

1. Introduction
The Wacker oxidation is a powerful synthetic transformation, which converts a terminal olefin to a methyl ketone via palladium catalysis, traditionally employing molecular oxygen as the terminal oxidant and a copper cocatalyst.1–3 Good functional group tolerance, ease of reaction, and the orthogonal reactivity of the substrate and product have led to the widespread application of this transformation in the industrial preparation of commodity chemicals, such as acetaldehyde, and in target directed synthesis.4 The most common system used is that initially reported by Clement and further advanced by Tsuji, employing a mixed DMF and water solvent system.2,3 In the Tsuji-Wacker oxidation, water is the source of the oxygen atom which is incorporated into the product, and molecular oxygen is the terminal oxidant (Figure 1).
Figure 1.

Oxidase type catalysis of the Wacker oxidation.
An alternative approach to the same transformation, which is less commonly employed for synthetic applications, utilizes an electrophilic metalloperoxide species, formed by metal activation of molecular oxygen or exogenous hydro- or alkylperoxides. These peroxymetallo reagents and/or catalysts are proposed to coordinate an olefin. Subsequently, the olefin inserts in a peroxymetallation step (Scheme 1).5 Early work in this field follows closely with similar metal-catalyzed peroxide-mediated epoxidation reactions.6,7 An advantage of this mechanistic manifold for affecting the transformation of terminal alkenes to methyl ketones seems to lie in the “preloaded” catalyst and the selective syn-metallation step. This is contrary to the Tsuji-Wacker oxypalladation step, which may occur in an anti fashion, but can be particularly dependent on the reaction conditions.8–16 Overall, some of the peroxymetallation reactions display good synthetic potential as they predominantly lead to single oxidation products via catalyst control, whereas the Tsuji-Wacker reaction is subject to substrate control (vide infra).
Scheme 1.

Activation of molecular oxygen leads to intermediates similar to those proposed when exogenous hydro- or alkylperoxides are used.
More recently, peroxide-mediated, ligand-modulated, palladium-catalyzed systems have been reported. These systems, which utilize commercially-available peroxides, allow for the efficient and selective oxidation of substrates, which would otherwise give mixtures of products when oxidized using Tsuji-Wacker conditions. The Wacker oxidation has been extensively reviewed,4,17–20 including instances which produce aldehydes;21 however, peroxide-mediated Wacker-type oxidations have not recently been reviewed and is the focus of the discussion herein.
2. Peroxide-Mediated Wacker-Type Oxidations
In peroxide-mediated Wacker-type oxidations, the source of the oxygen atom incorporated into the ketone product differs from that of the classical Wacker oxidation. In the Wacker oxidation, the oxygen atom arises from a molecule of water or hydroxide ion. Palladium is reduced to the zero oxidation state and is ultimately reoxidized to Pd(II) by molecular oxygen in an oxidase-type catalyst system (i.e. molecular oxygen is the terminal oxidant, but not the source of the oxygen atom incorporated into the product).1 In peroxide-mediated oxidations, an oxygen atom is incorporated from the terminal oxidant, either molecular oxygen or a peroxide (analogous to an oxygenase system).17,22–26 This key mechanistic discrepancy has been probed by isotopic labeling studies and will be discussed in the relevant sections.
A number of transition metals, including iridium,27 ruthenium,28,29 platinum,30 rhodium,5,6,20,22,24,26,29,31–46 and palladium,23,25,26,47–61 have been reported to either activate molecular oxygen or use exogenous peroxide to convert terminal olefins to methyl ketones. Significantly, the seminal systems involve rhodium, while palladium systems appear to have the greatest synthetic potential. In the next section, rhodium systems which utilize molecular oxygen will be discussed. This will be followed by a discussion of the use of palladium in conjunction with molecular oxygen or hydrogen peroxide; similar reactive intermediates should be shared by these systems. Finally, systems, which employ tert-butylhydroperoxide (TBHP) and palladium will be discussed, including their use in the oxidation of olefins that are challenging substrates in the Tsuji-Wacker oxidation.
2.1. Rhodium-Dioxygen
Isolated, or in situ generated, rhodium-dioxygen species are capable of oxidizing terminal olefins to methyl ketones. Read and coworkers reported the seminal work with rhodium cooxygenation of terminal olefins and Ph3P to methyl ketones and Ph3PO in 1972.31 Mimoun and coworkers significantly advanced the studies of related systems and developed a working mechanistic hypothesis.5,22,24 To this effect, a rhodium-catalyzed system has been developed, utilizing RhCl3 and Cu(ClO4)2(HMPA)4 in alcoholic solvents under an O2 atmosphere. It is proposed that Rh(III) oxidizes two equivalents of the alcohol (2 EtOH → 2 CH3COH) to give a Rh(I) species, which can then activate molecular oxygen to give a peroxorhodium(III) complex. Two moles of terminal olefin are then oxidized for each mole of O2 consumed. While this is the same stoichiometry observed in the classical Wacker oxidation, the oxygen atoms in the ketone product are proposed to arise from molecular oxygen via two distinct, but interdependent pathways (Scheme 2).
