

Abstract
The mammalian target of rapamycin (mTOR) pathway is an highly conserved signal transduction axis involved in many cellular processes, such as cell growth, survival, transcription, translation, apoptosis, metabolism, motility and autophagy. Recently, this signalling pathway has come to the attention of the scientific community owing to the unexpected finding that inhibition of mTOR by rapamycin, an antibiotic with immunosuppressant and chemotherapeutic properties, extends lifespan in diverse animal models. Moreover, rapamycin has been reported to rescue the cellular phenotype in a progeroid syndrome [Hutchinson–Gilford Progeria syndrome (HGPS)] that recapitulates most of the traits of physiological ageing. The promising perspectives raised by these results warrant a better understanding of mTOR signalling and the potential applications of mTOR inhibitors to counteract ageing‐associated diseases and increase longevity. This review is focused on these issues.
Keywords: ageing, Hutchinson–Gilford Progeria syndrome (HGPS), lamin A, mTOR inhibitors, rapamycin, progeria‐related diseases
Ageing and progeria‐related diseases
Ageing is a physiological process aimed at the homeostasis of tissues through removal of cells bearing damaged genome sections or nonfunctional organelles. The advantage of ageing is that the neoplastic transformation of tissues and organs is avoided. The side effects of ageing include a loss of stem cells and a reduction in extracellular components, which are also associated with inflammatory processes contributing to tissue degeneration. The ageing traits are recapitulated in the majority of the so‐called premature ageing (progeroid) syndromes, which are genetic diseases of variable severity, with onset in childhood or in the teens, and are described briefly in this section. Of note, all of the known progeroid syndromes are caused by defects in proteins that regulate chromatin dynamics and genome stability, from lamins to ReqQ helicases up to proteins directly involved in DNA damage repair. The role of mammalian target of rapamycin (mTOR) and mTOR inhibitors in the affected pathways is discussed in the section on mTOR molecular pathways in ageing.
LMNA‐linked progeroid syndromes
Progeric laminopathies are syndromic diseases associated with defects of the nuclear lamina protein lamin A/C. They represent a paradigm of age‐related diseases as they encompass most of the disorders linked to normal ageing. Hutchinson–Gilford Progeria syndrome (HGPS) is caused by heterozygous mutations of the LMNA gene encoding lamin A and lamin C. Patients are healthy at birth but rapidly develop a senescent phenotype, starting at a few months of age, and are typically affected by ageing disorders, including hair loss, articular defects, skin atrophy and rigidity, atherosclerosis and coronary heart disease leading to premature death, in the first or second decade. The recurrent mutation in HGPS patients is the silent G608G LMNA mutation, which activates a cryptic splice site, triggering the production of a truncated and farnesylated precursor of lamin A called progerin. Progerin is toxic to cells and causes nuclear dysmorphism and a severe loss of heterochromatin 1, mislocalization or loss of chromatin‐associated proteins such as the DNA‐bridging factor barrier‐to‐autointegration factor (BAF) and the DNA damage repair proteins poly(ADP‐ribose) polymerase 1 (PARP1) and p53 binding protein 1 (53BP1) 2, 3, 4, and accumulation of irreparable DNA damage 5. The same effects are observed in other progeroid syndromes featuring accumulation of farnesylated prelamin A, such as mandibuloacral dysplasia type A and B (MADA and MADB, respectively) 6, 7, 8, 9 and atypical Werner syndrome (A‐WS) 10, 11. MADA is a rare disease characterized by growth retardation, bone resorption at specific sites (including the clavicles, phalanges and mandible), mottled cutaneous pigmentation, skin rigidity, partial lipodystrophy and insulin resistance. Patients develop premature ageing traits in the first or second decade. Similar clinical signs are also observed in A‐WS and atypical progeria syndrome (APS), owing to mutations in the lamin A/C rod domain; these conditions do not necessarily feature accumulation of prelamin A and their pathogenetic pathways are still unclear 12. In all premature ageing syndromes caused by mutations in the LMNA gene, the central nervous system is spared, owing to physiological downregulation by microRNA‐9 controlling lamin A expression and its splicing isoform progerin in brain tissues 13, 14.
Cells from progeroid laminopathies are not only the best experimental model in which to test potential therapeutic approaches to these diseases, but also represent a powerful model for the study of the senescent phenotype associated with age‐related disorders. However, the involvement of lamin in mechanisms that favour longevity has been determined by studying cells and tissues from very old healthy subjects. In a recent study 15, it was demonstrated that the lamin A precursor (prelamin A) plays a key role in healthy ageing, as a master regulator of the recruitment of nuclear factors implicated in genome stability. It has also been demonstrated that the nuclear envelope acts as a sensor of stress conditions and drives chromatin dynamics (heterochromatin decondensation, recruitment of 53BP1, rapid repair of damaged DNA) aimed at cell survival and genome maintenance 15. Exacerbation of lamina remodelling as it occurs in progeroid laminopathies elicits the opposite, and deleterious, effects, mostly because of the accumulation of toxic prelamin A 4, 8, 16. Thus, comparative analysis of lamin A and prelamin A role in normal and pathological ageing processes may give new and relevant insights into the understanding of ageing pathways, including those involving mTOR signalling and autophagy, as detailed below.
Nucleotide excision repair (NER)‐linked progeroid syndromes
Other diseases featuring premature ageing traits are associated with defects in the DNA repair machinery caused by mutations in genes encoding NER proteins. NER is a multistep mechanism able to identify and restore nucleotidic changes due to ultraviolet (UV) rays or chemical compounds, modifying DNA structure 17. Mutations occurring to the proteins involved in this machinery are responsible for the onset of genetic disorders, and all feature in the development of cancers and increased sensitivity to light.
Xeroderma pigmentosum (XP) is an autosomal recessive genetic disorder involving eight gene variants, all featuring an impaired ability to repair cellular DNA damaged by UV light exposure from the sun. The signs of XP usually appear in infancy or early childhood, when affected children develop severe sunburn after spending a few minutes in the sun. People with XP have a greatly increased risk of developing multiple basal cell carcinomas (basaliomas) and other skin malignancies on the face, lips and eyelids during their lifetime. Death occurs early, owing to metastatic malignant melanoma and squamous cell carcinoma 18.
XP mutations can occur in the following genes: damage‐specific DNA binding protein 2 (DDB2), DNA excision repair crosscomplementation group 2, ‐3, ‐4, ‐5 (ERCC2, ERCC3, ERCC4, ERCC5), XPA and XP5 19. The products of these genes are all involved in the DNA damage repair pathway, based on the NER mechanism.
Cockayne syndrome (CS) is a rare autosomal recessive neurodegenerative progeroid pathology featuring clear premature ageing and severe light sensitivity 18. It is characterized by growth failure, impaired development of the nervous system, photosensitivity, eye disorders and premature ageing. It is often associated with a group of disorders called leukodystrophies, which are conditions characterized by the degradation of neurological white matter. Two types of CS have been described and occur as a result of mutations of different genes. Type I CS occurs following mutations in the DNA excision repair crosscomplementation group gene 8 (ERCC8), encoding the CS‐A protein. These mutations cause alternative splicing of the corresponding pre‐mRNA, leading to a nonfunctional protein 20. Type II CS is caused by mutations in the ERCC6 gene, encoding the CS‐B protein 21.
Trichothiodystrophy (TTD) is a rare autosomal recessive inherited disorder mostly characterized by brittle hair, due to a general sulfur deficiency, and by intellectual impairment 18. Mild cases of TTD may involve only the hair. However, more severe cases may cause delayed development, significant intellectual disability and recurrent infections; severely affected individuals may survive only up to infancy or early childhood. Approximately half of all patients with TTD have photosensitivity, which divides the classification of the disease into syndromes with or without photosensitivity 22, 23. In contrast to XP, patients affected by TTD do not show a higher risk of developing skin cancer.
RecQ‐associated progeria‐related syndromes
The family of RecQ proteins groups enzymes with ATP‐dependent helicase activity, required during the repair of damaged DNA to unwind the mutated double strand. Their actions prevent deleterious recombination and genomic instability 24; therefore, mutations in the corresponding genes are associated with an increased predisposition to cancer and ageing phenotypes.
Bloom syndrome (BS) is a rare autosomal recessive progeroid syndrome characterized by a striking genomic instability associated with a high risk of cancer. Its most prominent features include short stature and a rash on all areas of the skin that are exposed to the sun, developed in early age. Other clinical features include a high‐pitched voice; distinct facial features, including a long, narrow face, micrognathia, and prominent nose and ears; and changes to skin pigmentation, including hypopigmented and hyperpigmented areas 25. The genetic cause of BS is mutations in the RecQ2 gene, also known as the BLM gene, encoding the BS protein, which is one of the five RecQ DNA helicases identified so far. Two other RecQ‐linked genetic diseases are caused by defects in WRN and RecQ4 genes where WRN encodes for a protein responsible, when mutated for a progeroid syndrome: Werner's syndrome 26.
Werner's syndrome (WS) is a genetic disorder due to mutations in the WRN gene, which encodes a RecQ helicase called WRN protein. The clinical phenotype is almost identical to that observed in A‐WS syndrome linked to lamin A mutations 27. Affected individuals can exhibit growth retardation, short stature and signs of premature ageing, such as premature greying and loss of hair, wrinkles, a beaked nose, skin atrophy, loss of fat tissues and atherosclerosis 28. Interestingly, mutation of the WRN gene may cause a decrease in the stability of the corresponding mRNA. Mutations may also lead to the loss of the nuclear localization signal of the protein, leading to an aberrant accumulation of the WRN protein in the cytoplasm.
RecQ4 mutations cause Rothmund–Thomson syndrome (RTS). RTS patients exhibit some premature ageing symptoms similar to those of WS, including skin hyperpigmentation and widened blood capillaries, known as poikiloderma. Moreover, similar to progeric laminopathies, RTS patients present abnormal bone turnover but, unlike those affected by these syndromes, are predisposed to osteosarcoma.
DNA polymerase‐linked progeroid syndromes
The variant form of XP, known as XP‐V, is caused by mutations in the POLH (DNA polymerase eta) gene, which encodes a DNA polymerase; its mutation leads to an overall increase in UV‐dependent DNA damage, which ultimately causes the symptoms of XP 19.
Finally, mutations in the DNA polymerase delta p125 catalytic subunit (POLD1) have been recently linked to a form of mandibuloacral dysplasia, the mandibular hypoplasia, deafness, progeroid features and lipodystrophy syndrome (MDPL), which, from the clinical point of view, is identical to that caused by LMNA mutations but also features sensorineural deafness 29, 30. Human POLD1 modulates cell cycle progression and DNA damage repair 31. POLH, also known as DNA polymerase η, is involved in translesion DNA synthesis in the UV‐induced DNA damage response and acts downstream of the WRN protein 32.
mTOR signalling
mTOR pathway
The mTOR pathway is a highly conserved signal transduction axis involved in many cellular processes, such as cell growth, survival, transcription, translation, apoptosis, metabolism, motility, autophagy and ageing 33. This signalling pathway may be activated by various cellular stimuli, such as growth factors and cytokines, nutrients (e.g. glucose, amino acids) and inflammatory stimuli. Thus, considering the crucial role played by mTOR signalling in several physiological processes, it is evident that dysregulation of this cascade could have dramatic consequences. Indeed, it is well established that alterations in its activity are linked to the pathogenesis of a variety of human diseases, such as metabolic disorders (obesity and type 2 diabetes), cardiovascular and neurodegenerative diseases, age‐related disorders and cancer.
mTOR is a serine/threonine (Ser/Thr) kinase which belongs to the phosphoinositide kinase‐related family of protein kinases (PIKK) 34. In response to these growth signals, mTOR regulates cell growth and proliferation positively by promoting many anabolic processes and limiting catabolic processes such as autophagy 35 (Figure 1). mTOR collects input from several signal transduction networks, such as the phosphatidylinositol (PI) 3‐kinase (PI3K)/Akt and the Ras/Raf/mitogen‐activated protein kinase kinase (MEK)/extracellular signal‐regulated kinase (ERK). Moreover, recent studies have highlighted that mTOR also responds to inputs via the WNT and liver kinase B1 (LKB1)/AMP‐activated protein kinase (AMPK) signalling pathways 36. AMPK functions as a key sensor of intracellular energy balance: under metabolic stress conditions, LKB1 activates AMPK, which in turn directly inhibits mTOR 37.
