I: Prevalence and impact. J Am Acad Child Adolesc Psychiatry. 2003;42:915–922. doi: 10.1097/01.CHI.0000046892.27264.1A. [DOI] [PubMed] [Google Scholar]
- De la Chapelle A, Herva R, Koivisto M, Aula P. A deletion in chromosome 22 can cause DiGeorge syndrome. Hum Genet. 1981;57:253–256. doi: 10.1007/BF00278938. [DOI] [PubMed] [Google Scholar]
- De Ruiter K, Dekker M, Verhulst F, Koot H. Developmental course of psychopathology in youths with and without intellectual disabilities. J Child Psychol Psychiatry. 2007;48:498–507. doi: 10.1111/j.1469-7610.2006.01712.x. [DOI] [PubMed] [Google Scholar]
- Desmaze C, Scambler P, Prieur M, Halford S, Sidi D, Le Deist F, Aurias A. Routine diagnosis of DiGeorge syndrome by fluorescent in situ hybridization. Hum Genet. 1993;90:663–665. doi: 10.1007/BF00202489. [DOI] [PubMed] [Google Scholar]
- De Smedt B, Devriendt K, Fryns JP, Vogels A, Gewillig M, Swillen A. Intellectual abilities in a large sample of children with Velo-Cardio-Facial Syndrome: An update. J Intellect Disabil Res. 2007;51:666–670. doi: 10.1111/j.1365-2788.2007.00955.x. [DOI] [PubMed] [Google Scholar]
- Devriendt K, Fryns JP, Mortier G, van Thienen MN, Keymolen K. The annual incidence of DiGeorge/velocardiofacial syndrome. J Med Genet. 1998;5:789–790. doi: 10.1136/jmg.35.9.789-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiGeorge AM. Discussions on a new concept of the cellular basis of immunology. J Pediatr. 1965;67:907. [Google Scholar]
- Digilio MC, Marino B, Formigari R, Giannotti A. Maternal diabetes causing DiGeorge anomaly and renal agenesis. Am J Med Genet. 1995;55:410–513. doi: 10.1002/ajmg.1320550427. [DOI] [PubMed] [Google Scholar]
- Driscoll DA, Budarf ML, Emanuel BS. A genetic etiology for DiGeorge syndrome: Consistent deletions and microdeletions of 22q11. Am J Hum Genet. 1992;50:924–933. [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Salvin J, Sellinger B, McDonald-McGinn D, Zackai EH, Emanuel BS. Prevalence of 22q11 microdeletions in DGS and VCFS: Implications for genetic counseling and prenatal diagnosis. J Med Genet. 1993a;30:813–817. doi: 10.1136/jmg.30.10.813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Driscoll DA, Spinner NB, Budarf ML, McDonald-McGinn DM, Zackai EH, Goldberg RB, Shprintzen RJ, Saal HM, Zonana J, Jones MD, Mascarelo JT, Emanuel BS. Deletions and microdeletions of 22q11. 2 in Velo-Cardio-Facial syndrome. Am J Med Genet. 1993b;44:261–268. doi: 10.1002/ajmg.1320440237. [DOI] [PubMed] [Google Scholar]
- Duijff SN, Klaassen PW, de Veye HF, Beemer FA, Sinnema G, Vorstman JA. Cognitive development in children with 22q11. 2 deletion syndrome. Brit J Psychiat. 2012;200:462–468. doi: 10.1192/bjp.bp.111.097139. [DOI] [PubMed] [Google Scholar]
- Duijff SN, Klaassen PWJ, De Veye HFNS, Beemer FA, Sinnema G, Vorstman JA. Cognitive and behavioral trajectories in 22q11DS from childhood into adolescence: A prospective 6-year follow-up study. Res Dev Disabil. 2013;34:2937–2945. doi: 10.1016/j.ridd.2013.06.001. [DOI] [PubMed] [Google Scholar]
- Dykens EM. Psychopathology in children with intellectual disability. J Child Psychol Psychiatry. 2000;41:407–417. [PubMed] [Google Scholar]
