

Abstract
While adaptive mutations can confer proteins with new function via the introduction or optimization of reactive centers, or other structural changes, a role for the optimization of protein dynamics also seems likely but has been more difficult to evaluate. Antibody (Ab) affinity maturation is an example of adaptive evolution wherein the adaptive mutations may be identified and Abs may be raised to specific targets that facilitate the characterization of protein dynamics. Here, we report the characterization of three affinity matured Abs that evolved from a common germline precursor to bind the chromophoric antigen (Ag), 8-methoxypyrene-1,3,6-trisulfonate (MPTS). In addition to characterizing the sequence, molecular recognition, and structure of each Ab, we characterized the dynamics of each complex by determining their mechanical response to an applied force via 3-pulse photon echo peak shift (3PEPS) spectroscopy and deconvoluting the response into elastic, anelastic, and plastic components. We find that for one Ab, affinity maturation was accomplished via the introduction of a single functional group that mediates a direct contact with MPTS and which results in a complex with little anelasticity or plasticity. In the other two cases, more mutations were introduced, but none directly contact MPTS, and while their effects on structure are subtle, their effects on anelasticity and plasticity are significant, with the level of plasticity correlated with specificity, suggesting that the optimization of protein dynamics may have contributed to affinity maturation. A similar optimization of structure and dynamics may contribute to the evolution of other proteins.
Graphical Abstract
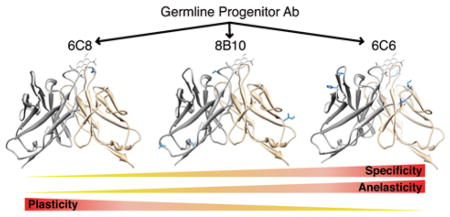
The evolution of novel protein function is a hallmark of all biological systems and a subject of intense interest. A challenge to characterizing the process is that it is typically difficult, if not impossible, to unambiguously determine the specific adaptive mutations that conferred a new function due to complex genetic interactions and the presence of the many neutral mutations that accumulate on the timescale of evolution. In addition, while it is obvious that mutations may confer new activities by installing or optimizing functionality, or by introducing other changes to the protein’s structure,1–3 dynamics may also be important. Indeed, it is dynamics that differentiates the limiting models of molecular recognition – flexibility is required for induced fit4- or conformational selection5–7- like recognition and rigidity is required for lock-and-key-like recognition.8 In addition, specificity is also an important selection pressure, and different levels of dynamics are inherently associated with different levels of specificity (just as flexibility allows for the adoption of structures involved in induced fit or conformational selection-like recognition, it will allow for the adoption of other conformations that recognize other targets). However, the characterization of protein dynamics is less straightforward than the characterization of structure, and the problem is further complicated by the fact that proteins have a vast number of internal motions, of which only a small subset is expected to contribute to a given function or to be subject to optimization during evolution.
Perhaps one of the most intuitive approaches to understanding the dynamics of any material is based on the response to an applied force.9–12 The resulting deformations may be characterized based on the timescale of their response (Figure 1A): elastic deformations recover on the timescale of bond vibrations and arise from motions within a single potential energy minimum (e.g. inertial side chain motions); anelastic deformations recover over time and arise from transitions between conformational substates separated by relatively low barriers (e.g. ring flips and backbone fluctuations); and plastic deformations, although often defined as permanent because their timescale of recovery exceeds that of the experiment, recover on the longest timescale13,14 and correspond to transitions between states separated by high energy barriers (e.g. larger loop motions and conformational changes). To apply the same approach to the study of protein dynamics, a useful timescale to differentiate anelastic and plastic deformations is nanoseconds, the lifetime of an initial encounter complex.15,16 Although this is a significantly shorter timescale than typically used with bulk materials, it provides a functionally useful differentiation of protein dynamics as the timescales of elastic and anelastic deformations are then fast, allowing them to compete with dissociation of the encounter complex and thereby facilitate induced fit-like recognition, while plastic deformations are slow and produce the sufficiently long-lived conformational heterogeneity that defines conformational selection-like recognition. When combined with the requirement of lock-and-key mechanisms for relatively small deformations of any kind, this choice of timescale makes it possible to associate each mechanism of molecular recognition with a specific profile of dynamics.
Figure 1.

Schematic representation of barrier crossings on a protein free-energy landscape (A) and peak shift decay (B) corresponding to elastic, anelastic, and plastic deformations. Coordinate Q represents a projection of all internal degrees of freedom of the system.
In principle, modern ultrafast nonlinear optical methods, such as 3-pulse photon echo peak shift (3PEPS) spectroscopy,17–19 are ideally suited to the characterization of protein dynamics because they reveal the response of a chromophore’s environment to the force exerted by an excitation-dependent change in the chromophore’s dipole moment. Briefly, in a 3PEPS experiment, two laser pulses are used to inscribe a phase grating in the sample, which then decays due to environmental fluctuations due to the relaxation of motions displaced by photoexcitation. This decay is monitored by applying a third pulse to the sample after a variable time delay that triggers rephasing of the chromophores whose environment remained unchanged, and thereby results in stimulated emission (the echo signal). The decay of the stimulated emission, measured as a decay in the peak shift of its integrated intensity relative to time zero,17 reflects the time scales and relative amplitudes of environmental dynamics in the vicinity of the chromophore. Decays on the femto- to nanosecond timescale may be attributed to elasticity and anelasticity (depending on their precise timescale), while slower motions are manifest as a static offset in the 3PEPS decay,19,20 indicating the presence of plasticity (Figure 1B). Moreover, while the technique is only sensitive to motions that are coupled to the chromophore’s transition dipole, if the chromophore can be associated specifically with the biologically relevant part of the protein (i.e. ligand or substrate binding site), then it enables the specific characterization of only those motions that are most likely to be biologically relevant.