Figure 1.
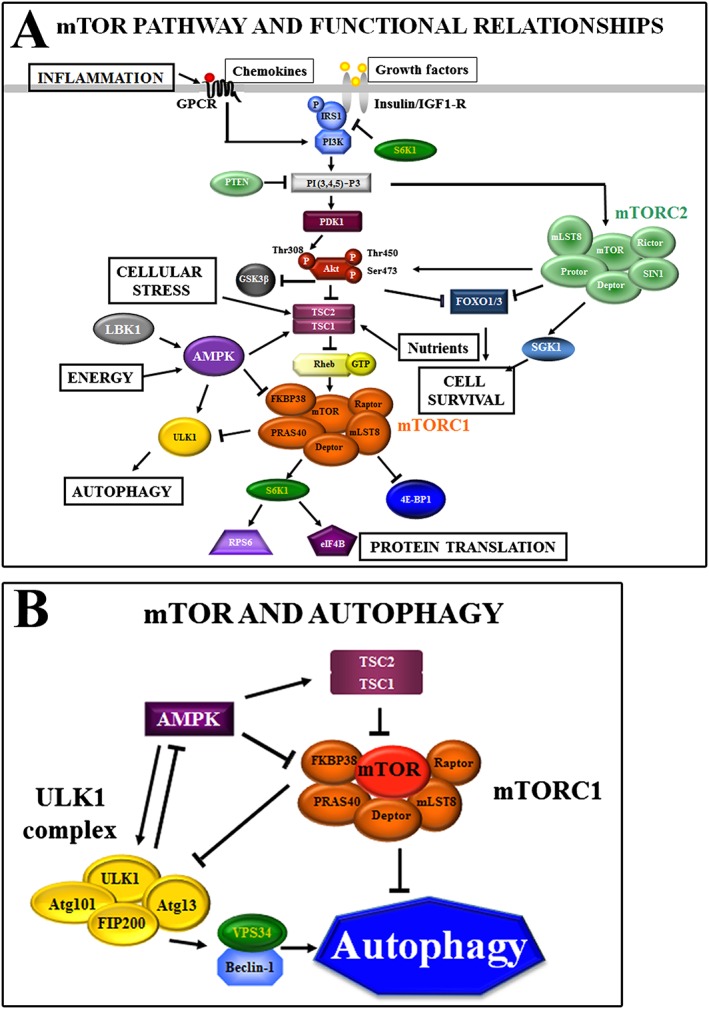
Cellular signalling upstream and downstream of mammalian target of rapamycin (mTOR) complexes. (A) mTOR pathways and functional relationships. mTOR forms two multiprotein complexes – mTOR complex (mTORC) 1 and mTORC2. The classical mTORC1‐positive inputs are growth factors, chemokines, nutrients and cell energy balance. Growth factors stimulate mTORC1 through the phosphatidylinositol (PI) 3‐kinase (PI3K)/Akt signalling pathway. Akt phosphorylates tuberous sclerosis complex 2 (TSC2). TSC2 is a GTPase‐activating protein that functions in association with TSC1 to inactivate the small G protein Rheb, which, in turn, upregulates mTORC1 activity. mTORC1 activity is required for the phosphorylation and subsequent activation of ribosomal S6 kinase 1 (S6K1), which regulates the elongation step of protein translation, ribosomal protein S6 (RPS6) and eukaryotic translation initiation factor 4B (eIF4B), ultimately promoting the initiation of translation and elongation. mTORC1 also phosphorylates and inactivates the translation inhibitor eIF4E‐binding protein 1 (4E‐BP1), which inhibits cap‐dependent translation. The mTOR pathway may be also regulated by the LKB1/AMP‐activated protein kinase (AMPK) pathway. AMPK phosphorylates TSC2, activating the TSC1/TSC2 complex and thus repressing mTORC1 activity. Moreover, AMPK phosphorylates Raptor, inducing Raptor and mTORC1 disassembly/inhibition. mTORC2 regulates cell survival through serum‐ and glucocorticoid‐activated kinase 1 (SGK1) and Akt. mTORC2 phosphorylates Akt at Ser473, priming Akt for further phosphorylation by phosphoinositide‐dependent kinase 1 (PDK1) at the Thr308 residue. Loss of phosphorylation at the Ser473 site, however, affects only some Akt substrates, such as Forkhead Homeobox type O (FOXO) transcription factors, but not TSC2, in response to growth factor signalling. mTORC2 also associates with actively translating ribosomes to phosphorylate cotranslationally Akt (at Thr450), which prevents the ubiquitination and degradation of Akt. mTORC2 is involved in the spatial control of cell growth via cytoskeletal regulation. (B) mTOR and autophagy. Under nutrient‐rich conditions, mTOR associates with and inhibits the UNC51‐like kinase (ULK) complex by phosphorylating autophagy‐related (ATG) 13 and ULK1. By contrast, stress signals or AMPK activation lead to mTORC1 dissociation from ULK1 and to the activation of the ULK complex, thereby triggering the autophagy machinery. Arrows indicate activating events, whereas perpendicular lines indicate inhibitory events. FIP200, focal adhesion kinase family‐interacting protein of 200 kD; FKBP38, FK‐506‐binding protein; GPCR, G protein‐coupled receptor; IGF1‐R, insulin‐like growth factor 1 receptor; GSK3β, glycogen synthase kinase 3β; IRS1, insulin receptor substrate 1; LKB1, liver kinase B1; mLST8, mammalian lethal with sec‐13 protein 8; PI(3,4,5)‐P3, phosphatidylinositol (3,4,5)‐trisphosphate; PRAS40, proline‐rich Akt substrate of 40‐kDa; PTEN, phosphatase and tensin homologue; SIN1, stress‐activated protein kinase‐interacting protein 1; VPS, vacuolar protein sorting
mTOR is the catalytic subunit of two functionally and structurally distinct multiprotein complexes: mTOR complex (mTORC) 1 and mTORC2, both of which are characterized by their different partner proteins and their substrate specificity 34.
mTORC1 is composed of mTOR, the regulatory associated protein of mTOR (Raptor), mammalian lethal with sec‐13 protein 8 (mLST8), proline‐rich Akt substrate of 40‐kDa (PRAS40), FK‐506‐binding protein (FKBP) 38, and dep domain containing mTOR‐interacting protein (Deptor) 34. The best known function of mTORC1 is the regulation of translation. It can phosphorylate components of the protein synthesis machinery, including p70 ribosomal S6 kinase 1 (S6K1) and the translation inhibitor, eukaryotic translation initiation factor 4E‐binding protein 1 (4E‐BP1) 38, leading to active translation of mRNAs involved in ribosome biogenesis 39.
mTORC2 comprises rapamycin‐insensitive companion of mTOR (Rictor), mLST8, stress‐activated protein kinase‐interacting protein 1 (SIN1), protein observed with Rictor (Protor) and Deptor, and is generally described as being insensitive to rapamycin/rapalogs. It is mainly activated by growth factors through PI3K/Akt, and controls several downstream AGC kinases such as Akt itself (phosphorylation on Ser473), serum‐ and glucocorticoid stimulated kinase (SGK) and protein kinase Cα (PKCα). It regulates cell proliferation but is also involved in the spatial control of cell growth via cytoskeleton regulation 40. Recently, it has been demonstrated that mTORC1, via activation of ribosome biogenesis and inhibition of autophagy‐mediated ribosome turnover, indirectly controls mTORC2 41 (Figure 1).
mTOR and autophagy
Through the phosphorylation of other targets, mTORC1 triggers metabolic changes such as mitochondrial biogenesis, the promotion of glycolysis, lipid biogenesis and the suppression of autophagy 33. Indeed, mTORC1 is a negative regulator of autophagy, a process required for the recycling of damaged organelles and for cellular adaptation to nutrient starvation, growth factor withdrawal and oxidative stress 35. Once activated, the autophagic machinery sequesters cytoplasmic components and, fusing with lysosomes, leads to the degradation of cell components and recycling of cellular building blocks. mTORC1 transduces anti‐autophagic signals by binding to the UNC51‐like kinase (ULK) multiprotein complex, which is essential for the initial steps of autophagy. mTORC1 directly phosphorylates ULK1 (which may also be a target for Akt following insulin stimulation), which in turn may regulate autophagy independently from mTORC1 function 42. Conversely, during nutrient deprivation or pharmacological mTORC1 inhibition, mTORC1 dissociates from the ULK1 complex, leading to active forms of ULK1.
By contrast, AMPK is an important positive regulator of the autophagic machinery. Under metabolic stress conditions, LKB1 activates AMPK, which directly phosphorylates ULK1 at multiple sites, thus upregulating ULK1 activity, and activates tuberous sclerosis complex 2 (TSC2), an indirect inhibitor of mTORC1 activity 43.
Following mTORC1 inhibition, derepressed ULK1, by binding to ATG14L (ATG ‐Autophagy related‐14 Like), is recruited to a molecular platform composed of vacuolar protein sorting 34 (Vps34) and beclin‐1. This leads to the phosphorylation of beclin‐1 on Ser 14, and activation of Vps34. VPS34 is the only class III PI3K member and plays an important role in regulating membrane trafficking and autophagy, releasing PI 3‐phosphate at the nascent autophagosome, the phagophore 44. In this condition, microtubule‐associated protein 1 light chain 3 (LC3), a structural protein found in the cytoplasm in its precursor form (LC3I) is cleaved, lipidated (i.e. coupled to phosphatidylethanolamine) and converted into its active autophagosomal, membrane‐bound form, LC3II 45.
Potential therapeutic effects of mTOR inhibitors
Considering the numerous functions played by mTORC1 and mTORC2 in several physiological processes, it is evident that dysregulation of this axis could have important negative effects. Indeed, upregulation of mTOR signalling has been found to be involved in the onset and progression of many human diseases, and mTOR is therefore regarded as a key target for innovative therapeutic strategies. In fact, several mTOR inhibitors have been released by pharmaceutical companies. Three classes of mTOR inhibitors have been developed: rapamycin/rapalogs that are allosteric mTORC1 inhibitors; ATP‐competitive, ‘active‐site’ mTORC1/mTORC2 inhibitors that target the catalytic site of mTOR; and dual PI3K/mTOR inhibitors that target both PI3K and mTORC1/mTORC2 (Figure 2).
Figure 2.
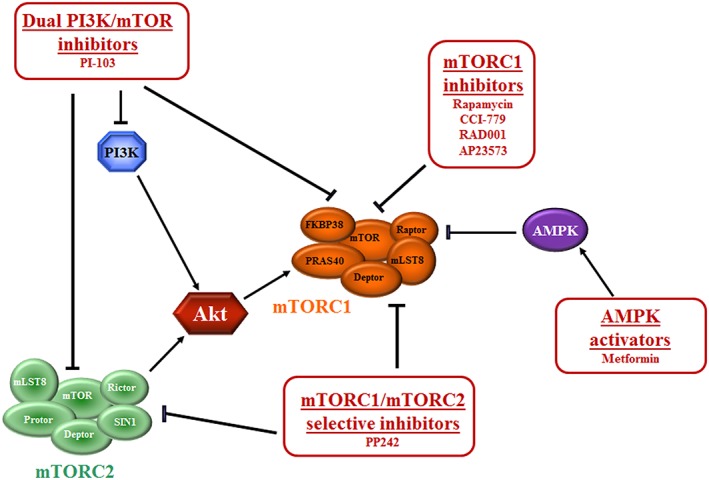
Mammalian target of rapamycin (mTOR) inhibitors. mTORC1 inhibitors are allosteric inhibitors that lead to the dissociation of Raptor from mTOR complex (mTORC) 1 and loss of contact between mTORC1 and its substrates. Dual phosphatidylinositol 3‐kinase PI3K/mTOR inhibitors target both PI3K and mTORC1/mTORC2, acting on the catalytic sites of PI3K and mTOR. mTORC1/mTORC2‐selective inhibitors target the catalytic site of the enzyme, thus acting on both mTORC1 and mTORC2. AMP‐activated protein kinase (AMPK) activator has a negative effect on mTORC1. Arrows indicate activating events; perpendicular lines indicate inhibitory events. FKBP38, FK‐506‐binding protein; mLST8, mammalian lethal with sec‐13 protein 8; PRAS40, proline‐rich Akt substrate of 40‐kDa
Rapamycin
Rapamycin (also known as sirolimus) is a natural compound produced by the bacterium Streptomyces hygroscopicus that acts as an allosteric mTORC1 inhibitor 46. It forms a gain‐of‐function complex with 12‐kDa FKBP12, which binds to the FKBP12/rapamycin‐binding (FRB) domain of mTOR only when mTOR is associated with other components of mTORC1. This complex leads to the dissociation of Raptor and loss of contact between mTORC1 and its substrates 47. Therefore, rapamycin does not directly target the mTOR catalytic site and does not affect mTORC2 activity, although it has been demonstrated that long‐term exposure can also affect mTORC2 assembly 48, 49.
