

Abstract
The analysis of linkage disequilibrium (LD) underpins the development of effective genotyping technologies, trait mapping and understanding of biological mechanisms such as those driving recombination and the impact of selection. We apply the Malécot-Morton model of LD to create additive LD maps that describe the high-resolution LD landscape of commercial chickens. We investigated LD in chickens (Gallus gallus) at the highest resolution to date for broiler, white egg and brown egg layer commercial lines. There is minimal concordance between breeds of fine-scale LD patterns (correlation coefficient <0.21), and even between discrete broiler lines. Regions of LD breakdown, which may align with recombination hot spots, are enriched near CpG islands and transcription start sites (P<2.2 × 10−16), consistent with recent evidence described in finches, but concordance in hot spot locations between commercial breeds is only marginally greater than random. As in other birds, functional elements in the chicken genome are associated with recombination but, unlike evidence from other bird species, the LD landscape is not stable in the populations studied. The development of optimal genotyping panels for genome-led selection programmes will depend on careful analysis of the LD structure of each line of interest. Further study is required to fully elucidate the mechanisms underlying highly divergent LD patterns found in commercial chickens.
Introduction
A detailed understanding of linkage disequilibrium (LD) structure is essential for designing single-nucleotide polymorphism (SNP) genotyping arrays, successful association mapping of genetic factors underlying traits of interest, establishing mechanisms of genetic recombination and elucidating patterns of selection and population structure. This is particularly true for agricultural species such as commercial chicken (Gallus gallus) where LD analysis has the potential to establish the genetic drivers of selection and therefore contribute to further commercial development of lines.
The chicken genome comprises macrochromosomes, intermediate chromosomes and microchromosomes. The macrochromosomes (GGA1–5) span 50–200 Mb, intermediate chromosomes (GGA6–10) range from 20 to 40 Mb and 28 microchromosomes (GGA11–38) that average ~12 Mb (Hillier et al., 2004; Megens et al., 2009; Schmid et al., 2015). The microchromosomes are characterised as having higher GC content, gene density and much higher recombination rates compared with macrochromosomes (~50–100 versus ~300 kb cM−1 in macrochromosomes). The latter may reflect the requirement for a minimum of at least one chiasma for each chromosome per meiosis and a higher density of cohesin-binding sites (Groenen et al., 2009).
Previous studies of LD in the chicken have established that the microchromosomes show reduced LD compared with macrochromosomes, and that these differences are almost completely explained by differences in the recombination rate (Megens et al., 2009). Studies of egg-laying chickens indicate higher levels of LD compared with broilers (Heifetz et al., 2005; Andreescu et al., 2007). Despite relatively low levels of LD in broilers, Andreescu et al. (2007) determined that there is significant overlap in LD for marker pairs across nine different commercial broiler lines.
LD maps constitute the LD analogue of the genetic linkage map and have been extensively utilised for human data (Maniatis et al., 2002; Tapper et al., 2005), and have also been previously applied to agricultural species (Khatkar et al., 2006). We here construct LD maps according to the Malécot-Morton model, using the program LDMAP (Maniatis et al., 2002; Tapper et al., 2005). This model is defined as:
where 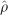 is the association between SNPs; the asymptote L is the ‘background' association between unlinked markers that is increased in small sample sizes and with residual population structure; M reflects association at zero distance with values ~1 consistent with monophyletic origin and <1 with polyphyletic inheritance; ε is the rate of LD decline; and d is the physical distance in kb between SNPs (Maniatis et al., 2002).
is the association between SNPs; the asymptote L is the ‘background' association between unlinked markers that is increased in small sample sizes and with residual population structure; M reflects association at zero distance with values ~1 consistent with monophyletic origin and <1 with polyphyletic inheritance; ε is the rate of LD decline; and d is the physical distance in kb between SNPs (Maniatis et al., 2002).
One LD unit (LDU, equal to εd) corresponds to the (highly variable) physical distance over which LD declines to ‘background' levels. Compared with the physical map, the LDU scale delimits regions of LD breakdown (that may represent recombination hot spots) as steps, and broader regions of low haplotype diversity (blocks) as plateaus. The overall LDU map length reflects time since an effective population bottleneck (Zhang et al., 2002; Service et al., 2006). Therefore, populations with shorter LDU maps have been founded more recently, experienced a more recent selective sweep or have a smaller effective population size compared with those with longer maps.
The close correspondence between LD patterns and much lower-resolution linkage maps suggest a dominant role for recombination in LD structure. However, unlike linkage maps that provide a low-resolution description of recombination over recent generations, LD maps are constructed from population data and reflect the historical impacts of recombination, mutation, selection and population history. LDU maps of commercial chicken lines may provide new insights into patterns of recombination and selection. Previous studies have begun to describe differences in recombination across Gallus genomes based on linkage and LD structure (Megens et al., 2009), and high-resolution genome-wide LD maps have the potential to yield further insights.
