

Abstract
The development of glycobiology relies on the sources of particular oligosaccharides in their purest forms. As the isolation of the oligosaccharide structures from natural sources is not a reliable option for providing samples with homogeneity, chemical means become pertinent. The growing demand for diverse oligosaccharide structures has prompted the advancement of chemical strategies to stitch sugar molecules with precise stereo‐ and regioselectivity through the formation of glycosidic bonds. This Review will focus on the key developments towards chemical O‐glycosylations in the current century. Synthesis of novel glycosyl donors and acceptors and their unique activation for successful glycosylation are discussed. This Review concludes with a summary of recent developments and comments on future prospects.
Keywords: catalysis, glycosylation reactions, promoters, protecting groups, stereoselectivity
1. Introduction
Carbohydrates, or sugars, have been deemed an important weapon in solving many of nature's mysteries. They are the most diverse and the most abundant biomolecules on earth. Apart from their energy‐storage functions, they were only assumed to serve as the structural and protective elements in the plant, bacterial, and animal cell walls.1 It was only in the later part of the 20th century that the involvement of carbohydrates in various other biological processes2 became apparent. The renaissance witnessed over the past few years in the field of glycobiology shows that glycoconjugates have enormous significance in various biochemical processes, such as molecular recognition, cell–cell interaction, immunological recognition, transmission of biological information, and so forth.3 Proper scientific knowledge of the mechanism of these biological processes, in turn, leads to the study of various new applications in biomedical research. These applications invariably lead to the advancement of modern day science, thereby increasing its fundamental interest to scientists.4 Thus, the demand for homogenous carbohydrate samples and their derivatives for biological research have increased extensively over the past decade.
In nature, carbohydrates exist as polysaccharides, glycoconjugates, or glycosides. But, the isolation of pure carbohydrate samples in sufficient amounts is difficult and cumbersome. In such cases, chemical syntheses of the relevant glyco‐structures become pertinent.5 For the chemical synthesis of complex carbohydrate molecules, the main challenge is to build glycosidic linkages connecting the monomeric units with proper stereo‐ and regiochemical orientation. Over the last few decades, many new and sophisticated procedures have been established for the successful synthesis of complex oligosaccharides. Thus, development of new methodologies for chemical glycosylation has emerged as an active area of research. More and more intriguing glycosidic bond syntheses are being standardized. However, achieving complete stereocontrol over glycosylation remains an eluding area of carbohydrate chemistry.6 Hence, optimizing the reaction conditions has long been the basic theme in carbohydrate synthesis.
Various types of novel glycosyl donors for glycosylation reactions have been synthesized and implemented.7, 8 Different activation strategies have been developed for the successful assembly of oligosaccharides from protected building blocks.9 Solid‐phase oligosaccharide synthesis has also made significant progress and is of high interest, as the process avoids the necessity to purify the intermediates.10 Automated oligosaccharide synthesis has also been developed, which has given a new edge in the field of synthetic oligosaccharide chemistry.11
Fundamental concepts of glycosylation have been covered in a wide range of Review articles.7 In this Review, we aim to indicate all of the different types of donors and their activation protocols, as well as the different types of O‐glycosylation processes developed after the onset of the 21st century. We will primarily stick to all newly developed methodologies of O‐glycoside formation reported in the 21st century.
2. General Aspects
2.1. Historical Background
Glycosylation, with all of its complexity, has long been a topic of research. The end of the 19th century witnessed the chemical formation of the first aryl and alkyl glycosidic bond formulated by Michael12 (p‐methoxy phenyl β‐d‐glucopyranoside) and Fischer13 (methyl α‐d‐glucopyranoside), respectively. Following their lead, Knoenigs and Knorr formulated a more controlled and modified glycosylation protocol.14 New methodologies are still being developed with more advanced variations in the mechanistic pathways. Later Zémplén and Gerecs,15 and subsequently Helferich and Wedermeyer,16 illustrated the principle of the metal(II) salt‐induced removal of leaving groups.17 With the general standardization of glycosyl protocols, efforts were made to control the stereochemical outcome of glycosylations. The requirement for the efficient syntheses of the two anomeric stereoisomers, 1,2‐cis and 1,2‐trans glycoside led to more advanced and detailed studies to introduce variation in the mechanistic pathways. The works of Lemieux et al.18 as well as Ness and Fletcher19 instigated the importance of the nature of the protecting groups, especially at the C‐2 position for its correlation with the stereoselective outcome of the glycosylations. This observation was further documented by Hashimoto et al and Fraser‐Reid et al.,20 whereby the concept of the ‘armed–disarmed’ approach was initiated, claiming ether linkage at the C‐2 position to be arming, leading to the 1,2‐cis glycoside, whereas an acyl linkage at the same position yielded the 1,2‐trans glycoside by virtue of the disarming property and neighboring group participation. Roy et al. introduced the ‘active’ and ‘latent’ thioglycoside donors that further expanded the concept of selective anomeric reactivity.21 The latent‐active glycosylation strategy was further utilized by Boons and Isles to form trisaccharide libraries, using vinyl and allyl donors with selective reactivity.22 Furthermore, Ogawa and co‐workers investigated the possibility of using orthogonal glycosyl donors and implemented the ‘orthogonal glycosylation strategy’ in the synthesis of various oligosaccharides.23
The search to find the ideal conditions for an effective glycoside formation revealed various modifications in the already established protocols. Varied promoter systems were implemented, newer activating groups established, and more versatile protecting groups were introduced. With further advanced control on the glycosidic linkages and reaction conditions, one‐pot glycosylation also came into vogue.24 More advances in computational chemistry and subsequent kinetic studies aided in solving the mystery behind the mechanism of glycosylation to a considerable degree. But, despite all of these efforts in the refinement of reaction conditions, the recognition of a single general procedure to describe chemical glycosylation in its entirety is yet to be accomplished.6
2.2. Anomeric Effect
It was between 1955 and 1958 that Edward and Lemieux first defined the ‘anomeric effect′, based on the stereochemistry of the C‐1 carbon of the pyranose ring, that is, the anomeric carbon.25, 26 It is also referred to as the Edward–Lemieux effect. It was originally defined as the tendency of an electronegative substituent attached to the anomeric carbon to reside in the axial position. However, further studies have revealed various physical interpretations behind it.
A popular and widely accepted theory is based on molecular orbital interaction, employing the hyperconjugation of the nonbonding electron pair on the ring oxygen atom with the vacant σ* orbital of the C−X bond, thereby stabilizing the axial configuration27 (Figure 1 A). However, extensive computational studies revealed that the energy due to hyperconjugation is not the bulk contributor in establishing the energy difference between the axial and equatorial configuration.28 Further, dipole moment theory26 says that, in the equatorial isomer, the dipoles of the heteroatoms are partially aligned, thereby repelling each other. But, in the axial configuration, the opposite orientation of the dipoles stabilizes the system by implicating a lower energy barrier (Figure 1 B). Studies have, thus, led to the hypothesis that the dipole–dipole interaction and electrostatic interaction form the bulk of the reasoning behind the conformational preferences of the carbohydrates; whereas, the hyperconjugation is only a minor factor. In view of the explanations, the axial stereoisomer represents the thermodynamically controlled product, whereas the equatorial isomer represents the kinetically controlled product (except for sugars with axial 2‐OH groups like d‐mannose and l‐rhamnose, where the axial product is both kinetically and thermodynamically controlled) (Figure 1 c).
Figure 1.
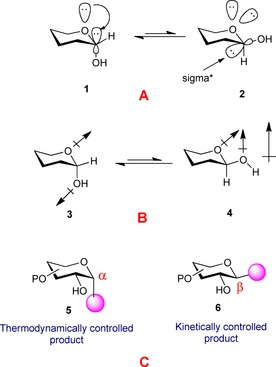
A) Explanation based on molecular orbital theory; B) explanation based on the dipole moment theory; C) the thermodynamically and kinetically controlled product.
2.3. Mechanistic Pathways
For proper, methodical chemical synthesis of glycosides, the most important phenomenon involved is called glycosylation. Chemical glycosylation is the coupling of the two monomeric sugar units, with each other or with other aglycons, through the formation of a new linkage, known as the glycosidic bond. The linkage may be of different types depending on the linking heteroatom. Herein, we primarily deal with O‐glycosides. However, other heteroatomic glycosides such as C, S, and N glycosides are also widely known in literature. Complete categorization of chemical glycosylation as an SN1 or SN2 reactions was tedious, as each theory brought forth many evidences in their support.29
For a more generalized mechanistic pathway (Scheme 1), the donor 7 is first pre‐activated with a leaving group attached to its anomeric hydroxyl group. The addition of an electrophilic promoter activates the leaving group of the donor to form complex 8. The next step initiates the formation of oxacarbenium ion 9 in its flattened half‐chair conformation. After this, nucleophilic attack by acceptor 10 occurs, thereby leading to the required glycoside formation 11. However, the attack of the acceptor can occur via two pathways, owing to the structural property of the oxacarbenium intermediate. The attack of the glycosyl acceptor from the bottom of the sugar ring leads to the formation of alpha (α) or 1,2‐cis glycoside 11 b; attack of the glycosyl acceptor from the top of the sugar ring yields beta (β) or 1,2‐trans glycoside 11 a. Furthermore, manipulation of the stability of the intermediate oxacarbenium ion remains responsible for the anomerization of the compound.30
Scheme 1.
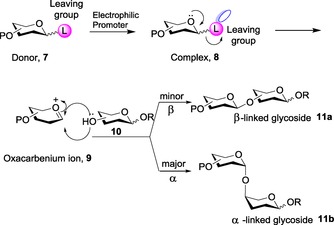
Mechanistic pathway depicting glycosylation.
Presence of a participating acyl group in the C‐2 position governs the stereochemistry of the glycosidic bond formed. The oxacarbenium ion formed owing to the departure of the leaving group further (Scheme 2) interacts with the acyl group in the C‐2 position to form the acyloxonium ion complex 14 by virtue of neighboring group participation.31 This enables the acceptor 10 to attack the anomeric carbon from only one side, that is, the 1,2‐trans side to form the 1,2‐trans glycoside 15.
Scheme 2.
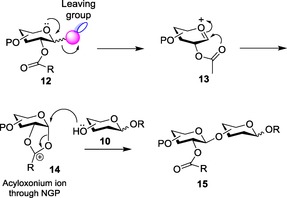
Mechanism for the formation of 1,2‐trans glycoside.
For the formation of 1,2‐cis glycoside 5, the presence of a non‐participating group is required in the C‐2 position of the donor molecule. Thus, increasing the stability of the oxacarbenium ion and its various electronic and steric factors may lead to the formation of the more thermodynamically controlled product, caused by the action of the anomeric effect described above (Figure 1 C).