One of the most remarkable examples of the evolution of molecular recognition is provided by the immune system, wherein antibodies (Abs) with specificity for virtually any foreign molecule (or antigen, Ag) are evolved within days from a limited set of precursors (i.e. germline Abs).21 The Ag binds within a combining site that is formed from loops of hypervariable sequence (or complementarity determining regions), three from the heavy chain (VH) and three from the light chain (VL) polypeptides (VH CDR 1–3 and VL CDR 1–3, respectively). A vast range of Ag may be recognized by a finite number of germline Abs because each Ab is polyspecific and able to recognize a broad range of targets with at least moderate affinity, thus triggering the introduction of adaptive mutations that optimize Ag recognition in a process known as affinity maturation.22–25 We26–28 and others1,2 have suggested that the polyspecificity present in the repertoire of germline Abs results from high levels of flexibility and conformational heterogeneity and that the evolution of specificity during affinity maturation results, at least in part, from conformational restriction. We have also suggested that a similar modulation of protein dynamics may contribute to the evolution of altered specificity with other proteins.26–28 Regardless, by evolving Abs to a chromophoric Ag, the chromophore is by definition bound in a biologically relevant manner, and adaptive mutations may be unambiguously identified and their biologically relevant effects on molecular recognition characterized via 3PEPS spectroscopy.
To study the evolution of molecular recognition, we have applied 3PEPS spectroscopy to Abs that were evolved to bind the chromophoric Ag fluorescein (Fl) or 8-methoxypyrene-1,3,6-trisulfonate (MPTS, Figure 2). We characterized panels of both anti-Fl and anti-MPTS Abs to examine the range of dynamics present within the different Abs.26,27,29,30 In the case of one anti-Fl Ab, we characterized dynamics as a function of affinity maturation and found that it reduced both anelasticity and plasticity, suggesting that Abs are indeed conformationally restricted during affinity maturation.28 Here, we report the identification of three novel anti-MPTS Abs, 6C8, 8B10, and 6C6. Interestingly, the three Abs were found to have diverged from a common ancestral germline, and thus any differences are by definition the result of the different mutations acquired. Characterization of the Abs revealed that while each evolved more optimal MPTS recognition, they did so via different mechanisms that are associated with rather different overall profiles of molecular recognition. Interestingly, for one Ab the unique profile of molecular recognition resulted from the introduction of a specific functional group, while for the other Abs the structural changes are more subtle and the differences in molecular recognition appear to have resulted at least in part from relatively larger changes in anelasticity and/or plasticity. The results suggest that the optimization of both structure and dynamics likely contribute to Ab affinity maturation, and by extension, to the evolution of novel function with other proteins.
Figure 2.
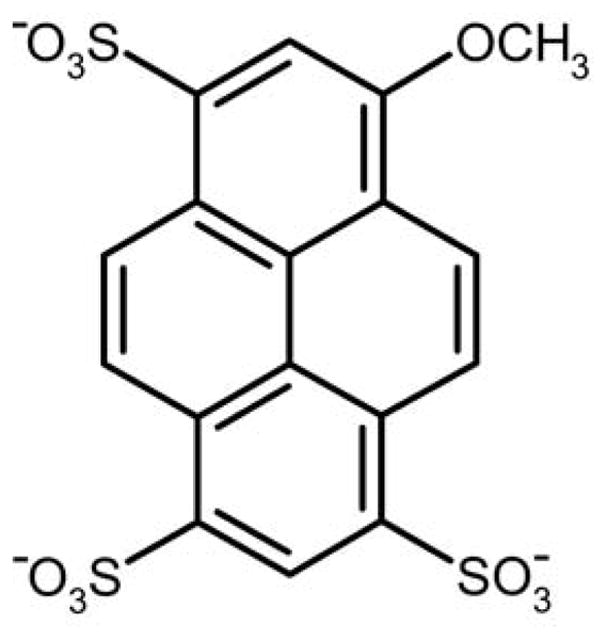
Structure of MPTS.
Materials and Methods
Generation and sequencing of anti-MPTS Abs
Abs were raised in Swiss Webster mice via immunization with MPTS conjugated to keyhole limpet hemocyanin (KLH); and monoclonal IgG antibodies were produced and purified from hybridoma supernatants by standard methods.31 To sequence the heavy-chain variable (VH) and light-chain variable (VL) gene regions, mRNA was extracted from fresh hybridoma cells (Qiagen RNeasy Kit) and used with oligonucleotide (dT)18 to synthesize cDNA (Superscript III Kit, Invitrogen). The VH and VL genes from the resulting cDNA were PCR-amplified using a specifically designed primer set for cloning mouse immunoglobulin regions32,33 and then sequenced. Nucleotide sequences of the VH and VL genes are provided in the Supporting Information.
Isothermal titration calorimetry (ITC)
Calorimetric titration of each Ab with MPTS in 1× PBS (phosphate buffered saline, pH 7.4) was performed at 20 °C, 25 °C, and 30 °C. For each ITC experiment, a 6 μM solution of antibody was titrated by injecting a 120 μM solution of MPTS. Each titration consisted of a preliminary 0.6 μL injection followed by 19 subsequent 2-μL additions, which were performed over 4 s periods at 120 s intervals. Thermodynamics parameters of Ag binding are provided in the Supporting Information.
ELISA procedure
Each well of a 96-well plate was coated with one of forty-five randomly selected proteins (see Table S3 of the Supporting Information) by addition of 100 μL of a stock solution and incubation at 4 °C overnight. The plates were then washed three times with 150 μL of 1× PBS. After washing, each well was blocked with 150 μL of 3% BSA in 1× PBS by incubating at 37 °C for 2 h. Next, 80 μL of a stock solution of each primary antibody (11.5 μg/mL for 6C8, 9.0 μg/mL for 8B10 and 12.3 μg/mL for 6C6) was added to each well, and the plates were incubated at 37 °C for 2 h. Wells were then washed an additional six times with 200 μL of ELISA washing buffer (0.05% Tween20 in 1× PBS). After the wash step, 100 μL of secondary antibody (1:2000 diluted chicken anti-mouse IgG Fc secondary antibody conjugated with HRP, Pierce/Thermo Scientific, IL, USA) was added into each well, and incubated at 37 °C for 2 h. After six additional washes with ELISA washing buffer, 200 μL of freshly prepared o-phenylenediamine (OPD) (Sigma-Aldrich, MO, USA)/H2O2 solution was added to each well. After 20 min, the reaction was quenched with 50 μL of 3M H2SO4, and absorbance at 492 nm was recorded (EnVision 2103 Multilabel Reader, Perkin Elmer). For quantification, a control well for each of the forty-five proteins was treated identically, with the exception that no primary Ab was added, and the absolute absorbance for the experimental well was calculated by subtraction. ELISA data for each protein and antibody was collected in triplicate.
