

Abstract
Purpose of review
The purpose of this review is to summarize recent observations on the role of lipoprotein(a) [Lp(a)] as a risk factor mediating cardiovascular disease.
Recent findings
Lp(a) is a highly prevalent cardiovascular risk factor, with levels >30 mg/dL affecting 20–30% of the global population. Up until now, no specific therapies have been developed to lower Lp(a) levels. Three major levels of evidence support the notion that elevated Lp(a) levels are a causal, independent, genetic risk factor for cardiovascular disease; epidemiologic studies and meta-analyses, genome wide association studies and mendelian randomization studies. Recent studies also have noted that individuals with low levels of Lp(a) are associated with a higher risk of incident type 2 diabetes mellitus, and conversely individuals with high levels have a lower risk, but this association does not appear to be causal. Novel therapies to lower Lp(a) include PCSK9 inhibitors and antisense oligonucleotides directly preventing translation of apolipoprotein(a) mRNA.
Summary
With this robust and expanding clinical database, a re-awakening of interest in Lp(a) as clinical risk factor is taking place. Trials are underway with novel drugs that substantially lower Lp(a) and may reduce its contribution to cardiovascular disease.
Keywords: lipoprotein(a), cardiovascular disease, aortic stenosis, diabetes mellitus, antisense oligonucleotides
Introduction
Lp(a) is becoming an established risk factor for cardiovascular disease (CVD), joining the ranks of the other 3 major lipid risk factors, including high levels of low density lipoprotein cholesterol (LDL-C) and triglycerides and reduced levels of high density lipoprotein cholesterol (HDL-C), as well as hypertension, smoking, and diabetes mellitus. This review will provide an update of the evidence base of Lp(a) as a causal, independent, genetic risk factor of CVD in this rapidly growing field, review its relationship to type 2 diabetes mellitus, describe recent studies with novel Lp(a) lowering agents to reduce CVD events, and provide insights into future studies and trials.
Lp(a) and CVD Risk
The etiologic role of Lp(a) in CVD is no longer debated due to recent landmark studies defining its relationship to CVD events. In aggregate, strong data exists in demonstrating that Lp(a) is a causal, independent genetic risk factor for CVD, and that this risk is curvilinearly related to CVD risk. For example, the Emerging Risk Factor Collaboration group performed a patient-level meta-analysis of long-term, prospective, primary prevention studies published between January 1970 and March 2009 evaluating Lp(a) risk in relation to CVD and all-cause mortality in 126,634 participants enrolled in 36 prospective studies totaling 1.3 million person-years of follow-up [1]. A total of 22,076 first-ever fatal or nonfatal vascular disease outcomes or nonvascular deaths were recorded, including 9336 coronary heart disease (CHD) events, 1903 ischemic strokes, 338 hemorrhagic strokes, 751 unclassified strokes, 1091 other vascular deaths, 8114 nonvascular deaths, and 242 deaths of unknown cause. The association of Lp(a) was 1.13 (1.09–1.18) per 1 SD with CHD risk, 1.10 (1.02–1.18) for ischemic stroke, 1.01 (0.98–1.05) for the aggregate of nonvascular mortality, and 1.00 (0.97–1.04) for cancer deaths, following adjustment for lipids and other conventional risk factors. The risk of CHD was flat until ~24 mg/dL, then rose continuously with increasing Lp(a), up to risk ratio of ~1.5 at 192 mg/dL (Figure 1A). These data clearly demonstrate that in subjects without prior CHD that Lp(a) is an independent predictor of CHD, even at modest levels of Lp(a) of >24 mg/dL, which is the approximately the 70th percentile population risk that encompasses approximately 2 billion people globally [2, 3].
Figure 1.
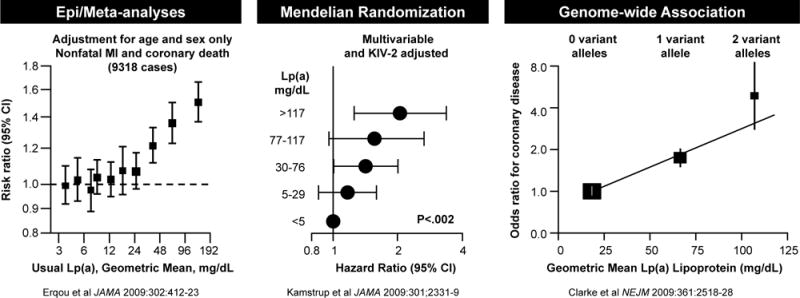
Three levels of evidence, epidemiological studies and meta-analyses, instrument variable analyses and genome-wide association studies, demonstrate the increasing risk for CVD due to elevated Lp(a). Reprinted with permission from Erqou et al [1], Kamstrup et al [5] and Clarke et al [6].
These findings are further supported by additional epidemiological as well as Mendelian randomization [4, 5] and genome wide association studies [6]. For example, Kamstrup et al [4] followed 9330 men and women for 10 years from the general population in the Copenhagen City Heart Study, of which 498 developed myocardial infarction (MI). They observed a stepwise increase in risk of MI with increasing levels of Lp(a), with no evidence of a threshold effect and Lp(a) levels >120 mg/dL predicting a 3–4-fold increase in risk of MI in the general population. In women, compared to levels <5 mg/dL (<22nd percentile), multifactorially adjusted hazard ratios (HR) for MI were 1.1 (0.6 to 1.9) for 5–29 mg/dL (22nd to 66th percentile), 1.7 (1.0 to 3.1) for 30–84 mg/dL (67th to 89th percentile), 2.6 (1.2 to 5.9) for 85–119 mg/dL (90th to 95th percentile), and 3.6 (1.7 to 7.7) for ≥120 mg/dL (>95th percentile). Equivalent values in men were 1.5 (0.9 to 2.3), 1.6 (1.0 to 2.6), 2.6 (1.2 to 5.5), and 3.7 (1.7 to 8.0).