Scheme 2.

Proposed catalytic cycle for rhodium-catalyzed oxidation of olefins.5
It is proposed that in the first stage of the catalytic cycle molecular oxygen is activated by the olefin complex A to the peroxorhodium(III) complex B, which upon insertion gives metallocycle C. Decomposition of C provides the first equivalent of ketone product and the rhodium oxo species D. Subsequent protonation gives E and coordination of another equivalent of olefin delivers F. A classical Wacker-like sequence is proposed to follow with syn-oxypalladation to give G, followed by β-hydride migration to give the second equivalent of ketone. Coordination of another alkene regenerates the Rh(I) species A.
Mimoun and coworkers report good methyl ketone selectivity for various simple alkyl olefin substrates. However, the conversion decreases with increasing hydrophobicity of the substrate (Figure 2). While some benzaldehyde product is detected from the oxidative cleavage of styrene, the main oxidation product is acetophenone.
Figure 2.

Oxidation of terminal olefins under RhCl3-Cu(ClO4)2 conditions.5
2.2. Palladium-O2 and Palladium-H2O2
Mimoun found that palladium-dioxygen complexes could convert terminal olefins to methyl ketones in the presence of a strong acid (Figure 3a) via formation of a palladium-hydroperoxide intermediate.23 This species is proposed to coordinate an olefin, followed by peroxymetallation, peroxide bond cleavage, and hydrogen atom shift to provide the methyl ketone product (Figure 3b). Isotopic labeling studies indicate that the oxygen atom incorporated in the ketone product arises from molecular oxygen, and not from adventitious water (Figure 3c). This reaction is stoichiometric in palladium, proceeding with concomitant oxidation of the phosphine ligands.
Figure 3.

a) The reaction of (Ph3P)2PdO2 with a strong acid and a terminal olefin. b) The proposed mechanism proceeds through a Pd–OOH species, and c) isotopic labeling indicates the source of the oxygen atom incorporated into the product.23
Molecular oxygen can insert into a palladium hydride species generated upon β-hydride elimination from primary or secondary alkoxides. Takehira and coworkers reported the oxidation of cyclohexene to cyclohexanone with cooxidation of ethanol to acetaldehyde (Figure 4a).52 Further, a system was developed, which utilizes CuCl2 in addition to palladium (Figure 4b).53,54 The authors suggest that a heterobimetallic Pd–Cu(I) species is formed and that the role of copper is as a transient oxygen carrier that facilitates the generation of a Pd–OOH species (Figure 4c). This is particularly interesting, considering the growing support for the role of copper in other Pd–Cu–O2 systems as more than a facilitation of the reoxidation of Pd(0) to Pd(II).16,62–64 In addition to oxidizable alcohols as viable solvents, THF and methyl ethyl ketone are competent solvents for the reported transformation. As suggested by the authors, and later supported by the findings of Cornell and Sigman,59 this is likely due to the ability of these molecules to form alkyl peroxides (Figure 4d), which can then act in a similar fashion to hydrogen peroxide. A related system is reported by Brégeault and coworkers utilizing BiCl3 and LiCl, also proposes the involvement of heterobimetallic complexes.48 However, olefin isomerization and other internal ketone isomers are observed as significant byproducts in this system.
Figure 4.

a) Cyclopentene is converted to cyclohexanone.52–54 b) The reaction is proposed to occur via alcohol oxidation to provide a Pd–H, into which molecular oxygen can insert. c) The authors suggest an operative Pd–Cu heterobimetallic species. d) THF and MEK are known to form peroxides, which may act in a similar fashion to H2O2.
Uemura and coworkers reported the catalytic oxidation of terminal olefins to methyl ketones in a process coupled to the oxidation of isopropanol, generating H2O2 and a proposed Pd–OOH intermediate (Scheme 3).51 As discussed previously, it is proposed that a Pd–H is generated upon oxidation of a sacrificial alcohol. Molecular oxygen can then insert into the Pd–H bond to give a Pd–OOH species, which can either coordinate an olefin and undergo peroxypalladation or react with another molecule of alcohol to give H2O2 and return the palladium to the alcohol oxidation pathway. Evidence for the generation of the Pd–OOH species is provided by an observed enhancement in the rate of O2 consumption when the reaction is performed in the presence of alkene as compared to identical conditions in the absence of alkene. The authors suggest that this is a result of a more rapid consumption of the putative Pd–OOH intermediate when alkene is present.
Scheme 3.