Thus, starting from Michael's vision of glycosylation, where the glycosyl acceptor was converted into its corresponding salt,12 to Fischer's use of hemiacetals as the donor,13 we have come a long way via Fraser‐Reid's idea of ‘armed–disarmed’ glycosylation.20b However, the details of the newer and the more developed concepts in decoding the mystery behind the mechanism of chemical glycosylations are beyond the scope of this Review.32
3. Glycosylations
3.1. Glycosyl Halides
3.1.1. Glycosyl Bromides and Chlorides
In 1901, Koenigs and Knorr14 and Fischer and Armstrong33 independently introduced the concept of glycosyl halides as donors, where glycosyl chlorides and bromides were reacted with alcohols in the presence of AgCO3 or Ag2O. Since then, there have been extensive studies on the preparation of glycosyl halides,34 including the treatment of free sugar units with AcBr35 (AcBr−AcOH),36 treatment of per‐acetylated sugars with HBr−AcOH37 (BiBr3−Me3SiBr),38 and subsequent conversion of the sugar hemiacetals under modified Mitsunobu conditions with PPh3/N‐halosuccinimide39 or PBr3.40 However, the harsh conditions in the established protocols often cause difficulties in extensive oligosaccharide synthesis, where acid‐labile protecting groups in substrates are a general phenomenon. Hence, in the search for more environmentally friendly and greener methodologies, photocatalytic synthesis came into vogue.41 Likewise, while Mizuno and co‐workers established the photo‐irradiative phase‐vanishing method for α‐glycosyl bromide synthesis42 by applying UV light (irradiation at 352 nm), Xue and co‐workers introduced the more versatile visible‐light‐mediated halide synthesis.43 However, in the latter case, an additional photocatalyst, tris(2,2′‐bipyridyl)ruthenium(II) chloride [Ru(bpy)3Cl2], was essential. Various protocols for the activation of glycosyl bromides are in use for the synthesis of 1,2‐cis glycosides with the help of Ag salts (AgOTf, Ag2CO3, AgClO4, etc.) or Hg salts [Hg(CN)2, HgBr2, HgCl2, etc.].44 The versatile use of Ag2O for glycosyl bromide activation was shown by Beejmohun et al. in the synthesis of monolignol glucosides.45
Owing to the restrictions offered by glycosyl halides in terms of their moisture stability and handling, glycosylations with glycosyl halides as donors require optimized low temperatures and inert conditions. Requirement of such conditions was usually managed with various limitations. However, the present century saw the emergence of room‐temperature ionic liquids (RTILs), which came as a ready solution for all of the constraints. Malhotra and co‐workers46 implemented a series of RTILs and molten halide salts for the glycosylation of acetabromo‐α‐d‐galactose with p‐nitrophenol. By virtue of their unique ionic combination and their probable role in the glycosylation pathway, ILs holds the promise of being an extensively used solvent and activator in the future. It has been shown that the activation of glycosyl bromides neat in ILs in the presence of AgCO3 gave β‐products in moderate yields.
In terms of reactivity, glycosyl bromides have lower reactivity than glycosyl iodides.47 However, glycosyl bromides exhibit less stability than glycosyl chlorides. In 2006, Oscarson and co‐workers reported the total synthesis of a tetrasaccharide O139 through the activation of glycosyl bromide over its corresponding thioethyl glycoside acceptor by using AgOTf as the promoter.48 This selective activation process, in turn, marks the initial steps towards more convenient one‐pot oligosaccharide synthesis. This method also promotes the selective activation of glycosyl chlorides with the help of AgOTf in the presence of thioglycosides.49 Recently, Nitz and co‐workers reported the activation of unprotected glycosyl chloride donors by using p‐toluene sulfonylhydrazide and it has been proven to be more reactive than its protected counterpart.50
3.1.2. Glycosyl Iodides
The limited shelf‐life of glycosyl iodides has always rendered them disadvantageous as donors for glycosylation. This restriction requires the in situ formation of glycosyl iodides and their subsequent involvement in glycosylation. In this context, many research groups have demonstrated reliable methods for generating glycosyl iodides from per‐O‐acetylated sugars through treatment of trimethylsilyl iodide (TMSI)51 or HI equivalents.52 The instability of TMSI was addressed by Koreeda and co‐workers who utilized the possibility of the synthesis of anhydrous HI by the reaction of solid iodine with a thiol component in an organic solvent.52a In line with this, Field and co‐workers also generated TMSI in situ by reacting hexamethyldisilane (HMDS) with iodine.53 The same group further modified their established procedures in 2004, by presenting the synthesis of per‐O‐acetylated glycosyl iodides 16 from unprotected free sugars 15 in significant yields (Scheme 3).54 This protocol, equipped with a no‐solvent system and inexpensive reagents, proved a versatile method, enabling the effective formation of glycosyl iodides.
Scheme 3.
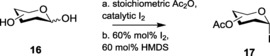
Preparation of glycosyl iodide from unprotected free sugars.
Field and co‐workers later studied the stability, reactivity, and characterization of glycosyl iodides as donors.55 Over extensive studies, it has been observed that, although glycosyl iodides are usually less effective for glycosylations, many research groups have illustrated its unique advantages over other glycosyl halides in terms of efficiency and stereospecificity.56 Thus, by using these glycosyl iodide donors, various glycosides have been synthesized. Here, the work of Gervay‐Hague and group deserve special mention. They have illustrated how glucosyl, galactosyl, and mannosyl iodides can react with oxa (20) and thio cycloalkanes to yield O‐glycosides, 21 and 22, respectively, with high β‐selectivity (Scheme 4).57 They also demonstrated the selectivity with decreasing temperature, where the β‐isomer was dominant at lower temperatures (Table 1).58 The most noteworthy point in this study was the realization that reactions did not require pre‐activation of the donor, which is exclusive for glycosyl iodides.
Scheme 4.
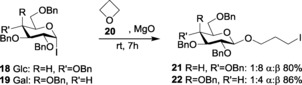
Synthesis of oxacycloalkane with glycosyl iodides.
Table 1.
Dependence of β‐selectivity with decreasing temperature.
| No. | Donor | Temp [°C] | Time | Yield [%] | α/β ratio |
|---|---|---|---|---|---|
| 1 | 18 | 40 | 2 h | 82 | 1:2 |
| 2 | 18 | 0 | 5 h | 76 | 1:5 |
| 3 | 18 | −60 | 3 days | 76 | 1:29 |
| 4 | 19 | 40 | 5 min | 83 | 1:4 |
| 5 | 19 | 0 | 30 min | 74 | 1:9 |
| 6 | 19 | −60 | 12 h | 73 | 1:50 |
Glycosyl iodides can also be selectively activated in the presence of other donor systems. In this respect, Gervay‐Hague and co‐workers demonstrated iterative oligosaccharide synthesis by selective activation of glycosyl iodides59 over per‐O‐acetylated glycosyl donors with TBAI/DIPEA. Selective activation of glycosyl iodides over thioglycosides was also implemented, where glycosides were formed though the activation of glycosyl iodides in the presence of triphenylphosphine oxide (Ph3PO).60 In another interesting example, Lam and Gervay‐Hague61 illustrated the double glycosylation of an orthogonally protected acceptor 23 with mannosyl iodide 24 by using AgOTf to yield the desired glycoside 25 (Scheme 5).
Scheme 5.
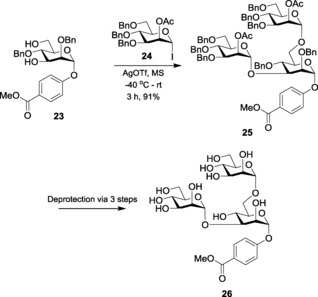
Synthesis of trimannoside (26) through double glycosylation.
Citing more studies in terms of glycoside synthesis,62 Castillon et al. converted per‐O‐acetylated iso‐globotrihexose to a glycosyl iodide and coupled it with stannylceramide in the presence of TBAI.63 In 2014, Gervay‐Hague and co‐workers activated glycosyl iodides with I2 to their corresponding reactive glycosyl donors and proceeded towards glycosylation to yield various glycoconjugates.64 Though there has been much study of glycosylations with typically benzyl‐protected glycosyl iodides,65 Stachulski and co‐workers presented a detailed study of glycosylation reaction of ‘disarmed’ glycosyl iodides with various types of alcohols promoted by the NIS–I2–TMSOTf system.66 Various types of ‘non‐heavy‐metal’ salts were also demonstrated as effective catalysts in the formation of α‐glycosides. Thus, it is evident that the promoter systems required for glycosyl iodides are widely varied. Glycosylations with glycosyl iodides have been reviewed extensively by Murakami et al.67
3.1.3. Glycosyl Fluorides
Mukaiyama et al.68 asserted the activation of glycosyl fluorides by SnCl2−AgClO4 and their role as glycosyl donors. Their versatility resides in their readily accessible nature and their higher thermal and chemical stability compared to other glycosyl halide counterparts. After the onset of the 21st century, the synthesis of glycosyl fluorides has been broadly developed and used for glycoside formation.69 The IPy2BF4‐catalyzed synthesis of glycosyl fluorides from thioglycosides was reported in 2007,70 which provided the best output being activated by TfOH, which mechanistically formed a co‐ordination complex with the pyridine moiety to liberate the iodonium species. More successful illustrations of glycosyl fluoride synthesis from thio‐, seleno‐, telluro‐, and n‐pentenyl glycosides have been reported.71 Kanie and co‐workers used N,N‐diethylaminosulfur trifluoride (DAST) in the absence of N‐bromosuccinimide (NBS).72 Dimethyl(methylthio)sulfoniumtrifluoromethanesulfonate (DMTS) was used as a substitute for NBS to give excellent yields. Glycosyl fluorides have also been widely used for intramolecular aglycon delivery (IAD) in the synthesis of β‐mannosides (Scheme 6).73
Scheme 6.
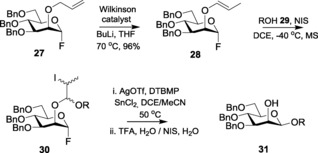
β‐Mannoside synthesis from glycosyl fluorides through IAD.