Instrumental variable analysis allows an assessment of potential causality by evaluating the role of genetic determinants in modifying plasma levels of quantitative traits than are then linked to CVD events. In this case, single nucleotide polymorphisms (snps) and kringle IV type 2 (KIV2) repeats in the LPA gene, which are fixed at conception and cannot be confounded by environmental factors and therefore rule out reverse causality, are the instrumental variables in this analysis. Furthermore, since Lp(a) levels are not significantly influenced by diet or exercise, they tend to minimally fluctuate around a pre-determined genetic level that makes this analysis similar to achieving different levels of Lp(a) over time following a therapeutic Lp(a)-lowering trial [7]. In a follow-up study, Kamstrup et al [5] further expanded this analysis in the Copenhagen City Heart Study, a prospective general population study with 16 years of follow-up (1991–2007, n = 8,637, 599 MI events); the Copenhagen General Population Study, a cross-sectional general population study (2003–2006, n = 29,388, 994 MI events); and the Copenhagen Ischemic Heart Disease Study, a case-control study (1991–2004, n = 2,461, 1231 MI events). They evaluated plasma Lp(a) levels, KIV2 isoforms, LPA snps and MIs adjudicated from 1976 through July 2007 for all participants. In the Copenhagen City Heart Study, multivariable-adjusted hazard ratios (HRs) for MI ranged from 1.2 (confidence interval [CI], 0.9–1.6; events/10 000 person-years, 59) for levels between the 22nd and 66th percentile to 2.6 (1.6–4.1; events/10 000 person-years, 108) for levels greater than the 95th percentile, respectively, compared to levels <22nd percentile (events/10 000 person-years, 55) (trend P < 0.001). Genetically elevated Lp(a) derived from all 3 cohorts were associated with an HR of 1.22 (1.09–1.37) per doubling of Lp(a) level on instrumental variable analysis, while the corresponding value for plasma Lp(a) levels on Cox regression was 1.08 (1.03–1.12) Figure 1B). These data are consistent with a causal association between elevated Lp(a) levels and increased risk of MI and make a strong rationale for Lp(a) lowering in general communities.
A variety of studies reported on genome-wide associations between LPA snps and CVD risk [6, 8–15]. For example, Clarke et al [6] measured 48,742 snps in 2100 candidate genes in 3145 case subjects with CAD and 3352 control subjects and as a replication in three independent populations of 4846 additional case subjects with CAD and 4594 control subjects. The LPA locus on 6q26–27 encoding Lp(a) had the strongest association with CAD, with snps rs10455872 and rs3798220 strongly associated with increased levels of Lp(a) and reduced copy number KIV2 repeats. rs10455872 had an odds ratio (OR) for CAD of 1.70 (1.49 to 1.95) and rs3798220 of 1.92 (1.48 to 2.49). A meta-analysis using a genotype score involving both LPA snps showed ORs for CAD of 1.51 (1.38 to 1.66) for one variant and 2.57 (1.80 to 3.67) for two or more variants (Figure 1C). After adjustment for Lp(a) levels, the association between the LPA genotype score and the risk of CAD was abolished. These findings of differences in LPA snps being associated with elevated Lp(a) level, which are then associated with increased risk of CAD with a gene-dose effect provide further support for a causal role of Lp(a) in mediating CAD.
The above studies were mainly in subjects without prior CHD and the question remains whether Lp(a) is a risk factor in mediating second CVD events, particularly where many risk factors are already addressed pharmacologically or through lifestyle changes. In 2014 O’Donoghue et al [16] reported data combined from 11 studies spanning two decades in 18,978 subjects. Subjects with Lp(a) levels in the highest quintile had an OR of 1.40 (1.15 to 1.71). However, there was significant between-study heterogeneity, so that when stratified on the basis of LDL-C, the association between Lp(a) and CVD events was significant when LDL-C was ≥130 mg/dl (OR: 1.46 (1.23 to 1.73, p<0.001), but was not statistically significance for studies with LDL-C <130 mg/dl (OR: 1.20 (0.90 to 1.60), p = 0.21).
Since that study, 3 large randomized trials reflecting a more modern experience, including JUPITER, LIPID and AIM-HIGH, reported that Lp(a) remains a predictor for CVD events, even in patients treated with potent statins reaching LDL-C concentrations as low as 62, 53 and 113 mg/dL, respectively (Figure 2).
Figure 2.
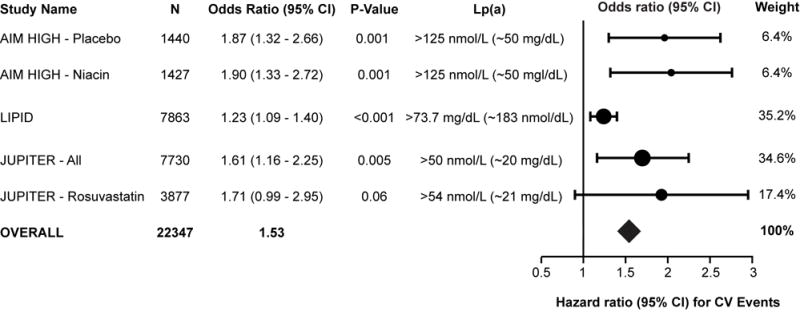
Demonstration of residual risk of elevated Lp(a) in the JUPITER, AIM-HIGH and LIPID trials, in setting of very aggressive statin therapy and well-controlled LDL-C levels. The Forest plot shows a study level meta-analysis of these 3 recent reports from these trials. Shown are baseline Lp(a) levels of AIM-HIGH niacin and placebo arms, baseline Lp(a) levels in the LIPID trial and baseline Lp(a) levels in the entire JUPITER arm and in the group receiving rosuvastatin and the hazard ratio of the highest quartile and recurrent CVD events.
