

Abstract
Flutamide (FLU), an oral, nonsteroidal antiandrogen drug used in the treatment of prostate cancer, is associated with idiosyncratic hepatotoxicity that sometimes causes severe liver damage, including cholestasis, jaundice, and liver necrosis. To understand the mechanism of toxicity, a combination of aryl hydrocarbon receptor (Ahr)-deficient (Ahr-/-) mice, primary hepatocytes, luciferase reporter gene assays, in silico ligand docking and ultra-performance chromatography-quadrupole time-of-flight mass spectrometry-based metabolomics were used. A significant increase of liver weights, and liver and serum bile acid levels was observed after FLU treatment, indicating hepatomegaly and disrupted bile acid homeostasis. Expression of the AhR gene battery was markedly increased in livers of wild-type mice Ahr+/+ treated with FLU, while no change was noted in Ahr-/- mice. Messenger RNAs encoded by AhR target genes were induced in primary mouse hepatocytes cultured with FLU, which confirmed the in vivo results. Ligand-docking analysis further predicted that FLU is an AhR agonist ligand which was confirmed by luciferase reporter gene assays. Multivariate data analysis showed that bile acids were responsible for the separation of vehicle- and FLU-treated Ahr+/+ mice, while there was no separation in Ahr-/- mice. Expression of mRNA encoding the bile acid transporter ABCC4 was increased and farnesoid X receptor signaling was inhibited in the livers of Ahr+/+ mice, but not in Ahr-/- mice treated with FLU, in agreement with the observed downstream metabolic alterations. These findings provide new insights into the mechanism of liver injury caused by FLU treatment involving activation of AhR and the alterations of bile acid homeostasis, which could guide clinical application.
Keywords: flutamide, aryl hydrocarbon receptor, bile acid homeostasis, ABCC4, metabolomics
Graphical abstract
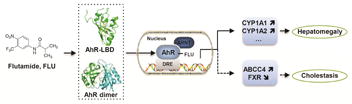
1. Introduction
Flutamide (FLU, 2-methyl-N-[4-nitro-3-(trifluoromethyl) phenyl] propanamide) is an oral, nonsteroidal antiandrogen, used since 1989 to treat advanced prostate cancer. Despite its efficacy, there is continues concern about idiosyncratic hepatotoxicity, which has an incidence of less than 1% to almost 10% [1]. In most cases, FLU-induced liver dysfunction is not acute but delayed with a latency period of approximately 4 months [2, 3]. FLU can result in cholestasis, jaundice, and liver necrosis, and has resulted in liver transplant and death [3]. As a result of this hepatotoxicity, the FDA placed a black box warning label on FLU in September 1999 [4, 5].
The aryl hydrocarbon receptor (AhR) is a ligand activated member of the basic helix-loop-helix, Per-ARNT-Sim (PAS) family of transcription factors that regulates various physiological and developmental processes. It is activated by a variety of xenobiotic compounds, most of which have lipophilic properties [6]. Upon ligand binding, the AhR translocates from the cytoplasm to the nucleus, dissociates from its cytoplasmic complex, and heterodimerizes with the aryl hydrocarbon receptor nuclear translocator (ARNT). The resulting AHR/ARNT heterodimer then binds directly to dioxin-responsive elements (DREs) of dioxin-responsive genes involved in xenobiotic metabolism that can influence the therapeutic activity and toxicity of a number of drugs, carcinogens, industrial compounds and environmental pollutants [7]. AhR regulates the expression of genes encoded for xenobiotic-metabolizing enzymes, including cytochromes P450 (CYP) 1A1, CYP1A2, and CYP1B1, UDP-glucuronosyltransferase 1A1 (UGT1A1), and NAD(P)H:quinone oxidoreductase 1 (NQO1) [8], as well as proteins involved in regulation of proliferation, differentiation, and apoptosis [9]. 2, 3, 7, 8-Tetrachlorodibenzo-p-dioxin (TCDD), 3-methylcholanthrene (3-MC), and omeprazole are classical high affinity AhR ligands [10, 11]. The toxicities induced by TCDD and many other dioxins are mediated by AhR [12, 13].
FLU, an isopropylanilide with nitro and trifluoromethyl substituents, is structurally distinct from classical AhR ligands and benzylimidazoles. However, FLU can induce CYP1A1 protein in ER-negative MDA-MB-468 cell lines, and CYP1B1 mRNA levels in both epidermal growth factor receptor 2 (ErbB2)-overexpressing BT474 and MDA-MB-468 breast cancer cell lines [14]. FLU also exhibited as an AhR agonist in rats [8].By using a compendium of gene expression profiles, FLU was shown to signal in rats similar to a classical AhR activator, but with additional effects on CYP2B and CYP3A that resembled the gene expression pattern of the atypical CYP1A inducers indole-3-carbinol and omeprazole [15]. However, there is no comprehensive study demonstrating that FLU is an AhR ligand and there is no direct evidence establishing that AhR activation mediates FLU-induced liver dysfunction. In the current study, FLU was shown to be an AhR ligand by the use of AhR-deficient (Ahr-/-) mice, primary hepatocyte cultures, luciferase reporter gene assays, and in silico ligand docking studies. In addition, prolonged treatment with FLU was found to disrupt bile acid homeostasis via modulation of the ATP binding cassette subfamily C member 4 (ABCC4) by AhR. These findings provide new evidence that activation of AhR impacts xenobiotic-induced liver injury and furthers the understanding of the role of the AhR-ABCC4 axis in bile acid homeostasis.
2. Methods
2.1. Animal experiments
Mouse experimental procedures, performed according to the National Institutes of Health (NIH) guidelines, were reviewed and approved by the National Cancer Institute Animal Care and Use Committee. The mice were housed in a specific pathogen-free environment controlled for temperature and light (25 °C, 12-hour light/dark cycle), and humidity (45–65%). Mice were treated humanely and with regard for the alleviation of suffering. Male 6- to 8-week-old mice on a C57BL/6N background were purchased from the Charles River and housed together in groups of five mice per cage. After acclimatization for 1 week in the NCI vivarium, the experiments were started. Male 8- to 10-week-old Ahr+/+ and Ahr-/- mice were on a mixed background (C57BL/6N × Sv/129) generated as previously described[16].
The study of FLU treatment consisted of three independent experiments. First, male C57BL/6N mice were randomly divided into two groups, 6 mice per group, and administered FLU (200 mg/kg, dissolved in corn oil) by gavage or with vehicle (corn oil) for 28 days. Second, male C57BL/6N mice were randomly divided into six groups and treated with the vehicle or FLU (200 mg/kg) for 3 days, 10 days, and 28 days, respectively for a time course study. Finally, a third experiment was performed to examine the contribution of AhR deficiency to the changes in liver injury induced by FLU treatment. Male littermate Ahr+/+ and Ahr-/- mice were randomly divided into eight groups and treated with the vehicle or FLU (200 mg/kg) for 3 days and 28 days, respectively. At the prescribed time points, the mice were killed after 4-hour fasting and blood collected immediately following CO2 asphyxiation, and centrifuged for 10 minutes at 8000 g at 4°C to isolate serum. Serum and livers were immediately frozen and kept at −80°C until analysis.
2.2. Histopathology and clinic biochemistry
Small blocks of liver tissue from all mice were immediately fixed in 10% neutral formalin, embedded in paraffin, and then stained with hematoxylin and eosin (H&E) followed by microscopic examination. Liver injury was further evaluated by measuring alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) in serum (Catachem In., Oxford, CT). Serum and liver total bile acid pool size was measured using the Catachem bile acid kit-C (H402-0C, Catachem In., Oxford, CT). Levels of triglycerides and free cholesterol in serum were measured using assay kits from Wako Diagnostics (Wako Diagnostics, Richmond, VA).
2.3. UPLC-ESI-QTOFMS analysis and multivariate data analysis
Ultra-performance liquid chromatography coupled quadrupole time-of-flight mass spectroscopy (UPLC-ESI-QTOFMS, ACQUITY UPLC®, Waters) was applied for metabolomic analysis as described [17]. Analytes of CA, α-MCA, β-MCA, γ-MCA, ω-MCA, DCA, CDCA, UDCA, HDCA, TCA, T- α-MCA, T-β-MCA, T-DCA, T-CDCA, T-UDCA, and T-HDCA in serum and liver extracts were quantified with TCA-d5 as internal standard. The results were calculated according to individual standard curves established as follows: areaanalyte/ areainternal.
2.4. RNA Isolation and quantitative real-time polymerase chain reaction (qPCR)
RNA was extracted from frozen liver tissue (20 mg) using TRIzol reagent (Invitrogen, Carlsbad, CA). cDNA was synthesized from 1 μg total RNA using qScript™ cDNA SuperMix (Quantabio, Beverly, MA). All the primer sequences for qPCR are listed in Table 1. Relative amount of mRNA of individual gene was calculated after normalizing to their corresponding Gapdh mRNA and the results expressed as fold change relative to the control group.
Table 1.
