

Abstract
Aims
The aim of the present study was to investigate the safety, tolerability, dose proportionality and relative bioavailability of tablet and oral solution formulations of BI 409306 in healthy male subjects, and to compare the safety and pharmacokinetics in subjects who were extensive metabolizers (EMs) or poor metabolizers (PMs) of cytochrome P450 (CYP)‐2C19.
Methods
The present randomized, double‐blind, placebo‐controlled, single‐centre study evaluated single rising doses of BI 409306 (0.5–500 mg) administered as a tablet or oral solution to EMs or PMs.
Results
Of 80 enrolled subjects (mean age 36.7 years), 79 (CYP2C19 EMs, 71; CYP2C19 PMs, eight) received treatment and completed the study. Adverse events (AEs) were mild to moderate in intensity. Overall, 17/71 (23.9%) EMs and 6/8 (75.0%) PMs experienced 28 and eight AEs, respectively, of which, 25 and seven AEs, respectively, were considered to be drug related. The most frequently reported AEs were nervous system and eye disorders; all occurred shortly (20–30 min) after administration and mostly resolved within 1–2 h. No serious AEs occurred. BI 409306 systemic absorption and elimination were rapid; peak plasma concentration (Cmax) was reached <1 h after drug administration, and the half‐life ranged from 0.99 h to 2.71 h. Both the tablet and oral solution resulted in similar exposures. In PMs, at dose levels of 10 mg and 100 mg, Cmax was 2.2–2.3‐fold higher, and the area under the plasma concentration–time curve over the time interval 0 extrapolated to infinity was 4.1–5.0‐fold higher compared with EMs.
Conclusions
In healthy male subjects, BI 409306 was generally safe and well tolerated, with rapid absorption and elimination. Systemic exposure was higher in CYP2C19 PMs than EMs at the same dose level.
Keywords: Alzheimer's disease, cognitive impairment, PDE9A, phosphodiesterase inhibitor, schizophrenia
What is Already Known about this Subject
Phosphodiesterase (PDE) inhibitors increase intracellular concentrations of second messengers, which are involved in learning and memory.
The potent and selective PDE9A inhibitor BI 409306 improved memory performance in preclinical studies.
This first‐in‐human study aimed to assess the safety, tolerability and pharmacokinetics of BI 409306 after a single dose.
What this Study Adds
BI 409306 showed good safety and tolerability in cytochrome P450 2C19 (CYP2C19)‐genotyped extensive metabolizer (EM) and poor metabolizer (PM) healthy male subjects. The highest tolerated dose was 350 mg.
BI 409306 was rapidly absorbed and eliminated. The increase in systemic exposure was slightly more than dose proportional.
PMs achieved a higher peak plasma concentration and area under the plasma concentration–time curve over the time interval 0 extrapolated to infinity than EMs.
Introduction
Both schizophrenia and Alzheimer's disease (AD) are characterized by abnormalities in glutamatergic neurotransmission related to N‐methyl‐D‐aspartate (NMDA) receptor hypofunction in cortical and hippocampal brain areas, besides dysfunction of dopaminergic or cholinergic transmitter systems 1, 2. These abnormalities are hypothesized to lead to cognitive impairment in schizophrenia and AD. NMDA receptor activation triggers a cascade of intracellular postsynaptic signalling events through elevation of second messengers [e.g. cyclic guanosine monophosphate (cGMP)], with subsequent activation of protein kinases leading to strengthening of synaptic plasticity as measured by long‐term potentiation (LTP), which is a key cellular process underlying learning and memory formation 3, 4, 5. Phosphodiesterase‐9 (PDE9) hydrolyses cGMP with the highest affinity among all PDEs, and it is expressed in cognition‐relevant brain regions—cortical and hippocampal areas—of rodents and humans 6, 7. Therefore, it is likely to be a significant determinant of intracellular basal cGMP levels in these brain regions. Consequently, inhibition of PDE9 should restore cGMP levels and thus NMDA receptor signalling to physiological levels and translate into memory improvement via strengthening of synaptic plasticity and LTP in patients (Figure 1).
Figure 1.
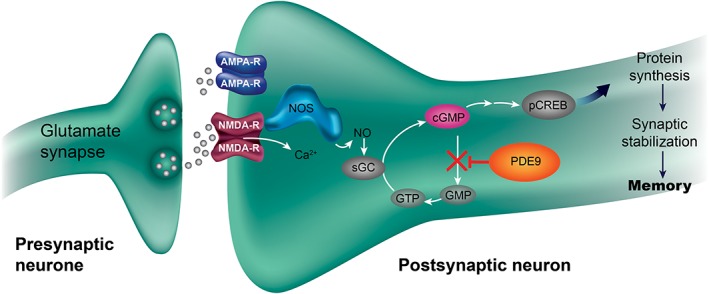
Putative mechanism of action of PDE9 inhibition for strengthening synaptic plasticity. AMPA‐R, α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor; cGMP, cyclic guanosine monophosphate; GMP, guanosine‐5′‐monophosphate; GTP, guanosine‐5′‐triphosphate; NMDA‐R, N‐methyl‐D‐aspartate receptor; NO, nitric oxide; NOS, nitric oxide synthase; pCREB, phosphorylated cyclic adenosine monophosphate response element binding protein; PDE9, phosphodiesterase 9; sGC, soluble guanylyl cyclase
In preclinical studies, PDE9 inhibition by the novel compound BI 409306 or other PDE9 compounds was shown to lead to strengthening of synaptic plasticity, as demonstrated by enhancement of LTP in rat hippocampal slices 8, 9, 10, 11. Moreover, administration of PDE9 inhibitors to rodents led to an increase in cGMP levels in the brain 9, 10, 12, 13 and improved cognitive performance in multiple rodent cognition tasks 9, 10, 12, 13, 14.