- Edelmann L, Pandita RK, Morrow BE. Low-copy repeats mediate the common 3-Mb deletion in patients with velo-cardio-facial syndrome. Am J Hum Genet. 1999;64:1076–8610. doi: 10.1086/302343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elstner CL, Carey JC, Livingston G, Moeschler J, Lubinsky M. Further delineation of the 10p deletion syndrome. Pediatrics. 1984;73:670–675. [PubMed] [Google Scholar]
- Emerson E. Prevalence of psychiatric disorders in children and adolescents with and without intellectual disability. J Intellect Disabil Res. 2003;47:51–58. doi: 10.1046/j.1365-2788.2003.00464.x. [DOI] [PubMed] [Google Scholar]
- Evers L, Curfs L, Bakker J, Boot E, da Silva Alves F, Abeling N, Bierau J, Drukker M, van Amelsvoort T. Serotonergic, noradrenergic and dopaminergic markers are related to cognitive function in adults with 22q11 deletion syndrome. Int J Neuropsychopharmacol. 2014;17:1159–1165. doi: 10.1017/S1461145714000376. [DOI] [PubMed] [Google Scholar]
- Feinstein C, Eliez S, Blasey C, Reiss AL. Psychiatric disorders and behavioral problems in children with velocardiofacial syndrome: usefulness as phenotypic indicators of schizophrenia risk. Biol Psychiatry. 2002;51:312–318. doi: 10.1016/s0006-3223(01)01231-8. [DOI] [PubMed] [Google Scholar]
- Fine SE, Weissman A, Gerdes M, Pinto-Martin J, Zackai EH, McDonald-McGinn DM, Emanuel BS. Autism spectrum disorders and symptoms in children with molecularly confirmed 22q11. 2 deletion syndrome. J Autism Dev Disord. 2005;35:461–470. doi: 10.1007/s10803-005-5036-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fryburg JS, Lin KY, Golden WL. Chromosome 22q11. 2 deletion in a boy with Opitz (G/BBB) syndrome. Am J Med Genet. 1996;62:274–275. doi: 10.1002/(SICI)1096-8628(19960329)62:3<274::AID-AJMG13>3.0.CO;2-H. [DOI] [PubMed] [Google Scholar]
- Fukushima Y, Ohashi H, Wakui K, Nishida T, Nakamura Y, Hoshino K, Ogawa K, Ohishi T. DiGeorge syndrome with del(4) (q21. 3q25): Possibility of the fourth chromosome region responsible for Di-George syndrome. Am J Hum Genet. 1992;51:A80. Abstract. [Google Scholar]
- Fung A, McEvilly R, Fong J, Silversides C, Chow E, Bassett A. Elevated prevalence of generalized anxiety disorder in adults with 22q11. 1 deletion syndrome. Am J Psychiatry. 2010;167:998. doi: 10.1176/appi.ajp.2010.09101463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giannotti A, Digilio MC, Marino B, Mingarelli R, Dallapiccola B. Cayler Cardiofacial syndrome and del 22q11: Part of CATCH22 phenotype. Am J Med Genet. 1994;30:807–812. doi: 10.1002/ajmg.1320530320. [DOI] [PubMed] [Google Scholar]
- Goodship J, Cross I, Scambler P, Burn J. Monozygotic twins with chromosome 22q11 deletion and discordant phenotype. J Med Genet. 1995;32:746–810. doi: 10.1136/jmg.32.9.746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodship J, Cross I, LiLing J, Wren C. A population study of chromosome 22q11 deletions in infancy. Arch Dis Child. 1998;79:348–351. doi: 10.1136/adc.79.4.348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottlieb S, Driscoll DA, Punnett HH, Sellinger B, Emanuel BS, Budarf ML. Characterization of 10p deletions suggests two non-overlapping regions contribute to the DiGeorge syndrome phenotype. (Letter) Am J Hum Genet. 1998;62:495–498. doi: 10.1086/301718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Eliez S, Thompson T, Hinard C, Penniman L, Feinstein C, Kwon, Jin, Jo B, Antonarakis SE, Morris MA, Reiss AL. COMT genotype predicts longitudinal cognitive decline and psychosis in 22q11.2 deletion syndrome. Nat Neurosci. 2005;8:1500–1502. doi: 10.1038/nn1572. [DOI] [PubMed] [Google Scholar]