List of qPCR mRNA primers.
| Gene | Forward primer sequence (5’→3’) | Reverse primer sequence (5’→3’) |
|---|---|---|
| Gapdh | AGGTCGGTGTGAACGGATTTG | TGTAGACCATGTAGTTGAGGTCA |
| Abcc2 | GTGTGGATTCCCTTGGGCTTT | CACAACGAACACCTGCTTGG |
| Abcc3 | GGGCTCCAAGTTCTGGGAC | CCGTCTTGAGCCTGGATAAC |
| Abcc4 | AGCTTCAACGGTACTGGGATA | TCGTCGGGGTCATACTTCTC |
| Ahr | AGCCGGTGCAGAAAACAGTAA | AGGCGGTCTAACTCTGTGTTC |
| Arnt | GACAGACCACAGGACAGTTCC | AGCATGGACAGCATTTCTTGAA |
| Bacs | TCTATGGCCTAAAGTTCAGGCG | CTTGCCGCTCTAAAGCATCC |
| Bat | GGAAACCTGTTAGTTCTCAGGC | GTGGACCCCCATATAGTCTCC |
| Bsep | TCTGACTCAGTGATTCTTCGCA | GTGTAGAGTGAAGTCCTCCTTAGC |
| Car | GCTCTTCCGGTCCCTAACC | GACAGAACGTAGTGTTGAGTGAG |
| Cdo | GGGGACGAAGTCAACGTGG | ACCCCAGCACAGAATCATCAG |
| Csd | CCAGGACGTGTTTGGGATTGT | ACCAGTCTTGACACTGTAGTGA |
| Cyp1a1 | GACCCTTACAAGTATTTGGTCGT | GGTATCCAGAGCCAGTAACCT |
| Cyp1a2 | AGTACATCTCCTTAGCCCCAG | GGTCCGGGTGGATTCTTCAG |
| Cyp1b1 | CACCAGCCTTAGTGCAGACAG | GAGGACCACGGTTTCCGTTG |
| Cyp2b10 | AAAGTCCCGTGGCAACTTCC | TTGGCTCAACGACAGCAACT |
| Cyp3a11 | CAGCTTGGTGCTCCTCTACC | TCAAACAACCCCCATGTTTT |
| Cyp7a1 | AACAACCTGCCAGTACTAGATAGC | GTGTAGAGTGAAGTCCTCCTTAGC |
| Cyp8b1 | CTAGGGCCTAAAGGTTCGAGT | GTAGCCGAATAAGCTCAGGAAG |
| Fxr | TGGGCTCCGAATCCTCTTAGA | TGGTCCTCAAATAAGATCCTTGG |
| Gsta1 | AAGCCCGTGCTTCACTACTTC | GGGCACTTGGTCAAACATCAAA |
| Hnf4 | GGTTTAGCCGACAATGTGTGG | TCCCGCTCATTTTGGACAGC |
| Mdr1 | CCCCCGAGATTGACAGCTAC | ACTCCACTAAATTGCACATTTCCTTC |
| Nqo1 | AGGATGGGAGGTACTCGAATC | AGGCGTCCTTCCTTATATGCTA |
| Ntcp | CAAACCTCAGAAGGACCAAACA | GTAGGAGGATTATTCCCGTTGTG |
| Pxr | GATGGAGGTCTTCAAATCTGCC | GGCCCTTCTGAAAAACCCCT |
| Shp | TCTGCAGGTCGTCCGACTATTC | AGGCAGTGGCTGTGAGATGC |
| Taut | GCACACGGCCTGAAGATGA | ATTTTTGTAGCAGAGGTACGGG |
| Ugt1a2 | ATGGACACGGGACTATGTGTG | CATGGGTAACACCAGCACTTTT |
2.5. Culturing of primary hepatocytes
Primary hepatocytes were isolated from C57BL/6N mice as previously reported [18] and seeded on 12-well plates. After starvation with FBS-free Williams’ Medium E for 2 h, the hepatocytes were exposed to 10 μM or 50 μM FLU or 2 μM 3-MC (positive control) for 24 h, and then collected and lysed for gene expression analysis.
2.6. Cell culture and luciferase assays
HepG2 and Hepa-1c1c7cells, obtained from the American Type Culture Collection (Manassas, VA) were cultured in a humidified atmosphere in 5% CO2 at 37 °C, in Dulbecco’s minimal essential medium (DMEM) complemented with nonessential amino acids, 10 % fetal bovine serum (Gemini Bio-Products, Woodland, CA) and 1% penicillin/streptomycin (Invitrogen, Carlsbad, CA). For the luciferase assays, pCMV6-XL4-AhR (human, OriGene Technologies, Rockville, MD), pcDNA3/βAhR (mouse) [19], and pGudLuc 6.1 (a DRE-driven luciferase reporter, kindly provided by Gary H. Perdew) plasmids were used. Cells were seeded into 24-well plates (8 × 104 cells/well). The plasmids were transfected using Lipofectamine® 3000 reagent (Thermo Fisher Scientific). Twenty-four hours after transfection, the cells were incubated with DMSO, 2 μM 3-MC, or 5 μM, 20 μM, and 100 μM FLU for 24 h. Luciferase activities were measured and plotted relative to the protein concentrations of the lysates.
2.7. Ligand Docking and binding energy calculations
The PAS domain of the AhR is the ligand-binding domain (LBD) [20-22]. Several PAS domain structures are currently available in the Protein Data Bank (PDB) [23] and the PAS domain structure of hypoxia-inducible factor 2R (HIF-2R) (PDB id:1p97) was selected for generating a homology model of the mouse AhR-LBD based on its structure similarity with AhR in this region. Each of the AhR ligand-binding domains were built using the homology modeling protocol that described previously [24]. Following development and refinement of the model, the mouse AhR-LBD homology model was analyzed to characterize the ligand-binding pocket. Then, the mouse AhR-LBD homology model was used to structurally characterize chemicals biological data available for the AhR. The lower equilibrium dissociation constant (Kd) were estimated according to the protocol described previous [25].
2.8. Statistical analysis
All the data are representative of at least three different experiments. Statistical analysis was performed using Prism version 6.0 (GraphPad Software, San Diego, CA) and power analysis was performed using StatMate version 2.0(GraphPad Software, San Diego, CA). The values are expressed as mean ± SEM. Values that fell more than two SD from the mean were considered as outliers and were excluded from statistical analyses. When comparing two groups, statistical significance was determined using two-tailed Student’s t-test. The data were analyzed by analysis of variance (ANOVA) followed by Fisher’s LSD test to examine the differences among more than two different groups. A value of P < 0.05 was considered statistically significant.
3. Results
3.1. Effects of FLU on liver in C57BL/6N mice
Mice treated with FLU (200 mg/kg body weight in corn oil) for 4 weeks exhibited mild histopathological changes in the liver with slight necrosis (Fig. 1A). Serum biochemistry including ALT, AST, ALP, and free cholesterol were mildly elevated between vehicle and FLU-treated mice (Table 2). Total bile acids in serum (Fig. 1B) and liver (Fig. 1C) were increased by almost 2 fold and 1.3 fold after FLU treatment, respectively, thus indicating mild cholestasis, although there is no obvious histological evidence for cholestasis. Furthermore, no difference in body weights was found (Table 2). However, significant increases were observed in liver weights (Fig. 1D) and liver/body weight ratios (Fig. 1E), indicative of hepatomegaly. Transcriptional targets of AhR (Cyp1a1, Cyp1a2, Cyp1b1, Nqo1, and Gsta1) and constitutive antigen receptor (CAR, Cyp2b10) were induced in the liver, while the targets of pregnane X receptor (PXR, Cyp3a11 and multi-drug resistance gene 1, Mdr1) were not changed (Fig. 1F-H). Cyp1a1 mRNA was induced by 1000 fold, and Cyp2b10 mRNA was elevated by 30 fold. These observations suggest that FLU is a potential AhR activator in mice and that AhR activation may be associated with FLU-induced liver enlargement. The contribution of CAR activation is much less when compared to AhR.
Fig. 1.
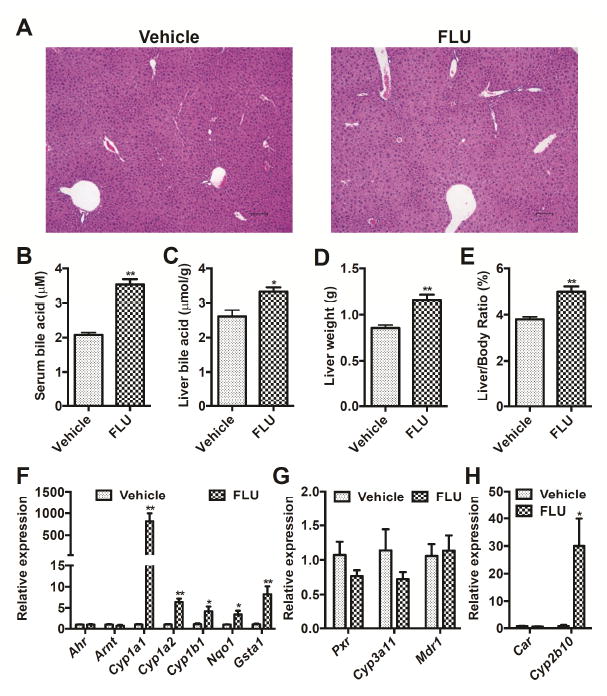
Xenobiotic response in mice after treatment with vehicle and FLU (200 mg/kg) for 28 days. (A) Light microscopic examination of H&E-stained liver sections. (B) Total bile acids in serum. (C) Total bile acids in liver. (D) Liver weight. (E) Liver weight /body weight ratio. (F) AhR battery gene expression in liver. (G) PXR battery gene expression in liver. (H) CAR battery gene expression in liver. Data are presented as mean ± SEM; n = 6/group. *P < 0.05, and ** P <0.01 versus vehicle group, by two-tailed Student’s t-test test.