In the present first‐in‐human phase I study, the safety and tolerability of single oral doses of BI 409306 (0.5–500 mg) were investigated in cytochrome P450 2C19 (CYP2C19)‐genotyped healthy male subjects. Secondary objectives of the study were to explore the dose proportionality and relative bioavailability of BI 409306 as a film‐coated tablet and an oral solution. As in vitro BI 409306 oxidative metabolism is dependent on CYP2C19, another secondary objective was to compare the safety and pharmacokinetic (PK) profiles of BI 409306 between CYP2C19 extensive metabolizers (EMs; 0.5–500 mg) and poor metabolizers (PMs; 10 mg and 100 mg).
Methods
Study design
The present randomized, double‐blind, placebo‐controlled, single rising‐dose, single‐centre, two‐part study was conducted in healthy male subjects (ClinicalTrials.gov: NCT01343706). Visits were conducted for screening (days −21 to −1), treatment with a single dose on day 1 (days 1–4) and end‐of‐trial examination (days 8–18; Figure 2). In part 1, 10 ascending dose levels of BI 409306 [0.5–500 mg; dose groups (DGs) 1–10] were sequentially investigated in subjects genotyped as homozygous CYP2C19 EMs (defined by the absence of the screened alleles *2 and *3), with at least 5 days between dose levels. One dose was tested in each group, and the decision to proceed to the next DG was based on safety, tolerability and the absence of dose‐limiting events in the previous DG. In DG11, 10 mg BI 409306 was evaluated in CYP2C19 PMs, defined as CYP2C19*2 /*2, CYP2C19*2 /*3 or CYP2C19*3 /*3, after tolerability of 100 mg dosing in EMs was confirmed. In part 2, re‐exposure for bridging tablet and solution formulations was evaluated in DG3 (5 mg) and DG6 (50 mg), respectively. Lastly, PMs from DG11 received re‐exposure with 100 mg. Doses used in this trial were based on subtherapeutic and estimated therapeutic ranges including a safety margin, and were derived based on the affinity of the compound for the target enzyme and allometric scaling of the PK profile observed in dogs. In each DG, only four subjects (three verum and one placebo) were treated on the same day, with a time interval of at least 1 h between drug administrations. If BI 409306 administration was found to be safe and well tolerated, the remaining four subjects from the respective DGs were treated 2 days later, with a time interval of at least 10 min. The next higher dose was administered at least 5 days later (based on the first subcohort of the previous group).
Figure 2.
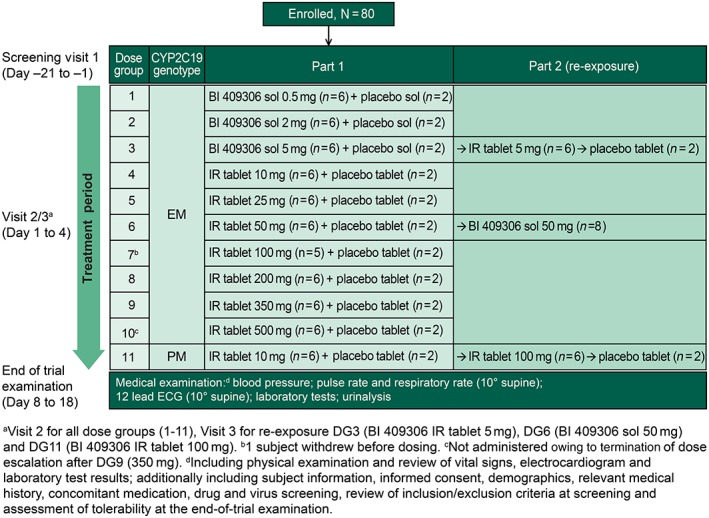
Study design. CYP, cytochrome P450; DG, dose group; ECG, electrocardiogram; EM, extensive metabolizers; IR, immediate release; PM, poor metabolizers; sol, oral solution
All subjects provided written informed consent to participate in the study. The clinical trial protocol and other documents were reviewed by the local independent ethics committee of the Physicians' Chamber of Rheinland‐Pfalz, Mainz, Germany. The study was conducted in compliance with the protocol, the principles laid down in the Declaration of Helsinki and International Conference on Harmonization Harmonized Tripartite Guidelines for Good Clinical Practice.
Treatments
In part 1, CYP2C19‐genotyped EMs were randomized on day 1, visit 2, to receive BI 409306 (six per DG) or placebo (two per DG) in a 3:1 ratio within each of 10 sequential DGs (Figure 3). After a ≥10‐h overnight fast, subjects in DG1, DG2 and DG3 received an oral solution (BI 409306 0.5–5 mg or placebo) and those in DG4 to DG10 received film‐coated tablets (BI 409306 10–500 mg or placebo). CYP2C19 PMs were randomized (3:1) to receive BI 409306 10 mg or placebo tablets (DG11). In part 2, subjects in DG3 (5 mg), DG6 (50 mg) and DG11 (PMs) were re‐exposed to treatment (5 mg in tablet form in DG3; 100 mg in tablet form in DG11; and 50 mg oral solution in DG6), with at least 5 days between treatments. The 100 mg re‐exposure dose for DG11 (PMs) was selected based on the results of safety and an interim PK analysis after 10 mg dosing (data not shown).
Figure 3.