The discovery of rapamycin immediately raised great interest in the scientific community for its numerous proprieties, e.g. as a powerful antibiotic, antiproliferative and immunosuppressant 50. In 1991, rapamycin was approved by the US Food and Drug Administration (FDA) for the prophylaxis of renal transplant rejection 51 and as a chemotherapeutic agent against renal carcinoma. Moreover, rapamycin has been used to inhibit restenosis after the implantation of stents during coronary angioplasty 52, 53, and it entered numerous clinical trials for conditions such as lymphangioleiomyomatosis 54 and tuberous sclerosis.
Rapalogs
The limitations in the solubility and pharmacokinetic properties of rapamycin led to the commercial development of new mTOR inhibitors, such as semi‐synthetic rapamycin analogues, named rapalogs or active site inhibitors. Rapalogs have an improved bioavailability when compared with rapamycin, and include CCI‐779 (temsirolimus or torisel), RAD001 (everolimus) and AP23573 (ridaforolimus), which have been, and continue to be, tested in a wide range of clinical trials for numerous chronic disease indications.
CCI‐779 is a rapamycin ester derivative that was approved by the FDA for the treatment of advanced‐stage renal cell carcinoma in 2007. In Europe, it is also approved for the treatment of mantle cell lymphoma 55. Unfortunately, in patients affected by advanced renal cell carcinoma, CCI‐779 has strong side effects, including hyperglycaemia, hyperlipidaemia, anaemia, peripheral oedema and dermatological adverse events 56.
RAD001 is another rapamycin analogue that has been approved by the FDA for patients affected by various malignancies, including renal cell carcinoma 57, subependymal giant cell astrocytoma 58 and progressive neuroendocrine tumours of pancreatic origin 59. The orally available RAD001 is more efficacious than rapamycin as it has a higher affinity for FKBP12 60.
More recently, two other classes of mTOR inhibitors, which target the catalytic site of mTOR, have been developed to overcome the side effects of rapalog ‘active‐site’ mTORC1/mTORC2 inhibitors.
PI3K and mTOR belong to the PIKK family of kinases, and share high sequence homology in their catalytic domains. Dual PI3K/mTOR inhibitors target the active sites of both the PI3K and mTOR, inhibiting the PI3K/Akt/mTOR pathway both upstream and downstream of Akt, thus avoiding the Akt activation following abolition of the mTORC1/S6K1/insulin receptor substrate 1 negative feedback loop, which is known to occur with rapalogs 61. The first compound of this class to be produced was the morpholinoquinazoline derivative PI‐103 62.
To reduce toxicity due to the use of these inhibitors, efforts were undertaken to design a new‐generation mTOR inhibitor (mTORC1/mTORC2‐selective inhibitor) that competes with ATP for binding to the kinase domain of mTOR, thus having a specific blocking effect on its kinase activity. In the same manner as with dual PI3K/mTOR inhibitors, acting on both mTOR complexes, mTORC1/mTORC2‐selective inhibitors display stronger effects than rapalogs in the treatment of a wide variety of cancers, and they might offer an efficient alternative to rapalogs. Their use also minimizes the reactivation feedback loops of Akt that are seen with rapalogs. The prototype of this class of drugs is PP242 63.
Recently, novel compounds that target mTOR indirectly are attracting attention for their better safety profiles, such as resveratrol (3,4′,5‐trihydroxy‐trans‐stilbene) and metformin (N,N‐dimethylbiguanide).
Resveratrol
Resveratrol is a natural polyphenol that shows numerous beneficial effects, acting as an antioxidant, anti‐inflammatory and anticancer drug. Moreover, it seems to have protective effects against a number of cardiovascular and neurodegenerative diseases 64. It has been reported that resveratrol can inhibit mTORC1 by blocking the interaction between Deptor and mTOR 65.
Metformin
Metformin belongs to the biguanide class of oral hypoglycaemic agents, which has been approved by the FDA for the treatment of type 2 diabetes since 1995. Intriguingly, metformin treatment has also been associated with a reduced risk of cancer 66 and of cardiovascular diseases 67. There is a growing body of evidence that metformin inhibits mTORC1 through a different mechanism, by activating AMPK. AMPK, in turn, blocks mTORC1 68 regulating it through Ras‐related GTP binding (Rag) GTPases 69 and indirectly activating regulated in development and DNA damage responses 1 (REDD1), a mTOR inhibitor that promotes TSC2 activity 70.
mTOR molecular pathways in ageing
Several processes downstream of mTOR signalling contribute to ageing, including the regulation of protein translation, autophagy, stem cell pool homeostasis, inflammation and cellular senescence (Figure 3).
Figure 3.
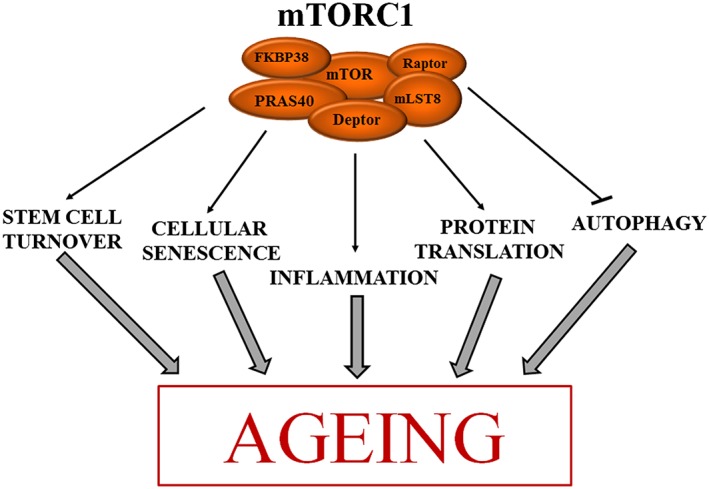
The mammalian target of rapamycin (mTOR) complex (mTORC1) signalling pathway contributes to ageing through various cellular processes. Active mTORC1 activates stem cell turnover, cellular senescence and protein translation (arrows), while it inhibits autophagy. These conditions contribute to the ageing of an organism. See text for details. FKBP38, FK‐506‐binding protein; mLST8, mammalian lethal with sec‐13 protein 8; PRAS40, proline‐rich Akt substrate of 40‐kDa
Protein translation
The inhibition of mTORC1‐dependent translation may extend lifespan by reducing the burden on the protein‐folding machinery, leading to improved protein quality. Reduced translation via knockout of S6K1 extends longevity and blocks the onset of age‐related pathologies 71. Moreover, genetic depletion of ribosomal proteins, as well as inhibition of translation initiation factors, can similarly extend lifespan in yeast and worms 72, 73.
Autophagy
Ageing is characterized by the accumulation of unprocessed material and by the decrease in autophagy. The anti‐ageing effects of rapamycin are also dependent on the induction of this catabolic mechanism in yeast, worms and flies 74, 75, 76. For instance, in Caenorhabditis elegans, a loss‐of‐function mutation in the insulin‐like growth factor (IGF) pathway causes autophagy, whereas inhibition of autophagy by mutation of essential Atg genes prevents the acquisition of longevity 77.
Stem cell pool turnover
mTORC1 is a key regulator of growth signals that drives the exit of tissue stem cells from quiescence. Stem cell activation contributes to tissue turnover but affects the stem cell pool, progressively eliciting stem cell niche depletion. Thus, rapamycin represents a tool for preserving the stem cell pool and, as a consequence, the functionality of tissues and organs over time. In support of this, six weeks of treatment with rapamycin in elderly mice improved the regenerative capacity of haematopoietic stem cells 78.
Regulation of the inflammatory response
Recent exciting advances in the study of the well‐defined mechanism of inflammaging 79 have suggested that tissue damage or stress trigger a specific secretory phenotype, starting from interleukin (IL) 1α production and nuclear factor‐kB activation 80. The environment elicited by cellular damage corresponds to a senescence‐associated secretory phenotype (SASP) and, unexpectedly, favours tissue repair and cellular senescence as a side effect 80, 81, 82. Cytokines produced during the inflammatory response signal through mTOR (Figure 3) either by activating PI3K, which targets mTORC2 complex, or by stimulating the p38/Erk pathway to activate mTORC1 83 Downstream of these biochemical pathways, cellular proinflammatory cytokines are mostly inhibited but ageing‐related molecules such as IL6 are activated 83. Thus, modulation of mTOR activity can affect inflammaging positively or negatively. It has been shown that rapamycin inhibits the expression of tumour necrosis factor and IL‐6, two main players in the ageing‐associated inflammatory response 83.
Cellular senescence
The mTOR pathway also drives cellular senescence 84, a cellular decline that follows development and maturation, characterized by loss of capacity to replicate without undergoing apoptosis and by a proinflammatory secretory phenotype (SASP) 85. In proliferating cells, mTOR drives cellular proliferation through activation of protein translation and modulation of nutrient sensing and the cellular response. Under stress conditions, the cyclin‐dependent kinase inhibitor p21 is upregulated to trigger cell cycle arrest and allow damage repair. In this context, mTOR contributes to converting irreversible cell cycle arrest to cellular senescence, an event referred to as geroconversion 86, 87. Geroconversion is a form of futile growth that occurs during cell cycle arrest. It leads to hypersecretory and hypertrophic cellular phenotypes and the activation of a specific inflammatory response, as also detailed above (Figure 3). In both human and rat cells, geroconversion 84 is suppressed by rapamycin and other rapalogs, maintaining cellular quiescence 88, 89. Recently, it has been demonstrated that low doses of dual mTORC1/mTORC2‐selective inhibitors (PP242 and Torin1) can also constrain cellular senescence 87.
mTOR inhibition to counteract ageing
There is an increasing interest in genetic and pharmacological mTOR inhibition aimed at extending longevity and improving the healthy lifespan.
The effects of mTOR inhibition in slowing down the ageing process have been demonstrated over the years in many animal models. Initial experimental data showed that reduced TOR activity in yeast, worms and flies may be sufficient to delay ageing 74, 90, 91.
Recent evidences suggested that mTOR plays the same important role in regulating lifespan in mammals. Indeed, Selman and colleagues demonstrated that deletion of S6K1 extends the median lifespan of mice and counteracts the onset of age‐related pathologies, such as bone, immune and motor dysfunction, as well as loss of insulin sensitivity 71. The effect on lifespan extension was also demonstrated in mice bearing heterozygous deletions of mTOR and mLST8, corroborating this hypothesis 49. Moreover, Wu and colleagues recently showed that, in a mTOR hypomorphic mouse model, there was an increase in median lifespan, associated with a reduction in the age‐dependent decline in tissue and organ functionality 92.
The specific effects of mTOR on lifespan have also been confirmed concurrently by pharmacological inhibitors. Rapamycin was the first mTOR inhibitor to be tested in mice, and was found to extend both mean and maximum lifespan 93.