Table 2.
Body weight and serum biochemistry in mice after treatment with vehicle and FLU (200 mg/kg) for 28 days.
| Vehicle | FLU | |
|---|---|---|
| Body weight (g) | 23.5 ± 0.431 | 23.9 ± 0.302 |
| ALT (U/L) | 110 ± 16.6 | 190 ± 20.1* |
| AST (U/L) | 125 ± 17.9 | 252 ± 43.7* |
| ALP (U/L) | 848 ± 53.8 | 1053 ± 54.9* |
| Triglyceride (mg/dL) | 75.8 ± 3.28 | 78.8 ± 4.39 |
| Free cholesterol (mg/dL) | 95.4 ± 5.22 | 127 ± 5.74** |
n = 6/group.
P < 0.05, and
P < 0.01 versus vehicle group, by two-tailed Student’s t-test test.
3.2. Time-dependent AhR activation by FLU
To further clarify the association between the development of hepatomegaly induced by FLU and the changes in AhR battery gene expression, mice were treated with FLU daily by intragastric gavage for 3, 10, and 28 days, respectively, and the changes in AhR and target gene expression were investigated. The hepatomegaly was induced by FLU as reflected by the increase in liver weights and liver/body weight ratios seen as early as 3 days after dosing, and further increased after 10 days and 28 days of treatment (Fig. 2A, B). Expression of AhR battery gene mRNAs levels were correspondingly increased in a time-dependent manner (Fig. 2C-G).
Fig. 2.
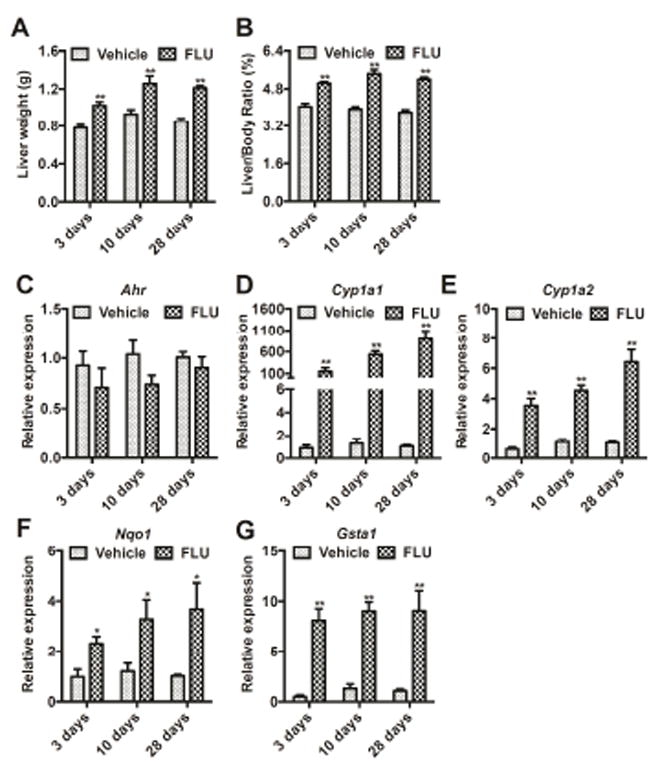
Time course of the response in mice after treatment with vehicle (coin oil) and FLU (200 mg/kg). (A) Changes in liver weights. (B) Changes in liver weight/body weight ratios. (C-G) Ahr (C) and battery genes, Cyp1a1 (D), Cyp1a2 (E), Nqo1 (F), and Gsta1 (G) mRNA expression. Data are presented as mean ± SEM; n = 6/group. *P < 0.05, and **P < 0.01 versus vehicle group, by two-tailed Student’s t-test test.
3.3. FLU regulates AhR battery genes expression in mouse hepatocytes
To investigate whether FLU directly activates the AhR signaling pathway in hepatocytes, the respective AhR battery gene mRNAs were quantitated in FLU-treated cultured mouse primary hepatocytes. FLU elevated Cyp1a1, Cyp1a2, Cyp1b1, Nqo1, and Gsta1 mRNA levels in hepatocytes at both 10 and 50 μM FLU (Fig. 3A). The activation levels at the concentration of 50 μM was similar to 3-MC at 2 μM. The concentrations of FLU and 3-MC applied were not toxic to primary hepatocytes as the cell viability indicated by CCK8 assays, was higher than 97%.
Fig. 3.
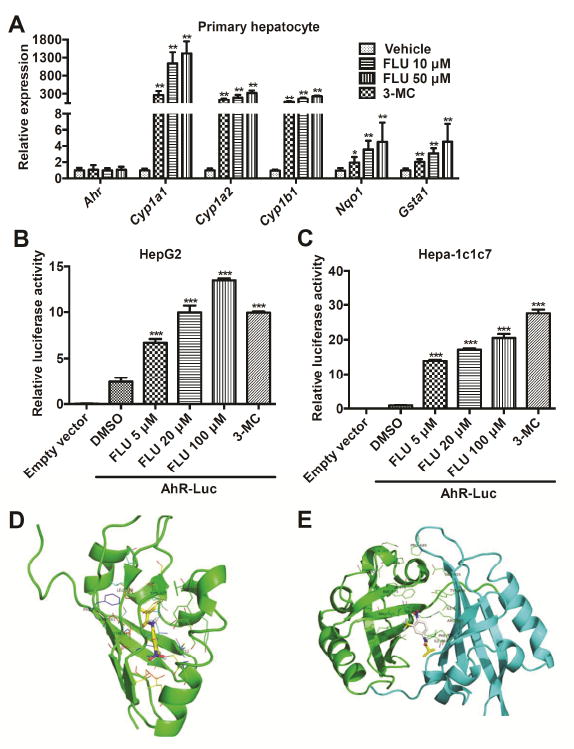
FLU stimulates mice AhR battery gene transcription. (A) qPCR analysis for AhR battery gene mRNA expression in mouse primary hepatocytes after FLU exposure. Significance was determined by two-tailed Student’s t-test test (*P < 0.05, **P < 0.01, versus vehicle group). (B and C) Luciferase assays for AhR activation in HepG2 (B) and Hepa-1c1c7 (C) cells. ***P < 0.001, compared with that of AhR-Luci+DMSO, by two-tailed Student’s t-test test. (D and E) Docking orientation of FLU into mouse AhR-LBD (PDB id: 4GHI) (D) and AhR dimer (PDB id: 4EQ1) binding pocket (ICM v3.5-1n, Molsoft) (E). The protein backbone is displayed as ribbon and colored by secondary structure. The residues are displayed as sticks and colored by atom type, with the carbon atoms in green. The ligands are displayed as sticks, colored by atom type, with carbon atoms in yellow or white, oxygen atom in red, fluorine atom in cyan, and nitrogen atom in blue.
3.4. Luciferase assay in HepG2 and Hepa-1c1c7 cell lines
To directly confirm that FLU can activate AhR, luciferase assays were performed. The AhR regulator element linked to the luciferase reporter was transiently co-transfected with empty vector or human AhR expression plasmid into HepG2 cells or mouse AhR expression plasmid into Hepa-1c1c7 cells. Cells were treated with increasing concentrations of FLU and activator 3-MC, as a positive control. AhR-dependent luciferase activity was increased in a dose dependent manner by FLU treatment with a significant activation at 5 μM or higher. The activation levels at the concentration of 20 μM in HepG2 cells and 100 μM in Hepa-1c1c7 cells were both of a similar magnitude as 3-MC at 2 μM (Fig. 3B, C).
3.5. Ligand-docking of FLU to the AhR
FLU was docked into the human AhR monomer and dimer models and the mouse AhR monomer model (Fig. 3D, E and Table 3). Kd values indicate a higher binding affinity between a particular ligand and AhR in this model. The Kd values obtained for TCDD, a classic AhR ligand, in human AhR-LBD, human AhR dimer, and mouse AhR-LBD were 1.77 × 10-5, 1.26 × 10-5, and 9.41 × 10-5 mol/L, respectively. The Kd values obtained for 3-MC, an AhR ligand used as a positive control in the cell studies, in human AhR-LBD, human AhR dimer, and mouse AhR-LBD were 7.32 × 10-6, 5.91 × 10-9, and 2.08 × 10-7 mol/L, respectively. The Kd values obtained for FLU binding in human AhR-LBD, human AhR-ARNT dimer, and mouse AhR-LBD were 1.05 × 10-5, 2.65 × 10-5, and 2.11 × 10-4 mol/L, respectively. These data confirmed that FLU is a novel AhR ligand and has similar binding affinity to human and mouse AhR as does TCDD, and also has a similar binding affinity to human AhR-LBD as does 3-MC, but much lower binding affinity to the human AhR-ARNT dimer and mouse AhR-LBD than 3-MC does.
Table 3.