Birds appear to lack the zinc-finger protein PRDM9 that is required for recombination hot spot localisation in humans and other mammals (Fumasoni et al., 2007; Oliver et al., 2009; Myers et al., 2010). Despite this, recent work by Singhal et al. (2015) has shown that hot spots are highly concordant between wild bird (finch) populations, apparently because of the concentration of recombination events near functional elements of the genome, namely CpG islands and transcription start sites (TSSs).
Here, we construct genome-wide LDU maps for three chicken breeds: broilers (BRO), white egg layers (WEL) and brown egg layers (BEL). We contrast the LD structure across the three breeds, and evaluate the consistency of LD patterns between breeds, potential recombination hot spots and differences between chromosome types and motifs underlying major features of the maps. In addition, we compare patterns between specific commercial lines (sub-populations) within breeds.
Materials and methods
Genotypic data used in this work are as reported in the validation populations of Kranis et al. (2013), with all genomic coordinates based on the Gallus_gallus-4.0 (galGal4) reference assembly. The samples described by Kranis et al. (2013) were supplied by Aviagen (Huntsville, AL, USA; broilers), Hy-line International (West Des Moines, IA, USA) and Lohmann of Synbreed Consortium (the white and brown egg layers). As described in this paper the variants genotyped were selected to provide an overall uniform distribution of allele frequencies, across the three breed types, best capturing the diversity of each. All of the genotype data used in this study are from individuals that share <80% identity by similarity genome-wide to minimise population substratification and have >95% genotyping completeness. Genotype assessment was performed using PLINK v1.07 (Purcell et al., 2007). Multidimensional scaling was undertaken using all autosomal markers in order to evaluate the population structure of the samples.
Once the analytical cohorts were defined, SNP marker filtering was undertaken independently for each cohort. Markers with <95% genotyping completeness, minor allele frequency<0.05 or Hardy–Weinberg equilibrium deviation P-value <0.001 were removed to leave a data set containing only common, high-quality markers. Within each breed the inbreeding coefficient (F) (Wright, 1922) was calculated using the PLINK program. LD maps were generated for the assembled autosomes GGA1–28 on filtered data according to the Malécot-Morton model using LDMAP (Morton et al., 2001; Kuo et al., 2007). Where necessary, filtered genotype data were split into ~25 000 marker segments (with 200 marker overlap) to allow for parallelised processing. Overlapping map segments were then trimmed of the terminal 25 markers, and merged to form complete, contiguous whole-chromosome LD maps for the assembled autosomes.
Comparisons of LD maps with existing linkage maps were made. The order of markers in linkage maps from Elferink et al. (2010) was revised in line with galGal4 from the native assembly based upon SNP positions within dbSNP (Sherry et al., 2001). Following transition to the galGal4 order, a small number of disordered markers in the linkage map, which distorted the cumulative map, were removed.
To compare map structure between breeds, we focussed on the macrochromosomes GGA1–5. These were chosen to avoid confounding factors such as potentially incomplete reference assemblies, as well as varying recombination rates for the microchromosomes (Hillier et al., 2004; Groenen et al., 2009; Elferink et al., 2010; Schmid et al., 2015). Spearman's rank correlation of LDU lengths for all 40 kb regions among the three breeds was calculated, after Rubin et al. (2010).
For finer-scale interrogation of the LDU length of 5 kb regions, we investigated the concordance seen for these regions with the longest LDU length. We performed these comparisons based upon LDU percentiles, as opposed to absolute LDU lengths, to allow for the differing global LD map lengths for the breeds. This analysis gives an indication of the extent to which narrow regions of intense LD breakdown (that may align with recombination hot spots) are shared between breeds/lines. A high degree of concordance between long LDU segments might suggest a high proportion of shared recombination hot spots between the samples considered. For comparison, we also compared human European and African populations (Pengelly et al., 2015), the two larger BRO2/3 lines, as well as BRO3a/b, a random bisection of the largest population, BRO3. As a final control, a randomised data set was used for which an equal number of 5 kb regions were randomly selected independently for each data set, and the concordance calculated; 100 pseudo-replicates were performed for each percentile cutoff.
For comparing LDU decline rates with genome features we focussed on the BRO data set as it has the largest sample size in the data set. GC content was calculated directly from the reference sequence for 5 kb regions, CpG islands were defined according to the UCSC (University of California, Santa Cruz) genome browser and Ensembl annotations were used to define transcripts (Karolchik et al., 2014). BEDTools was used to calculate the distance between elements and regions (Quinlan and Hall, 2010).
Results
Input data
The genotype data used in this work are as reported in the validation populations of Kranis et al. (2013), with ~1.8 million genotyped SNPs in total. Multidimensional scaling of all samples shows distinct clustering of breeds, though with three distinct population clusters within each breed, consistent with the three genotyped lines (Supplementary Figure S1). For initial LDU map construction, all lines of a breed were pooled. Sample groups used are described in Table 1. For clarity, we use BRO, BEL and WEL to refer to each pooled breed data set, and BRO2, BRO3 and so on to refer to a distinct line within breed where analysed.