Proposed linked catalytic cycles for alcohol oxidation and peroxide-mediated Wacker-type reactions sharing a Pd–OOH intermediate.51
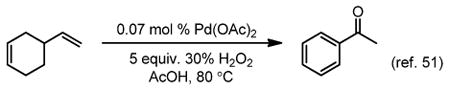
Roussel and Mimoun have reported a hydrogen peroxide-mediated system using very low loadings of palladium (0.07 mol %) to achieve good conversions of simple olefins to the corresponding methyl ketones with modest to good selectivity (Table 1).25 The byproducts were identified as 3- and 4-octanone, although they generally constituted a small percentage of the product mixture. Unfortunately, this reaction has the undesirable characteristic, from a safety standpoint, of catalytic H2O2 decomposition by palladium, which may be responsible for the lack of synthetic application. Additionally, over-oxidation was observed as a result of these conditions in the oxidation of 4-vinylcyclohex-1-ene to acetophenone (eq 1).51
Table 1.
Pd(OAc)2–H2O2 system.25

| |||
|---|---|---|---|
| Substrate | Solvent | % Conversiona | % 2-ketoneb |
| R = C6H13 | tBuOH | 89 | 82 |
| AcOH | 96 | 95 | |
| R = C8H17 | tBuOH | 90 | 80 |
| AcOH | 95 | 92 | |
| R = C10H21 | tBuOH | 89 | 75 |
| AcOH | 92 | 90 | |
|
|
tBuOH | 8 | 83 |
| AcOH | 100 | 85 | |
Determined by GC with o-dichlorobenzene as internal standard.
% selectivity for the methyl ketone product as compared to other products. Determined by GC.
 |
eq 1 |
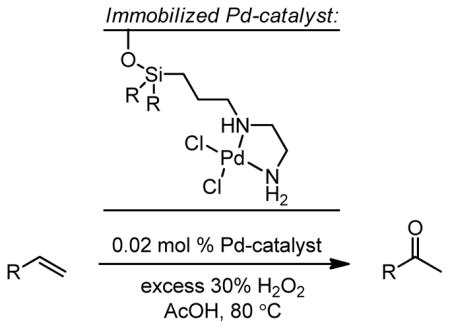
A montmorillonite silylpropylethylenediamine palladium complex has been reported by Choudary and coworkers, which converts terminal olefins to methyl ketones with short reaction times and very low catalyst loading (0.02 mol %) in the presence of H2O2 (Table 2).50 The catalyst was reported to retain full catalytic activity through four reuses.
Table 2.
Supported Pd–H2O2 system.50
| ||
|---|---|---|
| Substrate | Time (min) | % Yield |
| R = C4H9 | 45 | 96 |
| R = C6H13 | 60 | 92 |
| R = C8H17 | 60 | 92 |
| R = C10H21 | 60 | 90 |
|
|
55 | 94 |
| R = Ph | 30 | 98 |
2.3. Palladium-TBHP
Mimoun and coworkers were able to synthesize and isolate a series of tetrameric aceto-palladium-tert-butylperoxide complexes, which precipitated out of a solution of Pd(O2CR)2 in 80% tert-butylhydroperoxide.47 In particular, the active oxidant palladium tert-butylperoxide trifluoroacetate (PPT) was prepared. This complex stoichiometrically oxidizes 1-hexene to 2-hexanone in less than 10 minutes (based on palladium, Figure 5a). Additional support for TBHP as the source of the oxygen atom in the ketone product comes from the preparation of the stable peroxymercuration adduct 1, which upon transmetallation with Na2PdCl4 presumably provides the unstable pseudo-palladacyclic intermediate 2, which can decompose to provide acetophenone (Figure 5b).
Figure 5.

a) Stoichiometric oxidation of terminal olefins to methyl ketones by PPT. b) Transmetallation of peroxymercuration adduct with palladium to provide acetophenone.47
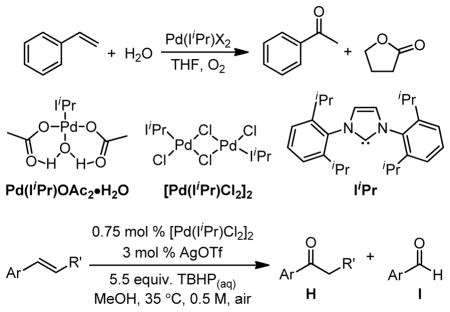
In an unanticipated result, Cornell and Sigman discovered a ligand-modulated peroxide-mediated Wacker-type oxidation, where TBHP was used in conjunction with an N-heterocyclic carbene ligand.59 While investigating a copper-free, direct O2 coupled Wacker oxidation, it was found that styrene (a classically challenging substrate for the Tsuji-Wacker oxidation)21,65 could be oxidized to acetophenone in THF solvent (Table 3). However, when the reaction progress was monitored by in situ FTIR spectroscopy, an extended induction period was observed. It was hypothesized that this was the result of a palladium-catalyzed oxidation of THF, as indicated by the observed formation of γ-butyrolactone. Instead of utilizing molecular oxygen, TBHP was found to be an efficient stoichiometric oxidant for the transformation of styrenes to acetophenone derivatives. Unfortunately, the synthetic utility of this system is limited to styrenyl substrates due to the propensity of the catalyst to isomerize alkenes and oxidize the resultant internal alkenes.66
Table 3.