In recent years, the National Institute of Ageing (NIA) Intervention Testing Program (ITP) embarked on a phase II study to test the possible clinical translational potential of rapamycin to delay ageing, and thus to extend the healthy lifespan in mice. Indeed, mTOR inhibition seems to be an innovative strategy for the treatment of a wide range of age‐associated conditions and diseases. The effects of rapamycin on lifespan extension have been reported when treatment is begun late in mouse life, at 20 months of age, in both genders 93. Slightly larger effects have been observed when the drug is administrated to young mice 94. Rapamycin's efficacy in mice has been also confirmed starting at four months of age and continued throughout life 95. It is interesting that rapamycin‐treated mice live three to four months longer (the equivalent of ten human years), with the same quality of life at the time of death, than mice that do not receive the drug 96.
Importantly, rapamycin delayed different hallmarks of ageing in mice, including cardiac hypertrophy, liver degeneration, and a decline in adrenal gland and endometrial function and tendon elasticity 97.
Various independent groups have reported that rapamycin also affects cognitive performance. Neff et al. revealed that learning and memory were improved after rapamycin treatment 98, while other independent groups found that rapamycin enhanced cognitive function in young adult mice and blocked age‐associated cognitive decline in older animals 99, 100. In addition, in transgenic mouse models of Alzheimer's disease (AD), rapamycin was able to delay the onset of the neurodegenerative disease, restoring memory 101, 102, 103.
In spite of the initial encouraging results on the anti‐ageing therapeutic potential of resveratrol in mice and primates 104, 105, recent studies have shown that this drug does not affect lifespan or longevity in male or female mice 94, or in a cohort of more than 800 people over the age of 65 years 106. Moreover, the use of resveratrol in progeric mice carrying an LMNA mutation did not improve the senescent phenotype 107.
With regard to metformin, evidence has shown that it extends healthy lifespan in worms 104, rats 108 and mice 105, 109, 110. In a promising NIA ITP study, long‐term treatment with low‐dose metformin in male mice, starting in middle age, showed a substantial extension of healthy lifespan and longevity 110.
Interestingly, a long‐term study in human patients, including the UK Prospective Diabetes Study, found that the treatment of diabetic patients with metformin reduced mortality from all causes, including diabetes‐related mortality, cancer and myocardial infarction 111, 112. All of these results are hopeful, given that metformin has been already demonstrated to be safe and effective in humans, and, thus, may represent a novel strategy to delay ageing and counteract age‐related diseases.
In mice, calorie restriction (CR) may counteract the onset of age‐related diseases such as stroke, diabetes, cancers, AD and Parkinson's disease (PD) 113. Clinical studies have also been carried out in humans, with promising results, showing that CR is linked to a decreased risk of cancer, diabetes and overall mortality 113. The positive effects of CR are attained, at least in part, through downregulation of the mTOR signalling pathway in yeast 114, worms and mice 115. Thus, mTOR inhibition by rapamycin or its analogues may mimic CR and counteract ageing processes.
mTOR inhibitors and progeria‐related disorders
Regulation of autophagy and protein accumulation
The importance of mTOR in ageing and age‐related diseases is mostly related to its function as a master regulator of autophagy. Increased basal autophagic activity has been reported in progeroid laminopathies 8, 116 and WS cells 117. Interestingly, as shown in Figure 4, increased autophagy observed in HGPS fibroblasts is coupled to a reduction in mTOR activity. Besides being related to alterations in mTOR signalling, the increased autophagy observed in progeria cells is also a response to the cellular accumulation of misfolded and/or aggregated proteins, typical of these cells 118. Although it is not an aspect common to all the progeroid syndromes studied so far, the mutated proteins responsible for the onset of the pathology, may be themselves targets of autophagic degradation. This has been demonstrated in MADA, for example, in which autophagy contributes, in part, to prelamin A degradation, thus reducing protein toxicity 8. However, progerin, the toxic prelamin A form that accumulates in HGPS, is minimally affected by the autophagic process under basal conditions 116, 119.
Figure 4.
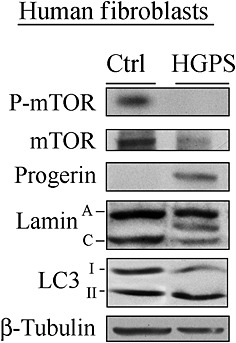
Undetectable mammalian target of rapamycin (mTOR) activity in Hutchinson–Gilford Progeria syndrome (HGPS) fibroblasts. HGPS fibroblasts at passage 20 were cultured and lysed, as described by Cenni et al 116. Immunoblotting was performed with the following antibodies: progerin (Enzo Life Sciences, Inc. Farmingdale, NY, USA.), phospho‐mTOR [P‐mTOR (Ser2448)], mTOR and microtubule‐associated protein 1 light chain 3 (LC3), respectively [#2971, #2972 and #4108 (Cell Signaling Technology Inc. Danvers, MA, USA)]. Lamin A/C (#6215, Santa Cruz Biotechnology, Inc. Dallas, TX, USA), β‐tubulin and flag (both from Sigma‐Aldrich, Saint Louis, MO, USA). The immunoblotted band between lamin A and lamin C bands corresponds to progerin
In many age‐related diseases, protein aggregate formation is a hallmark of the mutated cellular phenotype. Aside from the afore‐mentioned progeroid cells, neurodegenerative pathologies also show a clear accumulation of aggregates and highly insoluble structures both inside the cells and in the extracellular matrix, negatively affecting the regulation of the nervous connections 120. In AD, these aggregates consist of intracellular neurofibrillary tangles composed of hyperphosphorylated tau protein, and of extracellular plaques containing abnormally folded amyloid‐beta (Aβ) aggregates. Aβ is a short peptide obtained at the end of the pathological processing of the amyloid precursor protein, whose function is unclear but is thought to be involved in neuronal development. Aβ oligomers and plaques act by blocking proteasome function, inhibiting mitochondrial activity, altering intracellular calcium levels and stimulating inflammatory processes. Aβ also interacts with the signalling pathways that regulate the phosphorylation of the microtubule‐associated protein tau, which, in normal conditions, regulates axonal transport. Hyperphosphorylation of tau disrupts its normal function and leads to the accumulation of neurofibrillary tangles and toxic species of soluble tau. In addition, the degradation of hyperphosphorylated tau by the proteasome is inhibited by the actions of Aβ 121. In PD, the aggregates consist of α‐synuclein fibrils, which assemble to form lethal Lewy bodies in the nerve cells. In normal conditions, α‐synuclein is found mainly in the presynaptic terminals of neurones, where it regulates the neuronal Golgi apparatus and vesicle trafficking 122. Finally, in Huntington's disease (HD), toxic aggregates of mutated huntingtin (mHtt) are formed (see below). Wild‐type huntingtin is associated primarily with vesicles and microtubules, and is also involved in the upregulation of the expression of brain‐derived neurotrophic factor 123, 124.
mTOR inhibitors and lamin A/C‐dependent progeroid syndromes: HGPS and MADA
Our group has proved that, by promoting autophagy, rapamycin has a powerful negative effect on the expression level of progerin and farnesylated forms of prelamin A in both HGPS 4, 116, 119 and MADA cells 8. In 2011, we described that, by promoting the autophagic clearance of progerin and prelamin A, rapamycin treatment of HGPS fibroblasts rescues their chromatin phenotype, including histone methylation status and nuclear envelope protein distribution patterns 116. Importantly, rapamycin treatment did not affect lamin C protein levels but increased the relative expression of ZMPSTE24 (Zinc Metalloproteinase 24), which is the endoprotease involved in prelamin A maturation, suggesting that not only prelamin A degradation, but also prelamin A processing may be enhanced by drug treatment 116. Further, the results from Collins's laboratory showed that, by reducing progerin accumulation, the treatment of progeric cells with rapamycin reverses cellular phenotypes, such as nuclear blebbing, and delays the onset of cellular senescence 119.
Very recently, we demonstrated that all‐trans retinoic acid is synergistic with low‐dose rapamycin in reducing progerin accumulation in HGPS fibroblasts 4. This effect is obtained through transcriptional downregulation of LMNA and by mutated protein degradation, and elicits an improvement in heterochromatin organization and the DNA damage response, ultimately ameliorating cell cycle dynamics. These data are very promising, particularly because the low doses of the drugs required for this combined treatment avoid the potential side effects associated with chronic treatment.
Nuclear lamina turnover and protein modifications, e.g. occurring at cell division or during cellular differentiation, are mediated by several protein kinases that selectively lamin A/C and lamin B 125. For example, lamin A phosphorylation sites have been identified and related to myogenic differentiation 126. In addition, we have also reported that lamin A/C and nonfarnesylated prelamin A are phosphorylated by Akt1 at Ser404. This leads to lamin association to 14.3.3 proteins which is an event required for lamin A proteosomal degradation 127, 128, 129. Intriguingly, these mechanisms also affect prelamin A stability, both under physiological conditions and in premature ageing syndromes. For instance, mutated prelamin A accumulates in MADA cells, and prelamin A levels increase in vitro with the number of passages in culture and in vivo with the age of the patients, contributing to a worsening of the disease 6. Two forms of prelamin A accumulate in MADA cells 7. However, while nonfarnesylated prelamin A is, in part, degraded through autophagy, farnesylated prelamin A accumulates to toxic levels. This is because nonfarnesylated prelamin A is highly phosphorylated at Ser404, while the farnesylated form of the lamin A precursor is not, suggesting the existence of an objective hindrance to its lysosomal clearance 8. Nevertheless, in 2014, we demonstrated that the treatment of cultured MADA fibroblasts with rapamycin triggered the autophagic degradation of farnesylated prelamin A efficiently and selectively.
The treatment of progeroid cells with rapamycin elicited the recovery of several cellular parameters [8]. In particular, rapamycin treatment of MADA cells, which feature very low levels of the nicotinamide adenine dinucleotide‐dependent sirtuin 1 (SIRT‐1) in the nuclear matrix, reduced soluble SIRT‐1 levels, to the benefit of the insoluble matrix‐bound protein. Recovery of SIRT‐1 binding to the nuclear matrix is expected to improve enzyme activity and to be relevant to metabolic processes governed by SIRT‐1 130. Rapamycin treatment also led to the recovery of heterochromatin organization, as seen by the recovery of trimethylated Lysine (K) 9 on Histone 3, (usually indicated as trimethyl H3K9) distribution patterns and levels 8. The latter is a key objective of laminopathy treatment as heterochromatin loss and disorganization are a major feature of laminopathic cells 1, 4, 6, 131.
Farnesylated prelamin A accumulation observed in MADA cells is also responsible for the recruitment of the stress‐induced transcription factor Oct‐1 at the nuclear lamina. As a consequence of its sequestration, Oct‐1 activity is impaired. By reducing farnesylated prelamin accumulation, rapamycin administration was seen to elicit the release of Oct‐1, restoring its transcriptional activity. Finally, rapamycin treatment of MADA fibroblasts partially rescued cell cycle dynamics. In fact, although the doubling time of MADA fibroblasts was not significantly changed by rapamycin, a significant proportion of cells re‐entered the cell cycle following drug treatment, and the S‐phase length was restored 8.
We recently reported on the efficacy of rapamycin treatment in rescuing cell type‐specific defects associated with LMNA‐linked progeroid syndromes. In osteoblast‐like U2OS cells overexpressing a mutant form of lamin A found in MADA (LA‐R527H), osteoclast differentiation markers were affected 9. According to our evidence, this may have been due to the positive effect of the LA‐R527H mutant on the secretion of transforming growth factor‐β2, which, in turn, triggers osteoclastogenesis, an effect previously reported in primary MADA osteoblasts 132. A reduction in mutated prelamin A levels by rapamycin treatment reduced aberrant osteoclastogenesis 9. Thus, mTOR inhibitors may prove useful in the rescue of bone loss that affects MADA patients.
mTOR inhibitors and other progeria‐related disorders
As already stated, progeric laminopathies represent a paradigm of age‐related diseases as they summarize most of the disorders linked to normal ageing. Nonetheless, a number of pathologies feature some aspects of physiological ageing. A list of progeria‐related disorders for which mTOR inhibitor administration has been evaluated is presented below.