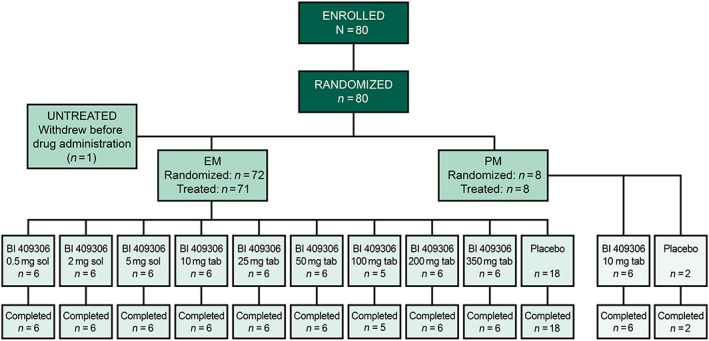
Patient disposition. EM, extensive metabolizers; PM, poor metabolizers; sol, oral solution; tab, tablet
Blinding and randomization
Subjects and investigators were blinded to treatment (BI 409306 or placebo) but were aware of dose levels. Randomization was accomplished using a validated system in which a pseudo‐random number generator and supplied seed number were used to ensure that the resulting allocation of medication numbers to treatment was reproducible and unpredictable.
Subjects
Healthy male volunteers, aged between 21 and 50 years, with a body mass index of 18.5–29.9 kg m−2, genotyped as homozygous CYP2C19 EMs and PMs, were included in the study. Key exclusion criteria included any findings from medical examination that showed a deviation from the normal range [including systolic blood pressure (BP) <120 mmHg and diastolic BP <80 mmHg, resting pulse rate (PR) 60–100 bpm, 12‐lead electrocardiogram (ECG) and clinical laboratory tests] and that were of clinical relevance; evidence of clinically relevant concomitant disease or central nervous system diseases; gastrointestinal, hepatic, renal, respiratory, cardiovascular, metabolic, immunological or hormonal disorders; chronic or relevant acute infections; surgery of the gastrointestinal tract (except appendectomy); and a history of orthostatic hypotension, fainting spells, blackouts or relevant allergies/hypersensitivity.
Study assessments
Primary assessments
Safety and tolerability were assessed by monitoring adverse events (AEs), clinical laboratory assessments, physical examination, vital signs (BP, PR, respiratory rate, oral body temperature and orthostatic test results), cardiac monitoring, 12‐lead ECG and Bond and Lader visual analogue scale (B&L VAS) scores. B&L VAS is a 16‐item, subject‐administered instrument that assesses mood using 100 mm scales anchored at either end by antonyms (e.g. alert–drowsy and calm–excited) 15. Investigators rated tolerability as ‘good’, ‘satisfactory’, ‘not satisfactory’ or ‘bad’ based on AEs and laboratory evaluation.
Secondary assessments
Blood samples for PK analyses were collected at 2 h before dosing, at 10, 20, 30 and 45 min and at 1, 1.5, 2, 2.5, 3, 4, 6, 8, 10, 12, 14 and 24 h after dosing. Urine was collected before dosing and in intervals up to 4, 8, 12 and 24 h after dosing. For DGs receiving ≥10 mg, blood and urine samples were also collected 48 h and 72 h after dosing. BI 409306 plasma and urine concentrations were determined by bioanalytical methods using high‐performance liquid chromatography coupled with tandem mass spectrometry (HPLC‐MS/MS) validated over the range 0.100–100 nM l−1 in plasma and 10.0–10 000 nM l−1 in urine. [13C2, D4]‐BI 409306 was used as an internal standard. Sample clean‐up was achieved by automated solid phase extraction in a 96‐well plate format. Chromatography was conducted on an analytical reversed‐phase HPLC column with gradient elution. The analytes were detected and quantified by HPLC‐MS/MS using electrospray ionization in the positive ion mode. PK parameters were determined from plasma concentration–time profiles and urinary excretion by noncompartmental methods and included the maximum measured concentration of BI 409306 in plasma (Cmax), time to Cmax (tmax), area under the plasma concentration–time curve of BI 409306 over the time interval 0 extrapolated to infinity (AUC0–∞), terminal half‐life (t1/2), and the amounts and fractions excreted over the various collection intervals (Aet1–t2 and fet1–t2).
Statistical analyses
Descriptive statistics were used for safety and PK assessments. The dose proportionality of Cmax, AUC0–∞, and Ae0–4 was determined in CYP2C19 EMs using a linear regression model; in CYP2C19 PMs, a mixed‐effects linear model was used; a 95% confidence interval (CI) was computed for the slope. Relative oral bioavailability (tablet vs. solution; intraindividual comparison) and genotype effect (CYP2C19 EM vs. CYP2C19 PM; interindividual comparison) were determined using analysis of variance of Cmax and AUC0–∞ on a log scale.
A sample size of eight subjects per DG (BI 409306, six; placebo, two) is commonly used in single rising‐dose studies and was considered sufficient for exploratory evaluation of single‐dose safety and PK. The safety population comprised all subjects who received at least one dose of the study drug. The PK analysis population included all evaluable subjects in the safety population. Evaluable subjects were those who had no important protocol violations relevant to the evaluation of PK, had no predose value greater than 5% of Cmax and who did not vomit or experience diarrhoea at or before two times the median tmax.
Results
Study population
Overall, 80 subjects were enrolled and randomized in the study; 79 were treated and completed the study (Figure 3). Of these, 71 were CYP2C19 EMs (DGs 1–9) and eight were CYP2C19 PMs (DG11). DG10 (500 mg) was not evaluated because dose escalation was terminated after DG9 (350 mg). The treated dataset for safety and PK comprised 79 subjects (one subject in DG7 withdrew consent before receiving the study drug). Demographics and baseline characteristics were similar across DGs. All subjects were Caucasian, and the mean [± standard deviation (SD)] age was 36.7 (±7.1) years (Table 1). No relevant concomitant medical conditions or concomitant therapies were reported.
Table 1.