- Gothelf D, Frisch A, Michaelovsky E, Weizman A, Shprintzen R. Velo-cardio-facial syndrome. J Ment Health Res Intellect Disabil. 2009;2:149. doi: 10.1080/19315860902756136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graham A. The development and evolution of the pharyngeal arches. J Anat. 2001;199:133–141. doi: 10.1046/j.1469-7580.2001.19910133.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green T, Gothelf D, Glaser B, Debbane M, Frisch A, Kotler M, Weizman A, Eliez S. Psychiatric disorders and intellectual functioning throughout development in velocardiofacial (22q11. 2 Deletion) Syndrome. J Am Acad Child Psy. 2009;48:1060–1068. doi: 10.1097/CHI.0b013e3181b76683. [DOI] [PubMed] [Google Scholar]
- Greenberg F, Crowder WE, Paschall V, Colon-Linares J, Lubianski B, Ledbetter DH. Familial DiGeorge syndrome and associated partial monosomy of chromosome 22. Hum Genet. 1984;65:317–319. doi: 10.1007/BF00291554. [DOI] [PubMed] [Google Scholar]
- Greenberg F, Elder FFB, Haffner P, Northrup H, Ledbetter DH. Cytogenetic findings in a prospective series of patients with DiGeorge anomaly. Am J Hum Genet. 1988;43:605–611. [PMC free article] [PubMed] [Google Scholar]
- Grossfeld PD, Mattina T, Lai Z, Favier R, Jones KL, Cotter F, Jones F. The 11q terminal deletion disorder: A prospective study of 110 cases. Am J Med Genet. 2004;129:51–61. doi: 10.1002/ajmg.a.30090. [DOI] [PubMed] [Google Scholar]
- Habel A, Herriot R, Kumararatne D, Allgrove J, Baker K, Baxendale H, Bu'Lock F, Firth H, Gennery A, Holland A, Illingworth C, Mercer N, Pannebakker M, Parry A, Roberts A, Tsai-Goodman B. Towards a safety net for management of 22q11. 2 deletion syndrome: Guidelines for our times. Eur J Pediatr. 2014;173:757–765. doi: 10.1007/s00431-013-2240-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder A, Jain M, Kabra M, Gupta N. Mosaic 22q11. 2 microdeletion syndrome: Diagnosis and clinical manifestations of two cases. Mol Cytogenet. 2008;1:18. doi: 10.1186/1755-8166-1-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward C, Sanborn K. Puberty and the emergence of gender differences in psychopathology. J Adolesc Health. 2002;30:49–58. doi: 10.1016/s1054-139x(02)00336-1. [DOI] [PubMed] [Google Scholar]
- Henry JC, Van Amelsvoort T, Morris RG, Owen MJ, Murphy DG, Murphy KC. An investigation of the neuropsychological profile in adults with velo-cardio-facial syndrome (VCFS) Neuropsychologia. 2002;40:471–478. doi: 10.1016/s0028-3932(01)00136-1. [DOI] [PubMed] [Google Scholar]
- Jansen PW, Duijff SN, Beemer FA, Vorstman JA, Klaassen PW, Morcus ME, Heineman-de Boer JA. Behavioral problems in relation to intelligence in children with 22q11. 2 deletion syndrome: A matched control study. Am J Med Gen A. 2007;143A:574–580. doi: 10.1002/ajmg.a.31623. [DOI] [PubMed] [Google Scholar]
