

Abstract
Aims
To prospectively select the dose of the paliperidone palmitate 3‐month (PP3M) formulation, using a pharmacometric bridging strategy based on the paliperidone palmitate 1‐month (PP1M) formulation previously approved for schizophrenia treatment.
Methods
Pharmacokinetic (PK) data from a 6‐month interim analysis of a single dose PP3M Phase I clinical trial was integrated with a previously developed PP1M population‐PK model. The model was updated to incorporate formulation as a covariate on absorption parameters and to explore the most critical design element of the Phase III study: the PP1M‐to‐PP3M dose multiplier for patients switching formulations. Plasma paliperidone concentrations were measured at predetermined intervals during Phase III, enabling comparison of the multiple‐dose PK between PP1M and PP3M. Exposure matching was assessed graphically to determine whether paliperidone plasma concentrations from the two formulations overlapped.
Results
Prospective steady‐state PK simulations revealed that a 3.5 multiple of the PP1M dose would yield a corresponding PP3M dose with comparable exposure. The prospective pharmacometric simulation and observed Phase III PK data agreed closely. Phase III results confirmed the hypothesis that efficacy of PP3M was noninferior to that of PP1M. The similarity in exposures between the two formulations was likely a key determinant of the equivalent efficacy between the two products observed in the Phase III study.
Conclusions
Successful prospective PP3M Phase III clinical trial dose selection was achieved through the use of pharmacometric bridging, without conducting a Phase II study and using only limited Phase I data for PP3M. We estimate that this strategy reduced development time by 3–5 years and may be applicable to other drug development projects.
Keywords: paliperidone palmitate, pharmacometric bridging, prospective validation
What is Already Known about this Subject
Two formulations of paliperidone for the treatment of schizophrenia were available globally until PP3M was introduced: an extended‐release oral formulation, and paliperidone palmitate 1‐month (PP1M) for intramuscular administration.
The new paliperidone palmitate 3‐month (PP3M) formulation is the first and only atypical antipsychotic for the treatment of schizophrenia that can be administered just four times a year.
What this Study Adds
Using model‐informed drug development principles, adaptations were implemented in the PP3M Phase I study based on analysis of emerging pharmacokinetic data; the Phase III program was started prior to the end of Phase I, and Phase II was skipped entirely.
We estimate that this strategy reduced development time by several years and is a successful example of using model‐based approaches to drive decision making in drug development.
Introduction
The steady rise in drug development costs and the small number of compounds that achieve regulatory approval is well recognized; model‐based drug development has been suggested as a strategy to decrease the attrition rate of compounds during development 1. This strategy can be especially helpful during life‐cycle management of antipsychotics administered as a new formulation that facilitates more convenient and potentially prolonged drug delivery.
Paliperidone is approved for the treatment of schizophrenia. Two formulations of paliperidone are available globally: an extended‐release (ER) oral formulation, and a long‐acting injectable (LAI) paliperidone palmitate 1‐month formulation (PP1M) for intramuscular administration 2. The LAI paliperidone palmitate 3‐month (PP3M), like PP1M, is an aqueous ER suspension. PP3M can be administered just four times a year 3, and this first‐of‐its‐kind dosing interval is an innovation in schizophrenia treatment. PP3M was approved by the Food and Drug Administration in May 2015 under Priority Review, a designation reserved for “a drug that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness.” In addition, the European Medicines Agency and Health Canada approved PP3M in May 2016 and June 2016, respectively.
For PP3M clinical development, it was hypothesized that efficacy depends on attaining target plasma concentrations of paliperidone that were observed with the previously‐approved formulations PP1M and paliperidone‐ER. The PP1M clinical programme involved a dedicated Phase II study 4, but a similar Phase II study was not conducted for PP3M based on the following rationale. Extensive Phase I, II and III studies with PP1M and also with paliperidone‐ER established the dose of each drug that is required to achieve therapeutic concentrations of paliperidone at steady state 5. Those PP1M and paliperidone‐ER exposure reference ranges served as the basis for the current PP3M bridging strategy. The PP3M Phase III trial, in part, served as the pharmacokinetics (PK) similarity study designed to confirm, through pharmacometric bridging, the dose of PP3M that would yield paliperidone exposure similar to that of PP1M and paliperidone‐ER. The PP3M Phase III data, in conjunction with modelling and simulation, effectively eliminated the need for a PP3M Phase II trial.