Patient demographics and baseline characteristics (safety population)*
| DG1 | DG2 | DG3 | DG4 | DG5 | DG6 | DG7 | DG8 | DG9 | DG11 | ||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (n = 18) | BI 409306 0.5 mg sol EM (n = 6) | BI 409306 2 mg sol EM (n = 6) | BI 409306 5 mg sol/tab EM (n = 6) | BI 409306 10 mg tab EM (n = 6) | BI 409306 25 mg tab EM (n = 6) | BI 409306 50 mg tab/sol EM (n = 6) | Placebo Tab 50 mg Sol EM (n = 2) | BI 409306 100 mg tab EM (n = 5) | BI 409306 200 mg tab EM (n = 6) | BI 409306 350 mg tab EM (n = 6) | BI 409306 10/100 mg tab PM (n = 6) | Total (n = 79) | |
| Mean (SD) age, years | 38.2 (5.6) | 36.5 (9.2) | 37.5 (6.5) | 35.7 (8.2) | 30.5 (5.9) | 32.2 (6.5) | 40.3 (6.1) | 37.5 (3.5) | 31.4 (7.3) | 38.5 (6.7) | 39.2 (8.8) | 39.5 (7.5) | 36.7 (7.1) |
| Mean (SD) BMI, kg m −2 | 25.0 (2.4) | 25.1 (1.7) | 26.9 (2.5) | 26.3 (1.5) | 24.6 (2.3) | 25.2 (2.2) | 27.0 (2.4) | 24.5 (0.8) | 26.1 (2.0) | 24.3 (2.6) | 25.6 (1.6) | 24.7 (2.4) | 25.4 (2.2) |
| CYP2C19‐predicted phenotype, n (%) | |||||||||||||
| UM | 7 (38.9) | 2 (33.3) | 4 (66.7) | 2 (33.3) | 3 (50.0) | 2 (33.3) | 4 (66.7) | 1 (50.0) | 4 (80.0) | 2 (33.3) | 3 (50.0) | 0 (0.0) | 34 (43.0) |
| EM | 8 (44.4) | 4 (66.7) | 2 (33.3) | 4 (66.7) | 3 (50.0) | 4 (66.7) | 2 (33.3) | 1 (50.0) | 1 (20.0) | 4 (66.7) | 3 (50.0) | 0 (0.0) | 36 (45.6) |
| PM | 2 (11.1) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 6 (100.0) | 8 (10.1) |
| NA | 1 (5.6) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (1.3) |
The table does not include DG10 (500 mg) as these subjects did not receive study drug because dose escalation was terminated after DG9 (350 mg). BMI, body mass index; CYP, cytochrome P450; DG, dose group; EM, extensive metabolizers; NA, not available; PM, poor metabolizers; SD, standard deviation; sol, oral solution; tab, tablet; UM, ultra‐rapid metabolizers.
Includes subjects who were dispensed study medication and documented to have taken ≥1 dose of study drug
Safety
AEs were reported for 23 (29.1%) subjects: 17 (23.9%) CYP2C19 EMs experienced 28 AEs (25 considered to be drug related), and six (75.0%) CYP2C19 PMs experienced eight AEs (seven considered to be drug related). All AEs were of mild‐to‐moderate intensity and short duration. The most frequently reported AEs were nervous system and eye disorders (Table 2); all occurred shortly after administration (20–30 min) and mostly resolved within 1–2 h. Commonly reported nervous system disorders included headache and somnolence. Headache was reported in three CYP2C19 EMs (one subject each in the 2 mg, 5 mg and 200 mg DGs) and four CYP2C19 PMs (two subjects each in the 10 mg and 100 mg DGs) treated with BI 409306. Somnolence was reported in two CYP2C19 EMs (one subject each in the 200 mg and 350 mg DGs). Commonly reported eye disorders included photopsia/flashing lights [five CYP2C19 EMs (100 mg, one; 200 mg, two; and 350 mg, two) and one CYP2C19 PM (100 mg)], photophobia/increased sensitivity to light [three CYP2C19 EMs (one subject each in the 100 mg, 200 mg and 350 mg DGs)], chromatopsia/change in colour perception [one CYP2C19 EM (350 mg) and one CYP2C19 PM (100 mg)] and blurred vision [one CYP2C19 EM (350 mg)] and trended towards dose dependency (intensifying in duration, intensity and frequency with dose escalation). No relevant abnormalities in laboratory results, ECG findings or vital signs were observed, with the exception of one episode of transient sinus tachycardia accompanied by moderate nausea in the 350 mg DG, which was of mild intensity and did not exceed 120 bpm. B&L VAS scores revealed no clinically relevant effects on subjects' mood, vigilance or stress levels (data not shown). Overall, tolerability was rated as ‘good’ in most [73 (92.4%)] subjects (‘satisfactory’, n = 5; ‘not satisfactory’, n = 1). No serious AEs were reported.
Table 2.
