

Abstract
It has historically been very difficult to conduct early phase drug studies in children for a number of reasons related to ethics, acceptability, rarity, standardization, end points, safety, dosing and feasibility. Over the past decade there have been a number of developments including novel clinical trial design, in silico pharmacology and microdosing that have significantly enhanced the ability of investigators to conduct early phase drug studies in children. While the evolution of drug therapy is creating a series of new challenges, there has never been a better time for conducting drug studies in children.
Keywords: children, clinical trials, drug development, early phase drug studies
Introduction
The current drug development process began after the Elixir of Sulfanilamide tragedy – in which a number of children died due to the use of diethylene glycol as a solvent – triggered the passage of the US Food and Drug Act in 1938 1. This was intended to ensure that drugs were safe and effective prior to approval for marketing. It should not be forgotten that the impetus for this Act was a therapeutic disaster largely involving children. One consequence of this was the current drug development process, which after pre‐clinical studies involves Phase I (first‐in‐man), Phase II (first‐in‐patient) and Phase III (comparison to standard therapy) studies (see Figure 1). An additional tragedy that spurred changes in drug regulation was the Thalidomide tragedy in the early 1960s that led to both the Kefauver‐Harris amendments to the Food and Drug Act in 1962 and the creation of national spontaneous reporting schemes for adverse drug reactions such as the Yellow Card Scheme in the UK 2, 3. While children have benefitted from the spontaneous reporting schemes, the other changes that were intended to provide safer drug therapy for children had quite the opposite effect. The Kefauver‐Harris amendments stated that, in order to be approved for marketing, a drug must not only be safe but also have substantial evidence of benefit under the conditions of use as defined in the product monograph, and provided powers for the Food and Drug Administration to enforce this. The result was that, rather than ensuring that well‐designed studies were conducted in children, product monographs simply stated, in more or less similar terms, that safety and efficacy of the drug in question had not been evaluated in children along with a legal disclaimer against use in children 2. This unintended consequence of a well‐meaning Act was best described by Shirkey who used the term “the therapeutic orphan” to describe that the majority of drugs on the market had no labelling for use in children – despite the fact that these drugs were commonly used, often as first line therapy, in children 2, 4.
Figure 1.
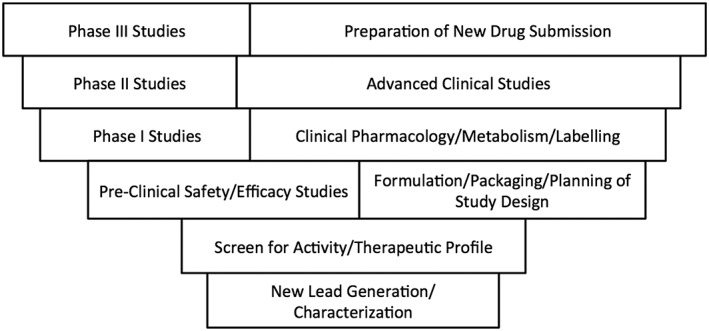
General overview of the drug development process. Modified from 6
Over the past 20 years there has been a concerted effort to address these problems, including development of national and international research networks to conduct drug research in children as well as changes in the drug approval process by drug regulatory authorities that have not only increased knowledge with respect to approved drugs but also have mandated inclusion of children in pre‐marketing studies of drugs likely to be used by children 5. Examples include the Best Pharmaceuticals for Children Act and the Pediatric Research Equity Act that became a permanent part of American law as part of the Congressional approval of the FDA Safety and Innovation Act in 2012 and the European equivalent, the Regulation on Medicines for Paediatric Use 5, 6. The regulatory agencies on both sides of the Atlantic have the ability – and indeed the mandate – to require companies making new drug submissions to provide a detailed plan (Pediatric Study Plan in the US, Paediatric Investigation Plan in Europe) for drugs likely to be used in children. New therapies that are likely to be useful in the treatment of children will therefore need to include children as part of their drug development plans 7, 8. This will create challenges as this has historically not been part of drug development planning for most new therapeutic entities, but there are also a number of advances that present opportunities to address this.