X-ray crystallography
Abs 8B10 (mouse, IgG2a, kappa) and 6C8 (mouse, IgG2b, kappa) IgG were purified from ascites fluid by standard methods and digested to Fab or F(ab′)2 with papain or pepsin, respectively. Papain (Sigma) was activated with 10 mM cysteine and 3 mM EDTA in 0.1 M sodium acetate (pH 5.5) at 37 °C for 15 min. Purified 8B10 IgG was digested with 2% (w/w) activated papain for 2 h at 37 °C, and the reaction was stopped by addition of iodoacetamide to a final concentration of 30 mM. The 6C8 IgG solution was adjusted to pH 3.5 with 1.0 M sodium acetate, and then digested with pepsin (Sigma, 4 % (w/w)) for 3 h at 37 °C. The reaction was stopped by addition of 3 M Tris to a final pH of 7. F(ab′)2 was then reduced to Fab′ with 15 mM cysteine, pH 7, for 2 h, and the reaction was stopped by adding iodoacetamide to a final concentration of 30 mM. In both cases, Fab or Fab′ was separated from Fc by affinity chromatography (HiTrap protein A; GE healthcare). Both Fabs were then further purified by size exclusion chromatography using a Superdex 200 16/60 column (GE Healthcare) in 20 mM Tris, 150 mM NaCl, pH 7.5.
In the same buffer used for size exclusion chromatography, Fabs were concentrated to 9.2 mg/mL and 4.8 mg/mL for 8B10 and 6C8 respectively, and then crystallized as follows. Aqueous MPTS (50 mg/mL) solution was added to Fab in a 10:1 (MPTS:Fab) molar excess and the Fab:MPTS complexes were screened for crystallization with the JCSG/IAVI/TSRI Crystalmation system (Rigaku) using the JCSG Core Suite (Qiagen) reagents kit followed by manual optimization of promising conditions. Crystals of the 6C8-MPTS complex were grown in sitting drops (0.4 μL protein plus 0.4 μL well solution) against 20 mM zinc acetate, 12% PEG 8000, 0.1 M MES, pH 6.5, at 4 °C. Crystals of the 8B10-MPTS complex were grown in sitting drops (0.4 μL protein plus 0.4 μL well solution) against 200 mM zinc acetate, 18% PEG 3350, at 20 °C. Crystals were cryoprotected by brief immersion in a mixture of 30% glycerol, 70% well solution (6C8-MPTS) or 30% ethylene glycol, 70% well solution (8B10-MPTS) followed by flash-cooling in liquid nitrogen. For both complexes, data were collected from a single crystal at APS beamline 23-ID-D. The 8B10 structure was determined by molecular replacement using Phaser34 and the coordinates of Fab 29G12 (PDB 1MEX) as a model, and the 6C8 structure was determined in a similar fashion using the 8B10 coordinates as a model. In both cases, the Fab search models were split into separate variable and constant domains to allow for variation in the Fab elbow angle. Structures were refined with Refmac535 and model building was performed with Coot.36 Both structures had one Fab-MPTS complex in the asymmetric unit. Data collection statistics and final refinement statistics are listed in Table 1, and a superposition of the two complexes is shown in Figure S5 in the Supporting Information. Coordinates and data have been deposited in the Protein Data Bank with deposition codes 4NJ9 for 8B10-MPTS and 4NJA for 6C8-MPTS.
Table 1.
X-ray Data Collection and Refinement Statistics
| 6C8-MPTS | 8B10-MPTS | |
|---|---|---|
| PDB ID | 4NJA | 4NJ9 |
| Beamline | APS 23-ID-D | APS 23-ID-D |
| Wavelength (Å) | 1.03318 | 1.03318 |
| Resolution (Å) | 42.60-2.20 (2.24-2.20) | 32.90-1.95 (1.98-1.95) |
| Space group | P21212, | P212121, |
| Cell dimensions (a,b,c (Å)) | 47.79, 92.66, 127.84 | 37.13, 58.44, 212.85 |
| # observations | 118,952 (5656) | 198,227 (9671) |
| # unique reflections | 28,547 (1414) | 35,255 (1727) |
| Completeness (%) | 97.2 (99.2) | 99.9 (100.0) |
| Rsym (%) | 7.5 (55.5) | 7.0 (66.8) |
| Rr.i.m (%) | 8.6 (63.8) | 7.7(73.7) |
| Rp.i.m (%) | 3.7(27.2) | 3.2(31.1) |
| < I>/<σ> | 18.3 (2.7) | 21.5 (2.2) |
| Refinement statistics (all refl. > 0.0 F/σF) | ||
|
| ||
| Resolution (Å) | 42.60-2.20 (2.26-2.20) | 32.90-1.95 (2.20-1.95) |
| # reflections (working set) | 27,076 (1742) | 33,113 (2167) |
| # reflections (test set) | 1446 (92) | 1752 (105) |
| Rwork (%) | 20.7 (34.2) | 21.4 (33.6) |
| Rfree (%) | 25.7 (38.8) | 26.3 (37.0) |
| Number of atoms (protein/MPTS/solvent) | 3338/30/151 | 3286/30/254 |
| Stereochemical parameters | ||
| Wilson B (Å2) | 34.1 | 27.4 |
| Average isotropic B (Å2) | 51.1 | 37.8 |
| All atoms | ||
| Average isotropic B (Å2) | 46.4, 56.4 | 36.9, 38.5 |
| Protein chains (L,H) | ||
| Average isotropic B (Å2) | 40.5 | 33.4 |
| MPTS | ||
| Average isotropic B (Å2) | 46.9 | 39.5 |
| Waters | ||
| rmsd bond lengths (Å) | 0.015 | 0.017 |
| rmsd bond angles (°) | 1.82 | 1.87 |
| Ramachandran plot distribution from Molprobity | 94.9% favored | 97.4% favored |
| 99.5% allowed | 100% allowed | |
| 0.5% disallowed | 0.0% disallowed | |
Rsym = Σ|Ii − <Ii>|/Σ|Ii|, where Ii is the scaled intensity of the ith measurement and <Ii> is the mean intensity for that reflection. Estimated Rr.i.m.=Rsym[N/(N−1)]1/2,where N=data multiplicity. Estimated Rp.i.m. = Rsym[1/(N−1)]1/2, where N=data multiplicity. Rcryst = Σ||Fobs| − |Fcalc||/Σ|Fobs|, where Fcalc and Fobs are the calculated and observed structure factor amplitudes, respectively. Rfree = as for Rcryst, but for 5.0% of the total reflections chosen at random and omitted from refinement.