Kd values of FLU, TCDD and 3-MC binding in the human and mouse AhR (unit, mol/L).
| Compound | AHR, human
|
AHR, human, dimer
|
AHR, mouse
|
|---|---|---|---|
| PDB id: 4GHI | PDB id: 4EQ1 | PDB id: 4ZP4 | |
| Ligands | 7.11×10-6 | 3.32×10-3 | 8.22×10-5 |
| FLU | 1.05×10-5 | 2.65×10-5 | 2.11×10-4 |
| TCDD | 1.77×10-5 | 1.26×10-5 | 9.41×10-5 |
| 3-MC | 7.23×10-6 | 5.91×10-9 | 2.08×10-7 |
3.6. FLU-induced hepatomegaly and bile acid disruption is AhR dependent
To determine if the effect of FLU on liver is attributable to AhR activation, FLU was administered to littermate Ahr-/- mice (200 mg/kg) for 4 weeks. Histology analysis revealed that mild necrosis induced by FLU in the Ahr+/+ mice was not evident in the Ahr-/- mice compared to the vehicle-treated mice (Fig. 4A). Liver weights (Fig. 4B) and liver/body weight ratios (Fig. 4C) were significantly increase in Ahr+/+ mice after FLU treatment, while Ahr-/- mice exhibited no change compared to the vehicle-treated Ahr+/+ mice. In agreement, the mRNA levels of AhR battery genes, including Cyp1a1, Cyp1a2, Nqo1, and Gsta1 were induced by FLU in the livers of Ahr+/+ mice, but were unaltered by FLU in Ahr-/- mice, except for Gsta1 which was slightly increased in the Ahr-/- livers (Fig. 4D). The increased total bile acids in both serum and liver by FLU treatment in the Ahr+/+ mice were not observed in the Ahr-/- mice (Fig. 4E, F). These results demonstrated that the hepatomegaly and alterations in bile acids induced by FLU are dependent on AhR.
Fig. 4.
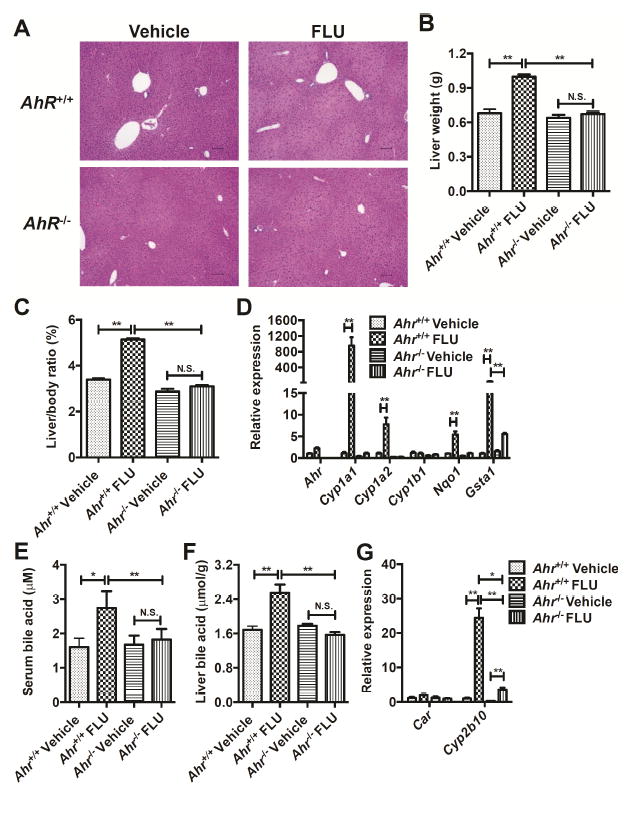
FLU alleviates hepatomegaly in Ahr-/- mice after treatment with vehicle or FLU (200 mg/kg) for 28 days. (A) Light microscopic examination of H&E-stained liver sections from mice. (B) Liver weights. (C) Liver/body weight ratios. (D) qPCR analysis for AhR battery genes mRNA expression in liver. (E) Total bile acids in serum. (F) Total bile acids in liver. (G) CAR battery gene expression in liver. Data are presented as mean ± SEM; n = 6/group. N.S., not significant. *P < 0.05, or **P < 0.01, by one-way ANOVA test.
To confirm the direct activation of AhR by FLU and not by other endogenous metabolites, FLU (200 mg/kg) was administered to littermate Ahr+/+ and Ahr-/- mice for 3 days. Similar results were exhibited as those of mice treated with FLU for 28 days. Liver weights (Fig. 5A) were significantly increased in the Ahr+/+ mice after FLU treatment, which was absent in the Ahr-/- mice. Similar trends were also observed in the liver/body weight ratios (Fig. 5B), but the results were not statistically significant (p = 0.07). Cyp1a1, Cyp1a2, Nqo1, and Gsta1 mRNA levels were significantly induced in the livers of Ahr+/+ mice, while Ahr-/- mice exhibited no change in the expression of these mRNAs after FLU treatment (Fig. 5C).
Fig. 5.
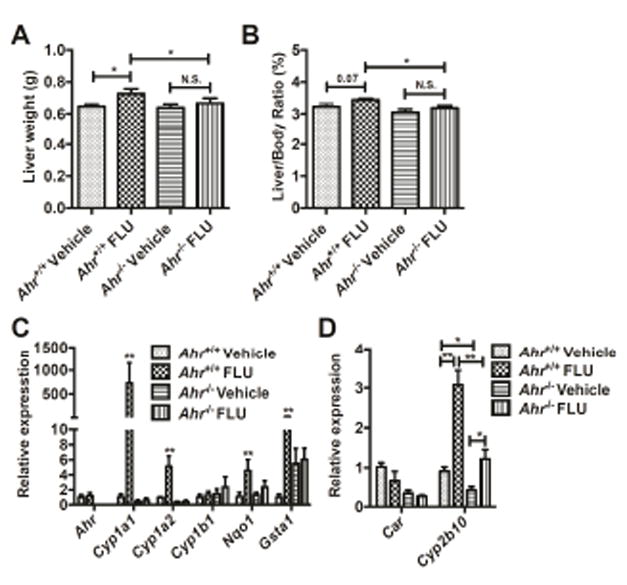
FLU alleviates hepatomegaly in Ahr-/- mice after treatment with vehicle and flutamide (200 mg/kg) for 3 days. A Liver weights. B Liver/body weight ratios. C qPCR analysis for AhR battery genes mRNA expression in liver. D CAR battery gene expression in liver. Data are presented as mean ± SEM; n = 6/group. N.S., not significant. *P < 0.05, or **P < 0.01, by one-way ANOVA test.
To further establish the connection between AhR and CAR activation by FLU, expression of Car and Cyp2b10 were determined in the livers of Ahr+/+ and Ahr-/- mice after prolonged FLU treatment. FLU increased Cyp2b10 mRNA level by 25 fold in the Ahr+/+ mice, while the expression of Cyp2b10 mRNA was 12-fold increased in the Ahr-/- mice (Fig. 4G). Similar results were obtained in the Ahr+/+ and Ahr-/- mice after a three-day treatment (Fig. 5D). These data suggest that activation of CAR by FLU may be partially mediated by AhR.
3.7. Multivariate data analysis and metabolite identification in Ahr+/+ mice and Ahr-/- mice
To further understand the association between AhR activation and FLU-induced liver dysfunction, metabolomic analysis was used to profile the metabolites in the serum from FLU-treated mice. Flutamide metabolites were excluded from the multivariate data analysis to ensure the separation was driven by the endogenous metabolite alterations. Unsupervised PCA was used to analyze the data sets from both vehicle- and FLU-treated groups. As expected, PCA modeling of mouse serum showed that samples from FLU treated Ahr+/+ mice on day 28 clustered away from the control samples, indicating significant metabolic changes between these two groups (Fig. 6A). Notably, they varied along the first component in the scores plot. These observations suggested that some metabolites were significantly modified by FLU treatment in the Ahr+/+ mice. In contrast, the samples from FLU treated Ahr-/- mice could not be separated from those of control mice (Fig. 6B), suggesting that similar metabolic disturbances were not present in FLU treated Ahr-/- mice.
Fig. 6.