For the next generation of oligosaccharide synthesis, selective activation of donors based on their activating groups is a major area of research. There has been much research aimed toward selective activation of glycosyl fluorides.74 Mukaiyama et al. selectively activated glycosyl fluorides over thioglycosides by using catalytic amounts of TfOH, HClO4, or C4F9SO3H.75 Interestingly, in a report by Yang and Yu in 2014,76 there is an illustration of the use of glycosyl fluoride as an acceptor in the presence of a thioglycoside donor for the synthesis of disaccharide in its fluoride form, which could subsequently be activated in the presence of a thioglycosides acceptor to form the trisaccharide. Glycosyl fluorides can also be activated in the presence of glycols with the help of Sn(OTf)2.77 Thus, these selective activation protocols are stepping stones for the formulation of more versatile iterative and automated oligosaccharide syntheses. Utilizing the different activation protocols, these donors have helped in the broader aspect of natural product syntheses.8
3.2. Thioglycosides
Thioglycosides are the most versatile glycosyl donor in oligosaccharide synthesis. Although the anomeric thio functionality is highly stable towards varied protecting‐group manipulations, it can be activated under extremely mild conditions and, hence, can be used in varied types of glycosylation conditions. The soft sulfur atom provides easy and selective reactivity with soft electrophiles. Various modifications have been made to the nature of thioglycosides over the years and many novel and mild promoter systems have been developed. The present century has seen modification in the use of harsh thiophilic promoters like Hg2+, Cu2+, and Ag+ salts to halonium ions, which have more thiophilic character. They revealed the promise of being activated under highly moderate conditions. Starting with the use of NBS78 to activate armed thioglycosides, much progress has been made in utilizing bromonium and iodonium salts for the activation process with promoters like NOBF4,79 NBS/TfOH,80 Br2/AgOTf,81 NIS/HOTf or AgOTf,82 IDCP (iodoniumdicollidine perchlorate),83 IDCT (iodonium sym‐collidiniumtriflate),84 I2,85 PhIO/Tf2O,86 and Ipy2BF4.70
In an attempt to develop more versatile promoter systems, Ye and co‐workers implemented the ‘soft’ nature of the bromonium species to report a bromodimethylsulfonium bromide (BDMS)/AgOTf87 system for the activation of thioglycosides. The low cost and easy accessibility of the BDMS (32) catalyst made it an effective glycosylating promoter (Figure 2). In a similar attempt, the same group went on to establish an O,O‐dimethylthiophosphonosulfenyl bromide (DMTPSB)–AgOTf88 (33) system for the pre‐activation of thioglycosides and subsequent glycosylations (Scheme 7 ).
Figure 2.
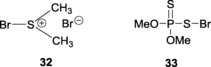
32: bromodimethylsulfonium bromide (BDMS); 33: O,O‐dimethylthiophosphonosulfenyl bromide (DMTPSB).
Scheme 7.

Synthesis of a disaccharide with DMTBSB/AgOTf as the promoter.
The presence of bromine was also shown to activate β‐ethyl thioglycoside 37 to provide disaccharide 39 as the exclusive α‐linked diastereomer (Scheme 8).89 Interestingly, the mechanistic rationale behind such activation lies in the in situ generation of glycosyl bromide, which acts as the glycosyl donor reacting with the acceptor.
Scheme 8.
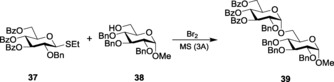
Glycosylation with Br2 as the promoter.
Similarly, Crich and Smith reported the use of S‐(4‐methoxyphenyl) benzenethiosulfinate (MPBT, 40) and trifluoromethanesulfonic anhydride (Tf2O)90, which react with each other to form an electrophile 41, aiding the in situ conversion of thioglycosides to their corresponding triflates. These glycosyl triflates were further implemented for the generation of the challenging β‐mannoside linkages (Scheme 9). 1‐Benzenesulfinyl piperidine (BSP) and Tf2O91 also served the same purpose, and it was further used for double glycosylation, using carbohydrate diol as the acceptor 43 (Scheme 10). Later, van der Marel and co‐workers proved the versatility of the system successfully applied the system.92
Scheme 9.
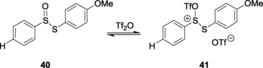
Activation of MPBT.
Scheme 10.
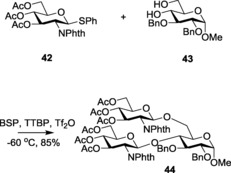
Example depicting double glycosylation.
Van der Marel et al.93 also introduced the diphenyl sulfoxide (DPS)–triflic anhydride combination for thioglycoside activation in β‐mannoside synthesis. They first activated a relatively armed thioglycoside 45 with the BSP/Tf2O activator system and coupled it with a thio acceptor 47 to yield disaccharide 48 in its thio form (Scheme 11 a). (N‐piperidino)phenyl(S‐thiophenyl)sulfide triflate (46) is formed as a by‐product. After quenching the by‐product with triethylphosphite, thio‐disaccharide 48 was activated with a more versatile DPS/Tf2O activator system for its coupling with another monosaccharide 49 for successful glycosylation (Scheme 11 b). This example extensively illustrates the difference in reactivity between the two activator systems. Similar pre‐activation of thioglycosides have also been achieved by benzenesulfinylmorpholine (BSM)–Tf2O94 as well as Me2S2–Tf2O,95 and was further utilized for glycosylation through both primary and secondary hydroxyl groups.
Scheme 11.
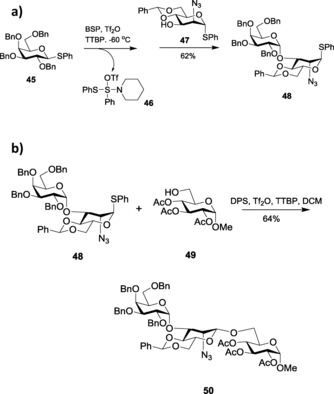
a) Pre‐activation of the donor. b) Subsequent glycosylation with DPS and TTBP as the promoter in the presence of Tf2O.
Thioglycosides, owing to their unique reactivity pattern, are promising candidates for one‐pot oligosaccharide synthesis. To establish this system, Wong and co‐workers implemented the novel reagent N‐(phenylthio)caprolactum for thioglycoside activation at room temperature.96 N‐(Phenylthio)caprolactam was first reacted with Tf2O, which further proceeded for thioglycoside activation. In a similar example, instead of Tf2O, TMSOTf was used for the pre‐activation of N‐(p‐methylphenylthio)‐ϵ‐caprolactam by Ghosh and co‐workers.97 They also reported the use of trichloroisocyanuric acid (TCCA),98 a readily available, inexpensive, and shelf‐stable reagent, in combination with a catalytic amount of TMSOTf for the efficient activation of the thioglycosides.
In 2000, Takeuchi et al.99 introduced the activation of disarmed thioglycosides through the combination of trityltetrakis(pentafluorophenyl)borate [TrB(C6F5)4], iodine, and DDQ. On a similar hypothesis, they also put forward another activator system, PhthNSEt in the presence of TrB(C6F5)4.100 The same complex was used in presence of both NIS and NBS for successful glycosylations.101
Various lanthanide(III) salts were also introduced for the activation of the thioglycoside donors. Employing the concept of lanthanide‐ion co‐ordination, Chung and Park reported a comparative study by using various lanthanide(III) salts to envisage the noncovalent approach of the IAD strategy in the process of the formation of β‐mannosides.102 The Lewis acidity and high co‐ordination numbers of the lanthanide salts were considered to be highly favorable for the desired co‐ordination. YbIII and EuIII were especially effective (Scheme 12) in the presence of NIS–TfOH as the activator system (Table 2).
Scheme 12.
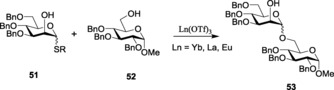
Glycoside formation with the help of lanthanide(III) salts.
Table 2.
The activation of glycosyl sulfoxide using different LaIII salts.
| No. | Ln[OTf]3 (equiv) | Activator | Solvent | Temp [°C] | Time | Yield [%] | α/β |
|---|---|---|---|---|---|---|---|
| 1 | Yb (1) | Tf2O | CH2Cl2 | −78→RT | 10 h | 63 | 1:2 |
| 2 | Yb (1) | Tf2O | CH3CN | −40→RT | 12 h | 63 | 1:2.8 |
| 3 | Yb (1) | NIS | CH2Cl2 | RT | 20 min | 47 | 1:1.5 |
| 4 | Yb (1) | NIS–TfOH (cat.) | CH2Cl2 | −78 | 8.5 h | 59 | 1:2.9 |
| 5 | Eu (1) | Tf2O | CH2Cl2 | −78→RT | 10 h | 76 | 1:1.5 |
| 6 | Eu (1) | Tf2O | CH3CN | −40→RT | 10 h | 82 | 1:4.3 |
| 7 | Eu (1) | NIS | CH2Cl2 | RT | 20 min | 48 | 1:2 |
| 8 | Eu (1) | NIS–TfOH (cat.) | CH2Cl2 | −78 | 6 h | 64 | 1:3 |
| 9 | La (1) | Tf2O | CH2Cl2 | −78→RT | 12 h | 51 | 1.7:1 |
Thus, NIS–NBS‐based activator systems that were introduced long ago saw a number of important modifications after the onset of the 21st century. Mukaiyama and co‐workers used TrOTf103 in conjunction with NIS for the convergent synthesis of oligosaccharides. Simultaneously, Li and co‐workers used Me3SiOTf with both NIS and NBS for the activation of phenyl and ethyl thioglycosides to introduce spacer arms.104 Moreover, the catalyst system involved for effective glycosylations demands highly moisture tolerant properties. In that respect, Mukherjee and Mukhopadhyay reported the use of La(OTf)3 as the Lewis acid activator with NIS for the activation of disarmed thioglycoside donors,105 which, by virtue of its solid nature and moisture‐tolerant capability, became more effective than other traditional reagents like TfOH or TMSOTf.106 Similarly, supported acid catalysts became interesting alternatives to traditional Lewis acid promoters. HClO4–silica was used for the activation of ‘disarmed’ thioglycosides in conjunction with NIS in 2005 by Field and co‐workers,107 which promised to be a better protic acid source under milder and safer reaction conditions. Later, Mukhopadhyay and co‐workers introduced a better and environmentally friendly H2SO4–silica system for the same purpose.108 The good availability of H2SO4–silica and its effective desiccant properties were reasons for its wide use for various bacterial and plant oligosaccharide syntheses in moderate to high yields (Figure 3).109 Specific acid‐washed molecular sieves (AW‐300 MS) along with NIS have also been shown to function as glycosylation promoters by Yao and Lee.110 Owing to its ease of handling and storage, and its capacity to scavenge water, AW‐300 MS proved beneficial as a Lewis acid in the NIS‐mediated glycosylation of thioglycosides.
Figure 3.