The conduct of early phase drug research is challenging at the best of times 9. There are a number of specific issues germane to enrolment of children in early phase drug trials and these will be considered in turn.
Ethics
The involvement of children in research studies has been hotly debated and this is a field in constant flux. Since the “Great Divide” after World War II that established the importance of research ethics and informed consent, there has been an on‐going debate as to the ethical issues involved in the participation of children in research 9, 10, 11. This has been true in the United States, Europe (ftp://ftp.cordis.europa.eu/pub/fp7/docs/ethical‐considerations‐paediatrics_en.pdf), Canada (http://www.pre.ethics.gc.ca/eng/policy‐politique/initiatives/tcps2‐eptc2/Default/) and many other jurisdictions 12. Historically there has been a pendulum swinging between a somewhat laissez faire approach to inclusion of children in drug studies with the arguably nihilistic view that children should not be involved in drug studies 9, 10, 11, 12. Over the past decade the argument has increasingly been made that drug studies in children are essential in order to provide evidence to guide safe and effective drug therapy and to facilitate the development of drugs for common and important paediatric disorders 13.
There are now ethical constructs that permit and even encourage involvement of children in drug research, notably when this will be of material benefit to children with disorders targeted by the agent in question. On‐going discourse has evolved to increasingly include discussion of the idea that involvement in research would not present more than minimal risk. While avoiding minimal risk usually means that, with the exception of children with cancer, it is unlikely children will be involved in Phase I studies, but children would certainly be ethically eligible for Phase II and Phase III studies (see Table 1). There have been increasing calls that ethical approval would require not only consent from parents but also assent from the children, certainly for adolescents 14, 15, 16. The question of how best to secure informed consent for drug research in adolescents remains problematic, in that in many jurisdictions minors can give informed consent for significant medical procedures – including those associated with significant risk – but often cannot themselves give consent for participation even in very low risk research studies. This remains an area of active debate and discussion.
Table 1.
Phases of pre‐clinical drug development in humans and examples in paediatrics
| Phase of drug development | Goal of studies | Examples in paediatric drug development |
|---|---|---|
| Phase I | First stages of drug testing in humans, typically conducted in health adult volunteers | Very rarely done in children with the exception of oncology drug (chemotherapy) and some drugs in neonatology (surfactant) |
| Phase II | First stages of drug testing for efficacy and safety, typically conducted in patients | Uncommon, and represent the first step for most drugs in terms of early phase studies in children. Regulatory advances have increased these studies for new drugs |
| Phase III | Effectiveness of the drug and the role in clinical practice, typically by comparison with “gold standard” therapy | Done at some times for drugs in children, most frequently for anti‐infectives and increasingly for other drug classes |
Acceptability
The issue of acceptability concerns questions for the child's family and also for clinicians, institutions and investigators. Historically it has been believed that parents are reluctant to enrol their children in clinical trials. Recent work has suggested that this may be more perception than reality 17, 18, 19. A multi‐centre study in France demonstrated that the refusal rate for clinical studies in children was related to the perceived burden on the family on the part of the paediatrician charged with enrolling patients in the study 20. This supports our finding in an earlier Anglo‐Canadian study that showed that paediatricians with limited training in ethics were very reluctant to enrol children in clinical trials 21. The degree of comfort of study personnel in working with children and families appears to be a key factor in the success or failure of drug studies in children with respect to enrolment or lack thereof. It is also increasingly evident that children are interested in being involved in studies for altruistic reasons with respect to the well‐being of other children.