In the AIM-HIGH (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes) trial [17], patients with prior CVD and low baseline levels of LDL-C were randomized to simvastatin plus placebo or simvastatin, plus extended-release niacin (ERN), with ezetimibe added as needed, in both groups, to maintain an on-treatment LDL-C 40–80 mg/dl. Baseline and on-study Lp(a) levels were predictive of CVD events in both simvastatin plus placebo (baseline HR: 1.24 [p = 0.002] and on-study HR: 1.21 [p = 0.017]) and the simvastatin plus ERN group (baseline HR: 1.25 [p = 0.001] and on-study HR: 1.18 [p = 0.028]) (Figure 2). The ERN modestly decreased Lp(a) 21%, but did not reduce CVD event in any quartile of Lp(a). However, it is noted that patients in this study were not selected to have elevated Lp(a) levels, and in fact median levels were 33.8 nmol/L (~13.5 mg/dL). Thus, the study was both underpowered for patients with elevated Lp(a) levels and also suggests that more potent Lp(a) lowering than 21% is needed to affect CVD outcomes.
In the LIPID (Long-Term Intervention with Pravastatin in Ischaemic Disease trial), the predictive value of Lp(a) and CHD outcomes during 6 years’ median follow-up were evaluated in 7863 patients with a prior CAD event following randomization to pravastatin or placebo. Baseline Lp(a) concentration was associated with total CHD events (P<0.001), total cardiovascular disease events (P=0.002), and coronary events (P=0.03), with greatest risk occurring at the upper decile of >73.7 mg/dL (Figure 2). For events after year 1, an increase in Lp(a) at 1 year was associated with adverse outcomes for total CHD events and total cardiovascular disease events (P=0.002 each), supporting measurement of Lp(a) for risk assessment of patients with known CHD.
In the JUPITER (Justification for the Use of Statins in Prevention: An Intervention Trial Evaluating Rosuvastatin) trial [18], baseline and on-treatment Lp(a) concentrations were assessed in 9612 multiethnic participants before and after treatment of rosuvastatin 20 mg/d or placebo, with outcomes reported for whites (n=7746). Baseline Lp(a) concentrations were independently associated with incident CVD (adjusted hazard ratio per 1-SD increment in Ln[Lp(a)], 1.18 (1.03–1.34); P=0.02), as were on-statin Lp(a) concentrations with residual risk of CVD (HR 1.27 (1.01–1.59); P=0.04) (Figure 2). Rosuvastatin significantly reduced incident CVD among participants with baseline Lp(a) ≥median (HR 0.62 (0.43–0.90) but numerically even more so in patients with Lp(a) <median (HR 0.46 (0.30–0.72), suggesting higher Lp(a) levels limit statin efficacy, as previously also shown in the 4S trial [19].
Lp(a) and Diabetes Mellitus
Elevated Lp(a) levels have generally [20, 21], but not universally [22, 23], been associated with increased CVD risk in both type 1 and type 2 diabetes mellitus. Interestingly, patients with diabetes have been shown to have lower Lp(a) levels in non-diabetics, both in patients with suspected CAD undergoing coronary angiography [22], as well as patients presenting with acute coronary syndromes [24]. This observation is unexplained, particularly since Lp(a) can act as an acute phase reactant in patients presenting with ACS [25].
Three studies have shown the lower Lp(a) levels are associated with increased risk of incident diabetes. The first observation was from the Women’s Health Study, studying 26,746 healthy US women (mean age 54.6 years) of which 1670 developed type 2 diabetes over a 13-year follow-up period [26]. Following multivariable adjustment Lp(a) was inversely associated with incident diabetes with HR for quintiles 2–5 vs. quintile 1 of 0.87 (0.75–1.01), 0.80 (0.68–0.93), 0.88 (0.76–1.02), and 0.78 (0.67–0.91). The HRs were 1.62 (0.91–2.89) for Lp(a) <10 mg/L and HbA1c <5%, 3.50 (3.06–4.01) for Lp(a) > or =10 mg/L and HbA1c 5% – <6.5%, and 5.36 (4.00–7.19) for Lp(a) <10 mg/L and HbA1c 5% – <6.5%.
The second study from the Copenhagen City Heart Study and the Copenhagen General Population Study evaluated 77,901 individuals had Lp(a) data, of whom 28,567 (36.7%) had Lp(a), KIV2 isoforms (sums of repeats) and snp rs10455872 measurements [27]. Low Lp(a) was associated with risk of type 2 diabetes with adjusted OR of 1.26 (1.09–1.45), 1.17 (1.01–1.36), 1.04 (0.90–1.21), and 1.05 (0.90–1.22), respectively, for quintiles 1–4, compared with quintile 5. High KIV2 sums of repeats were associated with risk of type 2 diabetes with OR 1.16, (1.05–1.28) for KIV2 quintile 5 versus quintiles 1–4 combined, but rs10455872 was not associated with risk of type 2 diabetes. For a halving of Lp(a) level, the causal OR for type 2 diabetes was 1.15 (1.05–1.27) for KIV2 sum of repeats and 0.99 (0.95–1.03) for rs10455872 genotype. This study suggests an observational association exists between low Lp(a) and incident diabetes but that the Mendelian randomization analysis does not suggest a causal relationship, except perhaps for the association with high isoform numbers.