In order to develop a formulation that yielded the target plasma concentrations over a three‐month period, formulation variables were optimized through a model‐based pharmacometric analysis. Altering these formulation variables prolonged the terminal half‐life and enabled shallower paliperidone concentration peaks with PP3M relative to PP1M. A critical component of our approach is the selection of an appropriate ‘dose multiplier’ for PP3M relative to PP1M. The dose multiplier is defined here as the multiple applied to the PP1M dose (that achieved clinical stability) to derive the equivalent PP3M dose that maintains similar paliperidone exposure after the switch from PP1M to PP3M. PP3M requires at least 4 months of adequate treatment with PP1M to achieve clinical stability, and thereafter, the dose‐multiplier is applied. When a dose multiplier of 3‐ or 3.5‐fold was used between PP1M and PP3M, paliperidone peak concentrations increased only about 2‐fold, and apparent half‐life was prolonged by ≥2‐fold after single dose administration 3, 6. This allowed PP3M to attain a peak/trough ratio comparable to that observed in the previously developed LAI PP1M and oral paliperidone‐ER formulations.
Integrated, lean and adaptive Phase I and III studies of PP3M were undertaken to study single‐ and multiple‐dose pharmacokinetics (PK) 3, 7, 8. The implementation of learn‐and‐confirm milestones allowed accelerated decision making. Pharmacometric analyses were performed to skip Phase II and start Phase III, using interim analysis (IA) of Phase I data instead of waiting for the conclusion of the Phase I/II programme, as is usually done during drug development. This report presents the pharmacometric bridging strategy from PP1M to PP3M, i.e., the prospective Phase III dose prediction based on the IA of single dose Phase I data, as well as its validation using Phase III data.
Materials and methods
Study protocols and amendments were reviewed by an Independent Ethics Committee or Institutional Review Board, as appropriate, for each site. The studies were conducted in compliance with the Declaration of Helsinki consistent with Good Clinical Practices and applicable regulatory requirements. Written informed consent was obtained from all patients before enrolment.
Patient populations
The patient populations in the PP3M Phase I and Phase III studies were similar 3, 7, 8; the Phase I study included patients with schizophrenia rather than healthy volunteers due to the long‐acting nature of the formulation. In the PP3M Phase I study, all patients were taking antipsychotic medication (but not risperidone or paliperidone), and the PP3M dose was in addition to their existing medication. Inclusion criteria for the Phase I and III studies were: adult patients (18–70 years old) of either sex with schizophrenia (Diagnostic and Statistical Manual of Mental Disorders, 4th edn). Patient demographics for the Phase I and III studies have been reported previously 3, 7, 8.
Phase I Study
A single‐dose Phase I PK/tolerability study 3 initially evaluated single intramuscular injections of PP3M using a dose range of 75–450 mg‐equivalent (mg‐eq.; 1.56 mg of paliperidone palmitate is 1 mg‐eq. of paliperidone). Patients were randomly assigned to one of five possible treatment groups, and an IA was conducted after at least 15 patients per treatment group had completed their 6‐month PK and safety evaluation. After the IA, and in parallel with the initiation of the Phase III studies, Phase I evaluation of safety at the highest dose (525 mg‐eq.) was started.
PP1M and PP3M models
The PP1M model structure, parameter estimates, code 2, 9, 10 and the PP3M model 11 have been described previously. The disposition of paliperidone after PP1M administration follows a one‐compartment model with first‐order elimination. The absorption sub‐model allows a fraction of the dose (F2) to enter relatively quickly into the central compartment via a zero‐order process, while the remaining fraction enters the systemic circulation after a certain lag time (t lag1) via a slow first‐order process with a rate constant k a that produces flip‐flop kinetics 10. Finally, the PP3M formulation was found to influence each of the three absorption parameters (F2, k a, t lag1), and therefore the apparent half‐life, C max and t max of paliperidone after PP3M administration.