In addition to individual investigators, the degree of comfort with drug studies in children varies considerably between institutions, sometimes with no clear link between experience in child health care and degree of comfort for recruiting children to drug studies. In this context, the creation of regional and national networks for children's research has been a great opportunity in terms of providing standards and resources to enhance the design and conduct of clinical research – including drug research – in children. An early example was the National Institutes of Health Pediatric Pharmacology Research Network linking research units throughout the United States, while a more recent example germane to the UK is the Medicines for Children Research Network created by the National Health Service which brought together expertise in paediatric drug studies across the UK 22. In the latter case this has been merged with the Paediatric Specialty Group to create a community of clinical practice that provides national research expertise in studies involving children, including drug studies (https://www.crn.nihr.ac.uk/children/). This creates the opportunity for shared expertise and more rapid translation of best practices.
Rarity
The issue of rarity speaks to a dichotomous reality in paediatric health care, in that many disorders are rare at any individual institution but are collectively reasonably common. We demonstrated this in a study of drug utilization in a cohort of one million Canadian children followed for a year, in which 70% of drug use was among 20% of children, these children representing a number of serious and chronic conditions 23. We also found that these children were mostly cared for in 16 academic health science centres across the country. Hence, while clinical trials of new therapies are clearly needed, it is difficult to use a single centre, and sample size has been a frequently cited problem for drug studies in children 6.
Thus, in addition to acceptance, the development of regional, national and international networks has been instrumental in providing mechanisms for timely recruitment of large numbers of patients using common instruments and with the evaluation of common outcomes. This has been most successful in paediatric haematology‐oncology and neonatology, as both fields have made considerable progress in assessing therapy and developing evidence‐based treatment protocols which have resulted in the survival of very small pre‐term infants and a very high rate of cure for many childhood cancers 6, 24, 25. Increasingly other groups – including academic general paediatricians and critical care paediatricians – are developing networks to apply the strength of synergy to problems within their care and research domains 26, 27. The existence and development of these networks provides a much improved platform to support drug research in children. Additionally, these networks can support highly specialized units such as Phase I units for childhood cancer, facilities that are uncommon but very important 28, 29.
Standardization, end points and safety
One of the key elements in drug research is the clinical trial. Since the first curative clinical trial – conducted in 1946–47 by the MRC Tuberculosis Research Unit to study streptomycin in the management of pulmonary tuberculosis – the randomized clinical trial has become a gold standard in the drug development process 6, 30, 31. For many years a randomized placebo‐controlled double blind clinical trial was considered essential to the drug development and approval process. This has been a problem in drug studies in children for several reasons 6. There have been issues with ethical approval and conduct as noted above. There has also been reluctance to use placebos during drug research in children. As the number of effective therapies has evolved, this question has become germane to clinical trials involving adults as well.
A key question is selection of a suitable end point, notably as this drives sample size and analysis strategy 6, 32. These end points may include biomarkers, the validity of which may not have been established in children. Development of valid and reproducible end points has become a research field in and of itself. An issue that complicates clinical trials in children is that many end points that are commonly used in adult clinical trials have not been validated in children – or indeed may not be possible 6. As an example, the evaluation of the efficacy of analgesic interventions in young children and infants was problematic as many validated instruments for the evaluation of pain involved self‐report, a problem for populations that are non‐verbal or who lack numerical literacy 33. However, great progress has been made over the past two decades in developing and validating end points that are relevant to and achievable in studies involving children. To return to the issue noted above, there have been a number of scales and observation tools developed that provide investigators with valid tools to study the effect of various interventions on pain on even the youngest of infants 33, 34.
In addition to progress in the selection of end points there have been a number of advances in the design of clinical trials for children (Figure 2). Analysis of clinical trials conducted in children has suggested that many designs used to date are associated with a significant risk of bias, notably with respect to sequence generation and allocation concealment 35. Also, the perception of lack of flexibility has historically been a problem for randomized drug studies in children 36. Over the past decade a number of novel trial designs have been developed to address these issues. One example is sequential design in which investigators conduct frequent analysis during subject enrolment to determine if the therapy of interest is superior 37. A type of sequential design suggested to be very useful for studies in children is adaptive design, in which planned interim analysis is used to inform modifications in trial design 38. This type of trial requires meticulous pre‐trial planning and consideration of issues such as blinded versus non‐blinded interim analysis 39. This can permit a trial to be stopped early in the case of an intervention that is either very effective or found to be ineffective, reducing the number of children needed for the trial. These trial designs may be unfamiliar to drug regulatory agencies and investigators planning on using them for early phase drugs studies are encouraged to discuss this with their respective drug regulatory agency.