- Jerome LA, Papaioannou VE. DiGeorge syndrome phenotype in mice mutant for the T-box gene, Tbx1. Nat Genet. 2001;27:286–291. doi: 10.1038/85845. [DOI] [PubMed] [Google Scholar]
- Jonas R, Montojo C, Bearden CE. The 22q11. 2 deletion syndrome as a window into complex neuropsychiatric disorders over the lifespan. Biol Psychiatry. 2014;75:351–360. doi: 10.1016/j.biopsych.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolin EM, Weller RA, Jessani NR, Zackai EH, Donald-McGinn DM, Weller EB. Affective disorders and other psychiatric diagnoses in children and adolescents with 22q11. 2 Deletion Syndrome. J Affect Disord. 2009;119:177–180. doi: 10.1016/j.jad.2009.02.016. [DOI] [PubMed] [Google Scholar]
- Jyonouchi S, McDonald-McGinn DM, Bale S, Zackai EH, Sullivan KE. CHARGE (coloboma, heart defect, atresia choanae, retarded growth and development, genital hypoplasia, ear anomalies/deafness) syndrome and chromosome 22q11. 2 deletion syndrome: A comparison of immunologic and nonimmunologic phenotypic features. Pediatrics. 2009;123:710–e871. doi: 10.1542/peds.2008-3400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kates W, Antshel K, Fremont W, Shprintzen R, Strunge L, Burnette C, Higgins A. Comparing phenotypes in patients with idiopathic autism to patients with velocardiofacial syndrome (22q11 DS) with and without autism. Am J Med Genet A. 2007;143A:2642–2650. doi: 10.1002/ajmg.a.32012. [DOI] [PubMed] [Google Scholar]
- Kelley RI, Zackai EH, Emanuel BS, Kistenmacher M, Greenberg F, Punnett HH. The association of the DiGeorge anomalad with partial monosomy of chromosome 22. J Pediatr. 1982;101:197–200. doi: 10.1016/s0022-3476(82)80116-9. [DOI] [PubMed] [Google Scholar]
- LaCassie Y, Arriaza MI. Letter to the editor. Opitz GBBB syndrome and the 22q11 deletion syndrome. Am J Med Genet. 1996;62:318. doi: 10.1002/(SICI)1096-8628(19960329)62:3<318::AID-AJMG21>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Lobdell DH. Congenital absence of the thyroid gland. Arch Pathol. 1959;67:412–418. [PubMed] [Google Scholar]
- Matsuoka R, Takao A, Kimura M, Imamura S, Kondo C, Joh-o K, Ikeda K, Nishibatake M, Ando M, Momma K. Confirmation that the conotruncal anomaly face syndrome is associated with a deletion within 22q11. 2. Am J Med Genet. 1994;53:285–289. doi: 10.1002/ajmg.1320530314. [DOI] [PubMed] [Google Scholar]
- Maughan B, Rowe R, Messer J, Goodman R, Meltzer H. Conduct disorder and oppositional defiant disorder in a national sample: Developmental epidemiology. J Child Psychol Psychiatry. 2004;45:609–621. doi: 10.1111/j.1469-7610.2004.00250.x. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Driscoll DA, Bason L, Christensen K, Lynch D, Sullivan K, Canning D, Zavod W, Quinn N, Rome J, Paris Y, Weinberg P, Clark BJ, Emanuel BS, Zackai EH. Autosomal dominant “Opitz” GBBB syndrome due to a 22q11. 2 deletion. Am J Med Genet. 1995;59:103–113. doi: 10.1002/ajmg.1320590122. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Low D, Zackai EH. Letter to the Editor: What's in a name? The 22q11. 2 deletion. Am J Med Genet. 1997a;72:247. [PubMed] [Google Scholar]