Oligosaccharides synthesized with H2SO4–silica as the promoter.
For expeditious oligosaccharide synthesis, various modifications in the protecting groups and the catalyst in the presence of NIS/NBS‐based promoters have been widely investigated. Périon et al. have used both Sn(OTf)2 and Cu(OTf)2 to perform critical glycosylation with the fucosyl donor by varying the protecting group in its C‐4 position.111 In another example, a stoichiometric amount of NBS was used with Bi(OTf)3 for the activation of alkyl and phenyl thioglycoside donors by Valerio et al.112
Introducing a novel promoter‐free glycosylation technique, Pohl and co‐workers efficiently used sub‐stoichiometric amounts of triphenyl bismuth ditriflate [Ph3Bi(OTf)2] to activate various thiopropyl glycosides.113 The highly robust nature and solubility of bismuth(V) salts made it a highly advantageous glycosylation catalyst, employing just catalytic amounts of the promoter. This glycosylation protocol opened more doors to investigate elegant glycosylations without the involvement of any co‐promoter. In sync, Vibhute et al. recently showed that only 3 mol % of AuCl3 catalyst was sufficient to trigger thioglycoside activation.114
Light‐induced activation of thioglycoside donors has also been investigated.115 In 2013, Bowers and co‐workers demonstrated the efficient use of visible light for mediating O‐glycosylation through the activation of thioglycosides.116 Ir[dF(CF3)ppy]2(dtbbpy)PF6 was used as the catalyst to oxidize an electron‐rich aryl thioglycoside, yielding an oxocarbenium‐ion intermediate. Then, effective glycosides were obtained in the presence of bromotrichloromethane and a non‐nucleophilic and protic solvent, hexafluoroisopropanol (HFIP) (Scheme 13). The proposed mechanism involved the unique visible‐light‐activated single‐electron oxidation of sulfur. In a similar mechanism, photosensitizer [Ru(bpy)3]2+ was used along with DTBMP for the activation of thioglucosides.117 Recently, glycosylations avoiding the use of photosensitizers were also reported, where UV irradiation was used to cleave the C−S bond118 and Cu(OTf)2 was oxidized to generate the species capable of undergoing glycosylation. In a similar proposal, O‐glycosylation of unprotected deoxythioglycosides was also performed with alcohols in the presence of DDQ under long‐wavelength UV irradiation through the single electron transfer (SET) mechanism.119 During the process, a boronic acid was used as a temporary protecting group for the glycosyl donors to prevent self‐coupling or formation of 1,6‐anhydro glycoside.
Scheme 13.

Light‐induced activation of thioglycoside donor.
Bennett et al. reported the use of air‐ and water‐stable iodonium salt phenyl(trifluoroethyl)‐iodoniumtriflimide to activate a wide array of both armed and disarmed thioglycosides for glycosylation at room temperature.120 A special advantage of this process is that low‐temperature reactions or additives used to remove water from the reaction, such as molecular sieves, are not necessary. Electrochemical O‐glycosylation of primary alcohols with thioglycoside donors have been reported by Nokamia and co‐workers121 in the presence of a small amount of sodium trifluoromethansulfonate as the supporting electrolyte (Scheme 14).
Scheme 14.

Electrochemical O‐glycosylation.
3.3. Glycosyl Imidates
Glycosyl imidates represent another versatile leaving group in addition to the thioglycosides, whose extensive use in the domain of oligosaccharide glycosylations is noteworthy.
3.3.1. O‐Imidates
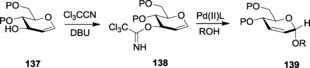
Since its inception by Schmidt et al. in 1980,122 glycosyl trichloroacetimidates and trifluoroacetimidates have become widely used glycosylating reagents. Glycosyl trichloroacetimidates can be readily prepared through the base‐promoted addition of an anomeric hydroxyl group to trichloroacetonitrile, using either inorganic (NaH, K2CO3, Cs2CO3, etc.) or organic (DBU) bases. Imidate‐based glycosylations require strong Lewis acids like TMSOTf,123 BF3⋅Et2O,124 TBDMSOTf,125 Tf2O,126 ZnBr2,127 or AgOTf128 and moisture‐stable activating reagents such as I2/Et3SiH.129
In special cases, such as fructofuranosides, where the synthesis of the trichloroacetimidate donor proved to be challenging, N‐phenyltrifluoroacetimidate was established as an efficient substitute.130 The leaving group showed effective α‐selectivity when performing glycosylations with various flavonoids.131 Their versatility has also been effectively established in the synthesis of kaempferol derivations by virtue of its subsequent activation with BF3 .Et2O.132 Versatility of BF3 .Et2O over TMSOTf for the activation of trichloroacetimidate or (N‐phenyl)trifluoroacetimidate donors, leading to the selective formation of 1,2‐trans glycosides in high yields, has been widely studied.133 However, with more recent studies in 2012, Cloninger and co‐workers have established that InIII salts serve as even better and more effective promoters than BF3⋅Et2O for performing glycosylations with glycosylacetimidate donors.134 To establish the novelty of the salts, InCl3, InBr3, and In(OTf)3 were reacted with various protecting‐group‐manipulated glycosyltrichloroacetimidate donors,134 and all of them led to the formation of the glycosylated products in high yields.
Use of iodine for the activation of armed and disarmed acetimidates was reported by Iadonisi and co‐workers.129 Addition of a catalytic amount of triethylsilane to trigger the in situ generation of HI was the key step for such an activation strategy. The same group previously135 established the use of catalytic amounts of Sm(OTf)3 to activate armed glycosyl trichloroacetimides. In analogous conditions, salts of other lanthanides like Sc(OTf)3, Tb(OTf)3, and Yb(OTf)3 were also used to activate trichloroacetimidate donors and promote glycosylations at room temperature.136 A catalytic amount of Yb(OTf)3 was also efficient for performing one‐pot glycosylation of glycosyltrichloro‐ and (N‐phenyl)trifluoroacetimidates.137
The similar activation properties of trichloro‐ and trifluoroacetimidates led to studies comparing their reactivities, which proved trichloroacetimidates to have more reactivity than glycosyl trifluoroactimidates.138 For instance, the readily accessible glucopyranose 60 was reacted with (N‐phenyl)trifluoroacetimidoyl chloride in the presence of a slight excess of K2CO3 in acetone to yield the trifluoroacetimidate derivative 61, which was then reacted with a trichloroacetimidate glycoside 62 in the presence of a catalytic amount of Yb(OTf)3, where the more reactive compound, 62, was readily consumed to yield disaccharide 63. In the same pot, acceptor 64 was added with extra catalyst at room temperature to give the required trisaccharide 65 (Scheme 15 a, b). To reduce the toxicity involved in the use of lanthanide salts, they also reported the alternate use of Bi(OTf)3 for the activation of perbenzylatedtrichloro (N‐phenyl)trifluoroacetimidate glycosyl donors.139
Scheme 15.
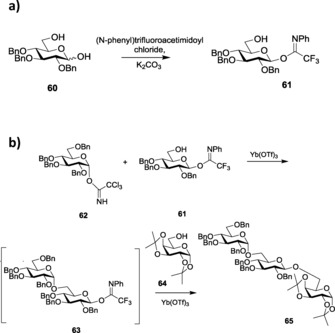
a) Synthesis of (N‐phenyl)trifluoroacetimide glycosyl donor. b) Glycosylations using O‐imidates.
Considering the high effectiveness of the catalyst in thioglycosides activation, Linhardt and co‐workers introduced easily accessible and environmentally benign HClO4–silica (optimized to 100:3 molar ratio) to perform the glycosylation with trichloroacetimidates (Scheme 16).140 This activation also allowed glycosylations with glycosides having various protecting‐group manipulations, giving excellent yields. In sync, Gildersleeve and co‐workers also used HClO4–silica for enhanced α‐selectivity.141 The moisture stability and easy handling of the catalyst made it highly useful in performing glycosylations with glycosyl trichloroacetimidates.
Scheme 16.
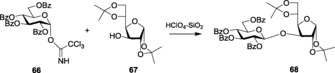
Glycosylation of trichloroacetimidate donor with HClO4−SiO2 as the promoter.
Similarly, acid‐washed molecular sieves (4 Å AW‐300 MS) have also been used to activate trichloroacetimidates along with the thioglycoside donors.142 Its moisture‐absorbent property was the key factor behind its successful use in glycosylations. The use of Amberlyst 15 H+ acidic resin has also been reported.143 β‐Mannosylation was also performed by Fukase and co‐workers with suitably protected N‐phenyl trifluoroacetimidatemannosyl pyranoside by utilizing TMSB(C6F5)4, which served as a dual Lewis acid/cation trap catalyst.144 Reactions with sensitive glycosyl trichloroacetimidates were achieved by Kunz et al.145 by using an Au catalyst, which provided high α‐diastereoselectivity. Subsequently, Vankar and co‐workers studied the application of gold catalysts in glycosylations and modified it to use AuCl3 and the AuCl3–phenylacetylene combination, which acted as good activators, both146 for armed and disarmed trichloroacetimidates. Notably, in this case, high β‐stereoselectivity was obtained. Recently, further investigation into gold catalysts by Peng and Schmidt has demonstrated the successful synthesis of glycosides with trichloroacetimidate donors147 via an intramolecular dual catalysis pathway, where AuCl3 acted as an initiator for the formation of catalyst–acceptor adducts. Interestingly, they obtained high β‐stereoselective products even with the use of armed glycoside donors.
A commercially available cationic PdII reagent, Pd(CH3CN)4(BF4)2, was used by Nguyen and co‐workers148 as the Lewis acid, because of its vacant d orbitals. The easily accessible Pd salt has, thus, been used for stereoselective glycosylations with trichloroacetimidate donors to give exclusive α‐products. The use of a larger mol % of the PdII source gave a higher yield, whereas the use of additives like DTBP with the same amount of PdII source did not show much difference in the yield (Table 3). To facilitate the synthesis of β‐glycoside products, Baati and co‐workers used TMSNTf2 as an efficient promoter in the glycosylation reaction when using per‐methacrylated Schmidt reagents.149
Table 3.
Efficiency of PdII salts in glycosylation.
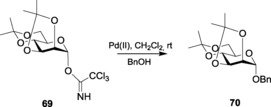
| Entry | Pd [II] source | PdII [mol %] | Additive | Time [h] | Yield [%] | α/β |
|---|---|---|---|---|---|---|
| 1 | Pd(CH3CN)4(BF4)2 | 3 | none | 5 | 70 | α only |
| 2 | Pd(CH3CN)4(BF4)2 | 5 | none | 3 | 85 | α only |
| 3 | Pd(CH3CN)4(BF4)2 | 5 | DTBP | 4 | 83 | α only |
Photoinduced reactions have attracted much interest in the last few years. Organic photoacids have been implemented for the activation of trichloroacetimidates under photoirradiation.150 An organic photoacid excited upon photoirradiation subsequently activates glycosyl imidates and facilitates coupling with corresponding alcohols as acceptors.