Recent findings have proven that the mTOR pathway is altered in cells from nevi and melanoma from XP patients with defective DNA repair. Masaki et al. 133 revealed that these cancerous cells have a very high frequency of UV‐type mutations in the phosphatase and tensin homologue (PTEN) tumour suppressor gene. PTEN is a phosphatase that removes 3′‐phosphate from PI(3,4,5)‐trisphosphate to yield PI(4,5)‐bisphosphate, thus switching off the PI3K/Akt/mTOR signalling pathway. XP‐induced mutation in the PTEN gene results in a loss of the protein activity, leading to a constitutive PI3K/Akt/mTOR cascade activation. The administration of PTEN/mTOR inhibitors in patients with these lesions may therefore offer an opportunity to prevent the development of melanoma 133, 134. It should be noted, however, that Populo and colleagues describe the therapeutic inhibition of mTOR to induce cell death and prevent the progression of melanomas, rather than to induce autophagy. trisphosphate [134].
Recent studies indicated that Cockayne's Syndrome group B protein, CSB, a protein involved in the DNA repair machinery that is mutated in CS, is enriched in the mitochondria. This suggests a role for CSB in mitochondrial DNA repair 135, 136 following oxidative stress. In CS cells, the lack of CSB leads to an increase in the release of reactive oxygen species, coupled with mitochondrial dysfunction and increased metabolism. In these cells, another effect of the impairment of autophagic processes, is a defective mithocondrial turnover. Scheibye‐Knudsen et al. demonstrated that, by promoting autophagy, rapamycin was able to promote mitochondrial turnover, rescuing the bioenergetic profile 137.
Talaei and colleagues have that fibroblasts from WS patients show a strong activation of the mTOR pathway, in spite of their high level of basal autophagy 117. This has been demonstrated by an increase in mTOR activity, as well as in S6K1 117. What is more, according to Talaei et al., the simultaneous activation of these pathways in WS cells may result in a signal that overrides suppression of autophagy by mTOR 117. Recent studies have found that WS cells also exhibit low levels of intracellular thiol groups, which are known to be antioxidants. This is probably responsible for the highly vulnerability of WS cells to hydrogen peroxide treatment, UV radiation and cytostatic drugs 138. Thiol groups, which are increased in sodium hydrosulfide treatment, were found to attenuate hydrogen peroxide‐induced DNA fragmentation in WS cells, suggesting a beneficial effect at the DNA level. In addition, sodium hydrosulfide treatment was also found to be responsible for an intense repression of the mTOR pathway 117. Although the molecular mechanism is still obscure, the inhibition of the mTOR pathway by thiol groups produces the same beneficial effects as rapamycin treatment – i.e. a decrease in the level of insoluble proteins and in the percentage of aggregates, a restored nuclear morphology and a normalized phenotype of WS cells 117.
Unfortunately, published data on possible alterations of the mTOR pathway, increased autophagy and the use of rapalogs as treatment for TTD or BS are still missing. As stated above, these progeroid syndromes are associated with deficiencies in DNA repair mechanisms that lead to highly damaged DNA, which is ultimately responsible for a high vulnerability to stress. At present, scientists believe that increasing the efficiency of DNA repair might be associated with an extended healthy lifespan 139, 140. In this context, mTOR inhibitors may not be useful as it has been reported that, by inhibiting p21 activity, they suppress the repair mechanisms [86].
mTOR inhibitors and other ageing‐related diseases
Mouse models of AD show increased activity of mTOR and its downstream kinases. This leads to an abnormal hyperphosphorylation of tau, the accumulation of which is toxic to cells. Nevertheless, recent findings support a role for Aβ aggregates as positive regulators of mTOR activity 101. By activating autophagy, the treatment of AD mice with rapamycin has been reported to reduce Aβ protein levels and reverse cognitive deficits 102, 141. Thus, mTOR inhibitors may represent a promising therapy for AD by reducing the hyperphosphorylation of tau, decreasing Aβ levels, and inducing autophagy 141.
Pathological protein deposits of α‐synuclein, Aβ and tau proteins can also be found in cells from patients with PD. This degenerative disorder also features the death of specific populations of cells, such as dopaminergic cells in the substantia nigra. According to recent theories, oxidative stress plays an important role in the evolution of some forms of PD, as the increase in oxidative stress precedes the signs of neuronal degeneration. Toxin accumulation and oxidative stress in PD are likely to be responsible for the activation of RTP801, a stress response protein that drives neuronal cell death by leading to the inactivation of the survival kinase Akt. Published evidence obtained from cell cultures and in vivo PD models suggests that the treatment of PD patients with rapamycin regulates RTP801expression negatively by reducing its stability, thereby protecting the brain against neuronal death 142.
Dopaminergic cell death in PD has been hypothesized also to be related to oxidative stress‐induced accumulation of nonfunctioning autophagosomes. By inducing autophagy, rapamycin was shown to restore lysosome levels and autophagosome clearance, attenuating dopaminergic cell death in both in vitro and in vivo models of PD 143. Moreover, rapamycin treatment has also been shown to counteract the accumulation of α‐synuclein aggregates in a mouse model of PD 144.
In HD, nerve cells appear full of aggregates made up of portions of mHtt, which is the product of HTT gene 145. The severity of this neurodegenerative disorder is related to the number of polyglutamine (polyQ) repeats generated by pathological HTT mutation. Accumulation of mHtt is highly toxic and is associated with neuronal death. In mouse models and in the brains of humans with HD, mTOR has been shown to be sequestered in polyQ aggregates. mTOR inhibitors can enhance autophagy and consequently reduce mutated‐huntingtin (mHtt) accumulation and neuronal death in cellular and animal models of HD 146, 147. Furthermore, it has been reported that rapamycin treatment can improve motor deficits in mouse models of HD 146.
On the whole, the administration of chemical compounds that act by inhibiting the mTOR pathway has led to excellent and promising results, as seen in primary cultures obtained from patients affected by progeria‐related syndromes and in cellular models reproducing these pathologies. However, it should be noted that, probably due to its immunosuppressant properties, the long‐term use of rapamycin is sometimes associated with many side effects (e.g. interstitial pneumonia, high levels of triglycerides, reduced wound healing and anaemia) which might also increase the risk of cancer and preclude its chronic use in age‐related diseases 148.
As the use of rapamycin monotherapy in a broad spectrum of diseases, especially in treating cancers, is limited due to its poor efficacy, it would be reasonable for rapamycin treatment of age‐related diseases to be combined with other compounds.
Perspectives
Several mTOR‐dependent processes are likely to contribute to the effects of mTORC1 inhibitors on ageing and diseases associated with premature ageing. Among those processes, activation of autophagy leading to the degradation of damaged organelles and misfolded or toxic proteins, as occurs in rapamycin‐treated cells from progeroid laminopathies, plays a beneficial role in counteracting cellular senescence. In this context, mTOR inhibition mimics and overcomes CR and its role in extending the lifespan. Moreover, hyperactivation of mTOR may be associated with inflammation, and mTOR inhibitors have anti‐inflammatory effects 81, 82. Thus, reduction of age‐associated inflammation 149 is another possible mechanism by which mTORC1 inhibition could slow ageing‐related pathologies in humans. However, a key issue is avoiding uncontrolled effects due to improper dosage of rapamycin or its analogues. The proposed combination with other drugs, such as retinoic acid or oxidative stress inhibitors, acting in synergy with mTOR inhibitors to activate longevity pathways is a promising perspective.
Competing Interest
All authors have completed the Unified Competing Interest form at www.icmje.org/coi_disclosure.pdf (available on request from the corresponding author) and declare: GL had support from Associazione Italiana Progeria Sammy Basso (A.I.Pro.Sa.B) and Fondazione Del Monte di Bologna e Ravenna for the submitted work; VC and GL had support from ‘fondi 5 per mille 2012 Istituto Ortopedico Rizzoli’.
The authors wish to thank all the members of the Italian Network for Laminopathies ( www.igm.cnr.it/1/laminopatie/ ). This work was supported by grants from: A.I.Pro.Sa.B. (‘Treatment of progeric mice with drugs that reduce progerin levels’; Grant to GL), Fondazione Del Monte Grant 20.07.2015 to GL and by ‘5 per mille 2012, funds from Rizzoli Orthopedic Institute to GL and VC.
Evangelisti, C. , Cenni, V. , and Lattanzi, G. (2016) Potential therapeutic effects of the MTOR inhibitors for preventing ageing and progeria‐related disorders. Br J Clin Pharmacol, 82: 1229–1244. doi: 10.1111/bcp.12928.
References
- 1. Columbaro M, Capanni C, Mattioli E, Novelli G, Parnaik VK, Squarzoni S, Maraldi NM, Lattanzi G. Rescue of heterochromatin organization in Hutchinson–Gilford progeria by drug treatment. Cell Mol Life Sci 2005; 62: 2669–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Capanni C, Cenni V, Haraguchi T, Squarzoni S, Schuchner S, Ogris E, Novelli G, Maraldi N, Lattanzi G. Lamin A precursor induces barrier‐to‐autointegration factor nuclear localization. Cell Cycle 2010; 9: 2600–10. [DOI] [PubMed] [Google Scholar]
- 3. Capanni C, Squarzoni S, Cenni V, D'Apice MR, Gambineri A, Novelli G, Wehnert M, Pasquali R, Maraldi NM, Lattanzi G. Familial partial lipodystrophy, mandibuloacral dysplasia and restrictive dermopathy feature barrier‐to‐autointegration factor (BAF) nuclear redistribution. Cell Cycle 2012; 11: 3568–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pellegrini C, Columbaro M, Capanni C, D'Apice MR, Cavallo C, Murdocca M, Lattanzi G, Squarzoni S. All‐trans retinoic acid and rapamycin normalize Hutchinson Gilford progeria fibroblast phenotype. Oncotarget 2015; 6: 29914–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Richard DJ, Cubeddu L, Urquhart AJ, Bain A, Bolderson E, Menon D, White MF, Khanna KK. hSSB1 interacts directly with the MRN complex stimulating its recruitment to DNA double‐strand breaks and its endo‐nuclease activity. Nucleic Acids Res 2011; 39: 3643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Filesi I, Gullotta F, Lattanzi G, D'Apice MR, Capanni C, Nardone AM, Columbaro M, Scarano G, Mattioli E, Sabatelli P, Maraldi NM, Biocca S, Novelli G. Alterations of nuclear envelope and chromatin organization in mandibuloacral dysplasia, a rare form of laminopathy. Physiol Genomics 2005; 23: 150–8. [DOI] [PubMed] [Google Scholar]