Figure 2.
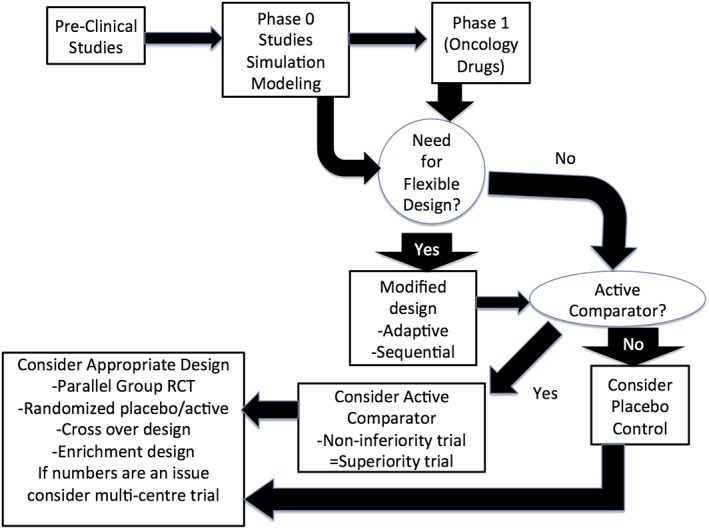
Considerations in planning the design of an early phase drug trial in children. A key aspect in planning relates to early decisions as to the need for a flexible design. Modified from 6
An additional issue of key importance in any clinical trial, and most certainly those involving drugs, is a robust and on‐going safety assessment 40. The drug approval process was designed to detect serious and common risks, and while this is generally the case, it is clear from such unfortunate events as that associated with the initial clinical trials of TGN1412 in the UK and fatty acid amide hydrolase (FAAH) inhibitors in France, that serious, even fatal, adverse effects still occur 41. This is of particular importance in the drug evaluation process for children as some common and important adverse drug effects are different in both incidence and manifestation in children than in adults 42, 43. The increase in interest in drug therapy for children over the past two decades has been accompanied by the development of new instruments both clinically and in vitro that offer considerable promise for more rapid and focused detection of adverse drug events in children, notably for novel therapeutic agents 44, 45.
An emerging area in clinical trial design with special relevance to children is the use of simulation and modelling early in the drug development process 46, 47. The value that simulation and modelling brings is the ability to factor in variables such as ontogeny of key pathways of drug clearance and data derived from adult studies to develop estimates for drug dosing which enables a clearer estimate of dose while reducing the requirement for additional studies 47. Simulation can also be used to provide firmer estimates of sample size and to illustrate the point at which increasing sample size does not significantly increase the precision of data gathered 47. The increasing sophistication of pharmacometrics in children also provides new opportunities.
Dosing and feasibility
There are feasibility issues with respect to involvement of children in early phase drug studies and first among them is dose selection. The issue of dose selection for Phase I trials is problematic at the best of times and the increasing percentage of biologicals as new therapeutic entities has only increased this problem. A recent review of failed paediatric drug development trials has suggested that in up to a quarter of trials that fail to establish efficacy or safety, the selection of the correct dose was a factor in this failure 48.
The conventional approach to developing dose considerations for children has been to extrapolate from adult dosing, often using techniques such as allometric scaling. This may be problematic in that the major issues in ontogeny directly impact on drug clearance, primarily in terms of a reduction in the capacity of children to clear drugs or drug metabolites, notably in infancy 49. While this is now well understood for drugs used in the first year of life, an under‐appreciated issue is that toddlers are notably more efficient in terms of oxidative metabolism, which may increase the risk for toxicity in drugs that undergo biotransformation to active metabolites 50.