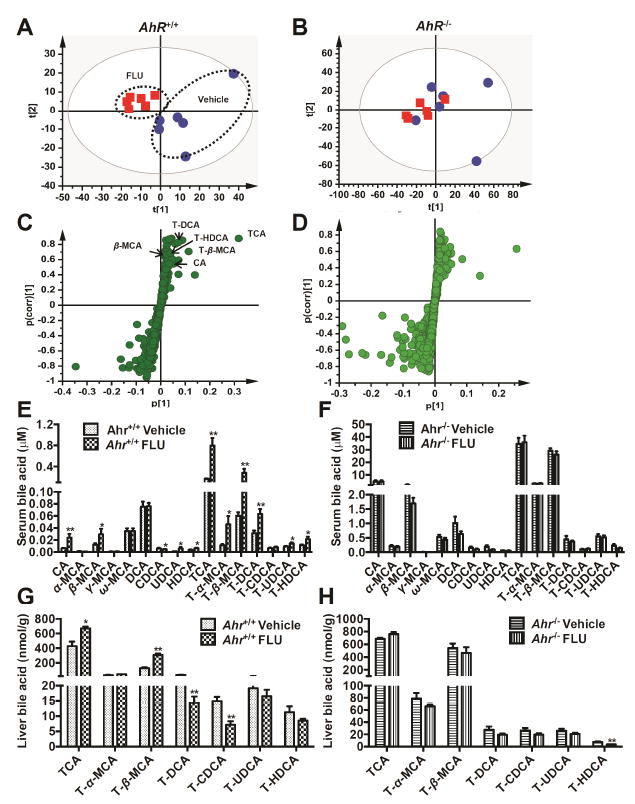
Multivariate data analysisand metabolite identification in Ahr+/+ and Ahr-/- mice by using UPLC ESI QTOFMS analysis. Ahr+/+ and Ahr-/- mice were treated with vehicle or FLU (200mg/kg) for 28 days. (A) Scores plot of serum metabolome in WT mice treated with vehicle (
 ) and FLU (
) and FLU (
 ) as determined by PCA. (B) Scores plot of serum metabolome in Ahr-/- mice dosed with vehicle (
) and FLU (
) as determined by PCA. (C) S-plot of OPLS-DA recognized serum metabolome in vehicle- and FLU-treated Ahr+/+ mice, in which identified metabolites were indicated. (D) S-plot of OPLS-DA recognized serum metabolome in vehicle- and FLU-treated Ahr-/- mice. Each point represents an individual mouse serum sample (A, B) and a unique ion (C, D). The t[1] and t[2] represent principal components 1 and 2, respectively. The p(corr)[1] represents the interclass difference, and p[1] represents the relative abundance of the ions. (E-H) Quantitation of bile acids in Ahr+/+ and Ahr-/- mice. (E) Individual bile acids in serum of Ahr+/+ mice. (F) Individual bile acids in serum of Ahr-/- mice. (G) Individual bile acids in livers of Ahr+/+ mice. (H) Individual bile acids in livers of Ahr-/- mice. Data are presented as mean ± SEM; n = 6/group. *P < 0.05, or **P < 0.01, versus vehicle group, by two-tailed Student’s t-test.
) as determined by PCA. (B) Scores plot of serum metabolome in Ahr-/- mice dosed with vehicle (
) and FLU (
) as determined by PCA. (C) S-plot of OPLS-DA recognized serum metabolome in vehicle- and FLU-treated Ahr+/+ mice, in which identified metabolites were indicated. (D) S-plot of OPLS-DA recognized serum metabolome in vehicle- and FLU-treated Ahr-/- mice. Each point represents an individual mouse serum sample (A, B) and a unique ion (C, D). The t[1] and t[2] represent principal components 1 and 2, respectively. The p(corr)[1] represents the interclass difference, and p[1] represents the relative abundance of the ions. (E-H) Quantitation of bile acids in Ahr+/+ and Ahr-/- mice. (E) Individual bile acids in serum of Ahr+/+ mice. (F) Individual bile acids in serum of Ahr-/- mice. (G) Individual bile acids in livers of Ahr+/+ mice. (H) Individual bile acids in livers of Ahr-/- mice. Data are presented as mean ± SEM; n = 6/group. *P < 0.05, or **P < 0.01, versus vehicle group, by two-tailed Student’s t-test.
After pattern recognition of serum samples in the vehicle- and FLU-treated groups, OPLS-DA was used to produce loading scatter S-plots (Fig. 6C). Each point represents an ion contributing to sample separation between groups. Ions that significantly contributed to separation between FLU-treated Ahr+/+ mice and the vehicle group increased in the first quadrant and decreased in the third quadrant. These ions are listed in Table 4 according to their correlations and abundance ranks after primary screening. Metabolites that had characteristics and accurate masses matching TCA, T-β-MCA, T-DCA, T-HDCA, β-MCA and CA were reanalyzed and compared with authentic standards using UPLC-ESI-QTOFMS system. Compound identities were confirmed by comparing retention times and fragmentation profiles of serum metabolites with authentic standards. Compared with the distribution in the Ahr+/+ mice, there were significantly fewer ions that were increased in the Ahr-/- mice (Fig. 6D).
Table 4.
Serum metabolites ions identified in the negative mode that were significantly altered in wild-type mice treated with flutamide for 28 days.
| P[1] | p(corr)[1] | RT(min) | m/z Found | Putative ion form | Elemental composition | Mass error (ppm) | Supposed identity |
|---|---|---|---|---|---|---|---|
| 0.32 | 0.88 | 10.58 | 514.2845 | [M-H]- | C26H45NO7S | 1.3 | TCA |
| 0.11 | 0.71 | 7.30 | 514.2890 | [M-H]- | C26H45NO7S | 10.3 | T-β-MCA |
| 0.07 | 0.8 | 13.99 | 498.2883 | [M-H]- | C26H45NO6S | -1.2 | T-DCA |
| 0.04 | 0.68 | 10.17 | 498.2891 | [M-H]- | C26H45NO6S | 0.4 | T-HDCA |
| 0.03 | 0.55 | 14.95 | 407.2789 | [M-H]- | C24H40O5 | -2.0 | CA |
| 0.02 | 0.66 | 12.69 | 407.2790 | [M-H]- | C24H40O5 | -1.7 | β-MCA |
To determine whether the composition of bile acids was affected by FLU exposure, levels of 16 bile acid metabolites were quantified by using UPLC-ESI-QTOFMS, according to a published method (Jiang et al. 2015). In serum, compared with vehicle-treated mice, FLU-treated Ahr+/+ mice had significantly higher levels of four unconjugated bile acids (CA, β-MCA, UDCA, and HDCA), and they also had significantly higher levels of most conjugated bile acids (TCA, T-α-MCA, T-β-MCA, T-DCA, T-UDCA, and T-HDCA) (Fig. 6E). However, the level of FXR agonist CDCA in serum was decreased significantly after FLU treatment (Fig. 6E). No change of bile acid concentrations was observed in serum of Ahr-/- mice after treated with the FLU for 28 days (Fig. 6F). In liver, the concentrations of unconjugated bile acids were very low and thus only the conjugated bile acids were measured. TCA and T-β-MCA, which are among the most abundant bile acids in mice, were significant increased by 1.6 fold and 2.5 fold after FLU treatment in the livers from Ahr+/+ mice respectively (Fig. 6G), but remained unchanged in the Ahr-/- mice (Fig. 6H). Although T-DCA and T-CDCA decreased in the Ahr+/+ mice, the relative abundance of these two bile acids were much lower than TCA and T-β-MCA. These observations further indicate that FLU affects bile acid homeostasis through an AhR-dependent mechanism. In addition, the TCA/T-β-MCA ratio was increased by 2 fold in serum but decreased in the liver after FLU treatment (Fig. 7), indicating that more FXR agonist TCA was transported outside of the hepatocytes leaving increased cellular levels of the FXR antagonist T-β-MCA.
Fig. 7.
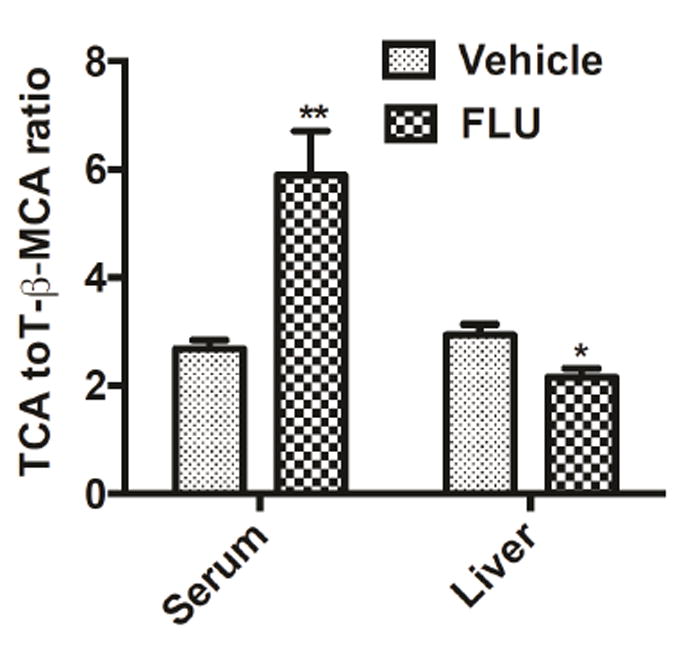
TCA/TβMCA ratios in the serum and livers of mice treated with vehicle or FLU (200 mg/kg) for 28 days. Data are presented as mean ± SEM; n = 6/group. *P < 0.05 versus vehicle group, by two-tailed Student’s t-test.
3.8. Effects of FLU on gene expression profiles and bile acid related genes
Farnesoid X receptor (FXR) serves as a bile acid sensor and plays a key role in regulation of bile acid homeostasis in enterohepatic circulation [26]. The question of whether altered bile acid profiles in FLU-treated Ahr+/+ mice were associated with changes in gene expression of enzymes involved in bile acid synthesis and transport was further addressed. FLU treatment (28 days) significantly down-regulated the expression of Fxr mRNA and its target gene small heterodimer partner (Shp) mRNA in the liver (Fig. 8A). The bile acid synthesis-related Cyp7a1 and Cyp8b1 mRNAs were not affected by FLU treatment (Fig. 8B). FLU has notable effects on the expression of bile acid transporters in the liver. The mRNA expression of bile salt export pump (BSEP) encoded by Abcb11, which transports bile acids from liver into the bile canaliculi, was significantly decreased by FLU, whereas the mRNA of Abcc4, the basolateral transporter, was markedly up-regulated by more than 10 fold (Fig. 8C). Different regulation of genes mediating bile acid homeostasis provides mechanistic evidence for the observation of elevated bile acid levels in serum and livers of the FLU-treated mice. In contrast, FLU-treated Ahr-/- mice did not exhibit significant changes in Fxr, Shp, Abcb11, and Abcc4 mRNA levels (Fig. 8D-F). These observations strongly indicate that FLU affects Abcc4 and the FXR signaling pathways through an AhR-dependent mechanism. However, only the expression of Abcc4 mRNA was induced by 2 fold after short-term FLU treatment for 3 days, and no significant change in Fxr, Shp, and Abcb11 mRNA levels was noted (Fig. 8G-I), thus suggesting that the FXR inhibition in the liver is a secondary event triggered by FLU.