- 7. Camozzi D, D'Apice MR, Schena E, Cenni V, Columbaro M, Capanni C, Maraldi NM, Squarzoni S, Ortolani M, Novelli G, Lattanzi G. Altered chromatin organization and SUN2 localization in mandibuloacral dysplasia are rescued by drug treatment. Histochem Cell Biol 2012; 138: 643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cenni V, Capanni C, Mattioli E, Columbaro M, Wehnert M, Ortolani M, Fini M, Novelli G, Bertacchini J, Maraldi NM, Marmiroli S, D'Apice MR, Prencipe S, Squarzoni S, Lattanzi G. Rapamycin treatment of mandibuloacral dysplasia cells rescues localization of chromatin‐associated proteins and cell cycle dynamics. Aging (Albany NY) 2014; 6: 755–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Evangelisti C, Bernasconi P, Cavalcante P, Cappelletti C, D'Apice MR, Sbraccia P, Novelli G, Prencipe S, Lemma S, Baldini N, Avnet S, Squarzoni S, Martelli AM, Lattanzi G. Modulation of TGFbeta two levels by lamin A in U2‐OS osteoblast‐like cells: understanding the osteolytic process triggered by altered lamins. Oncotarget 2015; 6: 7424–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Capanni C, Mattioli E, Columbaro M, Lucarelli E, Parnaik VK, Novelli G, Wehnert M, Cenni V, Maraldi NM, Squarzoni S, Lattanzi G. Altered pre‐lamin A processing is a common mechanism leading to lipodystrophy. Hum Mol Genet 2005; 14: 1489–502. [DOI] [PubMed] [Google Scholar]
- 11. Kandert S, Luke Y, Kleinhenz T, Neumann S, Lu W, Jaeger VM, Munck M, Wehnert M, Muller CR, Zhou Z, Noegel AA, Dabauvalle MC, Karakesisoglou I. Nesprin‐2 giant safeguards nuclear envelope architecture in LMNA S143F progeria cells. Hum Mol Genet 2007; 16: 2944–59. [DOI] [PubMed] [Google Scholar]
- 12. Garg A, Subramanyam L, Agarwal AK, Simha V, Levine B, D'Apice MR, Novelli G, Crow Y. Atypical progeroid syndrome due to heterozygous missense LMNA mutations. J Clin Endocrinol Metab 2009; 94: 4971–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Young SG, Jung HJ, Lee JM, Fong LG. Nuclear lamins and neurobiology. Mol Cell Biol 2014; 34: 2776–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nissan X, Blondel S, Navarro C, Maury Y, Denis C, Girard M, Martinat C, De Sandre‐Giovannoli A, Levy N, Peschanski M. Unique preservation of neural cells in Hutchinson–Gilford progeria syndrome is due to the expression of the neural‐specific miR‐9 microRNA. Cell Rep 2012; 2: 1–9. [DOI] [PubMed] [Google Scholar]
- 15. Lattanzi G, Ortolani M, Columbaro M, Prencipe S, Mattioli E, Lanzarini C, Maraldi NM, Cenni V, Garagnani P, Salvioli S, Storci G, Bonafe M, Capanni C, Franceschi C. Lamins are rapamycin targets that impact human longevity: a study in centenarians. J Cell Sci 2014; 127: 147–57. [DOI] [PubMed] [Google Scholar]
- 16. Lattanzi G. Prelamin A‐mediated nuclear envelope dynamics in normal and laminopathic cells. Biochem Soc Trans 2011; 39: 1698–704. [DOI] [PubMed] [Google Scholar]
- 17. Petruseva IO, Evdokimov AN, Lavrik OI. Molecular mechanism of global genome nucleotide excision repair. Acta Naturae 2014; 6: 23–34. [PMC free article] [PubMed] [Google Scholar]
- 18. Kraemer KH, Patronas NJ, Schiffmann R, Brooks BP, Tamura D, DiGiovanna JJ. Xeroderma pigmentosum, trichothiodystrophy and Cockayne syndrome: a complex genotype‐phenotype relationship. Neuroscience 2007; 145: 1388–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis 2011; 6: 70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Komatsu DE, Hadjiargyrou M. Activation of the transcription factor HIF‐1 and its target genes, VEGF, HO‐1, iNOS, during fracture repair. Bone 2004; 34: 680–8. [DOI] [PubMed] [Google Scholar]
- 21. Bregman DB, Halaban R, van Gool AJ, Henning KA, Friedberg EC, Warren SL. UV‐induced ubiquitination of RNA polymerase II: a novel modification deficient in Cockayne syndrome cells. Proc Natl Acad Sci U S A 1996; 93: 11586–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Itin PH, Sarasin A, Pittelkow MR. Trichothiodystrophy: update on the sulfur‐deficient brittle hair syndromes. J Am Acad Dermatol 2001; 44: 891–920 .quiz 21‐4 [DOI] [PubMed] [Google Scholar]
- 23. Stefanini M, Botta E, Lanzafame M, Orioli D. Trichothiodystrophy: from basic mechanisms to clinical implications. DNA Repair (Amst) 2010; 9: 2–10. [DOI] [PubMed] [Google Scholar]
- 24. Bachrati CZ, Hickson ID. RecQ helicases: suppressors of tumorigenesis and premature aging. Biochem J 2003; 374: 577–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kaneko H, Kondo N. Clinical features of Bloom syndrome and function of the causative gene, BLM helicase. Expert Rev Mol Diagn 2004; 4: 393–401. [DOI] [PubMed] [Google Scholar]
- 26. Hoeijmakers JH. DNA damage, aging, and cancer. N Engl J Med 2009; 361: 1475–85. [DOI] [PubMed] [Google Scholar]
- 27. Chen C, Contreras R. The bud scar‐based screening system for hunting human genes extending life span. Ann N Y Acad Sci 2004; 1019: 355–9. [DOI] [PubMed] [Google Scholar]
- 28. Navarro CL, Cau P, Levy N. Molecular bases of progeroid syndromes. Hum Mol Genet 2006; 15 (Spec No 2): R151–61. [DOI] [PubMed] [Google Scholar]
- 29. Weedon MN, Ellard S, Prindle MJ, Caswell R, Lango Allen H, Oram R, Godbole K, Yajnik CS, Sbraccia P, Novelli G, Turnpenny P, McCann E, Goh KJ, Wang Y, Fulford J, McCulloch LJ, Savage DB, O'Rahilly S, Kos K, Loeb LA, Semple RK, Hattersley AT. An in‐frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Genet 2013; 45: 947–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pelosini C, Martinelli S, Ceccarini G, Magno S, Barone I, Basolo A, Fierabracci P, Vitti P, Maffei M, Santini F. Identification of a novel mutation in the polymerase delta one (POLD1) gene in a lipodystrophic patient affected by mandibular hypoplasia, deafness, progeroid features (MDPL) syndrome. Metabolism 2014; 63: 1385–9. [DOI] [PubMed] [Google Scholar]
- 31. Song J, Hong P, Liu C, Zhang Y, Wang J, Wang P. Human POLD1 modulates cell cycle progression and DNA damage repair. BMC Biochem 2015; 16: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yoshimura A, Kobayashi Y, Tada S, Seki M, Enomoto T. WRNIP1 functions upstream of DNA polymerase eta in the UV‐induced DNA damage response. Biochem Biophys Res Commun 2014; 452: 48–52. [DOI] [PubMed] [Google Scholar]
- 33. Laplante M, Sabatini DM. mTOR signaling in growth control and disease. Cell 2012; 149: 274–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Memmott RM, Dennis PA. Akt‐dependent and ‐independent mechanisms of mTOR regulation in cancer. Cell Signal 2009; 21: 656–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Dunlop EA, Tee AR. mTOR and autophagy: a dynamic relationship governed by nutrients and energy. Semin Cell Dev Biol 2014; 36: 121–9. [DOI] [PubMed] [Google Scholar]
- 36. Shimobayashi M, Hall MN. Making new contacts: the mTOR network in metabolism and signalling crosstalk. Nat Rev Mol Cell Biol 2014; 15: 155–62. [DOI] [PubMed] [Google Scholar]
- 37. Martelli AM, Chiarini F, Evangelisti C, Ognibene A, Bressanin D, Billi AM, Manzoli L, Cappellini A, McCubrey JA. Targeting the liver kinase B1/AMP‐activated protein kinase pathway as a therapeutic strategy for hematological malignancies. Expert Opin Ther Targets 2012; 16: 729–42. [DOI] [PubMed] [Google Scholar]
- 38. Thoreen CC, Chantranupong L, Keys HR, Wang T, Gray NS, Sabatini DM. A unifying model for mTORC1‐mediated regulation of mRNA translation. Nature 2012; 485: 109–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chauvin C, Koka V, Nouschi A, Mieulet V, Hoareau‐Aveilla C, Dreazen A, Cagnard N, Carpentier W, Kiss T, Meyuhas O, Pende M. Ribosomal protein S6 kinase activity controls the ribosome biogenesis transcriptional program. Oncogene 2014; 33: 474–83. [DOI] [PubMed] [Google Scholar]
- 40. Oh WJ, Jacinto E. mTOR complex two signaling and functions. Cell Cycle 2011; 10: 2305–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zinzalla V, Stracka D, Oppliger W, Hall MN. Activation of mTORC2 by association with the ribosome. Cell 2011; 144: 757–68. [DOI] [PubMed] [Google Scholar]
- 42. Bach M, Larance M, James DE, Ramm G. The serine/threonine kinase ULK1 is a target of multiple phosphorylation events. Biochem J 2011; 440: 283–91. [DOI] [PubMed] [Google Scholar]
- 43. Dunlop EA, Tee AR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans 2013; 41: 939–43. [DOI] [PubMed] [Google Scholar]
- 44. Russell RC, Tian Y, Yuan H, Park HW, Chang YY, Kim J, Kim H, Neufeld TP, Dillin A, Guan KL. ULK1 induces autophagy by phosphorylating Beclin‐1 and activating VPS34 lipid kinase. Nat Cell Biol 2013; 15: 741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin‐like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell 2007; 130: 165–78. [DOI] [PubMed] [Google Scholar]
- 46. Heitman J, Movva NR, Hall MN. Targets for cell cycle arrest by the immunosuppressant rapamycin in yeast. Science 1991; 253: 905–9. [DOI] [PubMed] [Google Scholar]
- 47. Zhou H, Luo Y, Huang S. Updates of mTOR inhibitors. Anticancer Agents Med Chem 2010; 10: 571–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 2006; 22: 159–68. [DOI] [PubMed] [Google Scholar]
- 49. Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA. Rapamycin‐induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 2012; 335: 1638–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Chang JY, Sehgal SN, Bansbach CC. FK506 and rapamycin: novel pharmacological probes of the immune response. Trends Pharmacol Sci 1991; 12: 218–23. [DOI] [PubMed] [Google Scholar]
- 51. Kahan BD, Podbielski J, Napoli KL, Katz SM, Meier‐Kriesche HU, Van Buren CT. Immunosuppressive effects and safety of a sirolimus/cyclosporine combination regimen for renal transplantation. Transplantation 1998; 66: 1040–6. [DOI] [PubMed] [Google Scholar]
- 52. Serruys PW, Kutryk MJ, Ong AT. Coronary‐artery stents. N Engl J Med 2006; 354: 483–95. [DOI] [PubMed] [Google Scholar]
- 53. Woods TC, Marks AR. Drug‐eluting stents. Annu Rev Med 2004; 55: 169–78. [DOI] [PubMed] [Google Scholar]
- 54. McCormack FX, Inoue Y, Moss J, Singer LG, Strange C, Nakata K, Barker AF, Chapman JT, Brantly ML, Stocks JM, Brown KK, Lynch JP 3rd, Goldberg HJ, Young LR, Kinder BW, Downey GP, Sullivan EJ, Colby TV, McKay RT, Cohen MM, Korbee L, Taveira‐DaSilva AM, Lee HS, Krischer JP, Trapnell BC. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med 2011; 364: 1595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hess G, Herbrecht R, Romaguera J, Verhoef G, Crump M, Gisselbrecht C, Laurell A, Offner F, Strahs A, Berkenblit A, Hanushevsky O, Clancy J, Hewes B, Moore L, Coiffier B. Phase III study to evaluate temsirolimus compared with investigator's choice therapy for the treatment of relapsed or refractory mantle cell lymphoma. J Clin Oncol 2009; 27: 3822–9. [DOI] [PubMed] [Google Scholar]
- 56. Hudes G, Carducci M, Tomczak P, Dutcher J, Figlin R, Kapoor A, Staroslawska E, Sosman J, McDermott D, Bodrogi I, Kovacevic Z, Lesovoy V, Schmidt‐Wolf IG, Barbarash O, Gokmen E, O'Toole T, Lustgarten S, Moore L, Motzer RJ. Temsirolimus, interferon alfa, or both for advanced renal‐cell carcinoma. N Engl J Med 2007; 356: 2271–81. [DOI] [PubMed] [Google Scholar]
- 57. Motzer RJ, Escudier B, Oudard S, Hutson TE, Porta C, Bracarda S, Grunwald V, Thompson JA, Figlin RA, Hollaender N, Urbanowitz G, Berg WJ, Kay A, Lebwohl D, Ravaud A. Efficacy of everolimus in advanced renal cell carcinoma: a double‐blind, randomised, placebo‐controlled phase III trial. Lancet 2008; 372: 449–56. [DOI] [PubMed] [Google Scholar]