Summary of adverse events (safety population)*
| AE, n (%) | Placebo 1st (n = 20) | Placebo 2nd (n = 4) | BI 409306 | Total (n = 79) | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 mg sol EM (n = 6) | 2 mg sol EM (n = 6) | 5 mg sol EM (n = 6) | 5 mg tab EM (n = 6) | 10 mg tab EM (n = 6) | 25 mg tab EM (n = 6) | 50 mg tab EM (n = 6) | 50 mg sol EM (n = 8) | 100 mg tab EM (n = 5) | 200 mg tab EM (n = 6) | 350 mg tab EM (n = 6) | 10 mg tab PM (n = 6) | 100 mg tab PM (n = 6) | ||||
| Nervous system disorders | 1 (5.0) | 1 (25.0) | – | 2 (33.3) | – | 1 (16.7) | – | – | – | – | 2 (40.0) | 2 (33.3) | 1 (16.7) | 2 (33.3) | 2 (33.3) | 14 (17.7) |
| Headache | 1 (5.0) | – | – | 1 (16.7) | – | 1 (16.7) | – | – | – | – | – | 1 (16.7) | – | 2 (33.3) | 2 (33.3) | 8 (10.1) |
| Dizziness | – | 1 (25.0) | – | 1 (16.7) | – | – | – | – | – | – | – | – | – | – | – | 2 (2.5) |
| Paraesthesia | – | – | – | – | – | – | – | – | – | – | 1 (20.0) | – | – | – | – | 1 (1.3) |
| Parosmia | – | – | – | – | – | – | – | – | – | – | 1 (20.0) | – | – | – | – | 1 (1.3) |
| Presyncope | – | – | – | – | – | – | – | – | – | – | – | – | – | – | 1 (16.7) | 1 (1.3) |
| Somnolence | – | – | – | – | – | – | – | – | – | – | – | 1 (16.7) | 1 (16.7) | – | ‐ | 2 (2.5) |
| Eye disorders | – | – | – | – | – | – | – | – | – | – | 1 (20.0) | 3 (50.0) | 3 (50.0) | – | 2 (33.3) | 9 (11.4) |
| Photopsia | – | – | – | – | – | – | – | – | – | – | 1 (20.0) | 2 (33.3) | 2 (33.3) | – | 1 (16.7) | 6 (7.6) |
| Photophobia | – | – | – | – | – | – | – | – | – | – | 1 (20.0) | 1 (16.7) | 1 (16.7) | – | ‐ | 3 (3.8) |
| Chromatopsia | – | – | – | – | – | – | – | – | – | – | – | – | 1 (16.7) | – | 1 (16.7) | 2 (2.5) |
| Blurred vision | – | – | – | – | – | – | – | – | – | – | – | – | 1 (16.7) | – | – | 1 (1.3) |
| Cardiac disorders | – | – | – | – | – | – | – | – | 1 (16.7) | – | 1 (20.0) | – | 1 (16.7) | – | – | 3 (3.8) |
| Sinus tachycardia | – | – | – | – | – | – | – | – | – | – | 1 (20.0) | – | 1 (16.7) | – | – | 2 (2.5) |
| Extrasystoles | – | – | – | – | – | – | – | – | 1 (16.7) | – | – | – | – | – | – | 1 (1.3) |
| Palpitations | – | – | – | – | – | – | – | – | 1 (16.7) | – | – | – | – | – | – | 1 (1.3) |
| Gastrointestinal disorders | 1 (5.0) | – | – | 1 (16.7) | – | – | – | – | – | – | – | – | 2 (33.3) | 1 (16.7) | – | 5 (6.3) |
| Diarrhoea | 1 (5.0) | – | – | – | – | – | – | – | – | – | – | – | – | 1 (16.7) | – | 2 (2.5) |
| Dry mouth | – | – | – | – | – | – | – | – | – | – | – | – | 1 (16.7) | – | – | 1 (1.3) |
| Nausea | – | – | – | 1 (16.7) | – | – | – | – | – | – | – | – | 1 (16.7) | – | – | 2 (2.5) |
AE, adverse event; EM, extensive metabolizers; PM, poor metabolizers; sol, oral solution; tab, tablet
Includes all subjects dispensed study medication and documented to have taken ≥1 dose of study drug
PK results
In all the DGs, BI 409306 was rapidly absorbed within the first hour after oral administration and rapidly eliminated within 24 h following dose thereafter [geometric mean (gMean) t1/2 ranging from 0.99 h to 2.71 h in CYP2C19 EMs and PMs]; plasma concentration peaks were broader and declined more slowly in CYP2C19 PMs than EMs (Figure 4). Urinary excretion of BI 409306 was generally low (<2% of the dose in CYP2C19 EMs and up to 2.68% of the dose in CYP2C19 PMs). PK parameters are summarized in Table 3. Cmax and AUC0–∞ in CYP2C19 EMs increased slightly more than dose proportional over the entire dose range; the exponent β for the power model was 1.13 (95% CI: 1.06, 1.19) for AUC0–∞, 1.10 (95% CI: 1.03, 1.18) for Cmax and 1.05 (95% CI: 0.98, 1.12) for Ae0–4. Systemic exposure in CYP2C19 PMs increased similarly with dose.
Figure 4.
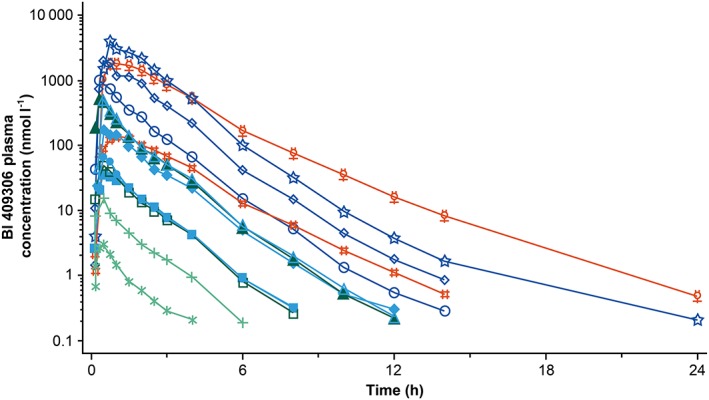
Geometric mean drug plasma concentration–time profiles after administration of 0.5–350 mg BI 409306 to cytochrome P450 2C19‐genotyped extensive metabolizers and poor metabolizers. EM, extensive metabolizers; PM, poor metabolizers.  0.5 mg solution EM (n = 6),
0.5 mg solution EM (n = 6),  5 mg solution EM (n = 6),
5 mg solution EM (n = 6),  10 mg tablet EM (n = 6),
10 mg tablet EM (n = 6),  50 mg tablet EM (n = 6),
50 mg tablet EM (n = 6),  100 mg tablet EM (n = 5),
100 mg tablet EM (n = 5),  350 mg tablet EM (n = 6),
350 mg tablet EM (n = 6),  100 mg tablet PM (n = 6),
100 mg tablet PM (n = 6),  2 mg solution EM (n = 6),
2 mg solution EM (n = 6),  5 mg tablet EM (n = 6),
5 mg tablet EM (n = 6),  25 mg tablet EM (n = 6),
25 mg tablet EM (n = 6),  50 mg solution EM (n = 8),
50 mg solution EM (n = 8),  200 mg tablet EM (n = 6),
200 mg tablet EM (n = 6),  10 mg tablet PM (n = 6)
10 mg tablet PM (n = 6)
Table 3.