3PEPS spectroscopy
All samples were prepared in 1× PBS (pH 7.4) using a stock solution of 2.9 mg/mL for 6C8, 2.6 mg/mL for 8B10, 3.9 mg/mL for 6C6, and 4.95 mM for MPTS. The required amount of MPTS and Ab (IgG) stock solutions were mixed such that a concentration of 700 μM of MPTS-Ab complex was achieved when the total solution was concentrated to the final volume of 100 μL. To ensure complete removal of unbound MPTS from the Ab-MPTS complexes, samples were washed by repeated dilution with 1× PBS and microfiltration using a 10 kDa MWCO centrifugal filter unit (Amicon Ultra, Millipore) until the filtrate was free of MPTS as determined by UV-Vis absorption. Absorption spectra of free and bound MPTS are shown in Figure S2 in the Supporting Information.
A detailed description of the experimental setup for 3PEPS has been reported previously.26 Briefly, the frequency-doubled pulses (~45 fs, 416 nm, 200 nJ) of the fundamental beam (836 nm) of a Ti:sapphire regenerative amplifier (Spitfire, Spectra Physics) was used as the excitation source. The excitation beams was split into three, roughly equal portions, arranged in an equilateral triangle and focused using a plano-convex lens (200 mm focal length) on the sample contained in a spinning cell with a path length of 250 μm. Each excitation pulse was ~20–25 nJ. The photon echo signals from the Ab-MPTS sample in the two phase-matched directions k1−k2+k3 and −k1+k2+k3 were detected with two large area avalanche photodiodes (Advanced Photonics). At least three independent experiments were performed for each Ab. The 3PEPS decays were fit using a model spectral density function formalism20 (for details see Supporting Information). Data fitting was performed on the 64-bit Linux cluster Garibaldi at The Scripps Research Institute using a custom suite of C programs developed in our laboratory based on the code provided by Dr. Delmar Larsen (University of California, Davis, CA). For representative 3PEPS fits and further experimental details see Supporting Information.
Results
Characterization of Ab evolution and molecular recognition
From a panel of anti-MPTS Abs, we identified three, 6C8, 8B10, and 6C6, that are homologous and share virtually identical VH recombination junctions,22,37,38 indicating that they arose from a common germline precursor (Figure 3). Thus, the sequence differences may be ascribed to affinity maturation. Consistent with the conclusion, the consensus sequence of the light and heavy chains are virtually identical to the germline genes IGKV3-5*01 F and IGHV1-34*01 F, respectively.39 This analysis reveals that Ab 6C8 evolved via a single VL mutation, I30N (IL30N); Ab 8B10 evolved via four mutations, NL76I, AH16T, DH85E, and IH100S; and Ab 6C6 evolved via five mutations, DL27cN, NL53I, EL55D, GH56D, and NH57H (Kabat numbering40).
Figure 3.

Amino acid sequence of the anti-MPTS Abs. CDRs are underlined. Kabat numbering is shown at the top.50 The consensus sequence is listed first with identical residues in the three the anti-MPTS Abs indicated with a dash.
To characterize the recognition of MPTS, isothermal titration calorimetry was used to determine the dissociation constant (KD) and binding enthalpy (ΔH°) for each sibling Ab, and these values were used to calculate the binding free energy (ΔG°) and entropy (ΔS°) (Table 2). Ab 6C6 binds MPTS with the highest affinity (~40 nM), resulting from a favorable ΔH° as well as a relatively favorable ΔS°, while Ab 8B10 binds MPTS with an intermediate affinity (~100 nM). Ab 6C8 binds MPTS with the lowest affinity (~600 nM) of the three siblings due to a less favorable ΔH°, despite a more favorable ΔS°.
Table 2.
Thermodynamic parameters at T = 298 K.
| KD (nM) | ΔG° (kcal/mol) | ΔH° (kcal/mol) | TΔS° (kcal/mol) | |
|---|---|---|---|---|
| 6C8 | 600 ± 100 | −8.5 ± 0.1 | −8.6 ± 0.04 | −0.1 ± 0.1 |
| 8B10 | 108 ± 8 | −9.6 ± 0.1 | −11.11 ± 0.01 | −1.5 ± 0.1 |
| 6C6 | 36 ± 7 | −10.2 ± 0.1 | −10.96 ± 0.03 | −0.7 ± 0.1 |
To more broadly characterize the molecular recognition of each Ab, we determined their relative affinities for forty-five randomly selected proteins via a standard ELISA (Figure 4). The broadest range of proteins is recognized by Ab 6C8. Compared to Ab 6C8, Ab 6C6 binds eight proteins more tightly, eleven with similar affinity, and twenty-six with lower affinity. In contrast, scarcely any of the proteins are recognized by Ab 8B10, which is thus apparently more specific for MPTS.
Figure 4.

Repertoire of proteins recognized by each anti-MPTS Ab as determined by ELISA. The 45 randomly selected proteins (see Table S2) are arranged according to the affinity with which they are bound by 6C8. Data were collected in triplicate and bars represent average absorbance normalized by Ab concentration.
