

Summary
This study describes variations in tumour growth patterns which occur when changes in the routes of inoculation and mouse strain are used to introduce tumours into established murine model systems that are known to vary in location and aggression. Intraperitoneal, subcutaneous, intravenous and hydrodynamic inoculations of B16F10 cells were compared among CD‐1, C57BL/6 and Balb/c mice. Most surprisingly, allogeneic tumour growth in Balb/c mice after intravenous and hydrodynamic inoculation of B16F10 cells was faster than tumour growth in the syngeneic C57BL/6 mice. These and other variations in the tumour growth patterns described here can help provide the researcher with more experimental control when planning to use the optimal tumour model for any particular study.
Keywords: B16F10, hydrodynamic injection, intravenous, lung metastasis, tumour growth, tumour location
Introduction
In vivo testing of cancer treatment is a necessary step in drug development, and employing an effective and relevant tumour model is essential for obtaining convincing results and determining accurate conclusions. For decades, murine models have been the most facile and efficient of the tumour models (Leenders et al. 2008). Tumours have been generated in vivo through carcinogenic (Imaoka et al. 2009; Ramos et al. 2012) or viral induction (Lewis et al. 2001), through genetic engineering to increase susceptibility to spontaneous tumour formation (Heyer et al. 2010; Politi & Pao 2011), and, most commonly, by inoculating mice with cancer cells that have been grown in vitro (Tsutsumi et al. 2009; Oliva et al. 2012).
Intraperitoneal (i.p.), subcutaneous (s.c.), intravenous (i.v.) and hydrodynamic (h.d.) (Liu et al. 1999; Zhang et al. 1999) injections are all viable routes for inoculation, and each produces a distinct pattern of tumour growth. Hydrodynamic injection is carried out by quickly pushing 1.6 ml (equal to the total blood volume of the mouse) of cells/solution through the tail vein (Liu et al. 1999; Zhang et al. 1999). Quickly injecting this large volume distends and allows efficient delivery to tissues, especially the liver. Thus, while the site of injection for both i.v. and h.d. inoculation is the tail vein, the sites of tumour growth are quite distinct (Kang et al. 2009; Li et al. 2011). In addition, another factor which may affect the fine tuning of murine tumour model growth patterns are differences between the susceptibility between growth patterns which can occur between the mouse strains themselves. For a given route of inoculation, CD‐1, C57BL/6 (C57), and Balb/c mice can generate significant differences in tumour growth rates and patterns, but there are few reports where this question specifically has been studied in detail. This report describes how differences in the strain or route of B16F10 murine melanoma inoculation can generate new and distinct tumour models for use in tumour characterization or therapeutic experiments.
Materials and methods
B16F10 murine melanoma cells were used for this study. Cells were maintained in DMEM with penicillin/streptomycin and 10% foetal bovine serum. Female CD‐1, C57 and Balb/c mice were purchased from the University of North Carolina's in‐house breeding facility.
CD‐1, C57 and Balb/c mice were ordered at the same time and were all aged 6 weeks at the time of inoculation. Four different inoculation routes were studied (n = 5 for each mouse strain and inoculation route, for a total of 12 different groups). All mice were inoculated on the same day with the same number, passage and population of cells. For i.v. injection, all three strains of mice were inoculated with 2 × 105 B16F10 cells in 200 μl PBS through the tail vein. For h.d. injection, all three strains of mice were inoculated with 2 × 105 B16F10 cells suspended in 1.6 ml of PBS by injecting the entire volume through the tail vein within a few seconds. For s.c. injection, all three strains of mice were inoculated with 2 × 105 B16F10 cells in 100 μl PBS. For i.p. injection, all three strains of mice were inoculated with 2 × 105 B16F10 cells in 200 μl PBS.
All mice were sacrificed 14 days after inoculation to allow quantitative comparisons between strains and routes of inoculation. Organs were fixed in 4% paraformaldehyde, and relevant organs were embedded in paraffin. These organs were sectioned and stained with H&E to visualize regions of tumour growth.
s.c. and i.p. tumour growth was quantified by dissecting and weighing the tumours. Lung tumour growth was measured by counting tumour nodules on each lung. Statistical analysis was performed using Student's t‐test and Holm–Sidak test to determine the statistical significance.
Ethical approval statement
All work performed on animals was in accordance with and permitted by the University of North Carolina Institutional Animal Care and Use Committee, protocol #14‐045.