- McDonald-McGinn DM, LaRossa D, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Wang P, Solot C, Schultz P, Lynch D, Bingham P, Keenan G, Weinzimer S, Ming JE, Driscoll D, Clark BJ, Markowitz R, Cohen A, Moshang T, Pasquariello P, Randall P, Emanuel BS, Zackai EH. The 22q11. 2 deletion: Screening, diagnostic workup, and outcome of results; report on 81 patients. Genet Test. 1997b;1:99–108. doi: 10.1089/gte.1997.1.99. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Kirschner R, Goldmuntz E, Sullivan K, Eicher P, Gerdes M, Moss E, Solot C, Wang P, Jacobs I, Handler S, Knightly C, Heher K, Wilson M, Ming JE, Grace K, Driscoll D, Pasquariello P, Randall P, Larossa D, Emanuel BS, Zackai EH. The Philadelphia story: The 22q11. 2 deletion: Report on 250 patients. Genet Couns. 1999;10:11–24. [PubMed] [Google Scholar]
- McDonald-McGinn DM, Tonnesen MK, Laufer-Cahana A, Finucane B, Driscoll DA, Emanuel BS, Zackai EH. Phenotype of the 22q11. 2 deletion in individuals identified through an affected relative: Cast a wide FISHing net! Genet Med. 2001;3:23–29. doi: 10.1097/00125817-200101000-00006. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Minugh-Purvis N, Kirschner RE, Jawad A, Tonnesen MK, Catanzaro JR, Goldmuntz E, Driscoll D, Larossa D, Emanuel BS, Zackai EH. The 22q11. 2 deletion in African-American patients: An underdiagnosed population? Am J Med Genet A. 2005a;134:242–246. doi: 10.1002/ajmg.a.30069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Gripp KW, Kirschner RE, Maisenbacher MK, Hustead V, Schauer GM, Keppler-Noreuil KM, Ciprero KL, Pasquariello P, Jr, LaRossa D, Bartlett SP, Whitaker LA, Zackai EH. Craniosynostosis: Another feature of the 22q11. 2 deletion syndrome. Am J Med Genet. 2005b;136:358–362. doi: 10.1002/ajmg.a.30746. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Sullivan KE. Chromosome 22q11. 2 Deletion Syndrome (DiGeorge syndrome/Velocardiofacial syndrome) Medicine. 2011;90:1–18. doi: 10.1097/MD.0b013e3182060469. [DOI] [PubMed] [Google Scholar]
- McDonald-McGinn DM, Fahiminiya S, Revil T, Nowakowska BA, Suhl J, Bailey A, Mlynarski E, Lynch DR, Yan A, Bilaniuk L, Sullivan KE, Warren ST, Emanuel BS, Vermeesch JR, Zackai EH, Jerome-Majewska LA. Hemizygous mutations in SNAP29 unmask autosomal recessive conditions and contribute to a typical findings in patients with 22q11. 2DS. J Med Genet. 2013;50(2):80–90. doi: 10.1136/jmedgenet-2012-101320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merikangas K, He J, Brody D, Fisher P, Bourdon K, Koretz D. 2010. Prevalence and treatment of mental disorders among US children in the 2001- NHANES. Pediatrics. 2004;125:75–81. doi: 10.1542/peds.2008-2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss EM, Batshaw ML, Solot CB, Gerdes M, McDonald-McGinn DM, Driscoll DA, Emanuel BS, Zackai EH, Wang PP. Psychoeducational profile of the 22q11.2 microdeletion: A complex pattern. J Pediatr. 1999;13(2):193–198. doi: 10.1016/s0022-3476(99)70415-4. [DOI] [PubMed] [Google Scholar]
- Murphy KC, Jones LA, Owen MJ. High rates of schizophrenia in adults with velo-cardio-facial syndrome. Arch Gen Psychiatry. 1999;56:940–945. doi: 10.1001/archpsyc.56.10.940. [DOI] [PubMed] [Google Scholar]