Characterization of Ab structure
Crystal structures of the 8B10-MPTS and 6C8-MPTS complexes were determined by molecular replacement to 1.95 Å and 2.20 Å resolution, respectively, with one Fab-MPTS complex per asymmetric unit. The two complexes are similar with RMSDs of only 0.42 Å for all Cα atoms within the variable domains (Figure 5). In each case, MPTS is bound in a similar fashion in a shallow pocket and buries ~230 Å2 and ~250 Å2 of MPTS and Ab surface area, respectively (MS, 1.7 Å probe radius),41 with ~55% of the buried Ab surface provided by the VL. The binding site is located on one face of VH CDR3, and is formed by GlyH97, TyrH98, GlyH99, SerH100, ArgH100a, GlyH100b, and TyrH100c, as well as TyrL28, IleL30, PheL32, ArgL50 and AsnL53 of VL CDR1 and CDR2. IleL30 and PheL32 form the floor of the binding pocket, while VH CDR3, ArgL50, and TyrL28 form a wall of the pocket that separates MPTS from the geometrical center of the Ab combining site. Two of the three MPTS sulfonate groups are oriented towards this wall, where a significant level of specificity appears to be mediated by five H-bonds, which are the only intermolecular H-bonds formed in the 8B10 and 6C8 complexes. Specifically, one sulfonate forms an H-bond with the backbone NH moieties of TyrH98 and GlyH99, and the side-chain of ArgL50, while the second sulfonate forms H-bonds with both the NH backbone and side-chain of ArgH100. In the 8B10 complex, the methoxy group and remaining sulfonate on the opposite side of MPTS are not engaged by the Ab. The IL30N somatic mutation in 6C8 is located in the floor of the pocket and introduces a side-chain H-bond with the third sulfonate group of MPTS, which results in the Ag being simultaneously engaged from opposite sides. A model for 6C6-MPTS was constructed using the 6C8-MPTS complex as a template (for details see Supporting Information). Comparison of the 6C6 model with the crystal structures revealed a similar binding pocket with the same five H-bonds observed in the 8B10 structure. As expected, the framework regions of all three complexes were superimposable with the largest differences observed in the CDR loops (Figure 5). Nonetheless, the differences were still small among the CDR loops that form the MPTS binding site (VL CDR1, VL CDR2, VL CDR3, and VH CDR3), and larger differences were observed only with the more distal VH CDR1 and VH CDR2 loops.
Figure 5.

Overlay of the 6C8 (blue) and 8B10 (red) crystal structures, and the 6C6 (grey) model structure based on the superposition of the constant regions.
Characterization of Ab dynamics
3PEPS characterization of each Ab-MPTS complex revealed four timescales of dynamics (Table 3 and Figure 6). Each complex showed a similar sub-100 fs decay that is attributed to elastic deformations associated with water molecules or charged protein side-chains. To fit this decay component, the frequency (ωBO) and damping constant (ΓBO) of a Brownian oscillator function were fixed to 550 fs and 320 cm−1, respectively, and the reorganization energy (λBO) was allowed to vary freely, yielding a value of ~400 cm−1 for each Ab-MPTS complex. Each complex also exhibited two ps timescale motions associated with anelastic deformations that were fit with a Kubo function. The faster of these decays is again similar among all the complexes, with time constants (τK1) of ~0.5 ps and reorganization energies (λK1) of ~90 cm−1, and is attributed to protein motions or to the motion of proximal water molecules, which are known to cause echo dephasing on this timescale.26 The slower ps decay is similar with 8B10 and 6C6 (τK2 = 4.7 ps and λK2 ~ 100 cm−1), but has a longer timescale (τK2 = 9.8 ps) and a lower amplitude (λK2 = 58 cm−1) with 6C8. Decays on this timescale are not observed for MPTS in solution and are thus attributed to protein motions.26 Finally, Ab 6C6 alone showed a longtime signal offset (Δinh = 138 cm−1), which corresponds to a decay on timescales longer than the experimental time window (~1 ns), and which is assigned to protein motions that interconvert different conformations of the protein, and thus to protein plasticity.28,29
Table 3.
Fit parameters for 3PEPS data
| elastic
|
anelastic
|
plastic
|
||||||
|---|---|---|---|---|---|---|---|---|
| λBO (cm−1) | ωBO (cm−1)a | ΓBO (cm−1)a | λK1 (cm−1) | τK1 (fs) | λK2 (cm−1) | τK2 (ps) | Δinh (cm−1) | |
| 6C8 | 430 ± 30 | 550 | 320 | 103 ± 8 | 500 ± 160 | 58 ± 14 | 9.8 ± 2.6 | 2 ± 1 |
| 8B10 | 414 ± 20 | 550 | 320 | 98 ± 7 | 470 ± 110 | 98 ± 18 | 4.7 ± 1.2 | 0 ± 1 |
| 6C6 | 420 ± 30 | 550 | 320 | 65 ± 36 | 580 ± 120 | 97 ± 7 | 4.7 ± 2.3 | 138 ± 10 |
Parameter fixed during fit.
Figure 6.

3PEPS decays for Abs 6C8, 8B10, and 6C6 (symbols, data points; lines, best fit to data).
Discussion
The hallmark of adaptive immunity is the evolution of Abs with exquisite specificity for virtually any foreign Ag from a relatively small number of precursor germline Abs. Interestingly, Abs 6C8, 8B10, and 6C6 diverged from a common germline Ab. While Ab 6C8 is only differentiated from the common germline precursor by a single mutation, suggesting that it was isolated during an early stage of its evolution, Abs 8B10 and 6C8 are differentiated by four and five mutations, respectively, which is more typical for Abs isolated via similar protocols.42–45 Regardless of their state of affinity maturation, the unique mutations in each Ab reveal that multiple pathways exist for the evolution of MPTS recognition, and the fact that each evolved from a common germline precursor allows for the differences between the Abs to be ascribed to their different pathways of evolution. Our analysis revealed that while each pathway optimized MPTS recognition, they did so in different ways, and as a result, each Ab possesses a rather different overall profile of molecular recognition. Nonetheless, a similar level of elasticity is observed for each Ab-MPTS complex, which is perhaps not unexpected since each has a similar disposition of charged protein side-chains and water molecules, and elasticity is largely insensitive to structural details.46 Thus, the differences in molecular recognition appear to result from changes in Ab structure, anelasticity, and/or plasticity.