Fig. 8.
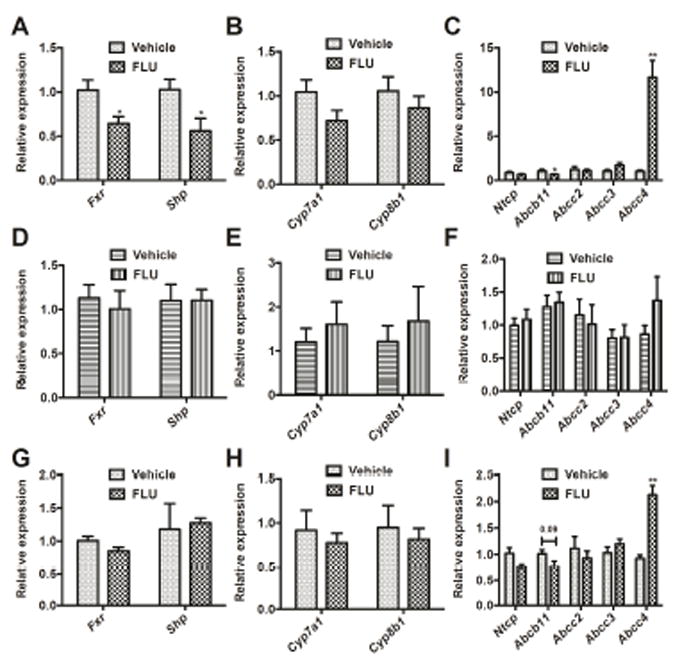
qPCR analysis for FXR and relative target gene expression mRNAs after FLU exposure. (A) qPCR analysis for Fxr and Shp mRNA expression in the livers of Ahr+/+ mice treated with FLU for 28 days. (B) qPCR analysis for bile acid synthesis mRNAs in the livers of Ahr+/+ mice treated with FLU for 28 days. (C) qPCR analysis for bile acid transporter mRNAs in the livers of Ahr+/+ mice treated with FLU for 28 days. (D) qPCR analysis for Fxr and Shp mRNA expression in the livers of Ahr-/- mice treated with FLU for 28 days. (E) qPCR analysis for bile acid synthesis mRNAs in the livers of Ahr-/- mice treated with FLU for 28 days. (F) qPCR analysis for bile acid transporter mRNAs in the livers of Ahr-/- mice treated with FLU for 28 days. (G) qPCR analysis for Fxr and Shp mRNA expression in the livers of Ahr+/+ mice treated with FLU for 3 days. (H) qPCR analysis for bile acid synthesis mRNAs in the livers of Ahr+/+ mice treated with FLU for 3 days. (I) qPCR analysis for bile acid transporter mRNAs in the livers of Ahr+/+ mice treated with FLU for 3 days. Data are presented as mean ± SEM; n = 6/group. *P < 0.05, or **P < 0.01, versus vehicle group, by two-tailed Student’s t-test.
4. Discussion
Clinical practice of FLU has been associated with hepatotoxicity, such as temporary elevations in transaminase markers in up to 62% of users and rare incidences of severe liver dysfunction [27, 28]. The common pattern of FLU-caused hepatotoxicity has been described as cholestatic hepatitis. However, the mechanism of FLU-induced liver injury remains obscure. The current study revealed that prolonged FLU treatment can induce cholestasis by modulating Abcc4 and inhibiting of FXR signaling; all of these events are AhR dependent.
Based on allometric scaling calculations, the FLU dose (200 mg/kg body weight) used in this study is equivalent to approximately 20 mg/kg body weight in humans. The total daily dose of FLU for adults is 750 mg (equivalent to 12.5 mg/kg body weight) in the clinic and thereby the dose used in the present study is within two fold of the therapeutic range in humans. Thus, the toxic responses observed in this study, including hepatomegaly and cholestasis, might be similar to that noted in patients.
Xenobiotics sometimes mimic small endogenous ligands, and thus can bind to a particular receptor either as an agonist or antagonist and disturb cellular homeostasis. In the current study, Cyp1a1 and Cyp1a2 mRNAs were markedly induced after FLU treatment in Ahr+/+ mice and not in Ahr-/- mice indicating that FLU-induced hepatomegaly is AhR dependent. The combination of induction study in primary hepatocytes, luciferase assays in the mouse cell line Hepa-1c1c7 transfected with mouse AhR and in silico ligand docking study with mouse AhR-LBD revealed that FLU is a mouse AhR ligand. Luciferase assays in the HepG2human cell line transfected with human AhR and in silico ligand docking analysis with human AhR-LBD and AhR dimer demonstrated that FLU is human AhR ligand. The potency of FLU towards both human and mouse AhR is identical, based on their identical Kd values calculated by ligand-docking. The human AhR and mouse AhR share limited (58%) transactivation domain sequence identity [29]. This finding suggests that AhR may differentially respond to ligand activation. The present results indicated that FLU may also act as an AhR agonist in humans. Furthermore, the primary route of FLU metabolism is 2-hydroxylation, by CYP1A1, CYP1A2, and CYP1B1 [30]. FLU-induced liver toxicity has also been linked to CYP1A-, CYP2C-, and CYP3A-mediated metabolic activation [31-33]. Increased CYP1A1 and CYP1A2 expression by AhR activation may alter the metabolic fate of FLU and its related toxic effects.
Ligand activation of AhR causes it to translocate from the cytoplasm to the nucleus, heterodimerize with ARNT, and regulates genes involved in xenobiotic metabolism [7]. After FLU treatment, the levels of Ahr and Arnt mRNA were not changed, suggesting that FLU only affects the AhR activity, but does not affect its transcription. Others reported that dehydroepiandrosterone can down-regulate CYP1A2 induction which requires the androgen receptor and increases the degradation rates of CYP1A2 mRNA by a post-transcriptional mechanism [34]. FLU is an androgen receptor antagonist and thus may abolish the dehydroepiandrosterone-mediated repression of CYP1A2 induction.
AhR activation was reported to up-regulate CAR activity in murine and human livers [35]. Although the expression of Cyp2b10 mRNA in the Ahr-/- mice was less than 50% of that in Ahr+/+ mice, FLU still can activate CAR in Ahr-/- mice, indicating that modulation of CAR signaling by FLU is both dependent and independent on AhR. Further studies on CAR activation are needed, especially with Car-null mice.
Cholestasis is defined as impairment of bile flow and retention of bile acids. FLU is a potent hepatotoxin, that can induce cholestatic jaundice in some patients [1]. Others found that FLU may cause cholestasis by inhibiting BSEP-mediated bile acid excretion [36]. Thus, bile acid metabolomics in serum and liver, and liver gene expression were performed. FLU significantly increased almost all the conjugated and unconjugated bile acids in serum, and TCA and T-β-MCA in the livers of Ahr+/+ mice, but not in the Ahr-/- mice. Taurine-conjugated bile acids constitute about 18% of total bile acids and 60% of taurine-conjugated bile acids are TCA and T-β-MCA in mouse [37]. The combination of elevated total bile acids, TCA and T-β-MCA observed indicated the occurrence of cholestasis by FLU, which was dependent on AhR.
The genes encoding bile acid synthesis enzymes and transporters are regulated by a complex network of transcriptional cascades, notably by the ligand-activated nuclear receptors FXR [38]. Certain bile acids including CDCA, DCA, and TCA are endogenous ligands for FXR [39] and T-β-MCA was found to be a natural FXR antagonist in mice [40, 41]. Transcriptional analysis revealed that the expression of Abcc4 mRNA was increased after both short-term (3 days) and long-term (28 days) treatment with FLU in the Ahr+/+ mice, but eased in Ahr-/- mice. ABCC4 is an ATP binding cassette transporter that facilitates the transport of bile salt conjugates from hepatocytes to blood [42]. The AhR binds to the proximal promoter of Abcc4 gene [43] and leads to induction of Abcc4 mRNA in the livers of male and female mice, but no change in Ahr-/- mice [44]. The AhR agonist TCDD significantly increased ABCC4 expression at both the mRNA and protein levels in HepG2 cells and human hepatocytes treated with TCDD [43]. The current metabolomics data suggest that increased ABCC4 expression by AhR activation may result in disruption of bile acid homeostasis. ABCC4 might have different substrate preferences towards different bile acids, leading to increase or decrease in bile acid metabolites that are FXR ligands. Consistently, the FXR antagonist T-β-MCA was found to be increased in liver compared to the FXR agonist TCA. Thereby, prolonged treatment with FLU inhibits FXR signaling in liver as the expression of FXR target genes Shp and Abcb11 were significantly decreased. Inhibition of BSEP (encoded by Abcb11) is known to induce cholestasis [45, 46]. Altogether, coordinated regulation of the ABCC4 may be directly mediated by AhR and lead to cholestatic liver injury. A recent study reported that 2,3,7,8-tetrachlorodibenzofuran (TCDF), a persistent environmental contaminant and AhR ligand, can also alter bile acid metabolism and inhibit FXR signaling [47]. Moreover, it should be noted that the bile acid pool compositions are inherently different in mice and humans. MCA is the primary bile acid in mice, whereas CDCA is the primary bile acid in humans. Concentrations of the FXR agonist CDCA and its taurine-conjugate T-CDCA were both decreased in mouse serum and liver, which may result in the suppressed FXR signaling. Thus, FLU may also induce cholestasis through the AhR-ABCC4-FXR axis in humans. However, the exact mechanism regarding AhR activation and FXR signaling requires additional investigation.