Geometric mean (and gCV%) pharmacokinetic parameters after single‐dose administrations of BI 409306
| BI 409306 | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 0.5 mg sol EM (n = 6) | 2 mg sol EM (n = 6) | 5 mg sol EM (n = 6) | 5 mg tab EM (n = 6) | 10 mg tab EM (n = 6) | 25 mg tab EM (n = 6) | 50 mg tab EM (n = 6) | 50 mg sol EM (n = 8) | 100 mg tab EM (n = 5) | 200 mg tab EM (n = 6) | 350 mg tab EM (n = 6) | 10 mg tab PM (n = 6) | 100 mg tab PM (n = 6) | |
| C max , nmol l −1 | 3.54 (34.5) | 18.8 (50.8) | 56.4 (36.4) | 55.9 (40.9) | 99.8 (79.1) | 300 (85.0) | 479 (67.0) | 613 (40.7) | 1370 (87.3) | 2950 (89.5) | 5540 (29.5) | 223 (38.1) | 3120 (31.4) |
| AUC 0–∞ , nmol·h l −1 | 3.95 (50.4) | 20.9 (41.3) | 76.7 (27.0) | 75.6 (32.2) | 95.9 (69.8) | 351 (49.4) | 539 (82.0) | 648 (42.9) | 1460 (68.5) | 3920 (44.4) | 7980 (17.1) | 476 (35.5) | 6060 (21.4) |
| * t max , h | 0.50 (0.33–0.50) | 0.33 (0.32–0.50) | 0.50 (0.33–0.75) | 0.63 (0.33–1.50) | 0.50 (0.33–0.75) | 0.50 (0.50–1.50) | 0.63 (0.33–0.75) | 0.33 (0.33–0.50) | 0.50 (0.33–0.50) | 0.63 (0.33–2.00) | 0.75 (0.50–2.00) | 0.75 (0.33–1.48) | 0.86 (0.50–2.00) |
| t 1/2 , h | 1.33 (25.5) | 0.995 (14.4) | 1.18 (28.1) | 1.16 (23.2) | 1.04 (21.7) | 1.23 (17.7) | 1.39 (14.8) | 1.34 (26.0) | 1.85 (22.0) | 2.18 (50.2) | 2.71 (16.5) | 1.83 (12.9) | 2.68 (29.3) |
| Cl F −1 , ml min −1 | 6770 (50.4) | 5130 (41.3) | 3490 (27.0) | 3540 (32.2) | 5580 (69.8) | 3810 (49.4) | 4970 (82.0) | 4130 (42.9) | 3660 (68.5) | 2730 (44.4) | 2350 (17.1) | 1120 (35.5) | 882 (21.4) |
| fe 0–4 , % | NC | 0.483 (46.5) | 0.583 (26.0) | 0.611 (24.2) | 0.461 (60.2) | NC | 0.523 (56.9) | 0.594 (48.1) | 0.597 (53.0) | 0.676 (48.8) | 0.786 (24.0) | 1.30 (14.3) | 1.78 (23.3) |
AUC0–∞, area under the concentration–time curve of the analyte in the plasma over the time interval 0 extrapolated to infinity; Cl F−1, total (apparent) clearance of the analyte in the plasma following extravascular administration; Cmax, maximum measured concentration of the analyte in the plasma; EM, extensive metabolizers; fe0–4, fraction of analyte eliminated in the urine in the time interval from 0 h to 4 h; gCV, geometric coefficient of variation; NC, not calculated; PM, poor metabolizers; sol, oral solution; tab, tablet; tmax, time from (last) dosing to the maximum measured concentration of the analyte in the plasma; t1/2, terminal half‐life of the analyte in the plasma.
Median and range (minimum to maximum)
Similar peak and total exposure was observed with tablets and oral solution; the AUC0–∞ ratio of tablet to solution was 98.5% (90% CI: 87.6%, 111%) for the 5 mg dose, and 84.2% (90% CI: 59.6%, 119%) for the 50 mg dose. Similar results were obtained for Cmax ratios. CYP2C19 PMs exhibited higher systemic exposure than CYP2C19 EMs; the ratio (CYP2C19 PM to EM) of Cmax and AUC0–∞ was 224% (90% CI: 125%, 401%) and 496% (90% CI: 291%, 843%), respectively, for the 10 mg dose, and 228% (90% CI: 124%, 421%) and 415% (90% CI: 254%, 677%), respectively, for the 100 mg dose.