In addition to trichloro‐ and N‐phenyl trifluoroacetimidate, trichloroacetyl carbamate glycosyl donors have also been reported to be effective glycosyl donors. Glycosyl trichloroacetyl carbamate synthesized upon reaction of the hemiacetal 71 with trichloroacetyl isocyanate at 0 °C was utilized as the donor for performing glycosylation with various acceptors, using a catalytic amount of TMSOTf as the promoter (Scheme 17).151 Many other electrophilic promoters have been used for the activation of such donors. Interestingly, Zn(OTf)2 was unable to activate the imidate donor, whereas the glycosylation with TfOH and Sc(OTf)3 was seen to proceed moderately. The same reaction was slow with other Lewis acids such as Mg(ClO4)2 and Cu(OTf)2, even at room temperature. However, the reaction provided high yields of glycosylated products with TMSClO4.152 Selective activations in different promoter systems invariably hold promise for the use of such imidate donors in iterative one‐pot glycoside synthesis.
Scheme 17.
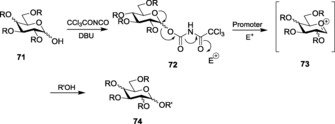
Synthesis of trichloroacetylcarbamate and its use in glycosylation.
O‐Glycosyl trichloro cyano acetimidates have also been popular as powerful glycosyl donors.153 To increase the donor properties of the glycosylimidate, one chlorine group of the trichloroacetinitrile was replaced with a more electronegative cyano group, that is, trichloromalonitrile was used as the activating agent.
3.3.2. S‐Imidates
Thioimidates have been developed as versatile building blocks for selective glycoside formation. Special mention is credited to Demchenko and co‐workers, who significantly contributed towards the development and establishment of various thioimidate donors.154
Demchenko and co‐workers introduced S‐benzoxazolyl (SBox) as a novel thioimidate donor.155 SBox proved to be a stable and efficient glycosyl donor for performing several types of glycosylations under mild conditions. Both 1,2‐cis and 1,2‐trans glycoside formations have been reported with SBox‐protected thioglycosides, 75 (Figure 4). The synthesis of SBox glycosides and the probable mechanism for its activation was reported by the same group.156 Standard thiophilic promoters like NIS/TfOH, AgOTf,157 Cu(OTf)2,158 MeOTf,159 and so forth have been used to activate this class of donors.160 SBox glycosides also showed selective activation over thioglycosides and pentenyl glycosides.157 Several glycosides with different protecting groups were also tested for activation,158a which revealed that per‐benzylated glycosides showed the optimum activation. Demchenko and co‐workers gave a detailed analysis about the effects and logistics behind variable activation patterns observed by armed and disarmed SBox glycosides. Armed glycosides could be chemoselectively activated over its disarmed counterpart161 to give trans‐glycosides. Oligosaccharides formed by using armed SBox donors showed high α‐ stereoselectivity.
Figure 4.
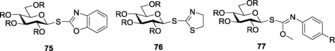
SBox (75), STaz (76), and SNea (77) glycosides.
However, SBox glycosides showed certain instability towards some extreme reaction conditions when being activated by triflic acid (TfOH). To bypass this limitation, Demchenko and co‐workers later introduced S‐thiazolinyl (STaz) glycosides, 76 (Figure 4), as building blocks for chemoselective and orthogonal oligosaccharide synthesis.162 SBox and STaz thioimidates were also implemented in solid‐phase glycosylation strategies (Scheme 18 a, b),163 where the thioimidates were utilized as donors for glycosylations with resin‐bound acceptors after being activated by promoters like AgOTf, TMSOTf, NIS/TMSOTf, MeOTf, and so forth.164 STaz glycosides proved to be highly stable towards extensive protecting‐group manipulations.165 These glycosides also showed activation in line with Fraser‐Reid's armed–disarmed activation strategy. Armed STaz glycosides could be chemoselectively activated over their disarmed counterparts.31b Demchenko and co‐workers also explored other analogs of silver salts to activate the STaz glycosides, and they successfully reported the application and versatility of silver tetrafluoroborate, AgBF4,166 as an excellent promoter for the activation of such donors. The more popular thiophilic promoter AgOTf was also found to be effective towards the activation of STaz donors.
Scheme 18.
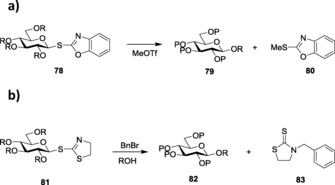
a) Activation of SBox glycosides. b) Activation of STaz glycosides.
S‐glycosyl O‐methyl phenylcarbamothioates (SNea carbamothioates) (77) (Figure 4) were introduced as the next‐generation glycosyl thioimidate donor.167 The SNea leaving group was illustrated to represent a bridging compound between acyclic thiocyanates (SCN) and cyclic SBox glycosides. These donors showed similar properties to SBox glycosides and extensive studies were performed to compare the activation properties of the two. Activation of SNea donors with promoters like Cu(OTf)2, AgOTf, or MeOTf afforded the α‐glycosides in significant yields.168 Other thioimidate S‐benzothiazolyl (SBaz) glycosides, 84 (Figure 5), have also been commonly used for glycosylation. This derivative was first introduced for glycosylation by Mukaiyama et al.169 In 2011, Hasty and Demchenko introduced another variant of thioglycoside donor, S‐benzimidazolyl (SBiz) glycosides, 85 (Figure 5), suitable for extensive oligosaccharide synthesis.170
Figure 5.

SBaz (84) and SBiz (85) glycosides.
Glycosyl dithiocarbamates (DTCs) have been used for glycosylations to give β‐selective products upon being activated by CuI or CuII triflates.171 They have been specifically used for glycosides with no protecting group in the C‐2 position.
3.4. Alkenyl Glycosides
Although thioglycosides are established as highly versatile donors that facilitate glycosylation reactions, the use of thioglycosides sometimes becomes highly unwanted because of the obnoxious smell of the thiols. Alkenyl glycosides fit perfectly in this void. Pent‐n‐enyl glycosides (NPGs) 86 pose as the most apt alternative in terms of their versatility and environmentally friendly nature. Similar to thioglycoside activation, activation of n‐pentenyl glycosides is achieved either by bromonium (NBS) or iodonium (NIS) ions. The subsequent cyclization leads to the transition state, that is, the tetrahydrofuranyl oxonium ion (88) (Scheme 19). The increased leaving‐group property of the tetrahydrofuran moiety 89 leads to the formation of the corresponding oxocarbenium ion, 90, which, upon addition of the attacking nucleophile (the acceptor 91), culminates in the proper glycosylated product 92.
Scheme 19.
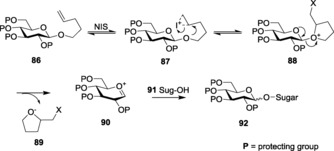
n‐Propargyl glycoside (NPG) activation.
The concept of activation of the pentenyl glycosides by Lewis acids was utilized by Hung and co‐workers in 2006172 for the introduction of 2‐allyloxyphenyl glycoside as an efficient donor system. This glycosyl donor could easily be activated by NIS/TfOH to give the product in moderate‐to‐high yields. The same group also introduced allylphenyl glycosides as a donor, which could be effectively activated by iodonium chloride (ICl)/AgOTf173 to produce the required glycosides. In 2009, gem‐2,2‐dimethyl and 2,3‐dimethyl 4‐pentenyl glycosides (Figure 6) were activated by cheaper and more versatile NBS promoter and claimed to undergo coupling 11‐ and 3‐times faster, respectively, than the master n‐pentenyl glycosides.174 The steric hindrance offered by the geminal methyl groups was assessed to trigger the ring‐closing step, causing more effective coupling.
Figure 6.

gem‐Dimethyl analogs of n‐pentenyl glycosides.
The present century has also seen a rise in the use of n‐pentenyl orthoesters 95 as a highly versatile donor system. Orthoesters can readily undergo electrophilic attack to form the furanylium intermediate 96 (Scheme 20). The enhanced leaving‐group property of the halomethyl furan moiety 89 aids in the easy conversion of intermediate 96 to 97. Furthermore, the attack of acceptor 98 effectively forms the required glycoside 99. Interestingly, the released moiety 89, which is highly non‐nucleophilic, fails to compete with the sugar nucleophile 98, which attacks the intermediate, dioxolenium ion 97, and forms the required glycoside 99.
Scheme 20.
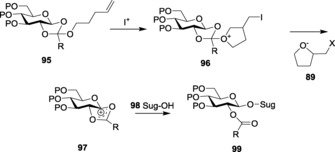
Mechanism of activation of n‐pentenyl orthoesters (NPOEs).
There have been various reports establishing the activation of n‐pentenyl orthoesters (NPOEs) by lanthanide triflates to form the glycosylated product. In an exemplary illustration, Fraser‐Reid and co‐workers showed that NPOE 100, upon being activated by NIS followed by Yb(OTf)3 (Scheme 21), produced highly regioselective glycosylated product 102 after reacting with the acceptor 101.175 The n‐pentenyl intermediate, 103, formed as a by‐product, however, remains inactivated. This n‐pentenyl glycoside 103 can be activated by Sc(OTf)3, which goes on to form the doubly glycosylated product 104 along with the normal glycoside102. Thus, NPOE glycosides prove to be better donors than n‐pentenyl glycosides, as they overcome reactivity restrictions faced in case of the latter.176
Scheme 21.
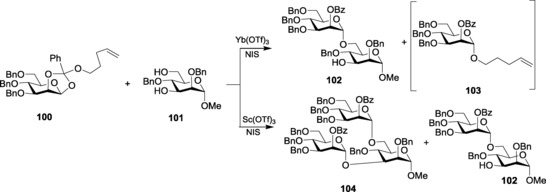
Different activation patterns of n‐pentenyl orthoesters.
The implementation of NPOEs for glycosylation was also evident in performing selective glycosylations with more than one free hydroxyl groups in the synthons. This facilitated the possibility of performing iterative glycosylations in the synthesis of larger oligosaccharide building blocks.177 In a comparative study, it was illustrated that n‐pentenyl glycosides gave a mixture of products, whereas NPOE yielded strictly the equatorial product in a clean reaction178 with a higher conversion ratio. Mathew et al. also demonstrated that NPOEs provided better yields upon being irradiated under microwave conditions.179 A detailed comparison between the activation of NPOEs under thermal and microwave conditions shows a distinct increase in the yield under the latter conditions. Microwave irradiation features the reaction mixture reaching the target temperature rapidly and maintaining the temperature throughout the entire reaction time, which becomes instrumental in obtaining better yields.