In conclusion, the present work revealed that FLU is able to activate AhR in the liver and in primary hepatocytes. In silico modeling was employed to fit the binding of FLU in the AhR-LBD, supporting the notion that FLU is an AhR ligand. Moreover, Ahr-null mice were used to show that FLU elevates liver injury in an AhR-dependent manner in vivo. Regulation of the Abcc4 by FLU may be mediated by AhR, potentially leading to FXR signaling inhibition and resulting in cholestatic liver injury. Activation of the AhR by FLU may represent a novel mechanism contributing to the FLU-induced liver injury. More broadly, the mechanism proposed in this study might apply to humans. However, the link between AhR activation and FXR inhibition and cholestasis is presumptive and needs further validation.
Acknowledgments
This study was supported by the National Cancer Institute Intramural Research Program and the National Natural Science Foundation of China [Grant No. 81473415 and 81373040]. X.G. was supported by Shanxi University. We thank Linda G. Byrd for assistance with the mouse studies.
Abbreviations
- FLU
flutamide
- AhR
aryl hydrocarbon receptor
- ARNT
aryl hydrocarbon receptor nuclear translocator
- CYP
cytochrome P450
- TCDD
2, 3, 7, 8-tetrachlorodibenzo-p-dioxin
- 3-MC
3-methylcholanthrene
- ABCC4
ATP-binding cassette sub-family C member 4
- MRP4
multidrug resistance-associated protein 4
- PAS
Per-ARNT-Sim
- DRE
dioxin-responsive elements
- LBD
ligand binding domain
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- ALP
alkaline phosphatase
- PXR
pregnane X receptor
- CAR
constitutive androstane receptor
- UGT1A1
UDP-glucuronosyltransferase 1A1
- NQO1, NAD(P)H
quinone oxidoreductase 1
- FXR
farnesoid X receptor
- SHP
small heterodimer partner
- BSEP
bile salt export pump
- CA
cholic acid
- α-MCA
α-muricholic acid
- β-MCA
β-muricholic acid
- γ-MCA
γ-muricholic acid
- ω-MCA
ω-muricholic acid
- DCA
deoxycholic acid
- CDCA
chenodeoxycholic acid
- UDCA
ursodeoxycholic acid
- HDCA
hyodeoxycholic acid
- TCA
taurocholic acid
- T-α-MCA
tauro-α-muricholic acid
- T-β-MCA
tauro-β-muricholic acid
- T-DCA
tauro deoxycholic acid
- T-CDCA
taurochenodeoxycholic acid
- T-UDCA
tauroursodeoxycholic acid
- T-HDCA
taurohyodeoxycholic acid
Footnotes
Conflict of interest
The authors declare that they have no conflict of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Contributor Information
Xiaoxia Gao, Email: gaoxiaoxia@sxu.edu.cn.
Cen Xie, Email: cen.xie@nih.gov.
Yuanyuan Wang, Email: ywang197@uthsc.edu.
Yuhong Luo, Email: yuhong.luo@nih.gov.
Tomoki Yagai, Email: tomoki.yagai@nih.gov.
Dongxue Sun, Email: dongxue.sun@nih.gov.
Xuemei Qin, Email: qinxm@sxu.edu.cn.
Kristopher W. Krausz, Email: krauszk@intra.nci.nih.gov.
Frank J. Gonzalez, Email: gonzalef@mail.nih.gov.
References
- 1.Wysowski DK, Fourcroy JL. Flutamide hepatotoxicity. The Journal of urology. 1996;155:209–212. [PubMed] [Google Scholar]
- 2.Manso G, Thole Z, Salgueiro E, Revuelta P, Hidalgo A. Spontaneous reporting of hepatotoxicity associated with antiandrogens: data from the Spanish pharmacovigilance system. Pharmacoepidemiol Drug Saf. 2006;15:253–259. doi: 10.1002/pds.1168. [DOI] [PubMed] [Google Scholar]
- 3.Thole Z, Manso G, Salgueiro E, Revuelta P, Hidalgo A. Hepatotoxicity induced by antiandrogens: A review of the literature. Urologia internationalis. 2004;73:289–295. doi: 10.1159/000081585. [DOI] [PubMed] [Google Scholar]
- 4.Takakusa H, Masumoto H, Yukinaga H, Makino C, Nakayama S, Okazaki O, Sudo K. Covalent binding and tissue distribution/retention assessment of drugs associated with idiosyncratic drug toxicity. Drug Metabolism and Disposition. 2008;36:1770–1779. doi: 10.1124/dmd.108.021725. [DOI] [PubMed] [Google Scholar]
- 5.Coe KJ. Metabolism and Cytotoxicity of the Nitroaromatic Drug Flutamide and Its Cyano Analog in Hepatocyte Cell Lines. ProQuest. 2008 [Google Scholar]
- 6.Sorg O. AhR signalling and dioxin toxicity. Toxicol Lett. 2014;230:225–233. doi: 10.1016/j.toxlet.2013.10.039. [DOI] [PubMed] [Google Scholar]
- 7.Flaveny CA, Murray IA, Perdew GH. Differential gene regulation by the human and mouse aryl hydrocarbon receptor. Toxicol Sci. 2010;114:217–225. doi: 10.1093/toxsci/kfp308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hu W, Sorrentino C, Denison MS, Kolaja K, Fielden MR. Induction of cyp1a1 is a nonspecific biomarker of aryl hydrocarbon receptor activation: results of large scale screening of pharmaceuticals and toxicants in vivo and in vitro. Molecular pharmacology. 2007;71:1475–1486. doi: 10.1124/mol.106.032748. [DOI] [PubMed] [Google Scholar]
- 9.Abel J, Haarmann-Stemmann T. An introduction to the molecular basics of aryl hydrocarbon receptor biology. Biol Chem. 2010;391:1235–1248. doi: 10.1515/BC.2010.128. [DOI] [PubMed] [Google Scholar]
- 10.Mimura J, Fujii-Kuriyama Y. Functional role of AhR in the expression of toxic effects by TCDD. Biochimica et Biophysica Acta (BBA)-General Subjects. 2003;1619:263–268. doi: 10.1016/s0304-4165(02)00485-3. [DOI] [PubMed] [Google Scholar]
- 11.Yoshinari K, Ueda R, Kusano K, Yoshimura T, Nagata K, Yamazoe Y. Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem Pharmacol. 2008;76:139–145. doi: 10.1016/j.bcp.2008.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Fernandez-Salguero PM, Hilbert DM, Rudikoff S, Ward JM, Gonzalez FJ. Aryl-hydrocarbon receptor-deficient mice are resistant to 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced toxicity. Toxicol Appl Pharmacol. 1996;140:173–179. doi: 10.1006/taap.1996.0210. [DOI] [PubMed] [Google Scholar]
- 13.Bunger MK, Moran SM, Glover E, Thomae TL, Lahvis GP, Lin BC, Bradfield CA. Resistance to 2,3,7,8-tetrachlorodibenzo-p-dioxin toxicity and abnormal liver development in mice carrying a mutation in the nuclear localization sequence of the aryl hydrocarbon receptor. J Biol Chem. 2003;278:17767–17774. doi: 10.1074/jbc.M209594200. [DOI] [PubMed] [Google Scholar]
- 14.Jin UH, Lee SO, Safe S. Aryl hydrocarbon receptor (AHR)-active pharmaceuticals are selective AHR modulators in MDA-MB-468 and BT474 breast cancer cells. J Pharmacol Exp Ther. 2012;343:333–341. doi: 10.1124/jpet.112.195339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.N SD, Coe Kevin J, Ulrich Roger G, He Yudong, Dai Xudong, Cheng Olivia, Caguyong Michelle, Roberts Chris J, Slatter J Greg. PProfiling the hepatic effects of flutamide in rats: a microarray comparison with classical aryl hydrocarbon receptor ligands and atypical cyp1a inducers. Drug Metab Dispos. 2006;34:1266–1275. doi: 10.1124/dmd.105.009159. [DOI] [PubMed] [Google Scholar]
- 16.Fernandez-Salguero P, Pineau T, Hilbert DM, McPhail T, Lee SS, Kimura S, Nebert DW, Rudikoff S, Ward JM, Gonzalez FJ. Immune system impairment and hepatic fibrosis in mice lacking the dioxin-binding Ah receptor. Science. 1995;268:722–726. doi: 10.1126/science.7732381. [DOI] [PubMed] [Google Scholar]
- 17.Jiang C, Xie C, Lv Y, Li J, Krausz KW, Shi J, Brocker CN, Desai D, Amin SG, Bisson WH, Liu Y, Gavrilova O, Patterson AD, Gonzalez FJ. Intestine-selective farnesoid X receptor inhibition improves obesity-related metabolic dysfunction. Nature communications. 2015;6:10166. doi: 10.1038/ncomms10166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Seglen PO. Preparation of isolated rat liver cells. Methods in cell biology. 1976;13:29–83. doi: 10.1016/s0091-679x(08)61797-5. [DOI] [PubMed] [Google Scholar]