Results and discussion
Tumour growth rates were compared among CD‐1, C57 and Balb/c mice 14 days after inoculation with 2 × 105 B16F10 cells. Five mice from each of the three mouse strains were inoculated via one of four different routes: intravenous (i.v.), hydrodynamic (h.d.), subcutaneous (s.c.) and intraperitoneal (i.p.). When the mice were inoculated i.v., all three strains of mice developed tumours specifically in their lungs, and the rates of tumour growth differed depending on mouse strain. Figure 1 shows that the C57 mice, the syngeneic hosts for B16F10 cells, rapidly developed tumours exclusively in their lungs. This result was expected, as this strain and cell line has long been used as a melanoma lung metastasis model (Lin et al. 1998; Peer & Margalit 2004). Unexpectedly, the tumour growth rate in the lungs of Balb/c mice after i.v. inoculation was significantly faster than that in C57 mice. This result was surprising because B16F10 inoculation in Balb/c mice should constitute a less robust allograft model when compared to the syngraft model in C57 mice. In contrast, the outbred and more heterozygous CD‐1 strain of mice developed lung tumours significantly more slowly than C57 mice.
Figure 1.
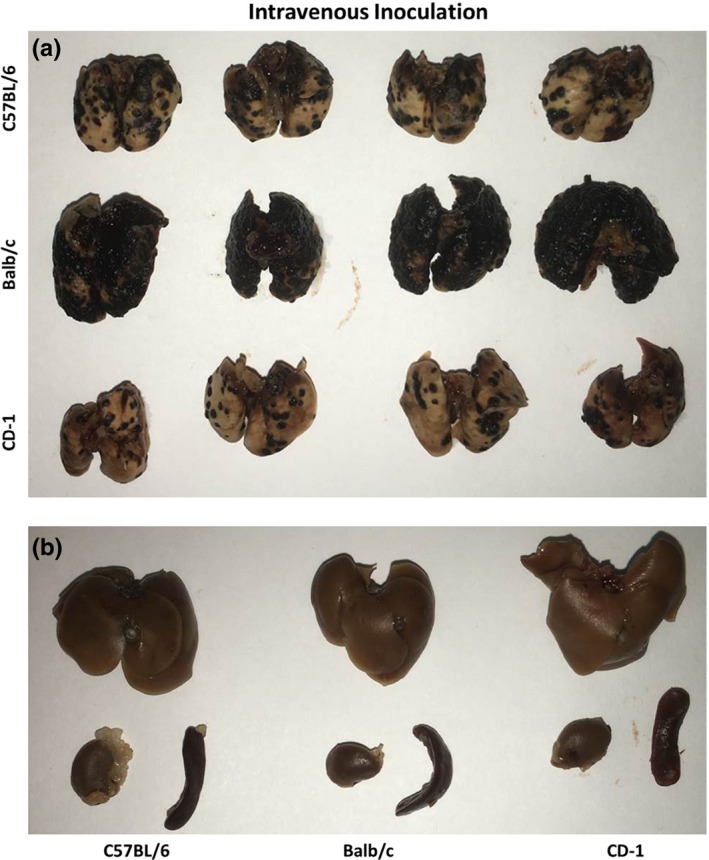
Tumour growth after i.v. inoculation in CD‐1, C57 and Balb/c mice. Fourteen days after i.v. inoculation of 2 × 105 B16F10 cells in 200 μl PBS, mice were sacrificed and their organs were fixed in 4% paraformaldehyde. (a) Tumour growth in lungs. Tumour growth in Balb/c lungs was much faster than that in C57 or CD‐1 lungs. (b) Representative liver, kidney and spleen from each strain showing lack of tumour growth outside of the lung.
Strain‐ and route‐dependent differences in tumour growth were also observed after h.d. inoculation (Figure 2). The large injection volume forced the injected tumour cells into several organs, especially the liver, generating tumours in the liver, lung, kidney, spleen and peritoneum. An overview of tumour growth patterns can be found in Table 1. The tumour growth rate in the lungs after h.d. inoculation was significantly slower than its rate after intravenous inoculation (Figure 3), most probably because the force of h.d. injection is sufficient to push the tumour cells through the lung capillaries and into downstream organs. Again, tumour growth in Balb/c mice was faster than that in C57 mice, although tumours were present in the liver, lung, kidney, spleen and peritoneum in all five mice per group. The tumour growth pattern after h.d. inoculation in both C57 and Balb/c mice was consistent with the literature describing growth in C57 mice (Kang et al. 2009; Li et al. 2011). Tumours in CD‐1 mice grew more slowly in the lung, but did not show many signs of growth in other organs after h.d. inoculation. The histological findings illustrated in Figure 4 show differences in lung and liver tumour burden after i.v. and h.d. inoculation respectively.