- Najmabadi H, Hu H, Garshasbi M, Zemojtel T, Abedini SS, Chen W, Hosseini M, Behjati F, Haas S, Jamali P, Zecha A, Mohseni M, Püttmann L, Vahid LN, Jensen C, Moheb LA, Bienek M, Larti F, Mueller I, Weiss-mann R, Darvish H, Wrogemann K, Hadavi V, Lipkowitz B, Esmaeeli-Nieh S, Wieczorek D, Kariminejad R, Firouzabadi SG, Cohen M, Fattahi Z, Rost I, Mojahedi F, Hertzberg C, Dehghan A, Rajab A, Banavandi MJ, Hoffer J, Falah M, Musante L, Kalscheuer V, Ullmann R, Kuss AW, Tzschach A, Kahrizi K, Ropers HH. Deep sequencing reveals 50 novelgenes for recessive cognitive disorders. Nature. 2011;478:57–63. doi: 10.1038/nature10423. [DOI] [PubMed] [Google Scholar]
- Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Neuropsychiatric disorders in the 22q11 deletion syndrome. Genet Med. 2001;3:79–84. doi: 10.1097/00125817-200101000-00017. [DOI] [PubMed] [Google Scholar]
- Niklasson L, Rasmussen P, Oskarsdottir S, Gillberg C. Autism, ADHD, mental retardation and behavior problems in 100 individuals with 22q11 deletion syndrome. Res Dev Disabil. 2009;30:763–773. doi: 10.1016/j.ridd.2008.10.007. [DOI] [PubMed] [Google Scholar]
- Olszewski AK, Radoeva PD, Fremont W, Kates WR, Antshel KM. Is child intelligence associated with parent and sibling intelligence in individuals with developmental disorders? An investigation in youth with 22q11. 2 deletion (velo-cardio-facial) syndrome. Res Dev Disabil. 2014;35:3582–3590. doi: 10.1016/j.ridd.2014.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oskarsdottir S, Vujic M, Fasth A. Incidence and prevalence of the 22q11. 2 deletion syndrome: A population-based study in Western Sweden. Arch Dis Child. 2004;89:148–151. doi: 10.1136/adc.2003.026880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papangeli I, Scambler P. The 2q11 deletion: DiGeorge and velocardiofacial syndromes and the role of T BX1. Interdiscip Rev Dev Biol. 2013;2:393–40310. doi: 10.1002/wdev.75. [DOI] [PubMed] [Google Scholar]
- Philip N, Bassett A. Cognitive, behavioural and psychiatric phenotype in 22q11. 2 deletion syndrome. Behav Genet. 2011;41:403–412. doi: 10.1007/s10519-011-9468-z. Review. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powis L, Oliver C. The prevalence of aggression in genetic syndromes: A review. Res Dev Disabil. 2014;35:1051–1071. doi: 10.1016/j.ridd.2014.01.033. [DOI] [PubMed] [Google Scholar]
- Ryan AK, Goodship JA, Wilson DI, Philip N, Levy A, Seidel H, Schuffenhauer S, Oechsler H, Belohradsky B, Prieur M, Aurias A, Raymond FL, Clayton-Smith J, Hatchwell E, McKeown C, Beemer FA, Dallapiccola B, Novelli G, Hurst JA, Ignatius J, Green AJ, Winter RM, Brueton L, Brøndum-Nielsen K, Scambler PJ. Spectrum of clinical features associated with interstitial chromosome 22q11 deletions: a European collaborative study. J Med Genet. 1997;34:798–804. doi: 10.1136/jmg.34.10.798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saldivar JS, Monroe T, Boomer T, Wardrop J, Jesiolowski J, Dharajiya N. Identification of 22q11 microdeletions by noninvasive prenatal testing (NIPT) - the clinical experience. Am J Ob Gyn. 2015;212:S178. [Google Scholar]