Table 1. Counts of chickens and SNPs remaining post filtering.
| BRO | BRO2a | BRO3 | BRO3a | BRO3b | BEL | WEL | ||
|---|---|---|---|---|---|---|---|---|
| Founders | Males | 58 | – | 50 | 26 | 24 | 12 | 8 |
| Females | 17 | – | 9 | 4 | 5 | 40 | 38 | |
| Total | 123a | 48 | 59 | 30 | 29 | 52 | 46 | |
| SNPs | Raw count | 966 355 | 790 531 | 789 359 | 692 467 | 778 135 | 891 200 | 691 954 |
| MAFb | 833 639 | 658 548 | 638 947 | 631 449 | 645 713 | 796 430 | 627 294 | |
| HWEc | 760 893 | 789 450 | 788 284 | 691 625 | 777 771 | 763 931 | 420 130 | |
| Missingnessd | 966 346 | 788 594 | 787 732 | 690 038 | 776 114 | 888 903 | 690 298 | |
| Final count | 630 435 | 655 905 | 636 535 | 628 382 | 643 554 | 667 605 | 354 737 |
Abbreviations: BEL, brown egg layers; BRO, broilers; HWE, Hardy–Weinberg equilibrium; MAF, minor allele frequency; SNP, single-nucleotide polymorphism; WEL, white egg layers.
Sex data unavailable for BRO2 line.
Markers with minor allele frequency <0.05 within the cohort excluded.
Markers with Hardy–Weinberg equilibrium deviation P-value <0.001 within the cohort.
Markers with >5% data missing excluded.
Global map properties
LD maps were generated for all assembled autosomal chromosomes for the three breeds (Supplementary Figure S2). A representative chromosomal LD map for GGA8 is shown in Figure 1. The physical map of the chromosome is represented on the x axis, whereas the y axis shows the LDU maps for each breed and the linkage map in centiMorgans (cM) (Elferink et al., 2010). As in human LDU and cM maps, there is a large central region showing little change in LDU or cM, consistent with the location of the submetacentric centromere, where recombination is suppressed and there is therefore intense LD (Tapper et al., 2005; Krasikova et al., 2006). Summary length statistics for all autosomes are shown in Table 2.
Figure 1.

Linkage and LD maps of GGA8 for all breeds. The broadly analogous structure of the linkage map and LD maps for the three populations can be seen. All maps contain a large plateau of ∼10 000 kb, corresponding to the centromere region. Overall length of the LD maps is inversely related to the strength of LD within a breed. BRO show the lowest LD overall reflecting relatively high haplotype diversity, whereas WEL show strongest LD and lowest population haplotype diversity. Note that there is no expectation that cM and LDU maps be of similar lengths, as the LDU map length is dependent upon population diversity.
Table 2. Physical, linkage and LDU map lengths for the three breeds.
| Linkage | BRO | BEL | WEL | |||||
|---|---|---|---|---|---|---|---|---|
| Chr | Length (Mb) | cM | LDU | LDU/cM | LDU | LDU/cM | LDU | LDU/cM |
| 1 | 195.2 | 413.5 | 713.7 | 1.7 | 612.4 | 1.5 | 363.8 | 0.9 |
| 2 | 148.8 | 281.3 | 452.1 | 1.6 | 462.8 | 1.6 | 207.8 | 0.7 |
| 3 | 110.4 | 236.9 | 439.2 | 1.9 | 359.4 | 1.5 | 186.3 | 0.8 |
| 4 | 90.2 | 195.2 | 309.8 | 1.6 | 277.1 | 1.4 | 164.1 | 0.8 |
| 5 | 59.5 | 154.4 | 198.6 | 1.3 | 207.1 | 1.3 | 126.3 | 0.8 |
| 6 | 34.9 | 93.8 | 171.9 | 1.8 | 114.7 | 1.2 | 81.4 | 0.9 |
| 7 | 36.2 | 103.1 | 150.3 | 1.5 | 153.1 | 1.5 | 76.3 | 0.7 |
| 8 | 28.7 | 96.6 | 134.5 | 1.4 | 112.2 | 1.2 | 57.0 | 0.6 |
| 9 | 23.4 | 88.1 | 123.4 | 1.4 | 101.4 | 1.2 | 61.0 | 0.7 |
| 10 | 19.9 | 80.6 | 97.4 | 1.2 | 77.0 | 1.0 | 65.3 | 0.8 |
| 11 | 19.3 | 64.0 | 73.9 | 1.2 | 85.4 | 1.3 | 34.5 | 0.5 |
| 12 | 19.9 | 69.1 | 101.4 | 1.5 | 88.4 | 1.3 | 51.6 | 0.7 |
| 13 | 17.7 | 62.7 | 76.3 | 1.2 | 85.7 | 1.4 | 38.5 | 0.6 |
| 14 | 15.1 | 67.4 | 65.9 | 1.0 | 68.1 | 1.0 | 46.2 | 0.7 |
| 15 | 12.6 | 53.6 | 61.6 | 1.1 | 54.6 | 1.0 | 24.8 | 0.5 |
| 16 | 0.5 | 59.1 | 2.0 | 0.0 | 2.3 | 0.0 | 1.1 | 0.0 |
| 17 | 10.3 | 50.9 | 65.7 | 1.3 | 52.6 | 1.0 | 24.5 | 0.5 |
| 18 | 11.2 | 51.7 | 56.4 | 1.1 | 53.9 | 1.0 | 31.1 | 0.6 |
| 19 | 10.0 | 52.3 | 67.9 | 1.3 | 57.6 | 1.1 | 31.3 | 0.6 |
| 20 | 14.2 | 55.1 | 61.5 | 1.1 | 58.6 | 1.1 | 31.1 | 0.6 |
| 21 | 6.8 | 56.9 | 56.8 | 1.0 | 51.1 | 0.9 | 29.8 | 0.5 |
| 22 | 4.1 | 56.4 | 21.9 | 0.4 | 21.7 | 0.4 | 9.5 | 0.2 |