Oxidation of styrenyl substrates with the Pd(NHC)–TBHP system.59
| |||||
|---|---|---|---|---|---|
| Ar | R′ | time (h) | % conversion | % yield | H:I |
| Ph | H | 24 | >99 | 75 | >130:1 |
| 2-MePh | H | 48 | >99 | 79 | 36:1 |
| 3-MePh | H | 32 | >99 | 83 | 22:1 |
| 4-MePh | H | 16 | >99 | 86 | 22:1 |
| 2,4,6-MePh | H | 24 | 95 | 71 | >150:1 |
| 3-CIPh | H | 48 | >98 | 80 | >150:1 |
| 3-NO2Pha | H | 24 | 90 | 79 | >150:1 |
| Phb | Ph | 48 | NA | 42c | 42:35 |
| Ph | Me | 48 | 97 | NA | 2.3:1d |
Conditions: 2.25 mol % [Pd(IiPr)Cl2]2, 12 mol % AgOTf.
Conditions: [alkene] = 0.3 M, 1.25 mol % [Pd(IiPr)Cl2]2, 4 mol % AgOTf, 35–50 °C.
Two equivalents of benzaldehyde produced via oxidative cleavage.
H is a 53:47 mixture of regioisomeric ketones.
Sigman and coworkers have further explored both copper-free Wacker oxidations, which utilize the bidentate amine ligand sparteine,16,66 as well as additional ligand-modulated TBHP-mediated Wacker-type oxidations.57,58,60,61 As a result of these investigations, a highly selective oxidation system was developed and will be discussed below.
Alkene substrates with adjacent heteroatoms have been shown to undergo anti-Markovnikov oxidation yielding aldehyde products (Figure 6a and 6b).21,67–70 This phenomenon has been reviewed21 and has also been exploited as a means to selectively prepare aldehydes.68–70 Since this outcome is thought to originate from a secondary coordination of the Lewis basic heteroatom to the electrophilic palladium, it was hypothesized that the proposed syn-peroxypalladation mechanism in combination with a bidentate amine ligand would leave only a single electrophilic alkene binding site (Figure 7a).
Figure 6.

a) Substrates with proximal heteroatoms can give aldehyde products under Tsuji-Wacker conditions.67,68 b) anti-Markovnikov oxypalladation leads to aldehyde product.
Figure 7.

a) Coordinatively saturated palladium and b) bidentate ligand evaluation.57
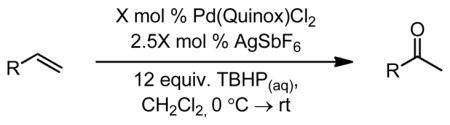
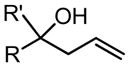
It was found that the Quinox ligand scaffold was uniquely suited for effective catalysis in this system (Figure 7b). Through empirical optimizations, a highly active catalyst system was developed, which oxidized terminal olefins selectively to their methyl ketone products (Table 4).57,58,61 The Quinox ligand is readily prepared from simple starting materials (Scheme 4)57,58 and is also commercially available. Substrates, such as protected allylic alcohols57 and amines,58 as well as unprotected homoallylic alcohols61 are selectively oxidized with catalyst-control using the Pd(quinox)-TBHP system. These results are in direct contrast to the observed results in the Tsuji-Wacker oxidation of these substrate classes (see Figure 6 and Table 5).21
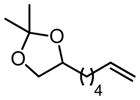
Table 4.
Scope of Pd(Quinox)Cl2–TBHP system.