The third study was in 8,490 participants from the European Prospective Investigation of Cancer (EPIC)-Norfolk cohort that included adults aged 40–79 years followed for an average 10 years, of which 593 participants developed incident type 2 diabetes [28]. Mendelian randomization with rs1045587 tested whether the association between Lp(a) levels and diabetes is causal based on EPIC-Norfolk combined with DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) data involving a total of 10,088 diabetes case participants and 68,346 control participants. In adjusted analyses, there was an inverse association between Lp(a) levels and diabetes with HR 0.63 (0.49–0.81), P trend = 0.003) comparing the top versus bottom quintile of Lp(a). In EPIC-Norfolk, a 1-SD increase in logLp(a) was associated with a lower risk of diabetes (OR 0.88 (0.80–0.95]. However, in Mendelian randomization analyses, a 1-SD increase in logLp(a) due to rs10455872, which explained 26.8% of the variability in Lp(a) levels, was not associated with risk of diabetes with OR 1.03 (0.96–1.10; P=0.41).
In summary these three prospective findings demonstrate a strong inverse association of Lp(a) levels with risk of incident diabetes. However, genetic variants that are strongly associated with increased Lp(a) levels do not appear to be associated with risk of diabetes. The mechanisms behind these observations are not clear, but imply reverse causality. Its possible that as patients develop metabolic syndrome and ultimately diabetes, that Lp(a) levels may fall. Possible etiologies may include the production of large VLDL particles that are less optimal for the assembly of Lp(a) due to its lipid content interfering with optimal initial interactions of apo(a) kringles repeats with apoB-100 [29]. This is supported by the general observation that Lp(a) levels are modestly (r=~0.20) inversely associated with triglyceride levels in all populations [24, 30]. There is also clinical and experimental data that insulin directly reduces Lp(a) levels via a negative response element pathway on the LPA gene [31, 32]. Thus, patients with hyperinsulinemia may actually develop a reduction of their basal Lp(a) levels. Finally, it is possible that high Lp(a) may have salutary effects on insulin resistance, but there is no evidence to suggest this at this time.
Current Therapies and Outcomes in Lp(a) Lowering
Table 1 show the major pharmaceuticals affecting Lp(a) levels. Although not well appreciated, many studies have shown that all statins may raise Lp(a) levels up to 10–50%. For example, high-dose atorvastatin [16] and rosuvastatin [33] both raise Lp(a) approximately 20–25%. This is not universal phenomenon, and is confounded by the variety of different assays as well as the lack of standardization of assays and calibrators measuring Lp(a). Additionally, low fat diets and garlic supplements have also been shown to increase Lp(a) levels [34].
Table 1.
Therapeutic agents that affect Lp(a) levels
| Changes in Lp(a) with therapeutic agents | |
|---|---|
|
| |
| Increase Lp(a) | Decrease Lp(a) |
| Statins (0–50%) | Clinically available |
| Ezetimibe (~20%) | LDL apheresis (acutely 60–80%, time-averaged 30–35%) |
| Low fat diets (20–30%) | Niacin (20–30%) |
| Garlic supplements | Mipomersen (20–40%) |
| IL-6 antagonists (30%) | |
| PCSK9 Inhibitors (20–40%) | |
| Aspirin | |
| Insulin | |
| Investigational | |
| Antisense to apo(a) (80–99%) | |
| CETP Inhibitors (20–35%) | |
| Other | |
| Thyroid analogues | |
| Oral estrogen/tamoxifen | |
| Anabolic steroids | |
| Neomycin | |
| N-acetylcysteine | |
| L-carnitine | |
A variety of clinically approved procedures and drugs have been shown to lower Lp(a). LDL apheresis lowers Lp(a) acutely 60–80%, but due to a fairly quick rebound, levels return to baseline within 1 week or so, leading to a time averaged reduction of 30–35%. There have been several studies evaluating events pre and post apheresis, and all have uniformly shown ~70–80% reduction in events [35]. Niacin lowers Lp(a) 20–30%, and the mechanism appears to be inhibition of apo(a) via cyclic AMP stimulated apo(a) transcription [36]. Niacin also reduces VLDL apoB-100 production [37]. Mipomersen, an antisense inhibitor to apoB-100 reduces Lp(a) a mean of 25% in patients with familial hypercholesterolemia and in non-familial hypercholesterolemia [38]. The mechanisms underlying this reduction are not understood, but may be related to lack of availability of apoB in the hepatocyte during time of Lp(a) assembly, as suggested by data in Lp(a)-transgenic mice treated with mipomersen [39].
Lp(a) is an acute phase reactant and patients with inflammatory conditions tend to have elevations in Lp(a). This is supported by recent data showing that treatment of patients with rheumatoid arthritis with IL-6 antagonists reduces Lp(a) by ~30% [40], which is supported by in vitro data that the LPA promoter contains an IL-6 response element. PCSK9 Inhibitors have also been shown to lower Lp(a) 20–40% [41, 42], but the mechanisms are not understood. Initial use of PCSK9 inhibitors will be limited to patients with familial hypercholesterolemia and otherwise high risk patients with CVD but will not be used in patients with isolated Lp(a) elevations and controlled LDL-C. Finally, there are data in a very small study in patients with CAD and cerebral infarction suggesting aspirin reduces Lp(a) levels, but this had not been confirmed and may have been confounded by resolution of the acute phase response in these patients. Interestingly, in a post-hoc analysis from the Women’s Health Study, although the overall trial was negative, women with elevated Lp(a) had significantly elevated CVD risk, but his risk was 2-fold lower in women randomized to aspirin [43]. Estrogen replacement also decreases Lp(a), and may also explain why post-menopausal women tend to have increases in Lp(a) relative to pre-menopausal status. In the Heart Estrogen/progestin replacement Study (HERS) trial, women randomized to estrogen/progestin therapy had an absolute 5.8 mg/dL reduction in Lp(a). In a randomized subgroup comparison, women with low baseline Lp(a) levels had less benefit from estrogen/progestin than women with high Lp(a) levels, suggesting a salutary effect on CHD risk mediated by elevated Lp(a) [44]. However, estrogen/progestin therapy is not used currently for cardiovascular risk prevention.