| 23 | 5.7 | 52.3 | 46.6 | 0.9 | 39.2 | 0.7 | 27.6 | 0.5 |
| 24 | 6.2 | 53.2 | 57.6 | 1.1 | 47.6 | 0.9 | 33.0 | 0.6 |
| 25 | 2.1 | 57.1 | 17.8 | 0.3 | 23.8 | 0.4 | 6.8 | 0.1 |
| 26 | 4.9 | 52.3 | 37.4 | 0.7 | 39.5 | 0.8 | 21.9 | 0.4 |
| 27 | 5.2 | 51.0 | 32.6 | 0.6 | 34.3 | 0.7 | 28.2 | 0.6 |
| 28 | 4.7 | 53.6 | 30.4 | 0.6 | 37.1 | 0.7 | 20.9 | 0.4 |
| Total | 917.7 | 2762.2 | 3724.6 | 1.3 | 3378.7 | 1.2 | 1881.7 | 0.7 |
Abbreviations: BEL, brown egg layers; BRO, broilers; Chr, chromosome; LDU, linkage disequilibrium unit; WEL, white egg layers.
LDU map lengths reflect haplotypic diversity within that population (Service et al., 2006; Pengelly et al., 2015) and can be compared with independent measures of population diversity such as F inbreeding coefficients. The mean F inbreeding coefficients are 0.21, 0.26 and 0.51 for the BRO, BEL and WEL populations, respectively, with the greater value for WEL indicating more limited genetic diversity within the population. In comparison, the ratio of LDU/Mb map lengths is also variable between breeds (3.50, 3.18 and 1.74 for BRO, BEL and WEL, respectively, across the autosomes; Table 2). This ranking of breeds by LDU length is consistent with the trend obtained from the F-statistic, in line with expectations. There is a general trend towards a lower LDU/cM ratio of map lengths for the smaller chromosomes (Figure 2), with a negative exponential relationship between length of chromosome in megabases and the LDU/cM ratio (r2>0.75, P<1.7 × 10−9 for all breeds).
Figure 2.

Relationship between physical chromosome length and LDU/cM ratio for all autosomes in the three breeds. There is a clear trend for the physically smaller chromosomes to exhibit lower LDU/cM ratios, with a negative exponential relationship. Lines indicate best fit for log10(length) vs LDU/cM.
LD map structure between breeds
To compare map structure between breeds, we focussed on the macrochromosomes GGA1–5. These were chosen because they are known to have more complete reference assemblies (compared with the microchromosomes), and have less variable recombination rates than the microchromosomes (Hillier et al., 2004; Groenen et al., 2009; Elferink et al., 2010; Schmid et al., 2015). Following map generation, we interrogated the fine map structure for the breeds to establish the extent to which patterns of LD are conserved between the breeds. There was a weak, though highly significant, correlation between breeds of LDU lengths of corresponding 40 kb regions (Spearman's ρ<0.21; P<2.2 × 10−16 for all pairwise comparisons; Figure 3). Narrow regions where there is strong breakdown of LD may align with recombination hot spots, for which there is a high degree of concordance across human populations (Jeffreys et al., 2004; Myers et al., 2005; Tapper et al., 2005; Pengelly et al., 2015). We investigated the extent to which regions with LD breakdown are conserved across the three breeds. In humans, recombination hot spots span just 1–2 kb (Jeffreys et al., 2004). To test concordance of narrow regions of LD breakdown we obtained LDU lengths within 5 kb windows; this window size was selected to allow for the incomplete resolution of array-based genotyping (Pengelly et al., 2015). We assessed whether regions with the longest LDU lengths within a breed were conserved between breeds. For pairwise comparisons between breeds, there is low concordance in the top LDU length percentile of 5 kb windows, with just ~5% concordance between breeds for the top 5th percentile (Figure 4). This is far lower than the concordance seen in humans, as well as that seen for the BRO3a/3b comparison that is derived from a random split of the BRO3 line.
Figure 3.

Comparison of LD breakdown intensity on GGA2 for the three breeds. The LDU/kb ratio is shown for sliding 40 kb windows. A common region of strong LD is shown at ~50 Mb, corresponding to the centromere. There are minimal other trends apparent in the localisation of LD intensities between breeds (ρ<0.21; P<2.2 × 10−16 for all pairwise comparisons).
Figure 4.