|
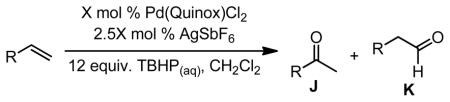
|
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
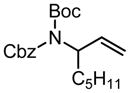
| Substrate | X | Time | % Yielda | ref. | Substrate | X | Time | % Yielda | J:K h | ref. |
|
|
|
|||||||||
| R = C8H17 | 2 | 20 min | 86 | 57 |
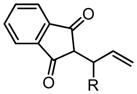
|
|||||
| R = HOC9H18 | 2 | 35 min | 98 | 57 | ||||||
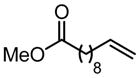
|
2 | 35 min | 87 | 57 | ||||||
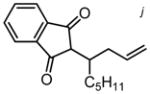
| R = C5H11 | 5i | 19 h | 91 | 96:4 | 58 | |||||
|
R = H | 5i | 18 h | 79 | >95:5 | 58 | ||||
| 2 | 30 min | 95 | 57 |
 j j
|
5i | 20 min | 82 | >95:5 | 58 | |
| R = ClC9H18 | 2 | 80 min | 89 | 57 | ||||||
| R = 4-MePh | 5 | 50 min | 88 | 57 | ||||||
| R = 3-NO2Ph | 5 | 17 h | 60 | 57 |
|
5 | 50 min | 81 | >95:5 | 58 |
| R = 4-BocNHPh | 5 | 60 min | 83 | 57 | ||||||
|
|
5 | 2.5 h | 74 | >95:5 | 58 | ||||
| PG = Acb | 5 | 20 h | 89 | 57 |
|
5 | 2.5 h | 95 | >95:5 | 58 |
| PG = Ac (98)c | 5 | 99 (98) | 57 | |||||||
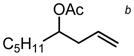
| PG = TBSd | 2 | 4.5 h | 77 | 57 | ||||||
| PG = CH2OCH2CH3 | 5 | 4 h | 81 | 57 |
|
5 | 2.5 h | 93 | >95:5 | 58 |
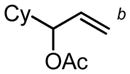 b b
|
5 | 17 h | 89 | 57 |
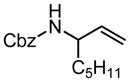
|
5 | 12 h | 74 | 90:10 | 58 |
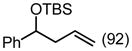 (92) (92) |
2 | 35 min | 92 | 57 | ||||||
| 2 | 99 (92) | 57 | ||||||||
 b b
|
3 | 3 h | 94 | 61 |
|
5 | 14 h | 76 | >95:5 | 58 |
|
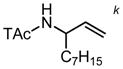 k k
|
5 | 23 h | 67 | >95:5 | 58 | ||||
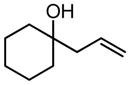
| R = C5H11 | 3 | 5 h | 81 | 61 | ||||||
| R = Ph | 3 | 24 | --e | 61 |
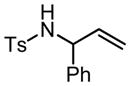
|
5 | 2 h | 90 | >95:5 | 58 |
| R = C4H9, R′ = Me | 5 | 5 h | 84 | 61 | ||||||
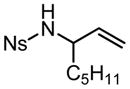
| R = Ph, R′ = Me | 10 | 8 h | 79f | 61 | ||||||
|
5 | 6.5 h | 71 | 61 |
|
5 | 4 h | 88 | >95:5 | 58 |
 b b
|
5 | 24 h | 57g | 61 |
 (99) (99) |
5 | 16 h | 69 (99) | >95:5 | 58 |
All yields represent average isolated yields of at least two reactions performed on >0.5 mmol scale unless otherwise noted.
Substrate added at room temperature.
Numbers in parentheses represent ee.
15 mol % AgSbF6 used.
Complex mixture, mostly recovered starting material.
Single experiment.
An average of 35% starting material was recovered.
Ratios determined by GC, 1H integrations, and/or yields of isolated products.
18 mol % AgSbF6 used.
Substrate added at 0 C.
An average of 12% starting material was recovered.
Scheme 4.

Synthesis of Quinox using ethanol amine or 2-chloroethylamine•HCl.57,58
Table 5.
Comparison of product distributions using either Tsuji-Wacker or Pd(Quinox)-TBHP conditions.
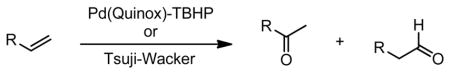
| ||||||
|---|---|---|---|---|---|---|
| Substrate | Tsuji-Wacker | ref. | Pd(Quinox)-TBHPa | ref. | ||
| % yield | ketone/aldehyde | % yield | ketone/aldehyde | |||
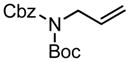
|
85b | 57:43 | 58 | 95 | >95:5 | 58 |
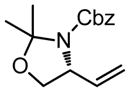
|
56b,c | 60:40 | 58 | 69 | >95:5 | 58 |
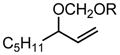
|
R = CH3 | R = CH2CH3 | ||||
| 65d | ~50:50 | 67 | 81 | >95:5 | 57 | |
|
90b | <1:99 | 68 | 91 | >96:4 | 58 |
For reaction conditions see table 4.
20 mol % PdCl2, 1 equiv. CuCl, DMF/H2O (7:1), O2, rt, 3 d.
32% recovered starting material.
10 mol % PdCl2, 1 equiv. CuCl, DMF/H2O (7:1), O2, 60 C, 24 h.