Fraser‐Reid and co‐workers also introduced mannopyranoside‐derived methyl orthoesters180 as possible donors for glycosylation. They underwent easy activation with the help of BF3⋅Et2O and demonstrated proceedings analogous to that of NPOEs. It was also observed that the iodonium‐ion activation and the stoichiometry of the Lewis acid had significant roles in the regioselectivity of the product formed.
3.5. Alkynyl Glycosides
Propargyl glycoside remains the most prominent alkynyl glycoside used in oligosaccharide synthesis to date. The versatility of the propargyl glycosides led to their introduction as a probable glycosyl donor suitable for oligosaccharide synthesis. The alkynophilicity of gold catalysts has been utilized by using AuIII halides as the catalyst to activate propargyl glycosides. Optimization of reaction conditions showed that, through AuCl3 activation, complete conversion of the starting material occurred at 60 °C after approximately 6 h. However, upon coupling with various aliphatic, alicyclic, steroidal, and sugar alcohols, the stereoselectivity of the product was a 1:1 α/β mixture.181 Other alkyne‐activating promoters like PtCl2, Co2(CO)8, and RuCl3, however, failed to give the product and resulted in decomposition of the donor. Building upon the concept of activation of propargyl glycosides with gold catalysts, the possibility of using the ‘armed–disarmed principle’ to build higher analogs of oligosaccharides was also explored. They coupled the armed per‐benzylated propargyl mannopyranoside 103 with disarmed per‐benzoylated mannopyranoside 104 to get the desired disaccharide 105 (Scheme 22 a), where the armed propargyl monosaccharide 103 acted as the donor, as anticipated.182 However, when the disarmed disaccharide formed, 105 was further utilized for sequential glycosylations by converting it to its armed counterpart 106; it was observed that instead of the desired trisaccharide, 107, disaccharide 108 was formed along with the anhydro sugar, 109 (Scheme 22 b). The reason behind such unnatural phenomenon was also explained by a mechanistic rationale (Scheme 23), stating the concept of ‘double‐activation’ of the armed glycosyl donor, 106, as the probable cause. Upon reacting with further disarmed aglycon, higher oligosaccharides were synthesized effectively. Hotha and co‐workers also reported glycosylation with propargyl glycosides at ambient temperature.183
Scheme 22.
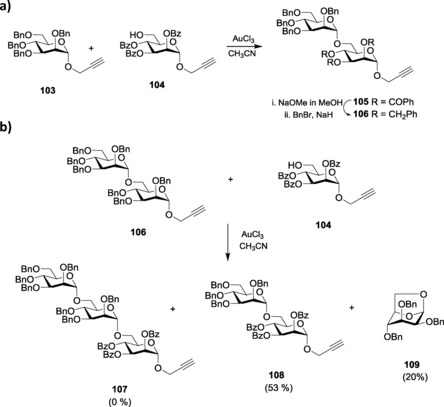
a) Selective glycosylation with propargyl glycosides. b) Unnatural inter‐glycosidic bond cleavage.
Scheme 23.
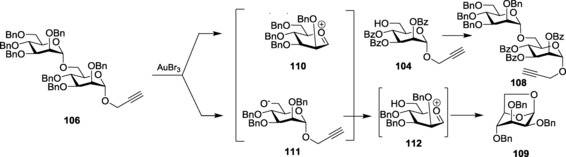
Mechanistic rationale.
Interestingly, gold catalyst AuBr3 alone led to a very slow reaction where propargyl furanosides were used as the donor, and the disaccharides were also formed in a low yield. Addition of AgOTf along with AuBr3, in turn, improved the reaction184 considerably, making it more spontaneous and high yielding. Benzylated propargyl furanoside113, acting as the armed donor, was also coupled with benzoylated propargyl pyranoside114 (Scheme 24) to give product 115 in line with the ‘armed–disarmed’ concept.
Scheme 24.
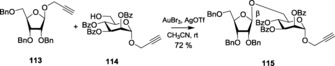
Glycosylation with propargyl furanosides.
Apart from propargyl glycosides, reports of the implementation of S‐but‐3‐ynyl glycoside donors and derivatives have also been revealed.185 These donors were used for gold‐catalyzed glycosylations for the synthesis of 2‐deoxy glycosides. Another set of alkynyl derivatives, o‐(methyltosylaminoethynyl)benzyl glycosides, was also developed as donors186 suitable for expeditious oligosaccharide synthesis and for the introduction of various aglycons.
Similar to the concept of alkenyl orthoesters, alkynyl orthoesters were developed as a versatile leaving group aiding in glycosylations. Propargyl orthoesters were glycosylated with various aglycons187 through gold (III) activation to give the highly stereoselective 1,2‐trans glycosylated product in moderate‐to‐high yields.188 However, Hotha and co‐workers revealed that the propargyl orthoesters, when coupled with carbohydrate‐derived secondary alcohols as the acceptor gave comparatively lower yields,189 when compared with coupling with primary carbohydrate alcohols. The same group also reported the selective activation of the propargyloxy group of the propargyl orthoesters in the presence of the competing propargyl glycosides by AuBr3 activation.190
With a similar intention to develop new and modified glycosylation strategies, Li et al. failed to activate 4‐pentynyl or 5‐hexynyl glycosides by gold(III) catalysis. To overcome the limitation, they introduced ortho‐alkynyl benzoate glycosides,191 a more active alkynyl group that can be activated by gold catalysts and illustrated the detailed mechanistic insight towards the glycosylation procedure.192 This protocol for activation has been implemented for the synthesis of various saponins193 and higher oligosaccharides.194 O‐Hexynyl benzoate glycosides 116 were glycosylated with various aglycons on being activated by Ph3PAuOTf at room temperature to get high β‐selective products (Scheme 25). The same alkynyl benzoate glycoside was also used for its coupling with acid alcohols.195 A chemoselective protocol has also been employed, where it was observed that Ph3PAuOTf activation in the presence of BF3⋅Et2O and DBU gave exclusive coupling with the acid alcohols, whereas the same coupling through Ph3PAuOTf in the presence of DTBP led to the exclusive formation of alcohol orthoester in high yields (Scheme 26). Further studies revealed that the loading of gold catalysts could be lowered to 0.01 equivalents or 0.5 mol % with the external assistance of an additional strong protic acid196 (10 mol % or 0.1 equiv HOTf).
Scheme 25.
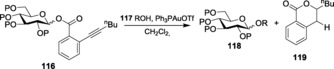
β‐Glycoside formation using o‐hexynyl benzoate glycosides.
Scheme 26.
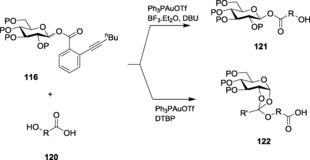
Chemoselective strategy to obtain different products from the same starting material.
There have been reports where glycosyl ortho‐alkynyl benzoates were efficiently activated by using cheap and easily available molecular iodine.197 The mild iodine was also capable of selectively activating these donors over thioglycosides, which helped in performing various sequential glycosylations (Scheme 27). Activation of alkynoate glycosides using Hg(OTf)2 as the promoter198 has also been reported.
Scheme 27.
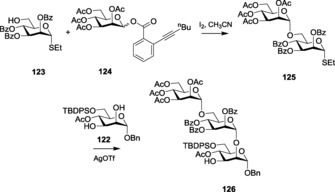
Synthesis of a trisaccharide with alkynoate glycosides.
A linear η2‐alkyne π complex of [Ph3PAu]+, namely AuI π‐bis‐(tert‐butyldimethylsilyl)acetylene triphenylphosphine,199was explored to activate ortho‐cyclopropylethynylbenzoate glycosides and subsequent glycosylations with both acceptors and non‐sugar aglycons. The same gold‐catalyzed activation of ortho‐alkynyl benzoate glycosides has been achieved by Seeberger and co‐workers in a continuous flow reactor.200 The procedure gave high yields of the glycoside products in much less time.
Yu and co‐workers introduced ortho‐alkynylphenyl thioglycosides,201 a similar glycosyl donor that could be activated by gold catalysts. Glycosylations with catalysts like [Btz−Au−PPh3]OTf and Ph3PAuOTf were seen to give the required product in excellent yields. Recently, Balamurugan and co‐workers introduced yet another easily accessible alkynyl leaving group, dipropargyl cyano acetate.202 per‐Acetylated glycosyl analogs with this leaving group were optimized for glycosylation with various sugar acceptors and non‐sugar aglycons by varying both the solvent and catalyst systems.
3.6. Glycals
Glycals are highly important components in carbohydrate chemistry that can be effectively used for the formation of 2,3‐unsaturated glycosides. The transformation of glycals 127 into unsaturated‐O‐glycosides 129 takes place in the presence of various catalysts like Lewis acids, metal salts, or other activating agents (Scheme 28). This methodology was first introduced by Ferrier203 and is more commonly identified as the Ferrier reaction.204 Catalysts represent the most important aspect of the Ferrier reaction, and various studies and investigations are being done to modify and improve the nature of the catalysts. The 21st century witnessed substantial modifications in the use of metal salts and metal triflates as catalysts for the coupling of the glycals.
Scheme 28.
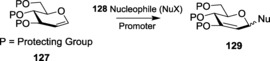
Ferrier reaction.
Starting with alkali metals, potassium dodecatungstocobaltate, K5CoW12O40⋅3 H2O (POM), was explored to understand the role of potassium as the catalyst. Various glycosylations with different acetylated glycals 130 and POM as the catalyst in varied solvents afforded α‐glycosides 132 as the major product in good‐to‐excellent yields (Scheme 29). Substantial optimization revealed acetonitrile as the best choice of solvent. Rafiee et al. also showed the probable mechanistic pathway that elegantly described the reason behind the effective use of perfect outer‐sphere one‐electron oxidants for such glycosylations.205 The catalyst showed high yields, even after being recovered four times, thus proving the high efficiency of the alkali metal as the catalyst.
Scheme 29.
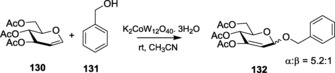
Use of potassium as the catalyst in the Ferrier(I) rearrangement.