Pairwise concordance of regions of LD breakdown (indicative of recombination hot spots) between populations. Shown is the proportion of regions in the top n percentile that intersect between the breeds. For pairwise comparisons between BRO/BEL/WEL ~5% of regions in the top 5 percentile are concordant. This proportion is also similar where two separate BRO lines (BRO2/BRO3) are compared. The low level of concordance between regions of LD breakdown, even between lines within breed, suggests there may be marked differences in haplotype structure as regions of LD breakdown align with increased haplotype diversity (Daly et al., 2001). When the largest population is bisected (BRO3a/b), this proportion is ~27%, still far lower than the equivalent comparison between European and African human populations (Pengelly et al., 2015). All comparisons show a greater proportion of concordance than the randomised data set, showing that the patterns are not wholly stochastic. Shaded regions indicate 95% confidence intervals of the trendlines, and concordance was calculated for 0.1% increments.
Characteristics of regions of LD breakdown
Despite the low concordance of the alignment of narrow regions of LD breakdown for interbreed comparisons for chickens, the concordance seen is approximately twofold greater than that expected by chance. Recombination intensity is known to be dependent on sequence context, and therefore hot spots are not expected to be randomly distributed (Groenen et al., 2009; Elferink et al., 2010). One key determinant of recombination rate, GC content (Groenen et al., 2009), was found to be significantly increased in 5 kb regions in the top first percentile of LDU length of any breed when compared with regions of zero LDU length in all breeds (42.0% GC vs 39.2% respectively, P<2.2 × 10−16). We further compared the LDU/kb ratio for 5 kb regions with their distance to the nearest CpG islands and TSS and found a highly significant, though weak, negative correlation in our data for both TSS and CpG islands, consistent with previous findings in finches (Figure 5) (Singhal et al., 2015).
Figure 5.

Association of LD breakdown with displacement from nearest functional element, namely CpG islands and TSS. There is a strong negative correlation between the distance from the functional elements and LDU/kb (ρ=−0.12 for CpG islands, ρ=−0.10 for TSS, P<2.2 × 10−16 for each comparison). Shown is the mean LDU/kb ratio for 5 kb bins, and shaded area indicates 95% confidence interval.
In order to better characterise the relationship between TSS and CpG islands with narrow regions of LD breakdown we constructed a 2 × 4 contingency table for 5 kb regions exhibiting limited LD breakdown (<0.003 LDU/kb) or intense LD breakdown (⩾0.003 LDU/kb) against whether the regions are within 125 kb of a CpG island, TSS, both or neither (Table 3). These values were selected based upon the approximate points of inflection in Figure 5. There was a highly significant deviation from the expected distributions under the null hypothesis (P=9.5 × 10−224, χ2 test). Regions within 125 kb of both a TSS and CpG island were 1.36-fold more likely to show strong LD breakdown; this is in excess of the additive enrichment for TSS solely and CpG island solely. This suggests that it is an interplay of factors that underlies the localisation of hot spots, with the location of CpG islands having the strongest effect in isolation.
Table 3. Odds ratios for recombination hot spot regions (LDU/kb ⩾0.003) being within 125 kb of genomic features.
| Feature | Odds ratio |
|---|---|
| Neither CpG nor TSS | 1.00 |
| CpG only | 1.12 |
| TSS only | 1.06 |
| Both CpG and TSS | 1.37 |
Abbreviations: LDU, linkage disequilibrium unit; TSS, transcription start site.
Discussion
The analysis of LD maps for the three breeds indicates extensive genome-wide LD. As one LD unit represents the distance over which LD declines to background levels, the genome-wide Mb/LDU ratio gives an indication of the average extent of LD, or ‘swept radius'. Average swept radii for the three breeds are 246 kb for BRO, 272 kb for BEL and 488 kb for WEL. In contrast, the corresponding figures for human populations are 55 kb for Europeans and 39 kb for Africans (Lau et al., 2007). Although estimates of swept radii are weakly influenced by SNP marker density (Pengelly et al, 2015), they provide an indication of, as expected, the strong LD found in commercial chicken populations that have been subject to intense selection. However, the profound differences in fine-scale LD structure we have identified are less expected. Although some large-scale genomic features such as centromeric regions, which typically have extensive and intense LD, are shared across breeds for some chromosomes (see, for example, Figure 1), there is relatively little concordance in genome-wide LD structure. The contours of the LD maps show many genome regions with widely divergent LD structure (Figure 3) and the overall correlation in LDU lengths of 40 kb windows is low (ρ=0.21). In contrast, the fine-scale LD structure between human populations is sufficiently concordant to support a merged ‘cosmopolitan' LD map that recovers 91–95% of the information within population-specific maps (Gibson et al., 2005).
The LDU/cM ratio of chromosome lengths is known to be virtually constant in human populations, strongly suggesting that recombination is the primary determinant of LD structure. However, for the three chicken breeds the linear relationship breaks down, with smaller chromosomes having a lower LDU/cM ratio (Figure 2). Similarly, Megens et al. (2009) found that recombination rates estimated from LD data were discordant with those obtained from the linkage map. Specifically, they found that the recombination frequency for two microchromosomes (GGA26 and GGA27) estimated from LD was only 2.8 times greater than that of macrochromosomes (GGA1 and GGA2) when the expectation from the linkage map was 4.5-fold greater recombination on the microchromosomes (Groenen et al., 2009). This discrepancy was attributed to biases in fitting a model using effective population sizes computed in physical rather than genetic distance windows.