Kinetic evidence and ligand modification studies support the hypothesis that a defined coordination sphere and syn-peroxypalladation are responsible for the excellent observed selectivity.60 The reaction was observed to show saturation kinetics in [TBHP], which is supportive of a mechanism in which palladium is “preloaded” with the peroxide. A hypothesis that the defined coordination sphere results from the electronic disparity between the ligand modules was supported by systematic modification of the quinox ligand electronics. The rate was observed to increase with addition of electron withdrawing groups to the quinoline ring (Figure 8a). Additionally, in a series of 4-trifluoromethylquinoline-2-pyridyl ligands, it was observed that more electron releasing groups on the pyridine ring (i.e. the more donating ligand module) also increased the rate of reaction (Figure 8b).
Figure 8.

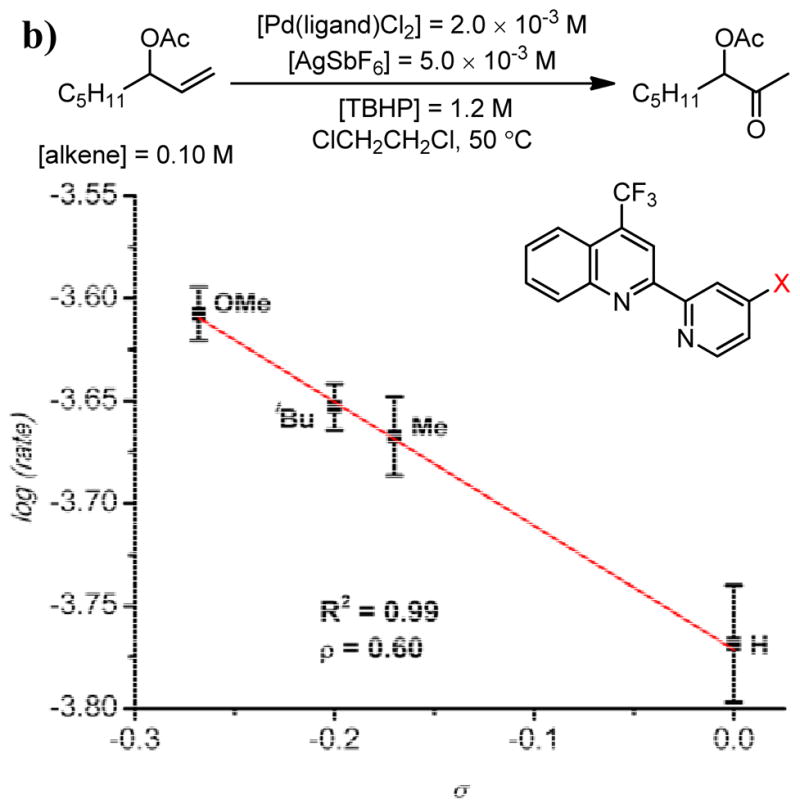
a) Hammett correlation for the log(rate) for a series of 4-substituted quinox ligands as a function of σp values. b) Hammett correlation for the log(rate) for a series of quinoline pyridyl ligands as a function of σp values.60
3. Summary and Outlook
The development of Wacker-type oxidations in which rhodium activates molecular oxygen has led to a number of systems, which utilize O2 insertion into palladium-hydrides. Similarly, hydrogen peroxide has been used as a stoichiometric oxidant in palladium-catalyzed systems. The catalytic decomposition of hydrogen peroxide to generate molecular oxygen by palladium and the lack of synthetic evaluation of these systems may be the reason that they have not seen broad synthetic application. The Pd(Quinox)-TBHP Wacker-type oxidation has shown to be highly selective for the oxidation of substrates, which are not selectively oxidized under Tsuji-Wacker conditions. Future work in this field should attempt to achieve the very low catalyst loadings reported in the Pd–H2O2 systems with the catalyst-controlled selectivity observed in ligand-modulated catalysis.
Acknowledgments
This work was supported by the National Institutes of Health (NIGMS RO1 GM63540).
Biographies
Brian W. Michel was born in Kirkland, Washington. He obtained a B.S. degree in chemistry in 2006 from Western Washington University, working on the enantioselective synthesis of heliannuols C and E under Professor James Vyvyan. In 2006 he joined the Department of Chemistry at the University of Utah, where he is currently working in Professor Matthew Sigman’s laboratory. He is currently working on research projects focused on the development of new catalytic oxidation reactions for targeted synthesis.
Matthew S. Sigman: Matt Sigman received a B.S. in chemistry from Sonoma State University in 1992 before obtaining his Ph.D. at Washington State University with Professor Bruce Eaton in 1996. He then moved to Harvard University to complete an NIH postdoctoral stint with Professor Eric Jacobsen. In 1999, he joined the faculty of the University of Utah where his research group has focused on the development of new synthetic methodology.