Prospective dose prediction
The 6‐month single dose PK data for PP3M was integrated with the PP1M data using a previously developed population‐PK model for PP1M 2. The update involved incorporation of formulation as a covariate on the absorption parameters in the PP1M model. The updated model was used to predict the multiple‐dose profile for PP3M, which allowed exploration of the most critical design element of the Phase III study, i.e. the PP1M‐to‐PP3M dose multiplier for patients switching formulations. The dose multiplier was estimated based on a sensitivity analysis where multipliers of 3:1, 3.5:1 and 4:1 between PP3M:PP1M were assessed via simulations. The expectation for the simulation results was that the paliperidone PK exposures with PP3M should not exceed the peak concentrations from the highest studied doses of oral paliperidone‐ER and LAI PP1M. Similarly, the PP3M paliperidone troughs should not be substantially lower than the PP1M paliperidone troughs at steady state as non‐inferiority would be tested against PP1M.
Evaluation of the dose multiplier suggested that a more than 3‐fold higher PP3M dose would be needed in the Phase III studies. Therefore, a higher PP3M dose of 525 mg eq. was chosen for Phase I safety evaluation through protocol amendment before proceeding to Phase III because this was the 3.5 multiple of the highest PP1M safe dose of 150 mg eq. The assessment of the PP3M 525 mg eq. dose included an evaluation of both safety and PK, and this additional cohort in the Phase I study was initiated in parallel with the start of the Phase III studies. Safety information (including injection site tolerability) from 10 patients who received the 525 mg eq. dose of PP3M for the first 3 months of the Phase I study was made available, allowing coverage for peak concentrations, before patients were switched from PP1M to their corresponding 3.5‐fold PP3M dosage in the Phase III studies.
Phase III study: Validation of dose predictions
One of the Phase III trials 8 was a non‐inferiority study where PP3M was tested as maintenance therapy in patients who were stabilized on PP1M during a 17‐week open‐label, flexible dose stabilization phase. Patients clinically stable at 17 weeks entered the 48‐week fixed dose double‐blind (DB) phase and were randomly assigned 1:1 to PP3M or PP1M. Dose selection for the PP3M arm was based on a 3.5‐fold multiplier of the last PP1M dose. Plasma paliperidone concentrations were measured at predetermined intervals during the DB phase of the Phase III study. This enabled comparison of the multiple‐dose PK between PP1M and PP3M upon completion of the Phase III study. Exposure matching between the two formulations was assessed visually to determine whether the paliperidone plasma concentrations from the two formulations overlapped. The prospective PK bridging and transition from PP1M to PP3M would be deemed successful if similar exposures and efficacy for the two formulations were observed in Phase III.
Results
Our approach to expediting the clinical development of PP3M is based on our previous experience with PP1M and oral paliperidone‐ER and is summarized in Figure 1. The IA of the Phase I data at 6 months suggested that changing the formulation variables altered the PK profile, which resulted in (i) slower drug release from the injection depot, (ii) a longer terminal half‐life and (iii) shallower paliperidone peaks. These findings would allow injections of relatively higher doses of PP3M once every quarter year 3. Steady‐state PK simulations (Figure 2, Table 1) revealed that a 3.5 multiple of the PP1M dose would yield a corresponding PP3M dose with comparable exposure. Doses up to 15 mg were tested in the oral paliperidone‐ER programme, and the simulated steady‐state peak concentrations at the highest PP3M dose (525 mg‐eq.) were similar to those predicted with 12–15 mg of oral paliperidone‐ER (data not shown).
Figure 1.

Flowchart summarizing the pharmacometric bridging exercise. Abbreviations: PP1M, paliperidone‐1 month; PP3M, paliperidone‐3 month; PK, pharmacokinetics
Figure 2.