The breakdown in linear correlation between LDU and cM map lengths could have one or more causes. The evidence suggests that the apparent historical recombination intensity (based on LD maps) is lower than current recombination intensity (based on the linkage map) for the smaller chromosomes. One possible explanation for the deviation from a linear relationship between LD and cM lengths is the intense selection that has underpinned the recent population history of these commercial chicken lines. Selection has been previously reported to distort LD-based maps of recombination (O'Reilly et al., 2008) because of the expected reduced haplotypic diversity at loci under purifying selection. This leads to an underestimation of recombination in these regions compared with the linkage map. As an extension of this, genes will be, on average, more conserved than intergenic regions and thus genes will have a reduced LDU/cM ratio, on average (Gibson et al., 2013). As the smaller chicken chromosomes have higher gene density, the impact of selection on reducing haplotypic diversity may have been more intense as compared with chromosomes with lower gene density. There is a strong exponential inverse correlation between chromosome size and gene transcripts per megabase (r2=0.745, P=3.42 × 10−9). The differential impact of selection on increased gene density for the smaller chromosomes may therefore at least partly explain the reduced LDU/cM ratio observed.
However, it is important to recognise that the reference genome sequence is incomplete for several chromosomes, particularly the microchromosomes (Schmid et al., 2015). As the construction of linkage maps requires lower density markers and measurable linkage extends much further than LD, the construction of a complete linkage map of a chromosome is less sensitive to the omission of small regions with missing or unreliable sequence. In contrast, LD is much shorter range and LD maps will be truncated in regions where SNP coverage is incomplete because of assembly gaps and poor SNP coverage. This is however unlikely to be the sole explanation for the lower LDU/cM ratios in the smaller chromosomes due to the close negative exponential relationship between the physical chromosome size and LDU/cM ratio, perhaps indicative of an underlying biological mechanism as opposed to solely a technical artefact because of the incomplete assemblies.
Our finding that the LD structure across the three breeds is highly discordant is in marked contrast to comparisons across human populations (Pengelly et al., 2015). Specifically, the pattern seen in humans of narrow regions of LD breakdown, which align with recombination hot spots and are highly concordant across populations, is not observed in chickens that show little concordance of similar regions across breeds. Comparisons between breeds, and even between lines within a breed (BRO2–BRO3, Figure 4) show alignment of such regions that is only slightly greater than ‘random'. Concordance within a random split of a sub-population (BRO3a–BRO3b) is much higher, but even then does not show the degree of alignment in the hot spot landscape of human European and African populations.
Although the different extent of genome-wide LD between the breeds has been known for some time (Aerts et al., 2007) and the LD pattern between white and brown egg layers has been recognised as clearly different (Qanbari et al., 2010), this is the among the first studies to recognise highly divergent fine-scale LD structure between breeds, as well as distinct lines of each breed. This finding has implications for trait mapping as it suggests that to ensure coverage, panels of tagging SNPs would be optimally selected only within the line of interest and that, unlike in human analyses, a ‘standard' linkage map may be less useful if it is not representative of the breed-specific recombination landscape.
Analyses of human LD maps have established that the recombination landscape can be recovered from LD structure (Jeffreys et al., 2004; Tapper et al., 2008; Myers et al., 2010). From the derived recombination landscape the chromatin-modifying zinc-finger protein PRDM9 was shown to regulate recombination at 40% of human hot spots by binding to a degenerate 13 base pair motif (Baudat et al., 2010; Myers et al., 2010). Remarkably, despite genomic similarity between humans and chimpanzees, there is virtually no sharing of recombination hot spot locations. Myers et al. (2010) found chimpanzee PRDM9 has a dramatically different predicted binding sequence to that of humans. PRDM9 sequences are known to exhibit extremely rapid evolution that explains lack of hot spot conservation in other species that have PRDM9. However, chicken genomes, along with all other avian genomes tested (48 species) appear to lack PRDM9 (Oliver et al., 2009; Singhal et al., 2015). Singhal et al. (2015) examined the genomes of the zebra finch (Taeniopygia guttata) and long-tailed finch (Poephia acuticauda) and found recombination hot spots are enriched near CpG islands and TSSs. In marked contrast to the lack of hot spot sharing between humans and chimpanzees, they found a high degree of sharing between these birds that were selected as showing divergence comparable to humans and chimpanzees. They argue that whereas the binding specificity of PRDM9-mediated recombination leads to rapid turnover and evolution of hot spots, association with functional genomic features, such as CpG islands and TSSs, leads to stasis in which there is conservation of hot spot locations over, potentially, tens of millions of years. However, in contrast, the evidence presented here suggests very limited sharing of regions of LD breakdown for the three chicken breeds based on LD structure.