Better appreciation of the role of ontogeny and disease enables an improved rationale for drug dosing in paediatric studies 51. A technique with considerable potential is microdosing, in which a pharmacokinetic study is performed using a subtherapeutic dose of a 14C labelled drug 52, 53, 54. This can be done as a “Phase 0” study prior to a Phase 1 or Phase 2 trial. Advances in analytical technology have been such that concerns about the volume of blood needed for pharmacokinetic studies – once a major concern in paediatrics – is now largely a historical curiosity except for very premature infants.
Once a dose has been selected, a consideration somewhat unique to paediatric drug studies is formulation. While drugs for adults – notably chronic therapy – are overwhelmingly given orally as tablets or capsules, the use of drugs in children must take into account the fact that medication naïve children under the age of 8 find it difficult to take tablets or capsules and even medication sophisticated children cannot reliably take medication in conventional tablets or capsules. The traditional approach to this problem has been to develop liquid formulations or to crush the tablets 55. Over the past decade there has been an explosion in the creation of novel dosing systems designed for children, work largely driven by developments in Europe and which offers great promise for making drug research – and drug therapy – much more practical for infants and small children 56.
Moving forward
While there have been many cultural, scientific and regulatory challenges that have made conducting early phase clinical trials in children difficult, developments over the past decade have addressed many of these issues and have provided the opportunity – indeed, in some cases the requirement – for the inclusion of children even in early stages of drug development. While there are new issues – such as the development of drugs for the neonate, the increasing appreciation of the importance of drug transporters in drug disposition in children and the complex issues raised by the increasing use of biologicals – that pose new and interesting challenges for paediatric pharmacy and clinical pharmacology – there has never been a more promising time for drug development in children 57, 58, 59, 60.
Competing Interests
There are no competing interests to declare.
Rieder, M. , and Hawcutt, D. (2016) Design and conduct of early phase drug studies in children: challenges and opportunities. Br J Clin Pharmacol, 82: 1308–1314. doi: 10.1111/bcp.13058.
References
- 1. Weinshilboum RM. The therapeutic revolution. Clin Pharmacol Ther 1987; 42: 481–484. [DOI] [PubMed] [Google Scholar]
- 2. Rieder MJ. If children ruled the pharmaceutical industry: the need for pediatric formulations. Drug News Perspect 2010; 23: 458–464. [DOI] [PubMed] [Google Scholar]
- 3. McLernon DJ, Bond CM, Hannaford PC, Watson MC, Lee AJ, Hazell L, et al. Adverse drug reaction reporting in the UK: a retrospective observational comparison of yellow card reports submitted by patients and healthcare professionals. Drug Saf 2010; 33: 775–788. [DOI] [PubMed] [Google Scholar]
- 4. Shirkey HC. Therapeutic orphans. J Pediatr 1968; 2: 119–120. [DOI] [PubMed] [Google Scholar]
- 5. European Union . Regulation (EC) No. 1901/2006 of the European Parliament and of the Council of 12 December 2006 on medicinal products for paediatric use and amending Regulation (EEC) No. 1768/92, Directive 2001/20/EC, Directive 2001/83/EC and Regulation (EC) No. 726/2004. 2007.
- 6. Council of Canadian Academies: The Expert Panel on Therapeutic Products for Infants, Children and Youth . Improving Medicines for Children in Canada. Ottawa: Council of Canadian Academies, 2014. Available at: http://www.scienceadvice.ca/uploads/eng/assessments%20and%20publications%20and%20news%20releases/therapeutics/therapeutics_fullreporten.pdf (last accessed 17 July 2016).