- 58. Krueger DA, Care MM, Holland K, Agricola K, Tudor C, Mangeshkar P, Wilson KA, Byars A, Sahmoud T, Franz DN. Everolimus for subependymal giant‐cell astrocytomas in tuberous sclerosis. N Engl J Med 2010; 363: 1801–11. [DOI] [PubMed] [Google Scholar]
- 59. Yao JC, Shah MH, Ito T, Bohas CL, Wolin EM, Van Cutsem E, Hobday TJ, Okusaka T, Capdevila J, de Vries EG, Tomassetti P, Pavel ME, Hoosen S, Haas T, Lincy J, Lebwohl D, Oberg K. Everolimus for advanced pancreatic neuroendocrine tumors. N Engl J Med 2011; 364: 514–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schuler W, Sedrani R, Cottens S, Haberlin B, Schulz M, Schuurman HJ, Zenke G, Zerwes HG, Schreier MH. SDZ RAD, a new rapamycin derivative: pharmacological properties in vitro and in vivo . Transplantation 1997; 64: 36–42. [DOI] [PubMed] [Google Scholar]
- 61. Dienstmann R, Rodon J, Serra V, Tabernero J. Picking the point of inhibition: a comparative review of PI3K/AKT/mTOR pathway inhibitors. Mol Cancer Ther 2014; 13: 1021–31. [DOI] [PubMed] [Google Scholar]
- 62. Fan QW, Knight ZA, Goldenberg DD, Yu W, Mostov KE, Stokoe D, Shokat KM, Weiss WA. A dual PI3 kinase/mTOR inhibitor reveals emergent efficacy in glioma. Cancer Cell 2006; 9: 341–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Apsel B, Blair JA, Gonzalez B, Nazif TM, Feldman ME, Aizenstein B, Hoffman R, Williams RL, Shokat KM, Knight ZA. Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases. Nat Chem Biol 2008; 4: 691–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Sun AY, Wang Q, Simonyi A, Sun GY. Resveratrol as a therapeutic agent for neurodegenerative diseases. Mol Neurobiol 2010; 41: 375–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Liu M, Wilk SA, Wang A, Zhou L, Wang RH, Ogawa W, Deng C, Dong LQ, Liu F. Resveratrol inhibits mTOR signaling by promoting the interaction between mTOR and DEPTOR. J Biol Chem 2010; 285: 36387–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Libby G, Donnelly LA, Donnan PT, Alessi DR, Morris AD, Evans JM. New users of metformin are at low risk of incident cancer: a cohort study among people with type two diabetes. Diabetes Care 2009; 32: 1620–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Giovannucci E, Harlan DM, Archer MC, Bergenstal RM, Gapstur SM, Habel LA, Pollak M, Regensteiner JG, Yee D. Diabetes and cancer: a consensus report. Diabetes Care 2010; 33: 1674–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Dowling RJ, Zakikhani M, Fantus IG, Pollak M, Sonenberg N. Metformin inhibits mammalian target of rapamycin‐dependent translation initiation in breast cancer cells. Cancer Res 2007; 67: 10804–12. [DOI] [PubMed] [Google Scholar]
- 69. Kalender A, Selvaraj A, Kim SY, Gulati P, Brule S, Viollet B, Kemp BE, Bardeesy N, Dennis P, Schlager JJ, Marette A, Kozma SC, Thomas G. Metformin, independent of AMPK, inhibits mTORC1 in a rag GTPase‐dependent manner. Cell Metab 2010; 11: 390–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Ben Sahra I, Regazzetti C, Robert G, Laurent K, Le Marchand‐Brustel Y, Auberger P, Tanti JF, Giorgetti‐Peraldi S, Bost F. Metformin, independent of AMPK, induces mTOR inhibition and cell‐cycle arrest through REDD1. Cancer Res 2011; 71: 4366–72. [DOI] [PubMed] [Google Scholar]
- 71. Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al‐Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ. Ribosomal protein S6 kinase one signaling regulates mammalian life span. Science 2009; 326: 140–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Hansen M, Taubert S, Crawford D, Libina N, Lee SJ, Kenyon C. Lifespan extension by conditions that inhibit translation in Caenorhabditis elegans . Aging Cell 2007; 6: 95–110. [DOI] [PubMed] [Google Scholar]
- 73. Steffen KK, MacKay VL, Kerr EO, Tsuchiya M, Hu D, Fox LA, Dang N, Johnston ED, Oakes JA, Tchao BN, Pak DN, Fields S, Kennedy BK, Kaeberlein M. Yeast life span extension by depletion of 60s ribosomal subunits is mediated by Gcn4. Cell 2008; 133: 292–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Bjedov I, Toivonen JM, Kerr F, Slack C, Jacobson J, Foley A, Partridge L. Mechanisms of life span extension by rapamycin in the fruit fly Drosophila melanogaster . Cell Metab 2010; 11: 35–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Rubinsztein DC, Marino G, Kroemer G. Autophagy and aging. Cell 2011; 146: 682–95. [DOI] [PubMed] [Google Scholar]
- 76. Kapahi P, Chen D, Rogers AN, Katewa SD, Li PW, Thomas EL, Kockel L. With TOR, less is more: a key role for the conserved nutrient‐sensing TOR pathway in aging. Cell Metab 2010; 11: 453–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B. Autophagy genes are essential for dauer development and life‐span extension in C. elegans . Science 2003; 301: 1387–91. [DOI] [PubMed] [Google Scholar]
- 78. Chen C, Liu Y, Zheng P. mTOR regulation and therapeutic rejuvenation of aging hematopoietic stem cells. Sci Signal 2009; 2: ra75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Franceschi C, Campisi J. Chronic inflammation (inflammaging) and its potential contribution to age‐associated diseases. J Gerontol A Biol Sci Med Sci 2014; 69 (Suppl. 1): S4–9. [DOI] [PubMed] [Google Scholar]
- 80. Yun MH, Davaapil H, Brockes JP. Recurrent turnover of senescent cells during regeneration of a complex structure. Elife 2015; 4: e05505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Laberge RM, Sun Y, Orjalo AV, Patil CK, Freund A, Zhou L, Curran SC, Davalos AR, Wilson‐Edell KA, Liu S, Limbad C, Demaria M, Li P, Hubbard GB, Ikeno Y, Javors M, Desprez PY, Benz CC, Kapahi P, Nelson PS, Campisi J. MTOR regulates the pro‐tumorigenic senescence‐associated secretory phenotype by promoting IL1A translation. Nat Cell Biol 2015; 17: 1049–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Serrano M. The inflammTORy powers of senescence. Trends Cell Biol 2015; 25: 634–6. [DOI] [PubMed] [Google Scholar]
- 83. Weichhart T, Hengstschlager M, Linke M. Regulation of innate immune cell function by mTOR. Nat Rev Immunol 2015; 15: 599–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Demidenko ZN, Blagosklonny MV. Growth stimulation leads to cellular senescence when the cell cycle is blocked. Cell Cycle 2008; 7: 3355–61. [DOI] [PubMed] [Google Scholar]
- 85. Campisi J, d'Adda di Fagagna F. Cellular senescence: when bad things happen to good cells. Nat Rev Mol Cell Biol 2007; 8: 729–40. [DOI] [PubMed] [Google Scholar]
- 86. Blagosklonny MV. Geroconversion: irreversible step to cellular senescence. Cell Cycle 2014; 13: 3628–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Leontieva OV, Demidenko ZN, Blagosklonny MV. Dual mTORC1/C2 inhibitors suppress cellular geroconversion (a senescence program). Oncotarget 2015; 6: 23238–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Demidenko ZN, Zubova SG, Bukreeva EI, Pospelov VA, Pospelova TV, Blagosklonny MV. Rapamycin decelerates cellular senescence. Cell Cycle 2009; 8: 1888–95. [DOI] [PubMed] [Google Scholar]
- 89. Blagosklonny MV, Hall MN. Growth and aging: a common molecular mechanism. Aging (Albany NY) 2009; 1: 357–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK. Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 2005; 310: 1193–6. [DOI] [PubMed] [Google Scholar]
- 91. Vellai T, Takacs‐Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F. Genetics: influence of TOR kinase on lifespan in C. elegans . Nature 2003; 426: 620. [DOI] [PubMed] [Google Scholar]
- 92. Wu JJ, Liu J, Chen EB, Wang JJ, Cao L, Narayan N, Fergusson MM, Rovira II, Allen M, Springer DA, Lago CU, Zhang S, DuBois W, Ward T, deCabo R, Gavrilova O, Mock B, Finkel T. Increased mammalian lifespan and a segmental and tissue‐specific slowing of aging after genetic reduction of mTOR expression. Cell Rep 2013; 4: 913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA. Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 2009; 460: 392–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Miller RA, Harrison DE, Astle CM, Baur JA, Boyd AR, de Cabo R, Fernandez E, Flurkey K, Javors MA, Nelson JF, Orihuela CJ, Pletcher S, Sharp ZD, Sinclair D, Starnes JW, Wilkinson JE, Nadon NL, Strong R. Rapamycin, but not resveratrol or simvastatin, extends life span of genetically heterogeneous mice. J Gerontol A Biol Sci Med Sci 2011; 66: 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Fischer KE, Gelfond JA, Soto VY, Han C, Someya S, Richardson A, Austad SN. Health effects of long‐term rapamycin treatment: the impact on mouse health of enteric rapamycin treatment from four months of age throughout life. PLoS One 2015; 10: e0126644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Richardson A. Rapamycin, anti‐aging, and avoiding the fate of Tithonus. J Clin Invest 2013; 123: 3204–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Li J, Kim SG, Blenis J. Rapamycin: one drug, many effects. Cell Metab 2014; 19: 373–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Neff F, Flores‐Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, Afonso LC, Aguilar‐Pimentel JA, Becker L, Garrett L, Hans W, Hettich MM, Holtmeier R, Holter SM, Moreth K, Prehn C, Puk O, Racz I, Rathkolb B, Rozman J, Naton B, Ordemann R, Adamski J, Beckers J, Bekeredjian R, Busch DH, Ehninger G, Graw J, Hofler H, Klingenspor M, Klopstock T, Ollert M, Stypmann J, Wolf E, Wurst W, Zimmer A, Fuchs H, Gailus‐Durner V, Hrabe de Angelis M, Ehninger D. Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest 2013; 123: 3272–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Halloran J, Hussong SA, Burbank R, Podlutskaya N, Fischer KE, Sloane LB, Austad SN, Strong R, Richardson A, Hart MJ, Galvan V. Chronic inhibition of mammalian target of rapamycin by rapamycin modulates cognitive and non‐cognitive components of behavior throughout lifespan in mice. Neuroscience 2012; 223: 102–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Majumder S, Caccamo A, Medina DX, Benavides AD, Javors MA, Kraig E, Strong R, Richardson A, Oddo S. Lifelong rapamycin administration ameliorates age‐dependent cognitive deficits by reducing IL‐1beta and enhancing NMDA signaling. Aging Cell 2012; 11: 326–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid‐beta, and tau: effects on cognitive impairments. J Biol Chem 2010; 285: 13107–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid‐beta levels in a mouse model of Alzheimer's disease. PLoS One 2010; 5: e9979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Majumder S, Richardson A, Strong R, Oddo S. Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 2011; 6: e25416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Onken B, Driscoll M. Metformin induces a dietary restriction‐like state and the oxidative stress response to extend C. elegans healthspan via AMPK, LKB1, and SKN‐1. PLoS One 2010; 5: e8758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Kovalenko IG, Poroshina TE, Semenchenko AV, Provinciali M, Re F, Franceschi C. Effect of metformin on life span and on the development of spontaneous mammary tumors in HER‐2/neu transgenic mice. Exp Gerontol 2005; 40: 685–93. [DOI] [PubMed] [Google Scholar]