We have established that the relationship between TSS and CpG islands with regions of LD breakdown/recombination hot spots, as identified in finches by Singhal et al. (2015), is also found in chickens. It is possible that although recombination in chickens is associated with these functional elements, this relationship has not resulted in convergence to the point where recombination hot spots are recognisably stable between lines. Perhaps the population history of the commercial lines studied has not been sufficiently long to allow for convergence of LD patterns. Furthermore, it is possible that small effective population sizes of these breeds also contributes to the observed limited convergence in LD structure. Further work, including the studying of outbred, wild Gallus populations, as well as whole-genome sequence-based LD mapping, would allow for further elucidation of mechanisms underlying the architecture of patterns of LD in chickens (Gheyas et al., 2015). Improvements to the reference assembly currently underway will provide more complete assemblies for the microchromosomes (Schmid et al., 2015), allowing for further investigation into differential properties of the chromosomes. It would be informative to extend these investigations to other agricultural species in order to determine whether this effect is specific to Gallus, or can also be observed in other selected avian, piscine and mammalian species.
LD maps have been shown to be of use in refining the order and orientation of contigs during compilation of reference genomes where there is ambiguity that may be of utility for enabling further improvements to the assembly (Ennis et al., 2001). LD maps, as described herein, are readily produced for any population for which genotype data are available, without requiring pedigree data. Given the high level of discordance in LD patterns between lines, data derived from the line of interest would provide far greater precision when utilised for trait mapping and other studies, as well as the optimisation of genotyping arrays to allow for maximal coverage for the minimal number of SNP markers for each line.
Data archiving
Genotype data used herein are available in the Dryad Digital Repository, http://dx.doi.org/10.5061/dryad.48gp0.
Acknowledgments
This work is supported by the UK Biotechnology and Biological Sciences Research Council (BBSRC) ‘Sparking Impact' scheme and by the University of Southampton, Faculty of Medicine Doctoral Training Fund. We acknowledge the use of the IRIDIS High Performance Computing Facility, and associated support services at the University of Southampton, in the completion of this work.
The authors declare no conflict of interest.
Footnotes
Supplementary Information accompanies this paper on Heredity website (http://www.nature.com/hdy)
Supplementary Material
References
- Aerts J, Megens HJ, Veenendaal T, Ovcharenko I, Crooijmans R, Gordon L et al. (2007). Extent of linkage disequilibrium in chicken. Cytogenet Genome Res 117: 338–345. [DOI] [PubMed] [Google Scholar]
- Andreescu C, Avendano S, Brown SR, Hassen A, Lamont SJ, Dekkers JCM. (2007). Linkage disequilibrium in related breeding lines of chickens. Genetics 177: 2161–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M et al. (2010). PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327: 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daly MJ, Rioux JD, Schaffner SF, Hudson TJ, Lander ES. (2001). High-resolutions haplotype structure in the human genome. Nat Genet 29: 229–232. [DOI] [PubMed] [Google Scholar]
- Elferink MG, van As P, Veenendaal T, Crooijmans RPMA, Groenen MAM. (2010). Regional differences in recombination hotspots between two chicken populations. BMC Genet 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ennis S, Collins A, Tapper W, Murray A, MacPherson JN, Morton NE. (2001). Allelic association discriminates draft orders. Ann Hum Genet 65: 503–504. [DOI] [PubMed] [Google Scholar]
- Fumasoni I, Meani N, Rambaldi D, Scafetta G, Alcalay M, Ciccarelli FD. (2007). Family expansion and gene rearrangements contributed to the functional specialization of PRDM genes in vertebrates. BMC Evol Biol 7: 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gheyas AA, Boschiero C, Eory L, Ralph H, Kuo R, Woolliams JA et al. (2015). Functional classification of 15 million SNPs detected from diverse chicken populations. DNA Res 22: 205–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson J, Tapper W, Ennis S, Collins A. (2013). Exome-based linkage disequilibrium maps of individual genes: functional clustering and relationship to disease. Hum Genet 132: 233–243. [DOI] [PubMed] [Google Scholar]
- Gibson J, Tapper W, Zhang W, Morton N, Collins A. (2005). Cosmopolitan linkage disequilibrium maps. Hum Genomics 2: 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groenen MAM, Wahlberg P, Foglio M, Cheng HH, Megens HJ, Crooijmans RPMA et al. (2009). A high-density SNP-based linkage map of the chicken genome reveals sequence features correlated with recombination rate. Genome Res 19: 510–519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heifetz EM, Fulton JE, O'Sullivan N, Zhao H, Dekkers JCM, Soller M. (2005). Extent and consistency across generations of linkage disequilibrium in commercial layer chicken breeding populations. Genetics 171: 1173–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillier LW, Miller W, Birney E, Warren W, Hardison RC, Ponting CP et al. (2004). Sequence and comparative analysis of the chicken genome provide unique perspectives on vertebrate evolution. Nature 432: 695–716. [DOI] [PubMed] [Google Scholar]