- 7. Hawcutt DB, Smyth RL. The new European regulation on pediatric medicines. Ped Drugs 2008; 10: 143–146. [DOI] [PubMed] [Google Scholar]
- 8. Olski TM, Lampus SF, Gherarducci G, Saint Raymond A. Three years of paediatric regulation in the European Union. Eur J Clin Pharmacol 2011; 67: 245–252. [DOI] [PubMed] [Google Scholar]
- 9. Matsui D, Kwan C, Steer E, Rieder MJ. The trials and tribulations of doing drug research in children. Can Med Assoc J 2003; 169: 1033–1034. [PMC free article] [PubMed] [Google Scholar]
- 10. Ackerman TF. The ethics of drug research in children. Paediatr Drugs 2001; 3: 29–41. [DOI] [PubMed] [Google Scholar]
- 11. Fernandez CF, Bioethics Committee . Ethical issues in health research in children. Paedietr Child Health 2008; 13: 707–712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Institute of Medicine (US) Committee on Clinical Research Involving Children . In: Ethical Conduct of Clinical Research Involving Children, eds Field MJ, Behrman RE. Washington, DC: National Academies Press, 2004. [PubMed] [Google Scholar]
- 13. Shaddy RE, Dennes C, the Committee on Drug and the Committee on Pediatric Research . Guidelines for the ethical conduct of studies to evaluate drugs in pediatric populations. Pediatr 2010; 125: 850–860. [DOI] [PubMed] [Google Scholar]
- 14. Hein IM, de Vries MC, Troost PW, Meynen G, van Goudoever JB, Lindauer RJL. Informed consent instead of assent is appropriate in children from the age of twelve: policy implications of new findings on children's competence to consent to clinical research. BMC Med Ethics 2015; 16: 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dove ES, Avard D, Black L, Knoppers BM. Emerging issues in paediatric health research consent forms in Canada: working towards best practices. BMC Med Ethics 2013; 14: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sibley A, Pollard AJ, Fitzpatrick R, Sheehan M. Developing a new justification for assent. BMC Med Ethics 2016; 17: 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pickler RH, Martin AT. Protection of children in research. J Pediatr Health Care 2010; 24: 66–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kaguelidou F, Amiel P, Blachier A, Iliescu C, Rozé JC, Tsimaratos M, et al. Recruitment in pediatric clinical research was influenced by study characteristics and pediatrician's perceptions: a multicenter study. J Clin Epidemiol 2013; 66: 1151–1157. [DOI] [PubMed] [Google Scholar]
- 19. Bang V, Mallad A, Kannan S, Bavdekar SB, Gogtay NJ, Thatte UM. Awareness about and views of parents on the off‐label drug use in children. Int J Risk Saf Med 2014; 26: 61–70. [DOI] [PubMed] [Google Scholar]
- 20. Blair JC, Povall A, Richardson P, Peak M. Parental attitudes to clinical studies in healthy children. Arch Dis Child 2015; 100: 1096–1097. [DOI] [PubMed] [Google Scholar]
- 21. Sammons HM, Malhotra J, Choonara I, Sitar S, Matsui D, Rieder MF. British and Canadian views on the ethics of paediatric clinical trials. Eur J Clin Pharmacol 2007; 63: 431–436. [DOI] [PubMed] [Google Scholar]
- 22. Cohen SN. The Pediatric Pharmacology Research Unit (PPRU) Network and its role in meeting pediatric labelling needs. Pediatr 1999; 104: 644–645. [PubMed] [Google Scholar]
- 23. Rieder MD, Matsui DM, MacLeod S. Myths and challenges – drug utilization for Canadian children. Paed Child Health 2003; 8: 7A. [Google Scholar]
- 24. Weiss AR, Nichols CR, Freyer DR. Enhancing adolescent and young adult oncology research within the national clinical trials network: rationale, progress and emerging strategies. Semin Oncol 2015; 42: 740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Profit J, Soll RF. Neonatal networks: clinical research and quality improvement. Semin Fetal Neonatal Med 2015; 20: 410–415. [DOI] [PubMed] [Google Scholar]