- Sandrin-Garcia P, Macedo C, Martell LR, Ramos ES, Guion-Almeida ML, Richieri-Costa A, Passos GA. Recurrent 22q11. 2 deletion in a sibship suggestive of parental germline mosaicism in velocardiofacial syndrome. Clin Genet. 2002;61:380–383. doi: 10.1034/j.1399-0004.2002.610511.x. [DOI] [PubMed] [Google Scholar]
- Sanlaville D, Etchevers HC, Gonzales M, Martinovic J, Clement-Ziza M, Delezoide AL, Aubry MC, Pelet A, Chemouny S, Cruaud C, Audollent S, Esculpavit C, Goudefroye G, Ozilou C, Fredouille C, Joye N, Morichon-Delvallez N, Dumez Y, Weissenbach J, Munnich A, Amiel J, Encha-Razavi F, Lyonnet S, Vekemans M, Attié-Bitach T. Phenotypic spectrum of CHARGE syndrome in fetuses with CHD7 truncating mutations correlates with expression during human development. J Med Genet. 2006;43:211–710. doi: 10.1136/jmg.2005.036160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scambler PJ, Carey AH, Wyse RKH, Roach S, Dumanski JO, Nordenskjold M, Williamson R. Microdeletion within 22q11 associated with sporadic and familial DiGeorge syndrome. Genomics. 1991;10:201–206. doi: 10.1016/0888-7543(91)90501-5. [DOI] [PubMed] [Google Scholar]
- Schneider M, Debbane M, Bassett AS, Chow EW, Fung WL, van den Bree M, Owen M, Murphy KC, Niarchou M, Kates WR, Antshel KM, Fremont W, McDonald-McGinn DM, Gur RE, Zackai EH, Vorstman J, Duijff SN, Klaassen PW, Swillen A, Gothelf D, Green T, Weizman A, Van Amelsvoort T, Evers L, Boot E, Shashi V, Hooper SR, Bearden CE, Jalbrzikowski M, Armando M, Vicari S, Murphy DG, Ousley O, Campbell LE, Simon TJ, Eliez S. International consortium on brain and behavior in 22q11. 2 deletion syndrome. Psychiatric disorders from childhood to adulthood in 22q11. 2 deletion syndrome: Results from the international consortium on brain and behavior in 22q11. 2 deletion syndrome. Am J Psychiat. 2014;171:627–639. doi: 10.1176/appi.ajp.2013.13070864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sedlakova E. Insufficiency of palatolaryngeal passage as a developmental disorder. Cas Lek Cesk. 1955;94:1304–1307. [PubMed] [Google Scholar]
- Shaikh TH, Kurahashi H, Saitta SC, Hu P, Rose BA, Driscoll DA, McDonald-McGinn DM, Zackai EH, Budarf ML, Emanuel BS. Chromosome 22-specific low copy repeats and the 22q11. 2 deletion syndrome: Genomic organization and deletion endpoint analysis. Hum Mol Genet. 2000;9:489–501. doi: 10.1093/hmg/9.4.489. [DOI] [PubMed] [Google Scholar]
- Shashi V, Keshavan MS, Howard TD, Berry MN, Basehore MJ, Lewandowski KE, Kwapil TR. Cognitive correlates of a functional COMT polymorphism in children with 22q11. 2 deletion syndrome. Clinical Genetics. 2006;69:234–238. doi: 10.1111/j.1399-0004.2006.00569.x. [DOI] [PubMed] [Google Scholar]
- Shashi V, Keshavan MS, Kaczorowski J, Schoch K, Lewandowski KE, McConkie-Rosell A, Hooper S, Kwapil TR. Socioeconomic status and psychological function in children with chromosome 22q11. 2 deletion syndrome: Implications for genetic counseling. J Genet Couns. 2010;19:535–544. doi: 10.1007/s10897-010-9309-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shprintzen RJ. Velo-cardio-facial syndrome: 30 Years of study. Dev Disabil Res Rev. 2008;14:3–10. doi: 10.1002/ddrr.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorensen KM, Agregaard P, Olesen C, Andersen PS, Larsen LA, Ostergaard JR, Schouten JP, Christiansen M. Detecting 22q11. 