Specific Antisense Therapy to Lower Lp(a)
Since Lp(a) is not an enzyme or receptor small molecules cannot inactivate it, and because it circulates in very large quantities (up to 500 mg/dL or up to 3 total grams for a person with 6 liters of plasma) it would likely require tens of grams of antibody to inactive it, making it highly expensive, impractical due to volume of dosing and the potential for immunological effects in clearing large amounts of immune complexes. An alternative approach in reducing Lp(a) levels would be required, one that prevents synthesis by the hepatocyte, its major (99%) site of synthesis. Early work using a antisense oligonucleotide (ASO) to human apo(a) led to the demonstration in 2011 that antisense oligonucleotides targeted to Lp(a) significantly lower Lp(a) and OxPL-apoB in Lp(a)-transgenic mice [45]. To optimize this for human applications, over 2200 second generation ASOs were tested for their ability to dose-dependently reduce apo(a) mRNA expression in transgenic LPA mouse primary hepatocytes. An optimal ASO designated as ISIS-APO(a)Rx was identified that binds to the exon 24–25 splice site of the mature human apo(a) transcript (Genbank Accession NM_005577.2) at position 3901–3920 bp [46]. KIV2 repeats are identical at the protein level, but are not conserved uniformly at the nucleotide level, which is why the drug only binds to a single splice junction with perfect complementarity.
ISIS-APO(a)Rx was evaluated in human volunteers and demonstrated a dose dependent mean Lp(a) reduction of 78% and maximal reduction of 92% in the highest doses, with similar effects on OxPL-apoB, [47] (Figure 3). ISIS-APO(a)Rx was equally effective across all isoforms and was not dependent on the baseline Lp(a) levels. A phase 2 study in 2 cohorts of patients with Lp(a) 50–175 mg/dL, representing >80th percentile, and >175 mg/dL, representing the >99th percentile of levels, is near completion (Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ISIS-APO(a)Rx in Patients With High Lipoprotein(a) - NCT02160899). Preliminary data reported orally at the American Heart Association meeting in 2015 demonstrates a mean of 71% reduction and maximum 94% reduction in Lp(a) levels. Finally, an optimized molecule, ISIS-APO(a)-LRx that also contains a GalNAc3 conjugate targeted to the hepatocyte asialoglycoprotein receptor to enhance potency [48], is currently being evaluated in healthy volunteers with elevated Lp(a) of >30 mg/dL (Safety, Tolerability, Pharmacokinetics, and Pharmacodynamics of ISIS-APO(a)-LRx in Healthy Volunteers With Elevated Lipoprotein(a) - NCT02414594). These trials are paving the way for a potential therapy in high risk populations in reducing Lp(a) levels, and in testing the clinical hypothesis that lowering Lp(a) will lower CVD risk and progression of aortic stenosis in broader populations.
Figure 3.
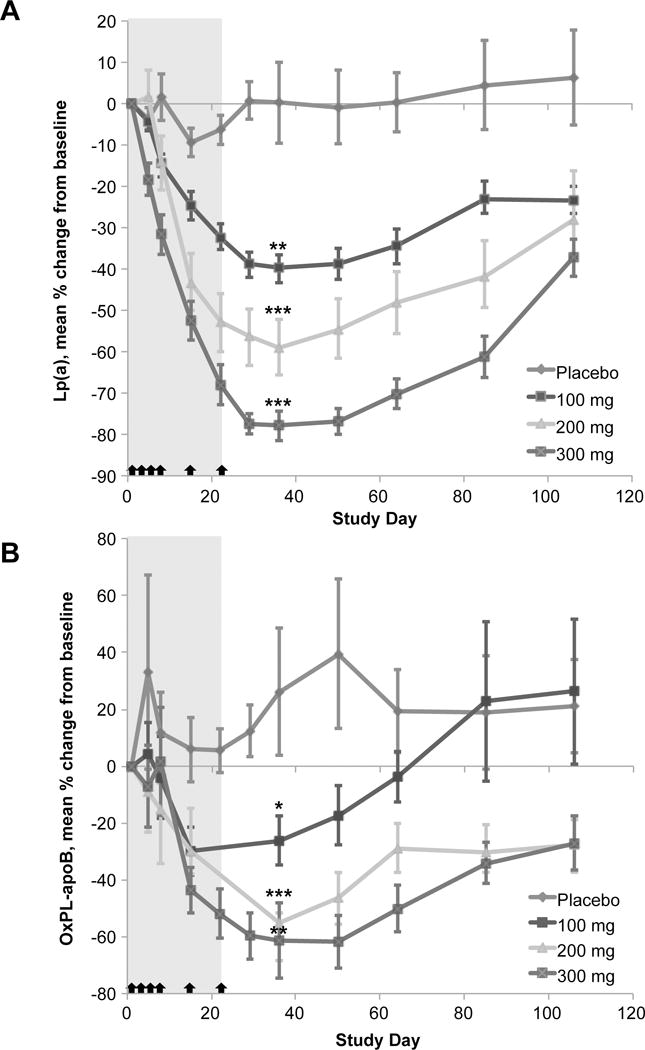
Mean percent change in Lp(a) and OxPL-apoB with time by treatment group in the multi-dose cohorts (A) Lp(a). (B) OxPL-apoB; the datapoints for days 22 and 29 in the 100 mg and 200 mg doses are not displayed in the graph (but were used in the statistical analysis) because the mean percent change values were skewed by two outliers with very low OxPL-apoB concentrations. The shaded area represents the dosing window and arrows indicate dosing at days 1, 3, 5, 8, 15, and 22. Lp(a)=lipoprotein(a). OxPL-apoB=oxidized phospholipids on apolipoprotein B. *p=0.020, †p≤0.008, ‡p≤0.001. P values are for the primary efficacy endpoint at day 36 as determined by the Exact Wilcoxon Rank Sum comparing ISIS-APO(a)Rx versus placebo. P values are only shown for the primary efficacy endpoint at day 36. Reprinted with permission from Tsimikas et al [47].