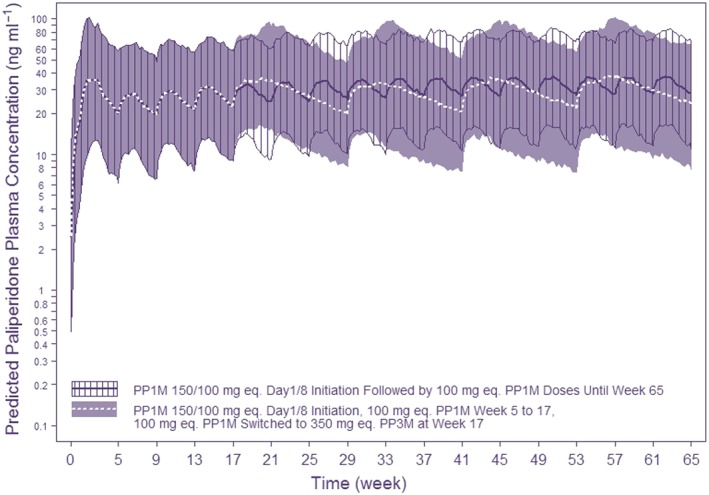
Simulated exposure using a 3.5‐fold dose multiplier between PP1M and PP3M based on a fit‐for‐purpose model built on single dose interim PK data from Phase I. This scenario simulates switching 100 mg‐eq. PP1M to 350 mg‐eq. PP3M vs. 100 mg‐eq. PP1M maintenance therapy. The lines/shaded‐hatched areas represent the model‐based median/90% prediction interval. 150/100 mg‐eq. injections on days 1/8 in the deltoid muscle is the recommended initiation regimen for PP1M. This was followed by 100 mg‐eq. PP1M on weeks 5, 9 and 13. Starting at week 17, a fixed dose of PP3M (350 mg‐eq.) or PP1M (100 mg‐eq.) were simulated
Table 1.
Difference in paliperidone pharmacokinetic (PK) metrics based on different dose multipliers between PP1M and PP3M (gluteal injections)
| Paliperidone PK metric | 3× dose multiplier | 3.5× dose multiplier | 4× dose multiplier | |||
|---|---|---|---|---|---|---|
| PP1M → PP3M (mg eq.) | Median difference in steady‐state exposure (ng ml−1)* | PP1M → PP3M (mg eq.) | Median difference in steady‐state exposure (ng ml−1)* | PP1M → PP3M (mg eq.) | Median difference in steady‐state exposure (ng ml−1)* | |
| Peak | 50 → 150 | −0.5 | 50 → 175 | 2.1 | 50 → 200 | 4.5 |
| 75 → 225 | −1.6 | 75 → 260 | 1.7 | 75 → 300 | 5.5 | |
| 100 → 300 | −3.0 | 100 → 350 | 1.6 | 100 → 400 | 5.9 | |
| 150 → 450 | −6.6 | 150 → 525 | −0.6 | 150 → 600 | 5.4 | |
| Trough | 50 → 150 | −3.7 | 50 → 175 | −2.1 | 50 → 200 | −0.4 |
| 75 → 225 | −5.8 | 75 → 260 | −3.3 | 75 → 300 | −0.7 | |
| 100 → 300 | −7.7 | 100 → 350 | −4.2 | 100 → 400 | −1.0 | |
| 150 → 450 | −11.5 | 150 → 525 | −6.6 | 150 → 600 | −2.1 | |
A negative value signifies that the median paliperidone peak or trough concentration is lower from PP3M compared to PP1M; a positive value signifies that the median paliperidone peak or trough concentration is higher from PP3M compared to PP1M.