- Jeffreys AJ, Holloway JK, Kauppi L, May CA, Neumann R, Slingsby MT et al. (2004). Meiotic recombination hot spots and human DNA diversity. Philos Trans R Soc Lond B Biol Sci 359: 141–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolchik D, Barber GP, Casper J, Clawson H, Cline MS, Diekhans M et al. (2014). The UCSC Genome Browser database: 2014 update. Nucleic Acids Res 42: D764–D770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khatkar MS, Thomson PC, Tammen I, Cavanagh J AL, Nicholas FW, Raadsma HW. (2006). Linkage disequilibrium on chromosome 6 in Australian Holstein-Friesian cattle. Genet Sel Evol 38: 463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kranis A, Gheyas AA, Boschiero C, Turner F, Yu L, Smith S et al. (2013). Development of a high density 600K SNP genotyping array for chicken. BMC Genomics 14: 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krasikova A, Deryusheva S, Galkina S, Kurganova A, Evteev A, Gaginskaya E. (2006). On the positions of centromeres in chicken lampbrush chromosomes. Chromosom Res 14: 777–789. [DOI] [PubMed] [Google Scholar]
- Kuo T-Y, Lau W, Collins AR. (2007). LDMAP: the construction of high-resolution linkage disequilibrium maps of the human genome. Methods Mol Biol 376: 47–57. [DOI] [PubMed] [Google Scholar]
- Lau W, Kuo TY, Tapper W, Cox S, Collins A. (2007). Exploiting large scale computing to construct high resolution linkage disequilibrium maps of the human genome. Bioinformatics 23: 517–519. [DOI] [PubMed] [Google Scholar]
- Maniatis N, Collins A, Xu CF, McCarthy LC, Hewett DR, Tapper W et al. (2002). The first linkage disequilibrium (LD) maps: delineation of hot and cold blocks by diplotype analysis. Proc Natl Acad Sci USA 99: 2228–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Megens H-J, Crooijmans RPMA, Bastiaansen JWM, Kerstens HHD, Coster A, Jalving R et al. (2009). Comparison of linkage disequilibrium and haplotype diversity on macro- and microchromosomes in chicken. BMC Genet 10: 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton NE, Zhang W, Taillon-Miller P, Ennis S, Kwok PY, Collins A. (2001). The optimal measure of allelic association. Proc Natl Acad Sci USA 98: 5217–5221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers S, Bottolo L, Freeman C, McVean G, Donnelly P. (2005). A fine-scale map of recombination rates and hotspots across the human genome. Science 310: 321–324. [DOI] [PubMed] [Google Scholar]
- Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS et al. (2010). Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327: 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver PL, Goodstadt L, Bayes JJ, Birtle Z, Roach KC, Phadnis N et al. (2009). Accelerated evolution of the Prdm9 speciation gene across diverse metazoan taxa. PLoS Genet 5: e1000753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly PF, Birney E, Balding DJ. (2008). Confounding between recombination and selection, and the Ped/Pop method for detecting selection. Genome Res 18: 1304–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pengelly RJ, Tapper W, Gibson J, Knut M, Tearle R, Collins A et al. (2015). Whole genome sequences are required to fully resolve the linkage disequilibrium structure of human populations. BMC Genomics 16: 666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purcell S, Neale B, Todd-Brown K, Thomas L, Ferreira MAR, Bender D et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 81: 559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qanbari S, Hansen M, Weigend S, Preisinger R, Simianer H. (2010). Linkage disequilibrium reveals different demographic history in egg laying chickens. BMC Genet 11: 103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin C-J, Zody MC, Eriksson J, Meadows JRS, Sherwood E, Webster MT et al. (2010). Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464: 587–591. [DOI] [PubMed] [Google Scholar]
- Schmid M, Smith J, Burt DW, Aken BL, Antin PB, Archibald AL et al. (2015). Third report on chicken genes and chromosomes 2015. Cytogenet Genome Res 145: 78–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Service S, DeYoung J, Karayiorgou M, Roos JL, Pretorious H, Bedoya G et al. (2006). Magnitude and distribution of linkage disequilibrium in population isolates and implications for genome-wide association studies. Nat Genet 38: 556–560. [DOI] [PubMed] [Google Scholar]
- Sherry ST, Ward MH, Kholodov M, Baker J, Phan L, Smigielski EM et al. (2001). dbSNP: the NCBI database of genetic variation. Nucleic Acids Res 29: 308–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal S, Leffler E, Sannareddy K, Turner I, Venn O, Hooper D et al. (2015). Stable recombination hotspots in birds. Science 350: 928–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper W, Collins A, Gibson J, Maniatis N, Ennis S, Morton NE. (2005). A map of the human genome in linkage disequilibrium units. Proc Natl Acad Sci USA 102: 11835–11839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tapper W, Gibson J, Morton NE, Collins A. (2008). A comparison of methods to detect recombination hotspots. Hum Hered 66: 157–169. [DOI] [PubMed] [Google Scholar]
- Wright S. (1922). Coefficients of inbreeding and relationship. Am Nat 56: 330. [Google Scholar]
- Zhang W, Collins A, Maniatis N, Tapper W, Morton NE. (2002). Properties of linkage disequilibrium (LD) maps. Proc Natl Acad Sci USA 99: 17004–17007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.