Conclusion
The field of Lp(a) is rapidly maturing and Lp(a) is now generally seen as a causal, independent, genetic risk factor for CVD. Lp(a) is highly prevalent and contributes to residual risk of CVD. The development of antisense oligonucleotides to lower Lp(a) to near normal levels will allow studies and trials of Lp(a) lowering that will hopefully reduce the risk of CVD and aortic stenosis mediated by this atherothrombotic risk factor.
Key points.
A large epidemiological and genetic database supports the conclusion that elevated Lp(a) levels causally mediate CVD.
Low Lp(a) levels are associated with incident type 2 diabetes mellitus, but this does not appear to be a causal mechanism and may instead imply reverse causality due to effects of excessive VLDL production and insulin on Lp(a) levels.
Niacin and PCSK9 inhibitors are the only viable options to lower Lp(a) levels in high risk patients at present, but specific trials of Lp(a) lowering with these agents in patients with elevated Lp(a) have not been performed.
The clinical development and on-going trials of antisense oligonucleotides specifically targeting Lp(a) has the ability normalize Lp(a) levels in almost all patients and to be a transformative therapy to reduce Lp(a)-contributable risk CVD.
Acknowledgments
I thank Tracy Reigle of Ionis Pharmaceuticals for preparation of the figures.
Financial support and sponsorship. NIH HL119828, HL055798, HL088093, HL106579, HL078610, HL124174.
Footnotes
Conflicts of interest. Dr. Tsimikas is a co-inventor of and receives royalties from patents owned by the University of California San Diego on oxidation-specific antibodies and has a dual appointment at UCSD and Ionis Pharmaceuticals, Inc.
References
- 1.Erqou S, Kaptoge S, Perry PL, et al. Lipoprotein(a) concentration and the risk of coronary heart disease, stroke, and nonvascular mortality. JAMA. 2009;302:412–423. doi: 10.1001/jama.2009.1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nordestgaard BG, Chapman MJ, Ray K, et al. Lipoprotein(a) as a cardiovascular risk factor: current status. Eur Heart J. 2010;31:2844–2853. doi: 10.1093/eurheartj/ehq386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White J, Varvel S, Tsimikas S. Abstract 14669: Prevalence of elevated Lp(a) Levels in 629,858 subjects from a referral laboratory population in the United States. Circulation. 2015;132:A14669. [Google Scholar]
- 4.Kamstrup PR, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. Extreme lipoprotein(a) levels and risk of myocardial infarction in the general population: the Copenhagen City Heart Study. Circulation. 2008;117:176–184. doi: 10.1161/CIRCULATIONAHA.107.715698. [DOI] [PubMed] [Google Scholar]
- 5.Kamstrup PR, Tybjaerg-Hansen A, Steffensen R, Nordestgaard BG. Genetically elevated lipoprotein(a) and increased risk of myocardial infarction. JAMA. 2009;301:2331–2339. doi: 10.1001/jama.2009.801. [DOI] [PubMed] [Google Scholar]
- 6.Clarke R, Peden JF, Hopewell JC, et al. Genetic variants associated with Lp(a) lipoprotein level and coronary disease. N Engl J Med. 2009;361:2518–2528. doi: 10.1056/NEJMoa0902604. [DOI] [PubMed] [Google Scholar]
- 7.Kronenberg F, Utermann G. Lipoprotein(a): resurrected by genetics. J Int Med. 2013;273:6–30. doi: 10.1111/j.1365-2796.2012.02592.x. [DOI] [PubMed] [Google Scholar]
- 8.Luke MM, Kane JP, Liu DM, et al. A polymorphism in the protease-like domain of apolipoprotein(a) is associated with severe coronary artery disease. Arterioscler Thromb Vasc Biol. 2007;27:2030–2036. doi: 10.1161/ATVBAHA.107.141291. [DOI] [PubMed] [Google Scholar]
- 9.Tregouet DA, Konig IR, Erdmann J, et al. Genome-wide haplotype association study identifies the SLC22A3-LPAL2-LPA gene cluster as a risk locus for coronary artery disease. Nat Genet. 2009;41:283–285. doi: 10.1038/ng.314. [DOI] [PubMed] [Google Scholar]
- 10.Ober C, Nord AS, Thompson EE, et al. Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J Lipid Res. 2009;50:798–806. doi: 10.1194/jlr.M800515-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lanktree MB, Anand SS, Yusuf S, Hegele RA. Comprehensive analysis of genomic variation in the LPA locus and its relationship to plasma lipoprotein(a) in South Asians, Chinese, and European Caucasians. Circ Cardiovasc Gen. 2010;3:39–46. doi: 10.1161/CIRCGENETICS.109.907642. [DOI] [PubMed] [Google Scholar]
- 12.Deo RC, Wilson JG, Xing C, et al. Single-nucleotide polymorphisms in LPA explain most of the ancestry-specific variation in Lp(a) levels in African Americans. PLoS One. 2011;6:e14581. doi: 10.1371/journal.pone.0014581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li Y, Luke MM, Shiffman D, Devlin JJ. Genetic variants in the apolipoprotein(a) gene and coronary heart disease. Circ Cardiovasc Genet. 2011;4:565–573. doi: 10.1161/CIRCGENETICS.111.959601. [DOI] [PubMed] [Google Scholar]