Figure 2.

Tumour growth after h.d. inoculation in CD‐1, C57 and Balb/c mice. Fourteen days after inoculation of 2 × 105 B16F10 cells in 1.6 ml PBS, mice were sacrificed and their organs were fixed in 4% paraformaldehyde. (a) Tumour growth in lungs. Tumour growth in Balb/c lungs was again faster than that in C57 or CD‐1 lungs, but lung tumour growth was overall slower than the lung tumour growth after i.v. inoculation. (b,c,d) Representative liver, kidney, spleen and intestines from C57, Balb/c and CD‐1 mice, respectively, showing broad tumour growth outside of the lung. Again, Balb/c mice showed faster tumour growth in liver than did C57 or CD‐1 mice.
Table 1.
Overview of tumour growth patterns across all tested strains and routes of inoculation
| Mice with tumours present in this organ | ||||||
|---|---|---|---|---|---|---|
| Liver | Spleen | Kidney | Lung | Peritoneum | s.c. | |
| Hydrodynamic | ||||||
| C57BL/6 | 5/5 | 5/5 | 5/5 | 5/5 | 4/5 | 0/5 |
| Balb/c | 5/5 | 5/5 | 5/5 | 5/5 | 5/5 | 0/5 |
| CD 1 | 0/5 | 0/5 | 1/5 | 5/5 | 1/5 | 0/5 |
| Intravenous | ||||||
| C57BL/6 | 0/4 | 0/4 | 0/4 | 4/4 | 0/4 | 0/4 |
| Balb/c | 0/4 | 0/4 | 0/4 | 4/4 | 0/4 | 0/4 |
| CD 1 | 0/4 | 0/4 | 0/4 | 4/4 | 0/4 | 0/4 |
| Intraperitoneal | ||||||
| C57BL/6 | 5/5 | 5/5 | 5/5 | 0/5 | 5/5 | 0/5 |
| Balb/c | 5/5 | 5/5 | 5/5 | 0/5 | 5/5 | 0/5 |
| CD 1 | 3/5 | 3/5 | 3/5 | 0/5 | 3/5 | 0/5 |
| Subcutaneous | ||||||
| C57BL/6 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 5/5 |
| Balb/c | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 5/5 |
| CD 1 | 0/5 | 0/5 | 0/5 | 0/5 | 0/5 | 5/5 |
Figure 3.
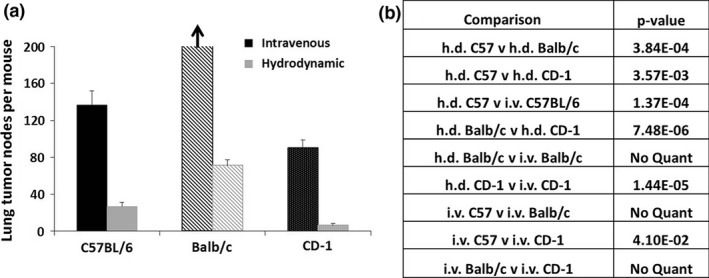
(a) Quantification of tumour nodes on lungs after i.v. or h.d. inoculation. Tumour nodes per mouse were counted and averaged. (b) Relevant P‐values were calculated using a Student's t‐test, and statistical significance was confirmed for all comparisons using the Holm–Sidak corrections. Because the tumour growth in Balb/c mice that received i.v. inoculation covered nearly the entire lung, tumour nodes were unable to be quantified, but the overall tumour burden in these mice was obviously much higher than in all other groups. P‐values were not calculated for comparisons between Balb/c i.v. and other groups.
Figure 4.