Discussion
In this first‐in‐human study evaluating the safety and PK of single rising doses of BI 409306 in healthy male subjects, BI 409306 was generally safe and well tolerated. BI 409306 was rapidly absorbed and eliminated, and had similar peak and total exposure, regardless of formulation. No notable safety findings or indications of safety risk were observed. VAS scores did not reveal any clinically relevant effects on mood, vigilance and stress levels. The most frequently reported AEs were eye disorders (photopsia, photophobia, chromatopsia and blurred vision) in EMs, and nervous system disorders (headache and somnolence) in PMs. All AEs were of mild‐to‐moderate intensity and resolved without sequelae. As there was a potential dose‐related trend in the occurrence of AEs, the maximum tolerated dose in CYP2C19 EMs was set at 350 mg. There is evidence that the occurrence of visual phenomena is linked to PDE9A action 16. The results of that study indicated that PDE9A modulates the processes within the cone pathway in the retina.
Mild AEs have been reported with another PDE9A inhibitor (PF‐04447943) being evaluated for the treatment of impaired cognition associated with AD 17, 18. Drug‐related AEs were found to be generally mild, with diarrhoea, headache and nausea most commonly reported in healthy volunteers 17. Similar results were observed in patients with mild‐to‐moderate probable AD. The incidence of severe AEs in PF‐04447943‐treated patients was comparable to that with placebo 18.
In the present study, the PK of BI 409306 were characterized by rapid absorption and elimination, in line with its PK properties in rodents 8, 12. A consistent, slightly more than dose‐proportional increase in total and peak systemic exposure was detected by inferential statistical analysis; however, the magnitude of the nonproportionality is likely to be negligible.
Subjects in the present study were prospectively genotyped as EMs or PMs of CYP2C19 enzymes. This was performed with the assumption that if the CYP2C19 loss‐of‐function variant resulted in a higher exposure to BI 409306, CYP2C19 PMs, who carry two nonfunctional alleles of the gene, would exhibit an altered profile. After administration of BI 409306 10 mg and 100 mg, peak and total exposure increased approximately 2.2–2.3‐fold and 4.1–5.0‐fold, respectively, when metabolism by CYP2C19 was depleted, suggesting possible differences in the metabolism of BI 409306 with increasing doses and status of metabolizing CYP2C19 enzymes. However, exposure in CYP2C19 PMs was still within the range achieved with the higher doses administered to CYP2C19 EMs. In addition, terminal t1/2 in CYP2C19 PMs was within the range observed with the higher doses administered to CYP2C19 EMs.
Tablet and oral formulations of BI 409306 had similar peak and total exposure following the 5 mg dose. After administration of 50 mg tablets, gMean AUC was reduced by approximately 16%, and gMean Cmax was reduced by approximately 20%. However, the two formulations were considered similar because sample size was small and visual intraindividual comparison of exposure parameters did not indicate any trend for differences.
Subjects and investigators were blinded to treatment (BI 409306 or placebo) but were aware of dose levels. This knowledge might have resulted in observer bias with regard to dose‐dependent and time effects, but sequential assessment of ascending doses was necessary to minimize subject risk. In addition, PK comparisons were exploratory in nature because of the small sample size.
In this first‐in‐human study, we focused on establishing safety and characterizing the PK and effects of different genetic variants of CYP2C19 phenotypes and drug transporters on the metabolism of BI 409306. Taking into account the unknown central nervous system side‐effect profile of BI 409306 in humans, and in order to avoid the undue risks to healthy volunteers by applying lumbar catheter placement, the pharmacodynamics of BI 409306 were further evaluated and proved in a subsequent study in healthy volunteers by monitoring changes in cGMP levels in the cerebrospinal fluid across therapeutic and supratherapeutic dose levels (25, 50, 100 and 200 mg) 19.
Overall, BI 409306 was generally safe and well tolerated up to 350 mg in CYP2C19 EM and up to 100 mg in CYP2C19 PM healthy male subjects, with no notable safety findings or indications of a safety risk. BI 409306 was rapidly absorbed and eliminated in all DGs. Cmax was approximately 2.2–2.3‐fold higher, and AUC0–∞ was approximately 4.1–5.0‐fold higher in CYP2C19 PMs compared with CYP2C19 EMs. The highest exposure was observed with BI 409306 350 mg, which was the highest dose investigated in the study.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: VM, KB, UF, AH, HZ‐G and MS had support from Boehringer Ingelheim for the submitted work; VM, KB, UF, AH, HZ‐G and MS were employees of Boehringer Ingelheim in the previous 3 years; no other relationships or activities that could appear to have influenced the submitted work.
We would like to thank Dr Holger Rosenbrock for his critical review of the manuscript and his valuable contribution pertaining to the mechanism of action of PDE9 inhibition, and Dr Frank Runge for developing the PK bioanalytical methods and generating concentration data. The authors meet the criteria for authorship as recommended by the International Committee of Medical Journal Editors. The authors received no direct compensation related to the development of the manuscript. Writing, editorial support and formatting assistance was provided by Suchita Nath‐Sain PhD, and Maribeth Bogush MCI, PhD, of Cactus Communications, which was contracted and funded by Boehringer Ingelheim for these services. Boehringer Ingelheim was given the opportunity to review the manuscript for medical and scientific accuracy as well as intellectual property considerations.
Contributors
All the authors contributed to conception and design, collection and assembly of data, data analysis and interpretation, and manuscript writing, and provided final approval of the manuscript.
Moschetti, V. , Boland, K. , Feifel, U. , Hoch, A. , Zimdahl‐Gelling, H. , and Sand, M. (2016) First‐in‐human study assessing safety, tolerability and pharmacokinetics of BI 409306, a selective phosphodiesterase 9A inhibitor, in healthy males. Br J Clin Pharmacol, 82: 1315–1324. doi: 10.1111/bcp.13060.