- 14.Do R, Willer CJ, Schmidt EM, et al. Common variants associated with plasma triglycerides and risk for coronary artery disease. Nat Genet. 2013;45:1345–1352. doi: 10.1038/ng.2795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Consortium CAD, Deloukas P, Kanoni S, et al. Large-scale association analysis identifies new risk loci for coronary artery disease. Nat Genet. 2013;45:25–33. doi: 10.1038/ng.2480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Donoghue ML, Morrow DA, Tsimikas S, et al. Lipoprotein(a) for risk assessment in patients with established coronary artery disease. J Am Coll Cardiol. 2014;63:520–527. doi: 10.1016/j.jacc.2013.09.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Albers JJ, Slee A, O’Brien KD, et al. Relationship of apolipoproteins A-1 and B, and lipoprotein(a) to cardiovascular outcomes: the AIM-HIGH trial (Atherothrombosis Intervention in Metabolic Syndrome with Low HDL/High Triglyceride and Impact on Global Health Outcomes) J Am Coll Cardiol. 2013;62:1575–1579. doi: 10.1016/j.jacc.2013.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Khera AV, Everett BM, Caulfield MP, et al. Lipoprotein(a) concentrations, rosuvastatin therapy, and residual vascular risk: an analysis from the JUPITER Trial (Justification for the Use of Statins in Prevention: an Intervention Trial Evaluating Rosuvastatin) Circulation. 2014;129:635–642. doi: 10.1161/CIRCULATIONAHA.113.004406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Berg K, Dahlen G, Christophersen B, et al. Lp(a) lipoprotein level predicts survival and major coronary events in the Scandinavian Simvastatin Survival Study. Clin Genet. 1997;52:254–261. doi: 10.1111/j.1399-0004.1997.tb04342.x. [DOI] [PubMed] [Google Scholar]
- 20.Hiraga T, Kobayashi T, Okubo M, et al. Prospective study of lipoprotein(a) as a risk factor for atherosclerotic cardiovascular disease in patients with diabetes. Diabetes Care. 1995;18:241–244. doi: 10.2337/diacare.18.2.241. [DOI] [PubMed] [Google Scholar]
- 21.Kollerits B, Auinger M, Reisig V, et al. Lipoprotein(a) as a predictor of cardiovascular disease in a prospectively followed cohort of patients with type 1 diabetes. Diabetes Care. 2006;29:1661–1663. doi: 10.2337/dc06-0546. [DOI] [PubMed] [Google Scholar]
- 22.Saely CH, Koch L, Schmid F, et al. Lipoprotein(a), type 2 diabetes and vascular risk in coronary patients. Eur J Clin Invest. 2006;36:91–97. doi: 10.1111/j.1365-2362.2006.01604.x. [DOI] [PubMed] [Google Scholar]
- 23.Qi Q, Workalemahu T, Zhang C, et al. Genetic variants, plasma lipoprotein(a) levels, and risk of cardiovascular morbidity and mortality among two prospective cohorts of type 2 diabetes. Eur Heart J. 2012;33:325–334. doi: 10.1093/eurheartj/ehr350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tsimikas S, Witztum JL, Miller ER, et al. High-dose atorvastatin reduces total plasma levels of oxidized phospholipids and immune complexes present on apolipoprotein B-100 in patients with acute coronary syndromes in the MIRACL trial. Circulation. 2004;110:1406–1412. doi: 10.1161/01.CIR.0000141728.23033.B5. [DOI] [PubMed] [Google Scholar]
- 25.Tsimikas S, Bergmark C, Beyer RW, et al. Temporal increases in plasma markers of oxidized low-density lipoprotein strongly reflect the presence of acute coronary syndromes. J Am Coll Cardiol. 2003;41:360–370. doi: 10.1016/s0735-1097(02)02769-9. [DOI] [PubMed] [Google Scholar]
- 26.Mora S, Kamstrup PR, Rifai N, et al. Lipoprotein(a) and risk of type 2 diabetes. Clinical chemistry. 2010;56:1252–1260. doi: 10.1373/clinchem.2010.146779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kamstrup PR, Nordestgaard BG. Lipoprotein(a) concentrations, isoform size, and risk of type 2 diabetes: a Mendelian randomisation study. The lancet Diabetes & endocrinology. 2013;1:220–227. doi: 10.1016/S2213-8587(13)70064-0. [DOI] [PubMed] [Google Scholar]
- 28.Ye Z, Haycock PC, Gurdasani D, et al. The association between circulating lipoprotein(a) and type 2 diabetes: is it causal? Diabetes. 2014;63:332–342. doi: 10.2337/db13-1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koschinsky ML, Marcovina SM. Structure-function relationships in apolipoprotein(a): insights into lipoprotein(a) assembly and pathogenicity. Curr Opin Lipidol. 2004;15:167–174. doi: 10.1097/00041433-200404000-00009. [DOI] [PubMed] [Google Scholar]
- 30.Tsimikas S, Clopton P, Brilakis ES, et al. Relationship of oxidized phospholipids on apolipoprotein B-100 particles to race/ethnicity, apolipoprotein(a) isoform size, and cardiovascular risk factors: results from the Dallas Heart Study. Circulation. 2009;119:1711–1719. doi: 10.1161/CIRCULATIONAHA.108.836940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rainwater DL, Haffner SM. Insulin and 2-hour glucose levels are inversely related to Lp(a) concentrations controlled for LPA genotype. Arterioscler Thromb Vasc Biol. 1998;18:1335–1341. doi: 10.1161/01.atv.18.8.1335. [DOI] [PubMed] [Google Scholar]