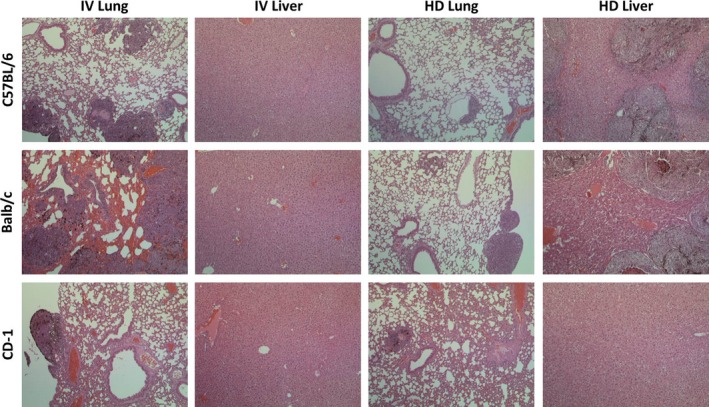
H&E‐stained sections of lung and liver in CD‐1, C57 and Balb/c mice. Tumour burden in Balb/c i.v. lungs is high, and the normal epithelium is surrounded with blood. No livers show tumour burden after i.v. inoculation, but there is a large burden in C57 and Balb/c tumours after h.d. inoculation.
Other routes of B16F10 inoculation were performed to investigate any other strain‐dependent tumour growth differences. Intraperitoneal tumour growth after i.p. injection was very fast in all three mouse strains, filling the peritoneal cavity and surrounding the intestines, spleen and kidneys, although the take rate in CD‐1 mice was only three of the five inoculated mice. There was no statistical difference between tumour growth rates among all three strains, and at the conclusion of the study, no tumours had yet metastasized to the lungs (data not shown). Subcutaneous tumour growth after s.c. injection was slowest in CD‐1 mice and significantly faster in C57 mice (Figure 5). Subcutaneous tumours in Balb/c mice grew more slowly than those in C57 mice, but not significantly so.
Figure 5.

Tumour growth after s.c. inoculation in CD‐1, C57 and Balb/c mice. Fourteen days after s.c. inoculation of 2 × 105 B16F10 cells in 100 μl PBS, mice were sacrificed, and tumours were harvested and weighed. (a) Relative s.c. tumour sizes in all three strains; (b) Average tumour weights; **P = 0.01.
The differences in tumour growth patterns shown here provide some interesting, albeit unexpected, results. The syngeneic nature of B16F10 inoculation in C57 mice is evidenced in its rapid tumour growth along all inoculation routes, but what is most interesting is the even faster tumour growth in Balb/c mice after i.v. and h.d. inoculation, even though the introduction of B16F10 cells generates an allogeneic model in which tumour growth should be suppressed by the host's immune system. The outbred CD‐1 mice did not show this increased tumour growth; their tumours grow more slowly than the tumours of both C57 and Balb/c mice.
The differences in the observed rate of tumour growth among CD‐1, C57 and Balb/c mice have generated many questions. It is simple enough to explain how the syngeneic C57 host may not mount a robust immune response against the injected B16F10 cells, resulting in a fast, widespread tumour growth among several routes of inoculation. Immune surveillance against B16F10 cells may also play a role in the slower tumour growth in the outbred CD‐1 mice. The most puzzling is the consistently faster tumour growth in Balb/c mice after i.v. and h.d. inoculation, both of which use injection via the tail vein as the route of inoculation. This unexpected result may have occurred at least in part because of inadequate rejection of the allogeneic cells by the Balb/c host. The cells may have overwhelmed the immune system in the Balb/c mice or induced a immune tolerance providing a niche for the tumour cells to grow quickly. Recent literature has shown that B16F10 cells produce anti‐inflammatory and immunosuppressive microvesicles containing phosphatidylserine and that these microvesicles contribute to increased tumour growth in Balb/c mice (Luize et al. 2009). While Balb/c and C57 mice were both used in this study, tumour growth in the different strains was not compared directly. Thus the nature of the immunological differences between the routes of innoculation, combined with mouse strain, should be taken into account when mice are used as animal models in the field of cancer therapy.
Conclusions
The differences in tumour growth among the different strains and inoculation routes implies that they constitute a series of independent B16F10 tumour models. Intravenous inoculation into C57 mice generates the expected fast‐growing lung metastasis model, while h.d. inoculation in C57 mice produces a model in which metastasis has spread throughout the body. Inoculations in Balb/c mice produce aggressive, allogeneic tumour models that might be used to study how immune contributions affect tumour growth, while the CD‐1 models provide less aggressive tumour growth that could be used to replicate slow‐growing tumours. The differences in growth speed will allow researchers to select a more or less aggressive tumour model that will best fit for their studies.