2 deletions by use of multiplex ligation-dependent probe amplification on DNA from neonatal dried blood spot samples. J Mol Diagn. 2010;12:147–151. doi: 10.2353/jmoldx.2010.090099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulik KK, Johnston MC, Daft PA, Russell WE, Dehart DB. Fetal alcohol syndrome and DiGeorge anomaly: Critical ethanol exposure periods for craniofacial malformations as illustrated in an animal model. Am J Med Genet Suppl. 1986;2:97–112. doi: 10.1002/ajmg.1320250614. [DOI] [PubMed] [Google Scholar]
- Swillen A, Devriendt K, Legius E, Eyskens B, Dumoulin M, Gewillig M, Fryns JP. Intelligence and psychosocial adjustment in velo-cardio-facial syndrome: A study of 37 children and adolescents with VCFS. J Med Genet. 1997;34:453–458. doi: 10.1136/jmg.34.6.453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swillen A, Devriendt K, Legius E, Prinzie P, Vogels A, Ghesquière P, Fryns JP. The behavioural phenotype in velo-cardio-facial syndrome(VCFS): From infancy to adolescence. Genet Couns. 1999;10:79–88. [PubMed] [Google Scholar]
- Swillen A, Devriendt K, Ghesquiere P, Fryns JP. Children with a 22q11 deletion versus children with a speech-language impairment and learning disability: Behavior during primary school age. Genet Couns. 2001;12:309–317. [PubMed] [Google Scholar]
- Tezenas Du Montcel ST, Mednizibal H, Ayme S, Levy A, Philip N. Prevalence of 22q11 microdeletion. J Med Genet. 1996;33:719. doi: 10.1136/jmg.33.8.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomita-Mitchell A, Mahnke D, Larson JM, Ghanta S, Feng Y, Simpson P, Broeckel U, Duffy K, Tweddell JS, Grossman WJ, Routes JM, Mitchell M. Multiplexed quantitative real-time PCR to detect 22q11.2 deletion in patients with congenital heart disease. Physiol Genomics. 2010;42A:52–60. doi: 10.1152/physiolgenomics.00073.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorstman JA, Morcus M, Duijff S, Klaassen P, Heineman-de Boer J, Beemer F, Swaab H, Kahn R, van Engeland H. The 22q11.2 deletion in children: High rate of autistic disorders and early onset of psychotic symptoms. J Am Acad Child Adolesc Psychiatry. 2006;45:1104–1113. doi: 10.1097/01.chi.0000228131.56956.c1. [DOI] [PubMed] [Google Scholar]
- Vorstman JA, Breetvelt E, Duijff S, Eliez S, Schneider M, Jalbrzikowski M, Armando M, Vicari S, Shashi V, Hooper S, Chow E, Fung An, Butcher N, Young D, McDonald-McGinn D, Vogels A, van Amelsvoort T, Gothelf D, Weinberger R, Weizman A, Klaassen P, Koops S, Kates W, Antshel K, Simon T, Ousley O, Swillen A, Gur R, Bearden C, Kahn R, Bassett AS the International 22q11. 2 Brain Behavior Syndrome Consortium. A cognitive decline precedes the onset of psychosis in patients with the 22q11. 2 deletion syndrome. JAMA. 2015 Feb 25; doi: 10.1001/jamapsychiatry.2014.2671. Epub ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zweier C, Sticht H, Aydin-Yaylagul I, Campbell CE, Rauch A. Human TBX1 missense mutations cause gain of function resulting in the same phenotype as 22q11. 2 deletions. Am J Hum Genet. 2007;80:510–710. doi: 10.1086/511993. [DOI] [PMC free article] [PubMed] [Google Scholar]