5. References and Notes
- 1.Smidt J, Hafner W, Jira R, Sieber R, Sedlmeier J, Sabel A. Angew Chem. 1962;74:93–102. [Google Scholar]
- 2.Clement WH, Selwitz CM. J Org Chem. 1964;29:241–243. [Google Scholar]
- 3.Tsuji J. Synthesis. 1984:369–384. [Google Scholar]
- 4.Takacs JM, Jiang X-t. Curr Org Chem. 2003;7:369–396. [Google Scholar]
- 5.Mimoun H, Perez Machirant MM, Seree de Roch I. J Am Chem Soc. 1978;100:5437–5444. [Google Scholar]
- 6.Mimoun H. J Mol Catal. 1980;7:1–29. [Google Scholar]
- 7.Mimoun H. Angew Chem. 1982;94:750–766. [Google Scholar]
- 8.Henry PM. J Org Chem. 1973;38:2415–2416. [Google Scholar]
- 9.Zaw K, Henry PM. J Org Chem. 1990;55:1842–1847. [Google Scholar]
- 10.Dumlao CM, Francis JW, Henry PM. Organometallics. 1991;10:1400–1405. [Google Scholar]
- 11.Francis JW, Henry PM. Organometallics. 1991;10:3498–3503. [Google Scholar]
- 12.Hamed O, Thompson C, Henry PM. J Org Chem. 1997;62:7082–7083. doi: 10.1021/jo971051q. [DOI] [PubMed] [Google Scholar]
- 13.Stille JK, Divakaruni R. J Organomet Chem. 1979;169:239–248. [Google Scholar]
- 14.Bäckvall JE, Heumann A. J Am Chem Soc. 1986;108:7107–7108. [Google Scholar]
- 15.Bäckvall JE, Bjöerkman EE, Pettersson L, Siegbahn P. J Am Chem Soc. 1984;106:4369–4373. [Google Scholar]
- 16.Anderson BJ, Keith JA, Sigman MS. J Am Chem Soc. 2010;132:11872–11874. doi: 10.1021/ja1057218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cornell CN, Sigman MS. Inorg Chem. 2007;46:1903–1909. doi: 10.1021/ic061858d. [DOI] [PubMed] [Google Scholar]
- 18.Tsuji J. Pure Appl Chem. 1999;71:1539–1547. [Google Scholar]
- 19.Feringa BL. Transition Met Org Synth. 1998;2:307–315. [Google Scholar]
- 20.Bortolini O, Di Furia F, Modena G, Seraglia R. J Mol Catal. 1984;22:313–317. [Google Scholar]
- 21.Muzart J. Tetrahedron. 2007;63:7505–7521. [Google Scholar]
- 22.Igersheim F, Mimoun H. J Chem Soc Chem Commun. 1978:559–560. [Google Scholar]
- 23.Igersheim F, Mimoun H. Nouv J Chim. 1980;4:711–713. [Google Scholar]
- 24.Igersheim F, Mimoun H. Nouv J Chim. 1980;4:161–166. [Google Scholar]
- 25.Roussel M, Mimoun H. J Org Chem. 1980;45:5387–5390. [Google Scholar]
- 26.Martin C, Faraj M, Martin J, Brégeault JM, Mercier J, Fillaux J, Dizabo P. J Mol Catal. 1986;37:201–212. [Google Scholar]
- 27.Atlay MT, Preece M, Strukul G, James BR. J Chem Soc Chem Commun. 1982:406–407. [Google Scholar]
- 28.Khan MMT, Rao AP. J Mol Catal. 1988;44:95–105. [Google Scholar]
- 29.Januszkiewicz K, Alper H. Tetrahedron Lett. 1983;24:5163–5164. [Google Scholar]
- 30.Strukul G, Ros R, Michelin RA. Inorg Chem. 1982;21:495–500. [Google Scholar]
- 31.Dudley C, Read G. Tetrahedron Lett. 1972:5273–5276. [Google Scholar]
- 32.James BR, Rempel GL. Can J Chem. 1968;46:571–574. [Google Scholar]
- 33.James BR, Kastner M. Can J Chem. 1972;50:1708–1712. [Google Scholar]
- 34.Read G, Walker PJC. J Chem Soc Dalton Trans. 1977:883–888. [Google Scholar]
- 35.Read G. J Mol Catal. 1978;4:83–84. [Google Scholar]
- 36.Tang R, Mares F, Neary N, Smith DE. J Chem Soc, Chem Commun. 1979:274–275. [Google Scholar]
- 37.Carlton L, Read G. J Mol Catal. 1981;10:133–141. [Google Scholar]
- 38.Nyberg ED, Pribich DC, Drago RS. J Am Chem Soc. 1983;105:3538–3544. [Google Scholar]
- 39.Dahlmann J, Hoeft E. Oxid Commun. 1983;5:391–403. [Google Scholar]
- 40.Dahlmann J, Hoeft E. Oxid Commun. 1983;5:405–414. [Google Scholar]