Identifying the variations in tumour models which could be generated by modifying mouse strain and route of inoculation might have many practical applications. The models described here provide some indication of the range of possible selection, but only compares the growth of one tumour line in a combination of three mouse strains and four routes of inoculation. If this effort is expanded to include other mouse strains and tumour lines, this could generate an extremely useful source to aid scientists in choosing the most effective model for their research.
Conflict of interest
No conflict of interest is involved in this research work.
Funding source
This work was supported by the National Cancer Institute – National Institutes of Health (5R01CA149387). Andrew Satterlee was supported by the NSF GRFP and by the Graduate School at the University of North Carolina at Chapel Hill.
Dedicated to the memory of Professor Feng Liu, PhD, 1955–2014, University of North Carolina at Chapel Hill.
References
- Heyer J., Kwong L.N., Lowe S.W. & Chin L. (2010) Non‐germline genetically engineered mouse models for translational cancer research. Nat. Rev. Cancer 10, 470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imaoka T., Nishimura M., Iizuka D. et al (2009) Radiation‐induced mammary carcinogenesis in rodent models: what's different from chemical carcinogenesis? J. Radiat. Res. 50, 281–293. [DOI] [PubMed] [Google Scholar]
- Kang J.H., Mori T., Niidome T., Katayama Y. (2009) A syngeneic hepatocellular carcinoma model rapidly and simply prepared using a hydrodynamics‐based procedure. Vet. J. 181, 336–339. [DOI] [PubMed] [Google Scholar]
- Leenders M.W., Nijkamp M.W. & Borel Rinkes I.H. (2008) Mouse models in liver cancer research: a review of current literature. World J. Gastroenterol. 14, 6915–6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis B.C., Chinnasamy N., Morgan R.A. & Varmus H.E. (2001) Development of an avian leukosis‐sarcoma virus subgroup A pseudotyped lentiviral vector. J. Virol. 75, 9339–9344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J., Yao Q. & Liu D. (2011) Hydrodynamic cell delivery for simultaneous establishment of tumor growth in mouse lung, liver and kidney. Cancer Biol. Ther. 12, 737–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin P., Buxton J.A., Acheson A. et al (1998) Antiangiogenic gene therapy targeting the endothelium‐specific receptor tyrosine kinase Tie2. Proc. Natl Acad. Sci. USA 95, 8829–8834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F., Song Y. & Liu D. (1999) Hydrodynamics‐based transfection in animals by systemic administration of plasmid DNA. Gene Ther. 6, 1258–1266. [DOI] [PubMed] [Google Scholar]
- Luize L.G., Chammas R., Monteiro R.Q., Moreira M.E. & Barcinski M.A. (2009) Tumor‐derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine‐dependent manner. Cancer Lett. 283, 168–175. [DOI] [PubMed] [Google Scholar]
- Oliva P., Decio A., Castiglioni V. et al (2012) Cisplatin plus paclitaxel and maintenance of bevacizumab on tumour progression, dissemination, and survival of ovarian carcinoma xenograft models. Br. J. Cancer 107, 360–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peer D. & Margalit R. (2004) Loading mitomycin C inside long circulating hyaluronan targeted nano‐liposomes increases its antitumor activity in three mice tumor models. International journal of cancer. Int. J. Cancer 108, 780–789. [DOI] [PubMed] [Google Scholar]
- Politi K. & Pao W. (2011) How genetically engineered mouse tumor models provide insights into human cancers. J. Clin. Oncol. 29, 2273–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos R.N., Oliveira C.E., Gasparoto T.H. et al (2012) CD25 + T cell depletion impairs murine squamous cell carcinoma development via modulation of antitumor immune responses. Carcinogenesis 33, 902–909. [DOI] [PubMed] [Google Scholar]
- Tsutsumi K., Yamaura T., Nakajima M., Honda T. & Kasaoka T. (2009) Silencing of focal adhesion kinase by tumor direct injection of small interfering RNA decreases in vivo tumor growth. Cancer Biol. Ther. 8, 1292–1299. [DOI] [PubMed] [Google Scholar]
- Zhang G., Budker V. & Wolff J.A. (1999) High levels of foreign gene expression in hepatocytes after tail vein injections of naked plasmid DNA. Hum. Gene Ther. 10, 1735–1737. [DOI] [PubMed] [Google Scholar]