Ab 6C8 has the lowest affinity for MPTS, but binds MPTS with the most favorable entropy, and of the three siblings it recognizes the broadest range of targets. The complex with MPTS shows no plasticity and has the lowest level of anelasticity of the siblings. Ab 6C8 acquired only the IL30N mutation during its evolution, and the structure reveals that this mutation installs an H-bond with the third sulfonate group of MPTS (Figure 7A). This intermolecular H-bond likely forces desolvation of both the Ab and MPTS upon complex formation, which accounts for the more favorable binding entropy. In addition, relative to the already present H-bonds, the new H-bond engages MPTS from the opposite side, which otherwise does not interact with the Ab. Thus, combined, the H-bonds act as a pincer that clamps MPTS in place, which likely accounts for the low levels of anelasticity and plasticity observed with this complex. The rigidity of the complex is clearly dependent on the presence of MPTS, and is not expected to be manifest in the free Ab, where increased rigidity might be expected to restrict molecular recognition. In fact, the high polyspecificity of Ab 6C8 suggests that the free Ab is more flexible than its siblings, thereby allowing it to accommodate a large range of targets via an induced-fit or conformational selection mechanism. This is consistent with 6C8 being only a single mutation removed from the presumably flexible and polyspecific germline Ab. Along with its modest affinity for MPTS, this suggests that this Ab was isolated at an early stage of affinity maturation, and that if affinity matured further, would have acquired more mutations that rigidified its binding site and increased its affinity and specificity for MPTS.
Figure 7.

View of the MPTS binding site in the crystal structures of Ab 6C8 (A) and 8B10 (B) and the model structure of Ab 6C6 (C). The VH and VL are shown in orange and light blue, respectively, side-chains of somatic mutations are shown in green, and waters in panels B and C are shown as blue spheres. Each Ab structure is viewed from an optimal perspective to visualize its unique set of somatic mutations.
Ab 8B10 has an intermediate affinity for MPTS but it is the most specific of the sibling Abs, showing little to no affinity for any of the randomly selected proteins. As with 6C8, the 8B10-MPTS complex shows no plasticity, but it does show significant anelasticity. Ab 8B10 evolved via the acquisition of four somatic mutations, NL76I, AH16T, DH85E, and IH100S, none of which directly contact MPTS. The residues at L76 and H85 are solvent exposed, and their mutation does not appear to introduce any structural changes relative to the other Abs. However, AH16T introduces a side-chain H-bond with the backbone carbonyl of LysH13 that appears to stabilize a β-turn involving residues H13 – H16. Residue H100 is located in a Gly-rich sequence of VH CDR3 and is flanked by residues that form H-bonds with the two proximal sulfonates of MPTS. The IH100S mutation of Ab 8B10 leads to the introduction of an ordered water molecule that is not present in the structure of the other Abs and which forms H-bonds that link the side chain and carbonyl backbone of SH100 with the side chains of ArgL96 and AspL94 (Figure 7B). While likely helping to order the combining site, the water-mediated H-bonds are expected to permit significant small scale motion, which may contribute to the anelasticity of the 8B10-MPTS complex. When comparing the 6C8 and 8B10 complexes, it is instructive to note that the dye coumarin 153 shows a similar increase in the timescale of diffusive motion upon a 4-fold increase in solvent viscosity (from 4.2 ps in CHCl3 to 10.7 ps in DMSO),47 suggesting that the differences in anelasticity of the two complexes are functionally significant. Nonetheless, the water-mediated interactions introduced are relatively directional and any larger scale motions would require their rupture, consistent with the absence of plasticity of the complex. Unlike Ab 6C8, the dynamics observed for the 8B10 complex are likely to be manifest in the free Ab, as none of the introduced mutations involve direct interactions with the Ag. Thus, the absence of plasticity likely explains the relatively high specificity of Ab 8B10 and suggests that this Ab recognizes MPTS with more of a lock-and-key-like mechanism.
Ab 6C6 has the highest affinity for MPTS, and it shows a relatively high level of polyspecificity compared to Ab 8B10 (although less so than Ab 6C8). Like the 8B10 complex, the 6C6–MPTS complex shows higher levels of anelasticity than the 6C8–MPTS complex, but the 6C6–MPTS complex is also the only complex to show a significant level of plasticity. Ab 6C6 evolved via the acquisition of five somatic mutations, DL27cN, NL53I, EL55D, GH56D, and NH57H, all of which are distal to the Ag (Figure 7C). The side-chain of the residue at position H57 is solvent exposed and unlikely to contribute to Ag binding. The DL27cN mutation appears to disrupt an H-bond with the backbone N of ArgL68, possibly stabilizing a water-mediated H-bond to GlyL29, whose flanking residues pack against the Ag. The NL53I somatic mutation increases packing interactions with TyrL49, which in turn packs with the side-chains of ArgL50 and TyrH100c. ArgL50 H-bonds with MPTS, and TyrH100c forms part of the floor of the binding site. TyrH100c also packs with the side-chain of residue L55, and the EL55D mutation appears to stabilize the unique orientation adopted by the TyrH100c side-chain. The GH56D mutation, located in VH CDR2, introduces an H-bond with the side-chain of TyrH50, which participates in a hydrophobic cluster that includes the side-chain of ArgH94. ArgH94 forms an H-bond with the backbone O of GlyH99, which forms part of the supporting structure behind the main walls of the Ag binding site. While all of these interactions may help to increase the affinity for MPTS, none directly contact the Ag, and because most involve less directional packing interactions, both small and larger scale distortions should be possible, accounting for the observed anelasticity, which is similar to that of Ab 8B10, and the observed plasticity, which is unique to this Ab. To gauge whether the level of plasticity is likely to be biologically relevant, we note that Δinh for chromophores in small molecule solvents is invariably zero, and can range up to only ~500 cm−1 in acrylic glasses, which possess perhaps the largest possible range of static environments.48,49 Thus, the variation in static inhomogeneity of the Ab-MPTS complexes is more than one third the difference between a free solvent and a glass environment, which implies that the differences in plasticity that they reflect are biologically significant. As with Ab 8B10, the dynamics observed for the 6C6 complex are likely to be manifest in the free Ab, as none of the introduced mutations involve interactions with the Ag. Thus, the high level of plasticity likely underlies the broad polyspecificity observed with this Ab, as it should facilitate the recognition of different targets with a conformational selection mechanism of molecular recognition.