- 41.Faraj M, Brégeault JM, Martin J, Martin C. J Organomet Chem. 1984;276:C23–C26. [Google Scholar]
- 42.Drago RS, Zuzich A, Nyberg ED. J Am Chem Soc. 1985;107:2898–2903. [Google Scholar]
- 43.Faraj M, Martin J, Martin C, Brégeault JM, Mercier J. J Mol Catal. 1985;31:57–71. [Google Scholar]
- 44.Bressan M, Morandini F, Morvillo A, Rigo P. J Organomet Chem. 1985;280:139–146. [Google Scholar]
- 45.Read G, Urgelles M. J Chem Soc Dalton Trans. 1985:1591–1596. [Google Scholar]
- 46.Read G. J Mol Catal. 1988;44:15–33. [Google Scholar]
- 47.Mimoun H, Charpentier R, Mitschler A, Fischer J, Weiss R. J Am Chem Soc. 1980;102:1047–1054. [Google Scholar]
- 48.Brégeault JM, Faraj M, Martin J, Martin C. New J Chem. 1987;11:337–343. [Google Scholar]
- 49.Derdar F, Martin J, Martin C, Brégeault JM, Mercier J. J Organomet Chem. 1988;338:C21–C26. [Google Scholar]
- 50.Rao YVS, Rani SS, Choudary BM. J Mol Catal. 1992;75:141–146. [Google Scholar]
- 51.Nishimura T, Kakiuchi N, Onoue T, Ohe K, Uemura S. J Chem Soc, Perkin Trans 1. 2000:1915–1918. [Google Scholar]
- 52.Takehira K, Hayakawa T, Orita H. Chem Lett. 1985:1835–1838.
- 53.Takehira K, Orita H, Oh IH, Leobardo CO, Martinez GC, Shimidzu M, Hayakawa T, Ishikawa T. J Mol Catal. 1987;42:247–255. [Google Scholar]
- 54.Takehira K, Hayakawa T, Orita H, Shimizu M. J Mol Catal. 1989;53:15–21. [Google Scholar]
- 55.Escola JM, Botas JA, Aguado J, Serrano DP, Vargas C, Bravo M. Appl Catal, A. 2008;335:137–144. [Google Scholar]
- 56.Escola JM, Botas JA, Vargas C, Bravo M. J Catal. 2010;270:34–39. [Google Scholar]
- 57.Michel BW, Camelio AM, Cornell CN, Sigman MS. J Am Chem Soc. 2009;131:6076–6077. doi: 10.1021/ja901212h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Michel BW, McCombs JR, Winkler A, Sigman MS. Angew Chem, Int Ed. 2010;49:7312–7315. doi: 10.1002/anie.201004156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cornell CN, Sigman MS. J Am Chem Soc. 2005;127:2796–2797. doi: 10.1021/ja043203m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Michel BW, Stephens LD, Sigman MS. J Am Chem Soc. In Revision. [Google Scholar]
- 61.McCombs JR, Michel BW, Sigman MS. J Org Chem. doi: 10.1021/jo200462a. In Revision. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hosokawa T, Takano M, Murahashi SI. J Am Chem Soc. 1996;118:3990–3991. [Google Scholar]
- 63.Hosokawa T, Nomura T, Murahashi SI. J Organomet Chem. 1998;551:387–389. [Google Scholar]
- 64.Jensen KH, Webb JD, Sigman MS. J Am Chem Soc. 2010;132:17471–17482. doi: 10.1021/ja108106h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wright JA, Gaunt MJ, Spencer JB. Chem Eur J. 2006;12:949–955. doi: 10.1002/chem.200400644. [DOI] [PubMed] [Google Scholar]
- 66.Cornell CN, Sigman MS. Org Lett. 2006;8:4117–4120. doi: 10.1021/ol061662h. [DOI] [PubMed] [Google Scholar]
- 67.Kang SK, Jung KY, Chung JU, Namkoong EY, Kim TH. J Org Chem. 1995;60:4678–4679. [Google Scholar]
- 68.Weiner B, Baeza A, Jerphagnon T, Feringa BL. J Am Chem Soc. 2009;131:9473–9474. doi: 10.1021/ja902591g. [DOI] [PubMed] [Google Scholar]
- 69.Friestad GK, Jiang T, Mathies AK. Org Lett. 2007;9:777–780. doi: 10.1021/ol063010z. [DOI] [PubMed] [Google Scholar]
- 70.Choi PJ, Sperry J, Brimble MA. J Org Chem. 2010;75:7388–7392. doi: 10.1021/jo1016585. [DOI] [PubMed] [Google Scholar]