References
- 1. Hu NW, Ondrejcak T, Rowan MJ. Glutamate receptors in preclinical research on Alzheimer's disease: update on recent advances. Pharmacol Biochem Behav 2012; 100: 855–62. [DOI] [PubMed] [Google Scholar]
- 2. Lin CH, Lane HY, Tsai GE. Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol Biochem Behav 2012; 100: 665–77. [DOI] [PubMed] [Google Scholar]
- 3. Bliss TV, Collingridge GL. A synaptic model of memory: long‐term potentiation in the hippocampus. Nature 1993; 361: 31–9. [DOI] [PubMed] [Google Scholar]
- 4. Cooke SF, Bliss TV. Plasticity in the human central nervous system. Brain 2006; 129: 1659–73. [DOI] [PubMed] [Google Scholar]
- 5. Reymann KG, Frey JU. The late maintenance of hippocampal LTP: requirements, phases, ‘synaptic tagging’, ‘late‐associativity’ and implications. Neuropharmacology 2007; 52: 24–40. [DOI] [PubMed] [Google Scholar]
- 6. Van Staveren WC, Steinbusch HW, Markerink‐Van Ittersum M, Repaske DR, Goy MF, Kotera J, et al. mRNA expression patterns of the cGMP‐hydrolyzing phosphodiesterases types 2, 5, and 9 during development of the rat brain. J Comp Neurol 2003; 467: 566–80. [DOI] [PubMed] [Google Scholar]
- 7. Lakics V, Karran EH, Boess FG. Quantitative comparison of phosphodiesterase mRNA distribution in human brain and peripheral tissues. Neuropharmacology 2010; 59: 367–74. [DOI] [PubMed] [Google Scholar]
- 8. Dorner‐Ciossek C, Giovannini R, Rosenbrock H. BI 409306, a novel phosphodiesterase 9A inhibitor, part I: potency, selectivity and in‐vitro functional characterization on synaptic plasticity. International Congress on Schizophrenia Research: Schizophrenia Bull 2015; 41: S31. [Google Scholar]
- 9. Hutson PH, Finger EN, Magliaro BC, Smith SM, Converso A, Sanderson PE, et al. The selective phosphodiesterase 9 (PDE9) inhibitor PF‐04447943 (6‐[(3S,4S)‐4‐methyl‐1‐(pyrimidin‐2‐ylmethyl)pyrrolidin‐3‐yl]‐1‐(tetrahydro‐2H‐pyran‐4‐yl)‐1,5‐dihydro‐4H‐pyrazolo[3,4‐d]pyrimidin‐4‐one) enhances synaptic plasticity and cognitive function in rodents. Neuropharmacology 2011; 61: 665–76. [DOI] [PubMed] [Google Scholar]
- 10. Kroker KS, Mathis C, Marti A, Cassel JC, Rosenbrock H, Dorner‐Ciossek C. PDE9A inhibition rescues amyloid beta‐induced deficits in synaptic plasticity and cognition. Neurobiol Aging 2014; 35: 2072–8. [DOI] [PubMed] [Google Scholar]
- 11. Kroker KS, Rast G, Giovannini R, Marti A, Dorner‐Ciossek C, Rosenbrock H. Inhibition of acetylcholinesterase and phosphodiesterase‐9A has differential effects on hippocampal early and late LTP. Neuropharmacology 2012; 62: 1964–74. [DOI] [PubMed] [Google Scholar]
- 12. Rosenbrock H, Marti A, Koros E, Runge F, Fuchs H, Giovannini R, et al. BI 409306, a novel phosphodiesterase 9A inhibitor, part II: in‐vivo characterization regarding target engagement and cognition tasks in rodents. Abstracts for the 15th International Congress on Schizophrenia Research. Schizophrenia Bull 2015; 41 (Suppl. 1): S36. [Google Scholar]
- 13. Kleiman RJ, Chapin DS, Christoffersen C, Freeman J, Fonseca KR, Geoghegan KF, et al. Phosphodiesterase 9A regulates central cGMP and modulates responses to cholinergic and monoaminergic perturbation in vivo . J Pharmacol Exp Ther 2012; 341: 396–409. [DOI] [PubMed] [Google Scholar]
- 14. van der Staay FJ, Rutten K, Barfacker L, Devry J, Erb C, Heckroth H, et al. The novel selective PDE9 inhibitor BAY 73‐6691 improves learning and memory in rodents. Neuropharmacology 2008; 55: 908–18. [DOI] [PubMed] [Google Scholar]
- 15. Bond A, Lader M. The use of analogue scales in rating subjective feelings. Br J Med Psychol 1974; 47: 211–8. [Google Scholar]
- 16. Dhingra A, Tummala SR, Lyubarsky A, Vardi N. PDE9A is expressed in the inner retina and contributes to the normal shape of the photopic ERG waveform. Front Mol Neurosci 2014; 7: 60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Evans RM, Nicholas T, Le V, Qiu R, Martin W, Martin D, et al. Safety and pharmacokinetics of PF‐04447943, a PDEA inhibitor, in single and multiple dose phase 1 studies in healthy volunteers. Alzheimers Dement 2010; 6: S135. [Google Scholar]
- 18. Schwam EM, Nicholas T, Chew R, Billing CB, Davidson W, Ambrose D, et al. A multicenter, double‐blind, placebo‐controlled trial of the PDE9A inhibitor, PF‐04447943, in Alzheimer's disease. Curr Alzheimer Res 2014; 11: 413–21. [DOI] [PubMed] [Google Scholar]
- 19. Boland K, Moschetti V, Dansirikul C, Pichereau S, Gheyle L, Sand M. Randomized, double‐blind, placebo‐controlled, parallel‐group proof of mechanism study to assess the pharmacokinetics and pharmacodynamic effect of different single oral doses of BI 409306 in healthy male volunteers. Abstracts for the 15th International Congress on Schizophrenia Research. Schizophrenia Bull 2015; 41 (Suppl. 1): S303. [Google Scholar]