The results demonstrate that a single progenitor Ab may be optimized by affinity maturation to better recognize MPTS via a variety of different pathways. With Ab 6C8, affinity maturation installed a single functional group that directly interacts with MPTS. While the affinity maturation of Abs 8B10 and 6C6 involved the introduction of more mutations, the effects on structure were more subtle, and the mutations have larger effects on anelasticity and plasticity. Moreover, that the less plastic Ab shows greater selectivity is consistent with the model that a restriction in plasticity is a critical component of affinity maturation as high specificity allows mature Abs to be produced at the high levels required to eradicate an infection without causing autoimmunity.26–28 Regardless, the data suggest that both structure and dynamics are affected by affinity maturation, and possibly that both contribute to the optimization of molecular recognition. A more direct and quantitative analysis of the optimization requires cloning, expression, and characterization of the germline Ab, which is currently limited by poor expression yields. While these studies employed a small molecule Ag, and small molecule and protein Ag often elicit different types of combining sites, it seems likely that the underlying mechanism(s) by which recognition is optimized may be similar. Moreover, a similar optimization of structure and dynamics likely contributes to the optimization of molecular recognition mediated by other proteins, and thus to the evolution of novel function, in general.
Supplementary Material
Acknowledgments
Funding
Funding was provided by the National Institute of Allergy and Infectious Disease (AI079319). Crystallographic data were collected at APS GM/CA that has been funded in whole or in part with federal funding from the National Cancer Institute (Y1-CO-1020) and the National Institute of General Medical Sciences (Y1-GM-1104). Use of the Advanced Photon Source was supported by the U.S. Department of Energy, Basic Energy Sciences, Office of Science, under contract No. DE-AC02-06CH11357.
We thank Julie Vanhasy and Kendra Pivaroff-Ward for Fab preparation and purification.
Footnotes
Supporting methods, tables, and figures; data analysis; nucleotide sequences of the Ab variable chain genes; supporting references. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Patten PA, Gray NS, Yang PL, Marks CB, Wedemayer GJ, Boniface JJ, Stevens RC, Schultz PG. The immunological evolution of catalysis. Science. 1996;271:1086–1091. doi: 10.1126/science.271.5252.1086. [DOI] [PubMed] [Google Scholar]
- 2.Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276:1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- 3.Ortlund EA, Bridgham JT, Redinbo MR, Thornton JW. Crystal structure of an ancient protein: evolution by conformational epistasis. Science. 2007;317:1544–1548. doi: 10.1126/science.1142819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Koshland DE., Jr Application of a theory of enzyme specificity to protein synthesis. Proc Natl Acad Sci USA. 1958;44:98–104. doi: 10.1073/pnas.44.2.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Foote J, Milstein C. Conformational isomerism and the diversity of antibodies. Proc Natl Acad Sci USA. 1994;91:10370–10374. doi: 10.1073/pnas.91.22.10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ma B, Shatsky M, Wolfson HJ, Nussinov R. Multiple diverse ligands binding at a single protein site: a matter of pre-existing populations. Protein Sci. 2002;11:184–197. doi: 10.1110/ps.21302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bosshard HR. Molecular recognition by induced fit: how fit is the concept? News Physiol Sci. 2001;16:171–173. doi: 10.1152/physiologyonline.2001.16.4.171. [DOI] [PubMed] [Google Scholar]
- 8.Fischer E. Einfluss der Configuration auf die Wirkung der Enzyme. Ber Dtsch Chem Ges. 1894;27:2985–2993. [Google Scholar]
- 9.Billington EW, Tate A. The Physics of Deformation and Flow. McGraw-Hill International; New York: 1981. pp. 1–626. [Google Scholar]
- 10.Marcinkowski MJ. Unified Theory of the Mechanical Behavior of Matter. Wiley; New York: 1979. pp. 1–260. [Google Scholar]
- 11.Soutas-Little RW. Elasticity. Dover Publications; New York: 1973. pp. 1–448. [Google Scholar]
- 12.Kachanov LM. Fundamentals of the Theory of Plasticity. Dover Publications; New York: 1971. pp. 1–510. [Google Scholar]
- 13.David L, Quinson R, Gauthier C, Perez J. The role of anelasticity in high stress mechanical response and physical properties of glassy polymers. Polymer Engineer Sci. 1997;37:1633–1640. [Google Scholar]
- 14.Oleynik E. Plastic deformation and mobility in glassy polymers. Prog Coll Pol Sci S. 1989;80:140–150. [Google Scholar]
- 15.Northrup SH, Erickson HP. Kinetics of protein-protein association explained by Brownian dynamics computer simulation. Proc Natl Acad Sci USA. 1992;89:3338–3342. doi: 10.1073/pnas.89.8.3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Camacho CJ, Kimura SR, DeLisi C, Vajda S. Kinetics of desolvation-mediated protein-protein binding. Biophys J. 2000;78:1094–1105. doi: 10.1016/S0006-3495(00)76668-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cho M, Yu JY, Joo T, Nagasawa Y, Passino SA, Fleming GR. The integrated photon echo and solvation dynamics. J Phys Chem. 1996;100:11944–11953. [Google Scholar]
- 18.de Boeij WP, Pshenichnikov MS, Wiersma DA. System-bath correlation function probed by conventional and time-grated stimulated photon echo. J Phys Chem. 1996;100:11806–11823. [Google Scholar]
- 19.Fleming GR, Cho MH. Chromophore-solvent dynamics. Annu Rev Phys Chem. 1996;47:109–134. [Google Scholar]
- 20.Mukamel S. Principles of Nonlinear Optical Spectroscopy. Oxford University Press; New York: 1995. [Google Scholar]
- 21.Landsteiner K. The Specificity of Serological Reactions. Dover; Mineola, NY: 1962. [Google Scholar]