- 26. Lannon CM, Peterson LE. Pediatric collaborative networks for quality improvement and research. Acad Pediatr 2013; 6: S69–S74. [DOI] [PubMed] [Google Scholar]
- 27. Zimmerman JJ, Anand KJ, Meert KL, Wilson DF, Newth CJ, Harrison R, et al. Research as a standard of care in the PICU. Pediatr Crit Care Med 2016; 17: e13–e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Morgenstern DA, Hargrave D, Marshall LV, Gatz SA, Barone G, Crowe T, et al. Toxicity and outcome of children and adolescents participating in Phase I/II trials of novel anticancer drugs: the Royal Marsden experience. J Pediatr Hematol Oncol 2014; 36: 218–223. [DOI] [PubMed] [Google Scholar]
- 29. Doussau A, Geoerger B, Jiménez I, Paoletti X. Innovations for Phase I dose‐findings in pediatric oncology clinical trials. Contemp Clin Trials 2016; 47: 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Metcalfe NH. Sir Geoffrey Marshall (1887–1982): respiratory physician, catalyst for anaesthesia development, doctor to both Prime Minster and King, and World War I barge commander. J Med Biogr 2011; 19: 10–14. [DOI] [PubMed] [Google Scholar]
- 31. Sacks H, Chalmers TC, Smith H Jr. Randomized versus historical controls for clinical trials. Am J Med 1982; 72: 233–240. [DOI] [PubMed] [Google Scholar]
- 32. Grimsrud KN, Sherwin CM, Constance JE, Tak C, Zuppa AF, Spigarelli MG, et al. Special population considerations and regulatory affairs for clinical research. Clin Res Regul Aff 2015; 32: 47–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Anand KJ, Aranda JV, Berde CB, Buckman S, Capparelli EV, Carlo WA, et al. Analgesia and anesthesia for neonates: study design and ethical issues. Clin Ther 2005; 27: 814–843. [DOI] [PubMed] [Google Scholar]
- 34. Manocha S, Taneja N. Assessment of paediatric pain: a critical review. J Basic Clin Physiol Pharmacol 2016; 27: 323–331. [DOI] [PubMed] [Google Scholar]
- 35. Hartling L, Hamm M, Klassen T, Chan A‐W, Meremikwu M, Moyer V, et al. Standard 2: Containing risk of bias. Pediatr 2012; 129: S124–S131. [DOI] [PubMed] [Google Scholar]
- 36. Chow SC, Change M. Adaptive design methods in clinical trials: a review. Ophanet J Rare Dis 2008; 3: 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Van der Lee JH, Wesseling J, Tanck MW, Offringa M. Sequential design with boundaries approach in pediatric intervention research reduces sample size. J Clin Epidemiol 2010; 63: 19–27. [DOI] [PubMed] [Google Scholar]
- 38. Van der Lee JH, Wesseling J, Tanck MW, Offringa M. Efficient ways exist to obtain the optimal sample size in clinical trials in rare diseases. J Clin Epidemiol 2008; 61: 324–330. [DOI] [PubMed] [Google Scholar]
- 39. Food and Drug Administration . Guidance for Industry: Adaptive Design Clinical Trials for Drugs and Biologics. Rockville, MD: FDA, 2010. [Google Scholar]
- 40. Bass AS, Hombo T, Kasai C, Kinter LB, Valentin JP. A historical view and vision into the future of the field of safety pharmacology. Handb Exp Pharmacol 2015; 229: 3–45. [DOI] [PubMed] [Google Scholar]
- 41. Kenter MJ, Cohen AF. The return of the prodigal son and the extraordinary development route of TGN1412 – lessons for drug development. Br J Clin Pharmacol 2015; 79: 545–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Rieder MJ. New ways to detect adverse drug reactions in pediatrics. Pediatr Clin North Am 2012; 59: 1071–1092. [DOI] [PubMed] [Google Scholar]