Three different dose multipliers of PP1M to PP3M were considered in the simulation analysis between PP3M and PP1M as part of the sensitivity analysis: 3.0:1, 3.5:1 and 4.0:1. The key PK metrics of interest were the differences in peak and trough paliperidone exposure between PP3M and PP1M at steady state with the three dose multipliers (Table 1). The multiplier of 3:1 predicted median paliperidone PP3M peaks and troughs that were lower than from PP1M, which could lead to lower efficacy for PP3M relative to PP1M. The dose multiplier of 3.5:1 offered an overall balance of peaks and troughs between the two formulations (i.e. the paliperidone peaks with PP3M were similar or slightly higher than those from PP1M), and the paliperidone troughs with PP3M were somewhat lower than those with PP3M. Simulated paliperidone concentrations with PP3M were more consistent with PP1M with a 3.5:1 multiplier, which could potentially lead to improved PP3M efficacy over the 3:1 multiplier. Finally, the dose multiplier of 4:1 in the simulations led to simulated higher peaks that were not bracketed by the highest tolerated doses of oral paliperidone‐ER. Moreover, the larger injection volume for 600 mg eq. dose of PP3M could preclude deltoid dosing with a 4:1 multiplier. Thus, based on this bridging exercise, the dose multiplier of 3.5:1 was recommended for the Phase III studies to ensure an acceptable balance of benefit vs. safety and tolerability risks when patients would be transitioned from PP1M to PP3M.
All PP3M doses, determined by a 3.5‐fold scaling of the prior PP1M dose, were generally tolerable in the Phase III studies 7, 8. The 3.5 dose multiplier led to observed plasma paliperidone concentrations that were consistent with those observed for PP1M (Figure 3) over a 3‐month period and were also in excellent agreement with the prospective predictions (Figure 2). This similarity in observed exposures between the two formulations is expected to be a major determinant of the equivalent efficacy between the two products. The Kaplan‐Meier estimate of the difference between PP3M and PP1M for patients who remained relapse free was 1.2% (95% confidence interval: −2.7%, 5.1%), with a lower confidence bound higher than the pre‐specified non‐inferiority margin of −15%. This verified the assumption that attaining similar concentrations of plasma paliperidone with PP3M (compared to PP1M) would preserve efficacy despite increasing the dosing interval from monthly with PP1M, to quarterly with PP3M. Safety findings for PP3M observed during the DB phase of Phase III studies were consistent with those observed in other clinical trials with paliperidone, and no new safety signals were detected 7, 8.
Figure 3.
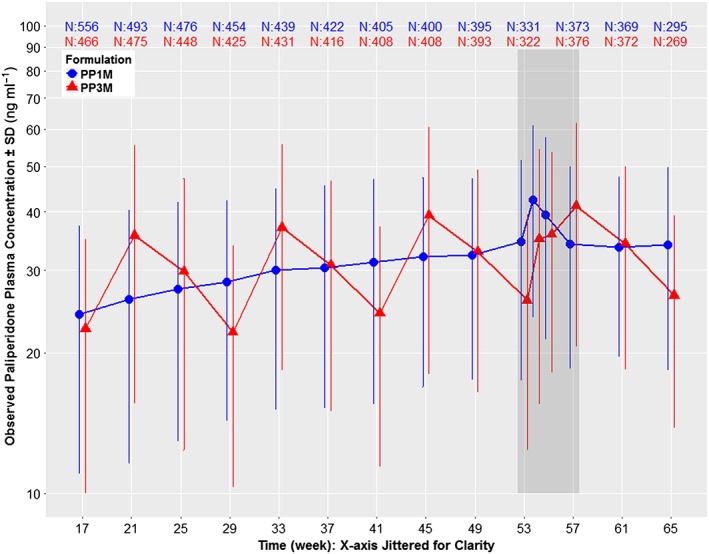
Dose normalized (100 mg‐eq. for PP1M and 350 mg‐eq. for PP3M) mean ± standard deviation (SD) semi‐logarithmic observed plasma concentration–time profiles of paliperidone comparing PP1M vs. PP3M in Phase III. Data are normalized to 100 mg‐eq. for PP1M and 350 mg‐eq. for PP3M as these two strengths represent the median doses for the two formulations in Phase III. The study design was as follows: open label phase with 150 mg‐eq. PP1M on day 1, 100 mg‐eq. PP1M on day 8, flexible dose of 50, 75, 100 or 150 mg‐eq. PP1M on week 5, week 9 and week 13 based on patient/physician preference, followed by a DB phase starting at week 17 with a fixed dose of PP3M (175, 263, 350 or 525 mg‐eq.) or PP1M (50, 75, 100 or 150 mg‐eq.). Data are presented only for the DB phase since dosing was fixed during this period, which allows dose normalization. The time period indicated by the dark grey shaded area between weeks 53 and 57 represents a phase of semi‐intensive PK sampling around steady‐state. Other than this semi‐intensive period, the PP1M samples were all collected at trough, but PP3M samples were not all trough
Discussion