- 19.Fukunaga BN, Hankinson O. Identification of a novel domain in the aryl hydrocarbon receptor required for DNA binding. J Biol Chem. 1996;271:3743–3749. doi: 10.1074/jbc.271.7.3743. [DOI] [PubMed] [Google Scholar]
- 20.Beischlag TV, Morales JL, Hollingshead BD, Perdew GH. The aryl hydrocarbon receptor complex and the control of gene expression. Critical Reviews™ in Eukaryotic Gene Expression . 2008;18 doi: 10.1615/critreveukargeneexpr.v18.i3.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Nguyen LP, Bradfield CA. The search for endogenous activators of the aryl hydrocarbon receptor. Chemical research in toxicology. 2007;21:102–116. doi: 10.1021/tx7001965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hankinson O. The aryl hydrocarbon receptor complex. Annual review of pharmacology and toxicology. 1995;35:307–340. doi: 10.1146/annurev.pa.35.040195.001515. [DOI] [PubMed] [Google Scholar]
- 23.Bisson WH, Koch DC, O’Donnell EF, Khalil SM, Kerkvliet NI, Tanguay RL, Abagyan R, Kolluri SK. Modeling of the aryl hydrocarbon receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands. J Med Chem. 2009;52:5635–5641. doi: 10.1021/jm900199u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cardozo T, Totrov M, Abagyan R. Homology modeling by the ICM method. Proteins: Structure, Function, and Bioinformatics. 1995;23:403–414. doi: 10.1002/prot.340230314. [DOI] [PubMed] [Google Scholar]
- 25.Schapira M, Totrov M, Abagyan R. Prediction of the binding energy for small molecules, peptides and proteins. Journal of Molecular Recognition. 1999;12:177–190. doi: 10.1002/(SICI)1099-1352(199905/06)12:3<177::AID-JMR451>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 26.Matsubara T, Li F, Gonzalez FJ. FXR signaling in the enterohepatic system. Molecular and cellular endocrinology. 2013;368:17–29. doi: 10.1016/j.mce.2012.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gomez JL, Dupont A, Cusan L, Tremblay M, Suburu R, Lemay M, Labrie F. Incidence of liver toxicity associated with the use of flutamide in prostate cancer patients. The American journal of medicine. 1992;92:465–470. doi: 10.1016/0002-9343(92)90741-s. [DOI] [PubMed] [Google Scholar]
- 28.Osculati A, Castiglioni C. Fatal liver complications with flutamide. Lancet. 2006;367:1140–1141. doi: 10.1016/S0140-6736(06)68498-5. [DOI] [PubMed] [Google Scholar]
- 29.Flaveny C, Reen RK, Kusnadi A, Perdew GH. The mouse and human Ah receptor differ in recognition of LXXLL motifs. Arch Biochem Biophys. 2008;471:215–223. doi: 10.1016/j.abb.2008.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rochat B, Morsman JM, Murray GI, Figg WD, McLeod HL. Human CYP1B1 and anticancer agent metabolism: mechanism for tumor-specific drug inactivation? Journal of Pharmacology and Experimental Therapeutics. 2001;296:537–541. [PubMed] [Google Scholar]
- 31.Fau D, Eugene D, Berson A, Letteron P, Fromenty B, Fisch C, Pessayre D. Toxicity of the antiandrogen flutamide in isolated rat hepatocytes. Journal of Pharmacology and Experimental Therapeutics. 1994;269:954–962. [PubMed] [Google Scholar]
- 32.Kang P, Dalvie D, Smith E, Zhou S, Deese A. Identification of a novel glutathione conjugate of flutamide in incubations with human liver microsomes. Drug Metab Dispos. 2007;35:1081–1088. doi: 10.1124/dmd.107.014860. [DOI] [PubMed] [Google Scholar]
- 33.Kang P, Dalvie D, Smith E, Zhou S, Deese A, Nieman JA. Bioactivation of flutamide metabolites by human liver microsomes. Drug Metab Dispos. 2008;36:1425–1437. doi: 10.1124/dmd.108.020370. [DOI] [PubMed] [Google Scholar]
- 34.Belic A, Toth K, Vrzal R, Temesvari M, Porrogi P, Orban E, Rozman D, Dvorak Z, Monostory K. Dehydroepiandrosterone post-transcriptionally modifies CYP1A2 induction involving androgen receptor. Chem Biol Interact. 2013;203:597–603. doi: 10.1016/j.cbi.2013.04.002. [DOI] [PubMed] [Google Scholar]
- 35.Patel RD, Hollingshead BD, Omiecinski CJ, Perdew GH. Aryl-hydrocarbon receptor activation regulates constitutive androstane receptor levels in murine and human liver. Hepatology. 2007;46:209–218. doi: 10.1002/hep.21671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Iwanaga T, Nakakariya M, Yabuuchi H, Maeda T, Tamai I. Involvement of bile salt export pump in flutamide-induced cholestatic hepatitis. Biol Pharm Bull. 2007;30:739–744. doi: 10.1248/bpb.30.739. [DOI] [PubMed] [Google Scholar]
- 37.Garcia-Canaveras JC, Donato MT, Castell JV, Lahoz A. Targeted profiling of circulating and hepatic bile acids in human, mouse, and rat using a UPLC-MRM-MS-validated method. J Lipid Res. 2012;53:2231–2241. doi: 10.1194/jlr.D028803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Eloranta JJ, Kullak-Ublick GA. Coordinate transcriptional regulation of bile acid homeostasis and drug metabolism. Archives of Biochemistry and Biophysics. 2005;433:397–412. doi: 10.1016/j.abb.2004.09.019. [DOI] [PubMed] [Google Scholar]
- 39.de Aguiar Vallim TQ, Tarling EJ, Edwards PA. Pleiotropic roles of bile acids in metabolism. Cell Metab. 2013;17:657–669. doi: 10.1016/j.cmet.2013.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sayin SI, Wahlstrom A, Felin J, Jantti S, Marschall HU, Bamberg K, Angelin B, Hyotylainen T, Oresic M, Backhed F. Gut microbiota regulates bile acid metabolism by reducing the levels of tauro-beta-muricholic acid, a naturally occurring FXR antagonist. Cell metabolism. 2013;17:225–235. doi: 10.1016/j.cmet.2013.01.003. [DOI] [PubMed] [Google Scholar]
- 41.Li F, Jiang C, Krausz KW, Li Y, Albert I, Hao H, Fabre KM, Mitchell JB, Patterson AD, Gonzalez FJ. Microbiome remodelling leads to inhibition of intestinal farnesoid X receptor signalling and decreased obesity. Nature communications. 2013;4:2384. doi: 10.1038/ncomms3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rius M, Hummel-Eisenbeiss J, Hofmann AF, Keppler D. Substrate specificity of human ABCC4 (MRP4)-mediated cotransport of bile acids and reduced glutathione. American Journal of Physiology-Gastrointestinal and Liver Physiology. 2006;290:G640–G649. doi: 10.1152/ajpgi.00354.2005. [DOI] [PubMed] [Google Scholar]
- 43.Xu S, Weerachayaphorn J, Cai SY, Soroka CJ, Boyer JL. Aryl hydrocarbon receptor and NF-E2-related factor 2 are key regulators of human MRP4 expression. Am J Physiol Gastrointest Liver Physiol. 2010;299:G126–135. doi: 10.1152/ajpgi.00522.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Aleksunes LM, Klaassen CD. Coordinated regulation of hepatic phase I and II drug-metabolizing genes and transporters using AhR-, CAR-, PXR-, PPARalpha-, and Nrf2-null mice. Drug Metab Dispos. 2012;40:1366–1379. doi: 10.1124/dmd.112.045112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stieger B, Fattinger K, Madon J, Kullak-Ublick GA, Meier PJ. Drug-and estrogen-induced cholestasis through inhibition of the hepatocellular bile salt export pump (Bsep) of rat liver. Gastroenterology. 2000;118:422–430. doi: 10.1016/s0016-5085(00)70224-1. [DOI] [PubMed] [Google Scholar]
- 46.Stieger B. Role of the bile salt export pump, BSEP, in acquired forms of cholestasis. Drug metabolism reviews. 2010;42:437–445. doi: 10.3109/03602530903492004. [DOI] [PubMed] [Google Scholar]
- 47.Zhang L, Nichols RG, Correll J, Murray IA, Tanaka N, Smith PB, Hubbard TD, Sebastian A, Albert I, Hatzakis E, Gonzalez FJ, Perdew GH, Patterson AD. Persistent Organic Pollutants Modify Gut Microbiota-Host Metabolic Homeostasis in Mice Through Aryl Hydrocarbon Receptor Activation. Environ Health Perspect. 2015;123:679–688. doi: 10.1289/ehp.1409055. [DOI] [PMC free article] [PubMed] [Google Scholar]