- 22.Tonegawa S. Somatic generation of antibody diversity. Nature. 1983;302:575–581. doi: 10.1038/302575a0. [DOI] [PubMed] [Google Scholar]
- 23.Berek, Milstein C. Mutation drift and repertoire shift in the maturation of the immune response. Immunol Rev. 1987;96:23–41. doi: 10.1111/j.1600-065x.1987.tb00507.x. [DOI] [PubMed] [Google Scholar]
- 24.Berek C, Milstein C. The dynamic nature of the antibody repertoire. Immunol Rev. 1988;105:5–26. doi: 10.1111/j.1600-065x.1988.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 25.Steele EJ, editor. Somatic Hypermutation in V-Regions. CRC Press; Boca Raton, FL: 1990. [Google Scholar]
- 26.Jimenez R, Case DA, Romesberg FE. Flexibility of an antibody binding site measured with photon echo spectroscopy. J Phys Chem B. 2002;106:1090–1103. [Google Scholar]
- 27.Jimenez R, Salazar G, Yin J, Joo T, Romesberg FE. Protein dynamics and the immunological evolution of molecular recognition. Proc Natl Acad Sci USA. 2004;101:3803–3808. doi: 10.1073/pnas.0305745101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zimmermann J, Oakman EL, Thorpe IF, Shi XH, Abbyad P, Brooks CL, Boxer SG, Romesberg FE. Antibody evolution constrains conformational heterogeneity by tailoring protein dynamics. Proc Natl Acad Sci USA. 2006;103:13722–13727. doi: 10.1073/pnas.0603282103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thielges MC, Zimmermann J, Yu W, Oda M, Romesberg FE. Exploring the energy landscape of antibody-antigen complexes: protein dynamics, flexibility, and molecular recognition. Biochemistry. 2008;47:7237–7247. doi: 10.1021/bi800374q. [DOI] [PubMed] [Google Scholar]
- 30.Adhikary R, Yu W, Oda M, Zimmermann J, Romesberg FE. Protein dynamics and the diversity of an antibody response. J Biol Chem. 2012;287:27139–27147. doi: 10.1074/jbc.M112.372698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harlow E, Lane D. Antibodies: a Laboratory Manual. Cold Spring Harbor Laboratory; Cold Spring Harbor, NY: 1988. pp. 371–396. [Google Scholar]
- 32.Sastry L, Alting-Mees M, Huse WD, Short JM, Sorge JA, Hay BN, Janda KD, Benkovic SJ, Lerner RA. Cloning of the immunological repertoire in Escherichia coli for generation of monoclonal catalytic antibodies: construction of a heavy chain variable region-specific cDNA library. Proc Natl Acad Sci USA. 1989;86:5728–5732. doi: 10.1073/pnas.86.15.5728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ulrich HD, Patten PA, Yang PL, Romesberg FE, Schultz PG. Expression studies of catalytic antibodies. Proc Natl Acad Sci USA. 1995;92:11907–11911. doi: 10.1073/pnas.92.25.11907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ. Phaser crystallographic software. J Appl Crystallogr. 2007;40:658–674. doi: 10.1107/S0021889807021206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- 36.Emsley P, Lohkamp B, Scott WG, Cowtan K. Features and development of Coot. Acta Crystallogr D Biol Crystallogr. 2010;66:486–501. doi: 10.1107/S0907444910007493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cunningham AJ. The Generation of Antibody Diversity: A New Look. Academic Press; London: 1976. pp. 1–211. [Google Scholar]
- 38.Alt FW, Blackwell K, Yancopoulos GD. Development of the primary antibody repertoire. Science. 1987;238:1079–1087. doi: 10.1126/science.3317825. [DOI] [PubMed] [Google Scholar]
- 39.Lefranc MP, Giudicelli V, Ginestoux C, Jabado-Michaloud J, Folch G, Bellahcene F, Wu Y, Gemrot E, Brochet X, Lane J, Regnier L, Ehrenmann F, Lefranc G, Duroux P. IMGT, the international ImMunoGeneTics information system. Nucleic Acids Res. 2009;37:D1006–1012. doi: 10.1093/nar/gkn838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kabat EA. Sequences of Proteins of Immunological Interest. U.S. Dept. of Health and Human Services., NIH; Bethesda, MD: 1983. pp. 1–323. [Google Scholar]
- 41.Connolly ML. The molecular surface package. J Mol Graph. 1993;11:139–141. doi: 10.1016/0263-7855(93)87010-3. [DOI] [PubMed] [Google Scholar]
- 42.McKean D, Huppi K, Bell M, Staudt L, Gerhard W, Weigert M. Generation of antibody diversity in the immune response of BALB/c mice to influenza virus hemagglutinin. Proc Natl Acad Sci USA. 1984;81:3180–3184. doi: 10.1073/pnas.81.10.3180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Berek C, Milstein C. The dynamic nature of the antibody repertoire. Immunol Rev. 1988;105:5–26. doi: 10.1111/j.1600-065x.1988.tb00763.x. [DOI] [PubMed] [Google Scholar]
- 44.Jacob J, Przylepa J, Miller C, Kelsoe G. In situ studies of the primary immune response to (4-hydroxy-3-nitrophenyl)acetyl. III The kinetics of V region mutation and selection in germinal center B cells. J Exp Med. 1993;178:1293–1307. doi: 10.1084/jem.178.4.1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lu YF, Cerny J. Repertoire of antibody response in bone marrow and the memory response are differentially affected in aging mice. J Immunol. 2002;169:4920–4927. doi: 10.4049/jimmunol.169.9.4920. [DOI] [PubMed] [Google Scholar]
- 46.Baker SP, Vinci RP, Arias T. Elastic and anelastic behavior of materials in small dimensions. MRS Bulletin. 2002;27:26–29. [Google Scholar]
- 47.Horng ML, Gardecki JA, Papazyan A, Maroncelli M. Subpicosecond measurements of polar solvation dynamics: coumarin 153 revisited. J Phys Chem. 1995;99:17311–17337. [Google Scholar]
- 48.Passino SA, Nagasawa Y, Joo T, Fleming GR. Three-pulse echo peak shift studies of polar solvation dynamics. J Phys Chem A. 1997;101:725–731. [Google Scholar]
- 49.Nagasawa Y, Passino SA, Joo T, Fleming GR. Temperature dependence of optical dephasing in an organic polymer glass (PMMA) from 300 K to 30 K. J Chem Phys. 1997;106:4840–4852. [Google Scholar]
- 50.Kabat EA, Wu TT. Identical V region amino acid sequences and segments of sequences in antibodies of different specificities. Relative contributions of VH and VL genes, minigenes, and complementarity-determining regions to binding of antibody-combining sites. J Immunol. 1991;147:1709–1719. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.