This report highlights the use of a pharmacometric bridging strategy to prospectively select the dose of PP3M for Phase III clinical trials, by leveraging the knowledge from PP1M experience, and using only limited single‐dose PP3M Phase I data without conducting a Phase II dose‐finding study. The validation of the pharmacometric bridging strategy is based upon the close agreement between the prospective predictions and observed Phase III results, which demonstrated a desirable and predictable PK profile for PP3M, as well as satisfactory efficacy and safety profiles. The once‐every‐3‐month dosing of PP3M is expected to be an advantage with respect to convenience, and for treatment of patients with schizophrenia who have difficulty with medication adherence 9.
The PP3M dosing interval raised significant development challenges. For example, single‐dose PK studies performed in schizophrenic patients (enrolment period ~1 year) required a PK follow‐up of 18 months 3. Conducting a typical programme with Phase I, II and III studies would have resulted in a long development path and significant costs. To expedite the development process, the PK model developed for PP1M 2 was updated to assist in the determination of the PP3M dose multiplier. This approach accelerated Phase III start‐up using partial single‐dose PK data, and further allowed the sponsor to investigate the 525 mg‐eq. dose in an additional Phase I panel. Applying modelling principles contributed to a lean development programme, which enabled the characterization of multiple‐dose PK directly in the Phase III studies. This strategy circumvented the need for a separate multiple‐dose PK study prior to Phase III, which would have delayed the start of Phase III and medication access for patients by several years. We estimate that this model‐based drug development approach reduced development time by 3–5 years. This study illustrates the successful application of a pharmacokinetic‐based bridging strategy guided by model‐based analysis, in lieu of conducting clinical studies, which can potentially be applied to other drug development programmes exploring novel formulations.
Registration
The studies on which these analyses are based are registered at www.clinicaltrials.gov (NCT01559272 and NCT01515423).
Previous publication
The primary data from the Phase I 3 and Phase III 7, 8 studies were previously published. The prospective prediction and validation data presented herein have not been presented or published previously.
Competing Interests
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: all authors had support from Janssen Research & Development for the submitted work; no other relationships or activities that could appear to have influenced the submitted work. All authors are employees of Janssen Research & Development, and all authors except Dr. Russu own Johnson & Johnson stocks.
The authors would like to thank Ms. Theresa Carneiro for assistance with manuscript formatting and submission.
Contributors
All authors met International Council of Medical Journal Editors criteria and all those who fulfilled those criteria are listed as authors. All authors had access to the study data, provided direction and comments on the manuscript, and made the final decision about where to publish these data and approved the final draft submission to the journal. Dr. Samtani provided the initial concept, contributed to interpretation of the data, performed the pharmacometric analysis, and developed the first draft of the manuscript; all authors contributed to the further development. Drs. Nandy, Vermeulen and Russu oversaw the pharmacometric analyses and provided interpretation of these data. Drs. Hough, Gopal, Savitz, Remmerie and Ravenstijn contributed to study design, analysis and interpretation of these data. Dr. Baum provided substantial contribution to the conception of the publication, helped draft the work and revise it critically for important intellectual content. Dr. D'hoore led the complex formulation development from initiation through all phases of development. All authors provided intellectual contributions to the development of this manuscript.