- 32.Neele DM, de Wit EC, Princen HM. Insulin suppresses apolipoprotein(a) synthesis by primary cultures of cynomolgus monkey hepatocytes. Diabetologia. 1999;42:41–44. doi: 10.1007/s001250051110. [DOI] [PubMed] [Google Scholar]
- 33.Capoulade R, Chan KL, Yeang C, et al. Oxidized phospholipids, lipoprotein(a), and progression of calcific aortic valve stenosis. J Am Coll Cardiol. 2015;66:1236–1246. doi: 10.1016/j.jacc.2015.07.020. [DOI] [PubMed] [Google Scholar]
- 34.Taleb A, Witztum JL, Tsimikas S. Oxidized phospholipids on apolipoprotein B-100 (OxPL/apoB) containing lipoproteins: A biomarker predicting cardiovascular disease and cardiovascular events. Biomarkers Med. 2011;5:673–694. doi: 10.2217/bmm.11.60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leebmann J, Roeseler E, Julius U, et al. Lipoprotein apheresis in patients with maximally tolerated lipid-lowering therapy, lipoprotein(a)-hyperlipoproteinemia, and progressive cardiovascular disease: Prospective observational multicenter study. Circulation. 2013;128:2567–2576. doi: 10.1161/CIRCULATIONAHA.113.002432. [DOI] [PubMed] [Google Scholar]
- 36.Chennamsetty I, Kostner KM, Claudel T, et al. Nicotinic acid inhibits hepatic APOA gene expression: studies in humans and in transgenic mice. J Lipid Res. 2012;53:2405–2412. doi: 10.1194/jlr.M029769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ooi EM, Watts GF, Chan DC, et al. Effects of extended-release niacin on the postprandial metabolism of Lp(a) and apoB-100-containing lipoproteins in statin-treated men with type 2 diabetes mellitus. Arterioscler Thromb Vasc Biol. 2015 doi: 10.1161/ATVBAHA.115.306136. [DOI] [PubMed] [Google Scholar]
- 38.Santos RD, Raal FJ, Catapano AL, et al. Mipomersen, an antisense oligonucleotide to apolipoprotein B-100, reduces lipoprotein(a) in various populations with hypercholesterolemia: results of 4 phase III trials. Arterioscler Thromb Vasc Biol. 2015;35:689–699. doi: 10.1161/ATVBAHA.114.304549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Merki E, Graham MJ, Mullick AE, et al. Antisense oligonucleotide directed to human apolipoprotein B-100 reduces lipoprotein(a) levels and oxidized phospholipids on human apolipoprotein B-100 particles in lipoprotein(a) transgenic mice. Circulation. 2008;118:743–753. doi: 10.1161/CIRCULATIONAHA.108.786822. [DOI] [PubMed] [Google Scholar]
- 40.Muller N, Schulte DM, Turk K, et al. IL-6 blockade by monoclonal antibodies inhibits apolipoprotein (a) expression and lipoprotein (a) synthesis in humans. J Lipid Res. 2015;56:1034–1042. doi: 10.1194/jlr.P052209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Desai NR, Kohli P, Giugliano RP, et al. AMG145, a monoclonal antibody against proprotein convertase subtilisin kexin type 9, significantly reduces lipoprotein(a) in hypercholesterolemic patients receiving statin therapy: an analysis from the LDL-C assessment with proprotein convertase subtilisin kexin type 9 monoclonal antibody inhibition combined with statin therapy (LAPLACE)-thrombolysis in myocardial infarction (TIMI) 57 trial. Circulation. 2013;128:962–969. doi: 10.1161/CIRCULATIONAHA.113.001969. [DOI] [PubMed] [Google Scholar]
- 42.Raal FJ, Giugliano RP, Sabatine MS, et al. Reduction in lipoprotein(a) with PCSK9 monoclonal antibody evolocumab (AMG 145): a pooled analysis of more than 1,300 patients in 4 phase II trials. J Am Coll Cardiol. 2014;63:1278–1288. doi: 10.1016/j.jacc.2014.01.006. [DOI] [PubMed] [Google Scholar]
- 43.Chasman DI, Shiffman D, Zee RY, et al. Polymorphism in the apolipoprotein(a) gene, plasma lipoprotein(a), cardiovascular disease, and low-dose aspirin therapy. Atherosclerosis. 2009;203:371–376. doi: 10.1016/j.atherosclerosis.2008.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shlipak MG, Simon JA, Vittinghoff E, et al. Estrogen and progestin, lipoprotein(a), and the risk of recurrent coronary heart disease events after menopause. JAMA. 2000;283:1845–1852. doi: 10.1001/jama.283.14.1845. [DOI] [PubMed] [Google Scholar]
- 45.Merki E, Graham M, Taleb A, et al. Antisense oligonucleotide lowers plasma levels of apolipoprotein (a) and lipoprotein (a) in transgenic mice. J Am Coll Cardiol. 2011;57:1611–1621. doi: 10.1016/j.jacc.2010.10.052. [DOI] [PubMed] [Google Scholar]
- 46.Graham MJ, Viney N, Crooke R, Tsimikas S. Antisense inhibition of apolipoprotein(a) to lower plasma lipoprotein(a) levels in humans. J Lipid Res. 2015 doi: 10.1194/jlr.R052258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsimikas S, Viney NJ, Hughes SG, et al. Antisense therapy targeting apolipoprotein(a): a randomised, double-blind, placebo-controlled phase 1 study. Lancet. 2015;386:1472–1483. doi: 10.1016/S0140-6736(15)61252-1. [DOI] [PubMed] [Google Scholar]
- 48.Prakash TP, Graham MJ, Yu J, et al. Targeted delivery of antisense oligonucleotides to hepatocytes using triantennary N-acetyl galactosamine improves potency 10-fold in mice. Nucleic acids research. 2014;42:8796–8807. doi: 10.1093/nar/gku531. [DOI] [PMC free article] [PubMed] [Google Scholar]