- 106. Semba RD, Ferrucci L, Bartali B, Urpi‐Sarda M, Zamora‐Ros R, Sun K, Cherubini A, Bandinelli S, Andres‐Lacueva C. Resveratrol levels and all‐cause mortality in older community‐dwelling adults. JAMA Intern Med 2014; 174: 1077–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Strandgren C, Nasser HA, McKenna T, Koskela A, Tuukkanen J, Ohlsson C, Rozell B, Eriksson M. Transgene silencing of the Hutchinson–Gilford progeria syndrome mutation results in a reversible bone phenotype, whereas resveratrol treatment does not show overall beneficial effects. FASEB J 2015; 29: 3193–205. [DOI] [PubMed] [Google Scholar]
- 108. Anisimov VN. Metformin for aging and cancer prevention. Aging (Albany NY) 2010; 2: 760–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Anisimov VN, Berstein LM, Egormin PA, Piskunova TS, Popovich IG, Zabezhinski MA, Tyndyk ML, Yurova MV, Kovalenko IG, Poroshina TE, Semenchenko AV. Metformin slows down aging and extends life span of female SHR mice. Cell Cycle 2008; 7: 2769–73. [DOI] [PubMed] [Google Scholar]
- 110. Martin‐Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye‐Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin MJ, Schwab M, Pollak M, Zhang Y, Yu Y, Becker KG, Bohr VA, Ingram DK, Sinclair DA, Wolf NS, Spindler SR, Bernier M, de Cabo R. Metformin improves healthspan and lifespan in mice. Nat Commun 2013; 4: 2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Scarpello JH. Improving survival with metformin: the evidence base today. Diabetes Metab 2003; 29: 6S36–43. [DOI] [PubMed] [Google Scholar]
- 112. Dowling RJ, Goodwin PJ, Stambolic V. Understanding the benefit of metformin use in cancer treatment. BMC Med 2011; 9: 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Longo VD, Mattson MP. Fasting: molecular mechanisms and clinical applications. Cell Metab 2014; 19: 181–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Wei M, Fabrizio P, Hu J, Ge H, Cheng C, Li L, Longo VD. Life span extension by calorie restriction depends on Rim15 and transcription factors downstream of Ras/PKA, Tor, and Sch9. PLoS Genet 2008; 4: e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Cheng CW, Adams GB, Perin L, Wei M, Zhou X, Lam BS, Da Sacco S, Mirisola M, Quinn DI, Dorff TB, Kopchick JJ, Longo VD. Prolonged fasting reduces IGF‐1/PKA to promote hematopoietic‐stem‐cell‐based regeneration and reverse immunosuppression. Cell Stem Cell 2014; 14: 810–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Cenni V, Capanni C, Columbaro M, Ortolani M, D'Apice MR, Novelli G, Fini M, Marmiroli S, Scarano E, Maraldi NM, Squarzoni S, Prencipe S, Lattanzi G. Autophagic degradation of farnesylated prelamin A as a therapeutic approach to lamin‐linked progeria. Eur J Histochem 2011; 55: e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Talaei F, van Praag VM, Henning RH. Hydrogen sulfide restores a normal morphological phenotype in Werner syndrome fibroblasts, attenuates oxidative damage and modulates mTOR pathway. Pharmacol Res 2013; 74: 34–44. [DOI] [PubMed] [Google Scholar]
- 118. Lopez‐Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell 2013; 153: 1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Cao K, Graziotto JJ, Blair CD, Mazzulli JR, Erdos MR, Krainc D, Collins FS. Rapamycin reverses cellular phenotypes and enhances mutant protein clearance in Hutchinson–Gilford progeria syndrome cells. Sci Transl Med 2011; 3: 89ra58. [DOI] [PubMed] [Google Scholar]
- 120. Ross CA, Poirier MA. Protein aggregation and neurodegenerative disease. Nat Med 2004; 10 (Suppl.): S10–7. [DOI] [PubMed] [Google Scholar]
- 121. Tseng BP, Green KN, Chan JL, Blurton‐Jones M, LaFerla FM. Abeta inhibits the proteasome and enhances amyloid and tau accumulation. Neurobiol Aging 2008; 29: 1607–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Cooper AA, Gitler AD, Cashikar A, Haynes CM, Hill KJ, Bhullar B, Liu K, Xu K, Strathearn KE, Liu F, Cao S, Caldwell KA, Caldwell GA, Marsischky G, Kolodner RD, Labaer J, Rochet JC, Bonini NM, Lindquist S. Alpha‐synuclein blocks ER–Golgi traffic and Rab1 rescues neuron loss in Parkinson's models. Science 2006; 313: 324–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Gauthier LR, Charrin BC, Borrell‐Pages M, Dompierre JP, Rangone H, Cordelieres FP, De Mey J, MacDonald ME, Lessmann V, Humbert S, Saudou F. Huntingtin controls neurotrophic support and survival of neurons by enhancing BDNF vesicular transport along microtubules. Cell 2004; 118: 127–38. [DOI] [PubMed] [Google Scholar]
- 124. Zuccato C, Ciammola A, Rigamonti D, Leavitt BR, Goffredo D, Conti L, MacDonald ME, Friedlander RM, Silani V, Hayden MR, Timmusk T, Sipione S, Cattaneo E. Loss of huntingtin‐mediated BDNF gene transcription in Huntington's disease. Science 2001; 293: 493–8. [DOI] [PubMed] [Google Scholar]
- 125. Buendia B, Courvalin JC, Collas P. Dynamics of the nuclear envelope at mitosis and during apoptosis. Cell Mol Life Sci 2001; 58: 1781–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Cenni V, Sabatelli P, Mattioli E, Marmiroli S, Capanni C, Ognibene A, Squarzoni S, Maraldi NM, Bonne G, Columbaro M, Merlini L, Lattanzi G. Lamin A N‐terminal phosphorylation is associated with myoblast activation: impairment in Emery–Dreifuss muscular dystrophy. J Med Genet 2005; 42: 214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Cenni V, Bertacchini J, Beretti F, Lattanzi G, Bavelloni A, Riccio M, Ruzzene M, Marin O, Arrigoni G, Parnaik V, Wehnert M, Maraldi NM, de Pol A, Cocco L, Marmiroli S. Lamin A Ser404 is a nuclear target of Akt phosphorylation in C2C12 cells. J Proteome Res 2008; 7: 4727–35. [DOI] [PubMed] [Google Scholar]
- 128. Marmiroli S, Bertacchini J, Beretti F, Cenni V, Guida M, De Pol A, Maraldi NM, Lattanzi G. A‐type lamins and signaling: the PI 3‐kinase/Akt pathway moves forward. J Cell Physiol 2009; 220: 553–61. [DOI] [PubMed] [Google Scholar]
- 129. Bertacchini J, Beretti F, Cenni V, Guida M, Gibellini F, Mediani L, Marin O, Maraldi NM, de Pol A, Lattanzi G, Cocco L, Marmiroli S. The protein kinase Akt/PKB regulates both prelamin A degradation and Lmna gene expression. FASEB J 2013; 27: 2145–55. [DOI] [PubMed] [Google Scholar]
- 130. Liu B, Ghosh S, Yang X, Zheng H, Liu X, Wang Z, Jin G, Zheng B, Kennedy BK, Suh Y, Kaeberlein M, Tryggvason K, Zhou Z. Resveratrol rescues SIRT1‐dependent adult stem cell decline and alleviates progeroid features in laminopathy‐based progeria. Cell Metab 2012; 16: 738–50. [DOI] [PubMed] [Google Scholar]
- 131. Columbaro M, Mattioli E, Schena E, Capanni C, Cenni V, Levy N, Navarro CL, Del Coco R, Squarzoni S, Camozzi D, Hutchison CJ, Wehnert M, Lattanzi G. Prelamin A processing and functional effects in restrictive dermopathy. Cell Cycle 2010; 9: 4766–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132. Avnet S, Pallotta R, Perut F, Baldini N, Pittis MG, Saponari A, Lucarelli E, Dozza B, Greggi T, Maraldi NM, Capanni C, Mattioli E, Columbaro M, Lattanzi G. Osteoblasts from a mandibuloacral dysplasia patient induce human blood precursors to differentiate into active osteoclasts. Biochim Biophys Acta 1812; 2011: 711–8. [DOI] [PubMed] [Google Scholar]
- 133. Masaki T, Wang Y, DiGiovanna JJ, Khan SG, Raffeld M, Beltaifa S, Hornyak TJ, Darling TN, Lee CC, Kraemer KH. High frequency of PTEN mutations in nevi and melanomas from xeroderma pigmentosum patients. Pigment Cell Melanoma Res 2014; 27: 454–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Populo H, Soares P, Lopes JM. Insights into melanoma: targeting the mTOR pathway for therapeutics. Expert Opin Ther Targets 2012; 16: 689–705. [DOI] [PubMed] [Google Scholar]
- 135. Aamann MD, Sorensen MM, Hvitby C, Berquist BR, Muftuoglu M, Tian J, de Souza‐Pinto NC, Scheibye‐Knudsen M, Wilson DM 3rd, Stevnsner T, Bohr VA. Cockayne syndrome group B protein promotes mitochondrial DNA stability by supporting the DNA repair association with the mitochondrial membrane. FASEB J 2010; 24: 2334–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Kamenisch Y, Fousteri M, Knoch J, von Thaler AK, Fehrenbacher B, Kato H, Becker T, Dolle ME, Kuiper R, Majora M, Schaller M, van der Horst GT, van Steeg H, Rocken M, Rapaport D, Krutmann J, Mullenders LH, Berneburg M. Proteins of nucleotide and base excision repair pathways interact in mitochondria to protect from loss of subcutaneous fat, a hallmark of aging. J Exp Med 2010; 207: 379–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Scheibye‐Knudsen M, Ramamoorthy M, Sykora P, Maynard S, Lin PC, Minor RK, Wilson DM 3rd, Cooper M, Spencer R, de Cabo R, Croteau DL, Bohr VA. Cockayne syndrome group B protein prevents the accumulation of damaged mitochondria by promoting mitochondrial autophagy. J Exp Med 2012; 209: 855–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Saito H, Moses RE. Immortalization of Werner syndrome and progeria fibroblasts. Exp Cell Res 1991; 192: 373–9. [DOI] [PubMed] [Google Scholar]
- 139. de la Rosa J, Freije JM, Cabanillas R, Osorio FG, Fraga MF, Fernandez‐Garcia MS, Rad R, Fanjul V, Ugalde AP, Liang Q, Prosser HM, Bradley A, Cadinanos J, Lopez‐Otin C. Prelamin A causes progeria through cell‐extrinsic mechanisms and prevents cancer invasion. Nat Commun 2013; 4: 2268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Baker DJ, Dawlaty MM, Wijshake T, Jeganathan KB, Malureanu L, van Ree JH, Crespo‐Diaz R, Reyes S, Seaburg L, Shapiro V, Behfar A, Terzic A, van de Sluis B, van Deursen JM. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nat Cell Biol 2013; 15: 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Wong M. Mammalian target of rapamycin (mTOR) pathways in neurological diseases. Biochem J 2013; 36: 40–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Malagelada C, Jin ZH, Jackson‐Lewis V, Przedborski S, Greene LA. Rapamycin protects against neuron death in in vitro and in vivo models of Parkinson's disease. J Neurosci 2010; 30: 1166–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Dehay B, Bove J, Rodriguez‐Muela N, Perier C, Recasens A, Boya P, Vila M. Pathogenic lysosomal depletion in Parkinson's disease. J Neurosci 2010; 30: 12535–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Liu K, Shi N, Sun Y, Zhang T, Sun X. Therapeutic effects of rapamycin on MPTP‐induced Parkinsonism in mice. Neurochem Res 2013; 38: 201–7. [DOI] [PubMed] [Google Scholar]
- 145. Rubinsztein DC. How does the Huntington's disease mutation damage cells? Sci Aging Knowledge Environ 2003; 2003: PE26. [DOI] [PubMed] [Google Scholar]
- 146. Ravikumar B, Vacher C, Berger Z, Davies JE, Luo S, Oroz LG, Scaravilli F, Easton DF, Duden R, O'Kane CJ, Rubinsztein DC. Inhibition of mTOR induces autophagy and reduces toxicity of polyglutamine expansions in fly and mouse models of Huntington disease. Nat Genet 2004; 36: 585–95. [DOI] [PubMed] [Google Scholar]
- 147. Sarkar S, Rubinsztein DC. Huntington's disease: degradation of mutant huntingtin by autophagy. FEBS J 2008; 275: 4263–70. [DOI] [PubMed] [Google Scholar]
- 148. Bove J, Martinez‐Vicente M, Vila M. Fighting neurodegeneration with rapamycin: mechanistic insights. Nat Rev Neurosci 2011; 12: 437–52. [DOI] [PubMed] [Google Scholar]
- 149. Castellani GC, Menichetti G, Garagnani P, Giulia Bacalini M, Pirazzini C, Franceschi C, Collino S, Sala C, Remondini D, Giampieri E, Mosca E, Bersanelli M, Vitali S, Valle IF, Lio P, Milanesi L. Systems medicine of inflammaging. Brief Bioinform 2015. Aug 24. [DOI] [PMC free article] [PubMed] [Google Scholar]