Samtani, M. N. , Nandy, P. , Ravenstijn, P. , Remmerie, B. , Vermeulen, A. , Russu, A. , D'hoore, P. , Baum, E. Z. , Savitz, A. , Gopal, S. , and Hough, D. (2016) Prospective dose selection and acceleration of paliperidone palmitate 3‐month formulation development using a pharmacometric bridging strategy. Br J Clin Pharmacol, 82: 1364–1370. doi: 10.1111/bcp.13050.
References
- 1. Lalonde RL, Kowalski KG, Hutmacher MM, Ewy W, Nichols DJ, Milligan PA, et al. Model‐based drug development. Clin Pharmacol Ther 2007; 82: 21–32. [DOI] [PubMed] [Google Scholar]
- 2. Samtani MN, Vermeulen A, Stuyckens K. Population pharmacokinetics of intramuscular paliperidone palmitate in patients with schizophrenia: a novel once‐monthly, long‐acting formulation of an atypical antipsychotic. Clin Pharmacokinet 2009; 48: 585–600. [DOI] [PubMed] [Google Scholar]
- 3. Ravenstijn P, Remmerie B, Savitz A, Samtani MN, Nuamah I, Chang CT, et al. Pharmacokinetics, safety, and tolerability of paliperidone palmitate 3‐month formulation in patients with schizophrenia: a phase‐1, single‐dose, randomized, open‐label study. J Clin Pharmacol 2016; 56: 330–339. [DOI] [PubMed] [Google Scholar]
- 4. Kramer M, Litman R, Hough D, et al. Paliperidone palmitate, a potential long‐acting treatment for patients with schizophrenia. Results of a randomized, double‐blind, placebo‐controlled efficacy and safety study. Int J Neuropsychopharmacol 2010; 13: 635–647. [DOI] [PubMed] [Google Scholar]
- 5. Samtani MN, Gopal S, Gassmann‐Mayer C, Alphs L, Palumbo JM. Dosing and switching strategies for paliperidone palmitate: based on population pharmacokinetic modelling and clinical trial data. CNS Drugs 2011; 25: 829–845. [DOI] [PubMed] [Google Scholar]
- 6. Cleton A, Rossenu S, Crauwels H, Berwaerts J, Hough D, Gopal S, et al. A single‐dose, open‐label, parallel, randomized, dose‐proportionality study of paliperidone after intramuscular injections of paliperidone palmitate in the deltoid or gluteal muscle in patients with schizophrenia. J Clin Pharmacol 2014; 54: 1048–1057. [DOI] [PubMed] [Google Scholar]
- 7. Berwaerts J, Liu Y, Gopal S, Nuamah I, Xu H, Savitz A, et al. Efficacy and safety of the 3‐month formulation of paliperidone palmitate vs. placebo for relapse prevention of schizophrenia: a randomized clinical trial. JAMA Psychiatry 2015; 72: 830–839. [DOI] [PubMed] [Google Scholar]
- 8. Savitz A, Xu H, Gopal S, Nuamah I, Ravenstijn P, Janik A, et al. Efficacy and safety of paliperidone palmitate 3‐month formulation for patients with schizophrenia: A randomized, multicenter, double‐blind, noninferiority study. Int J Neuropsychopharmacol 2016; 19: pii: pyw018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Samtani MN, Sheehan JJ, Fu DJ, Remmerie B, Sliwa JK, Alphs L. Management of antipsychotic treatment discontinuation and interruptions using model‐based simulations. Clin Pharmacol 2012; 4: 25–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gopal S, Gassmann‐Mayer C, Palumbo J, Samtani MN, Shiwach R, Alphs L. Practical guidance for dosing and switching paliperidone palmitate treatment in patients with schizophrenia. Curr Med Res Opin 2010; 26: 377–387. [DOI] [PubMed] [Google Scholar]
- 11. Magnusson MO, Samtani MN, Plan EL, Jonsson EN, Rossenu S, Vermeulen A. Population pharmacokinetic modeling of paliperidone palmitate 3‐month formulation. PAGE. Abstracts of the Annual Meeting of the Population Approach Group in Europe. Available at http://www.page‐meeting.org/?abstract=3408 (last accessed 8 July 2016).