Proceeding to the more versatile transition metals, iron(III) chloride was established as an efficient catalyst for Ferrier rearrangement by Bettadaiah and Srinivas,206 where it was used for the glycosylation of tri‐O‐acetyl‐d‐glucal with butan‐1‐ol, yielding the α‐isomer as the major product. Among the reaction conditions tested, refluxing acetonitrile was found to be best. In such optimized conditions, the loading of the catalysts could be lowered to 0.001 mol equivalents.207 FeCl3 has also been used in ionic‐liquid medium. The versatility of ionic liquid resides in the phenomenon of their adjustable solubility. [bmim]Cl–xFeCl3‐based ionic liquid was prepared by mixing anhydrous FeCl3 with 1‐butyl‐3‐methylimidazolium chloride, [bmim]Cl. This ionic medium was further utilized as the catalyst for the reaction of glucals with various alcohols (Scheme 30).
Scheme 30.
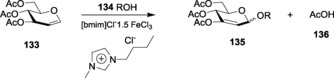
Ionic‐liquid‐induced Ferrier glycosylation.
A comparative assessment has also been shown for the Ferrier reaction by using CAN (ceric ammonium nitrate) and Fe2(SO4)3⋅xH2O, respectively.208 The latter was shown to yield exclusive 2,3‐unsaturated‐α‐glucosides with both per‐acetyl and per‐benzyl glucals. From the schematic study with various iron(III) salts by Wang and Chen in 2012, it is evident that ferric triflate is the most versatile iron‐based Lewis acid for performing the glycosylation of peracetyl glycals with alcohols in terms of selectivity and yield (Table 4). The use of only 10 mol % of the catalyst in dichloromethane as the solvent gave a maximum of 87 % yield with the α‐isomer as the major product.209
Table 4.
Comparative study of FeIII catalysts in Ferrier rearrangements.
| No. | Catalyst | Time [min] | Yield [%] | α/β |
|---|---|---|---|---|
| 1 | FeCl3 | 120 | 31 | 5:1 |
| 2 | Fe2(SO4)3 | 120 | negligible | nil |
| 3 | Fe(NO3)3 | 120 | 41 | 9:1 |
| 4 | Fe(OTf)3 | 30 | 88 | 10:1 |
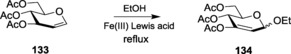
The inexpensive ZnCl2 was also shown to catalyze Ferrier glycosylation when using SN1 active nucleophiles. The catalyst provided good‐to‐excellent yields upon reaction of 3,4,6‐tri‐O‐acetyl‐d‐glucal 133 with allylic, benzylic, and tertiary alcohols in dichloromethane medium.206 Liu and co‐workers illustrated that ZnCl2 impregnated with activated alumina can be optimally utilized for the preparation of 2,3‐unsaturated glycosides. They have shown the use of this catalyst system both in the presence and absence of solvent system.210 The same catalyst system was also used in the subsequent year for Ferrier aza‐glycosylation.211 More recently, Zn(OTf)3 has been estimated to be a cheaper, milder, and more efficient catalyst for the stereoselective synthesis of α‐glycosides with a variety of O and S nucleophiles adorned with a wide range of functionalities.212 In 2014, Srinivas and co‐workers reported the use of the otherwise unreactive ZnBr2 in the α‐glycosylation reaction of 3,4,6‐tri‐O‐acetyl glucal 133 with a variety of primary, secondary, tertiary, allyl, and benzyl alcohols under microwave conditions.188
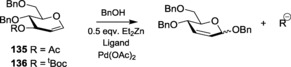
In another noteworthy set of experimental results, Et2Zn was used to activate the alcoholic acceptor. However, the synthesis of 2,3‐unsaturated glycosides was achieved by reacting the substituted glucal with the Zn‐salt‐activated alcohol in the presence of a Pd catalyst.213 Special consideration was given to the fact that, in such reactions, the anomeric stereoselectivity was governed by the reagent and solvent system rather than the protecting group. Addition of di(tert‐butyl)‐2‐biphenylphosphine, DTBBP yielded the β‐product, whereas the employment of P(OMe)3 produced the α‐isomer as the major product (Table 5). Nguyen and co‐workers also implemented the use of PdII/L system for the direct construction of the α‐glycoside from the glycals through the formation of glycal trichloroacetimidates (Scheme 31).214 Following extensive work with several ligand systems, Pd(PhCN)2Cl2 was reported to give the maximum stereoselectivity in the presence of DTTBP providing the α‐isomer in 84 % yield. Furthermore, many derivatives of 2,3‐unsaturated glycosides have been prepared by using the same concept of implementing a PdII/L catalyst to activate the glycal π system.215 Recently, Liu and co‐workers employed decarboxylative strategy by using Pd2(dba)3 as the catalyst and DtBPF [1,1′‐bis(di‐tert‐butylphosphino)ferrocene] as the ligand in the presence of the base, Cs2CO3, for the formation of 1,2‐unsaturated O‐glycosides.216 Higher base loading (2.0 equiv) led to the effective formation of the β‐glycoside. Excellent α‐selectivity was obtained by coupling carbohydrate acceptors with glycals when being activated by ReV‐oxo complex, ReOCl3(SMe2)(Ph3PO).217
Table 5.
Difference in stereoselectivity in Pd‐catalyzed glycosylation of glycals.
| No. | Compound | Ligand | Yield [%] | α/β |
|---|---|---|---|---|
| 1 | 135 | 15 % DTBBP | 92 | <1:25 |
| 2 | 135 | 30 % P(OMe)3 | 90 | 7:1 |
| 3 | 136 | 15 % DTBBP | 92 | <1:25 |
| 4 | 136 | 30 % P(OMe)3 | 90 | 5:1 |
Scheme 31.
PdII catalyzed conversion of glycal to the glycoside.
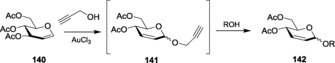
Similarly, with other variety of donors, the high alkynophilicity and oxophilicity of Au catalysts were utilized to perform Ferrier glycosylation. Later, depending on the same phenomenon, Balamurugan and Koppolu used AuCl3 as the catalyst under mild reaction conditions to synthesize 2,3‐unsaturated glycoside with high α‐stereoselectivity.218 When glycal 140 was reacted with propargyl alcohol in the presence of AuCl3 in acetonitrile medium, intermediate 141 was formed in situ, which was subsequently subjected to glycosylation with acceptors to give the product in moderate‐to‐good yield with the α‐isomer predominating (Scheme 32). Thus, wide studies of glycosylations with gold catalysts have established their versatility in the successful formation of products with a wide range of nucleophiles.
Scheme 32.
AuCl3‐catalyzed propargylation and subsequent glycosylation.
Studies have also been done with other transition‐metal salts. Copper(II) triflate has been reported to give 2,3‐unsaturated product under milder conditions.219 Kashyap and co‐workers introduced RuCl3⋅3 H2O as an efficient Lewis acid catalyst for such reactions.220 Niobium(V) chloride was also implemented as an efficient catalyst for the synthesis of unsaturated glycosides from per‐O‐acetylated glucal221 by virtue of its high stability, moisture tolerance, and economic viability. In a corresponding report, the phenomenon of water solubility of scandium(III) triflate was utilized to perform Ferrier glycosylations in aqueous medium. In non‐aqueous medium, only 5 % w/w of Sc(OTf)3 proved to be adequate to complete the reaction within a maximum time period of 3.5 h with a wide variety of aryl and alkyl alcohols.222 A variety of yttrium salts were also explored, of which Y(OTf)3 was found to be the most efficient, 10 mol % of which was optimum to bring the reaction to completion.223 Among other transition‐metal salts, TiCl3(OTf),224 organozinc reagents,225 and zirconium(IV) chloride226 have also been used as highly efficient catalysts for type I Ferrier rearrangement.
Proceeding to the use of lanthanides as catalysts for Ferrier rearrangements, Yadav et al. illustrated Dy(OTf)3‐catalyzed glycosylation of glycals with alcohols, phenols, and hydroxy‐α‐amino acids in 1‐butyl‐3‐methylimidazolium hexafluorophosphate, ([bmim]PF6) ionic liquid.227 High yields of unsaturated glycopyranosides with the α‐isomer predominating were obtained in mild, solvent‐free conditions by glycosylation of glycals mediated by inexpensive, versatile, and environmentally friendly lanthanum(III) nitrate catalyst.228 However, this catalyst failed to provide a significant yield with solvents like CH2Cl2, THF, diethyl ether, and so forth, but on using nitromethane as the solvent, the yield improved significantly (Table 6).229 The good stability of the catalyst and milder reaction conditions made Er(OTf)3 one of the most efficient catalysts for triggering Ferrier glycosylation (Scheme 33). In almost all the cases, the α‐anomer was found to be predominant. A recent addition in Ferrier glycosylation is the electrochemical generation of zirconium catalyst, which provided a novel protocol for the effective glycosylation of acetylated glycals.230
Table 6.
Comparative study of glycosylations with glucals in various solvents.
| No. | Solvent | Time [h] | Yield [%] |
|---|---|---|---|
| 1 | CH2Cl2 | 48 | 25 |
| 2 | Et2O | 48 | 15 |
| 3 | THF | 48 | 10 |
| 4 | CH3CN | 48 | 35 |
| 5 | CH2Cl2(dry) | 12 | 46 |
| 6 | Et2O | 12 | 25 |
| 7 | CH3CN (dry) | 12 | 58 |
| 8 | CH3NO2 (dry) | 2 | 90 |
| 9 | CH3NO2 (dry) | 6 | 90 |
Scheme 33.
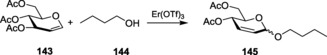
The use of erbium triflate as a catalyst.