- 43. Lathyris D, Panagiotou OA, Baltogianni M, Ioannidis JP, Contopoulos‐Ioannidis DG. Safety of medical interventions in children versus adults. Pediatr 2014; 133: e666–e673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Gallagher RM, Kirkham JJ, Mason JR, Bird KA, Williamson PR, Nunn AJ, et al. Development and inter‐rater reliability of the Liverpool adverse drug reaction causality assessment tool. PLoS One 2011; 6: e28096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Elzagallaai AA, Rieder MJ. In vitro testing for diagnosis of idiosyncratic adverse drug reactions: implications for pathophysiology. Br J Clin Pharmacol 2015; 80: 889–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stockmann C, Barrett JS, Roberts JK, Sherwin C. Use of modeling and simulation in the design and conduct of pediatric clinical trials and the optimization of individualized dosing regimens. CPT Pharmacometrics Syst Pharmacol 2015; 4: 630–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Vinks AA, Emoto C, Fukuda T. Modeling and simulation in pediatric drug therapy: application of pharmacometrics to define the right dose for children. Clin Pharmacol Ther 2015; 98: 298–308. [DOI] [PubMed] [Google Scholar]
- 48. Momper JD, Mulugeta Y, Burckart CJ. Failed pediatric drug development trials. Clin Pharmacol Ther 2015; 98: 245–251. [DOI] [PubMed] [Google Scholar]
- 49. Kearns GL, Abdel‐Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology – drug disposition, action and therapy in infants and children. N Engl J Med 2003; 349: 1157–1167. [DOI] [PubMed] [Google Scholar]
- 50. Hanly L, Chen N, Rieder M, Koren G. Ifosfamide nephrotoxicity in children: a mechanistic basis for pharmacologic prevention. Expert Opin Drug Saf 2009; 8: 155–156. [DOI] [PubMed] [Google Scholar]
- 51. Kearns GL. Selecting the proper pediatric dose: it is more than size that matters. Clin Pharmacol Ther 2015; 98: 238–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Roth‐Cline M, Nelson RM. Microdosing studies in children: a US regulatory perspective. Clin Pharmacol Ther 2015; 98: 232–233. [DOI] [PubMed] [Google Scholar]
- 53. Turner MA, Mooij MG, Vaes WHJ, Windhorst AD, Hendrikse NH, Knibbe CA, et al. Pediatric microdosing and microtracer studies using 14C in Europe. Clin Pharmacol Ther 2015; 98: 234–237. [DOI] [PubMed] [Google Scholar]
- 54. Burt T, Yoshida K, Lappin G, Vuong L, John C, de Wildt SN, et al. Microdosing and other Phase‐0 clinical trials: facilitating translation in drug development. Clin Transl Sci 2016; 9: 74–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nunn T, Williams J. Formulation of medicines for children. Brit J Clin Pharmacol 2005; 59: 674–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ivanovska V, Rademaker CMA, van Dijk L, Mantel‐Teeuwisse AK. Pediatric drug formulations: a review of challenges and progress. Pediatr 2014; 113: 361–372. [DOI] [PubMed] [Google Scholar]
- 57. Allegaert K, van den Anker J. Neonatal drug therapy: the first frontier of therapeutics for children. Clin Pharmacol Ther 2015; 98: 288–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Brouwer KLR, Aleksunes LM, Brandys B, Giacoia GP, Knipp G, Lukacova V, et al. Human ontogeny of drug transporters: review and recommendations of the Pediatric Transporter Working Group. Clin Pharmacol Ther 2015; 98: 266–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Kim T, Han N, Sohn M, Oh JM, Lee EK, Ji E, et al. Pharmacogenomic biomarker information in FDA‐approved paediatric drug labels. Basic Clin Pharmacol Toxicol 2015; 116: 438–444. [DOI] [PubMed] [Google Scholar]
- 60. Stachnik J, Gabay M. Biologics in pediatrics In: Safe and Effective Medicines for Children: Pediatric Studies Conducted Under the Best Pharmaceuticals for Children Act and the Pediatric Research Equity Act, eds Field M, Boat T. Washington, DC: National Academy of Science, 2012; 285–320. [PubMed] [Google Scholar]