Basic metals, such as aluminum, have also been used as catalysts. The use of aluminum triflate as a potential catalyst in Ferrier reaction has also been widely studied. In 2002, it was revealed that tri‐O‐benzyl‐d‐glucal catalyzed by Al(OTf)3 gave both 2‐deoxy glycosides and Ferrier‐rearranged pseudoglycal products at 60 and 0 °C, respectively.231 The versatility of the aluminum salt was verified when a wide range of acceptors were used ranging from alkyl alcohols to phenolic alcohols. Al(OTf)3 was also used to show the temperature dependence in Ferrier rearrangement. Williams and co‐workers noticed that different temperatures yielded different products upon reacting glycals with alcohols in the presence of an Al catalyst.231 At 60 °C, 2‐deoxy‐1‐O‐glycosides were obtained, whereas on lowering the temperature to 0 °C, Ferrier product 2,3‐unsaturated‐O‐glycosides were obtained. Both products were obtained in high α‐selectivity. Sequential glycosylation has also been illustrated on being catalyzed by AlCl3.232 In this reaction, derivatives of 3‐hydroxy galactal 146 were reacted with diethyl phosphorochloridite and Et3N, which provided the corresponding 3‐O‐diethoxyphosphanyl‐d‐galactal, 147. It subsequently underwent Ferrier rearrangement with various nucleophiles in the presence of AlCl3 to give the required 2,3‐unsaturated glycosides 148 (Scheme 34). Indium is another basic metal that has been frequently reported to catalyze the Ferrier reaction with glycals. In an interesting finding, Boga and Balasubramanian reported a Ferrier glycosylation where glycals were reacted with varied nucleophilic acceptors catalyzed by 20 mol % of highly anhydrous InCl3.233a They, however, continued the reaction for a longer time to observe the conversion of O‐glycoside to the corresponding C‐glycoside.233 It was found to provide a better yield compared to the other metal‐based catalysts. Unactivated glycals with no protecting group in the C‐3 position have also been successfully activated to undergo Ferrier rearrangement by InCl3.234 In a similar protocol, basic BiCl3 235 has also been used. Bismuth(III) triflate was implemented as a probable catalyst for Ferrier rearrangement both in the presence and absence of silica.236 Various types of alkyl and aromatic alcohols have been illustrated to undergo the glycosylation to stereoselectively produce the α‐isomer as the major product in a considerably high yield.
Scheme 34.
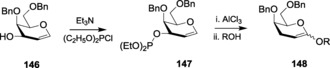
Glycosylation induced by diethyl phosphorochloridite.
Addition of glycols to the glycals through Ferrier glycosylation using tellurium salts237 was also reported for the expeditious synthesis of various macromolecules. Thus, metal‐catalyzed Ferrier reaction has been extensively studied in the present century. However, more protocols to promote the formation of β‐selective products are still in demand.
Advancing from metal‐based Ferrier glycosylations, immediate interest is directed towards the importance of the development of metal‐free strategies. In 2008, De et al. showed the effectiveness of metal‐free synthesis of 2,3‐unsatrated glycosides from corresponding glycals.238 They showed that the glucal reacted with various nucleophilic alcohols in a greater amount (20 equiv) to give the corresponding 2,3‐unsaturated product without the addition of any metal or Lewis acid catalyst, provided HFIP is used. The solvent‐free rearrangement reaction was further facilitated by the use of Montmorillonite K‐10 as the catalyst.239 Application of microwave heating was also reported to be advantageous over standard heating conditions, leading to less effective reaction time. In both cases, exclusive α‐products were obtained with a cleaner and milder method. Montmorillonite K‐10 clay‐supported Bi(OTf)3 240 has also been illustrated to achieve the synthesis of various types of sialic acid through Ferrier transformation. Other versatile catalyst systems, for example HClO4–silica,241 CF3SO3H–silica,242 NaHSO4–silica,243 and H2SO4—silica,244 have been used for the synthesis of a wide range of pseudoglycosides. In the context of organic acid catalysts, when acetic acid failed to yield any desired unsaturated glycoside, Liu and co‐workers decided to test the effectiveness of camphorsulfonic acid (CSA)245 as a probable catalyst for triggering the formation of pseudoglycosides. Both (R) and (S)‐CSA, in their optically pure forms, were reported to give the desired product, the (S)‐isomer being the more efficient one.
Various other catalyst systems have been developed over the decade, promoting the formation of 2,3‐unsaturated O‐ and C‐glycosides from glycals. Notable ones are cyanuric chloride‐catalyzed Ferrier reaction,246 bromodimethylsulfonium (BDMS)‐mediated rearrangement,247 and 3,5‐dinitrobenzoic acid (DNBA)‐mediated synthesis of O and S‐glycoside.248 The incorporation of potassium alkynyltrifluoroborate group to the glucal has also been implemented by BF3⋅Et2O‐mediated249 Ferrier glycosidation.
The vinyl oxirane chemistry was implemented by Crotti and co‐workers who mastered the synthesis of the oxirane or epoxide from the corresponding glycal and further used it for subsequent catalyzed or uncatalyzed250 glycosylation with a wide range of nucleophiles under different reaction conditions.251 This glycosylation protocol led to yield the β‐isomer as the major product, whereas the use of benzene as the solvent gave the β‐product exclusively.252 However, the synthesis of α‐derivatives has also been reported by using the same protocol.253 The addition of organolithium reagents254 onto the oxiranes by using the same procedure also gave efficient reactions to give the desired product.
3.7. Deoxy Sugars
3.7.1. 2‐Deoxy Sugars
2‐Deoxy sugars with the OH group in the C‐2 position replaced by a hydrogen atom are widely distributed in various natural products. The wide biological importances of the 2‐deoxy sugars have made them a highly novel synthon in chemical glycosylations. However, the formation of glycosidic linkages with these 2‐deoxy glycosides pose quite a challenging task, owing to the absence of any directing group in the C‐2 position. Moreover, the absence of any electron‐withdrawing group in the strategic C‐2 position makes the moieties highly acid labile, leading to hydrolysis of the compound. Various analyses have been performed over the years to control the stereoselectivity of the glycosides formed at the anomeric position of the 2‐deoxy sugars. Many of the earlier developed protocols have been extensively discussed in various Reviews.255
Employing direct methodology for the stereoselective synthesis of natural 2‐deoxy glycosides, Lear et al.256 established the use of the mild activating system, AgPF6/DTBMP (2,6‐di‐tert‐butyl‐4‐methylpyridine). Amongst the various Ag catalysts, this promoter system was found to give the maximum α‐stereoselectivity under optimized conditions. Recently, in the entire domain of glycosylation, pre‐activation of the donors is much in demand. In the case of glycals, this has also been established to give better products. As an illustration, Ye and co‐workers first activated 2‐deoxy thioglycosides 149 by using BSM−Tf2O.257 Subsequently, addition of acceptor 151 to the same reaction mixture gave the 1,2‐cis‐glycosylated product 152 with high stereoselectivity (Scheme 35). 2,6‐Dideoxy thiosugars 153 were also observed to give similar results. In a side reaction, when the mixture of donor and acceptor was directly exposed to the promoter system, the yield diminished drastically, indicating the importance of pre‐activation of the donor.
Scheme 35.
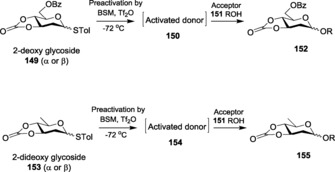
Glycosylations based on pre‐activation protocol with 2‐deoxy and 2‐dideoxy glycosides.
For the synthesis of higher analogs of 2‐deoxy oligosaccharides, Issa and Benett established the use of potassium hexamethyldisilazane (KHMDS)/p‐toluenesulfonic anhydride258 as an effective promoter system, leading to the exclusive synthesis of β‐linked 2‐deoxy‐sugar disaccharides from hemiacetal donors. The reaction was presumed to go through a SN2 pathway via an in situ glycosyl sulfonate intermediate to accomplish a highly β‐stereoselective glycosylation.
Glycosylation of glycals mediated by metal catalysts is, in itself, a wide area of research. Zhu and co‐workers investigated the role of 2‐deoxy S‐but‐3‐ynyl thioglycosides as an effective glycoside donor185 for performing O‐glycosylation through the homogeneous cationic gold(I)‐catalyzed selective activation of the alkyne group attached with the more nucleophilic thio functionality. The reaction conditions were optimized by the use of 5 mol % (4‐CF3−Ph)3PAuCl along with 10 mol % AgOTf, which gave almost quantitative yield. Coming to organoboron‐catalyzed glycosylations, per‐acetylated 2‐deoxy as well as 2,6‐dideoxy chlorides were reported to show high regio‐ and β‐stereoselectivity on activation with diphenylborinate 156 (Figure 7) in moderate‐to‐good yields. However, the requirement of a more nucleophilic acceptor was essential and, hence, sugar diols were employed, limiting the scope of such glycosylations259 with the halide counterparts. Polymer‐assisted activation of 2‐deoxy thioglycosides has also been investigated by Kirschning and co‐workers.260 Polymer‐supported haloate(I) complexes 157 and 158 were used to activate the thioglycosides for the subsequent formation of higher oligosaccharides.
Figure 7.
156: Borinic acid pre‐catalyst; 157, 158: Polymer‐supported haloate complexes.
Remote participation of certain protecting groups to facilitate the glycosidic bond formation with 2‐deoxy thioglycosides has also been studied.261 Mong and co‐workers have shown the effects of picoloyl (Pico) group, illustrating their role for the glycosylation262 with deoxy glycoside donors. Thioglycosides with the picolyl protecting group at the C‐6 position, on being activated with NIS/TMSOTf, was seen to give significant β‐stereoselectivity. Introduction of S‐(phenylthiomethyl)benzyl moiety at the C‐6 position of 2‐deoxy or 2,6‐dideoxy glycosyl donors was also responsible for the synthesis the α‐isomer.263
3.7.2. 2‐Amino‐2‐deoxy Sugars
By virtue of their structures and reactivities, 2‐amino‐2‐deoxy sugars are widely distributed in various living organisms as glycoconjugates.2, 264 Previous Reviews265 have widely explained the strategy behind the introduction of the participation and non‐participating group in the C‐2 position to direct the glycosylation to the required stereochemistry. 2‐Amino‐2‐deoxy sugars have been widely used in oligosaccharide syntheses.266 Repeating sequence of α‐GalNAc was found to be effectively synthesized by the introduction of oxazolidinone group.267 The N‐acetyloxazolidinone‐protected thioglycosides both as the glycosyl donor 159 and acceptor 160 was effective (Scheme 36) when activated with Ph2SO−Tf2O promoter system to give exclusive α‐stereoselective product 161 in good yields. The synthesis of various important core glycoside parts of GPI anchors, heparin, heparin sulfate, and heparosan had also been successfully accomplished by Hung and co‐workers.268 In 2008, anhydrous FeCl3‐catalyzed direct formation of α‐N‐acetyl‐glycosaminides was also successfully achieved by Wei et al.269 and it was effectively implemented in the large‐scale preparation of fluorogenic TN‐antigen probes.
Scheme 36.
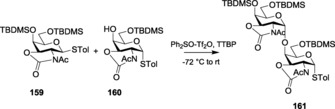
The use of N‐acetyloxazolidinone protected thioglycosides as both donor and acceptor.
Recently, glycosides with a phthalimido group in the C‐2 position were found to be activated by the hypervalent iodine compound, phenyliodine bis(trifluoroacetate) (PIFA) in the presence of triflic acid.270 Proper optimization of the reaction conditions of this metal‐free glycosylation afforded the coupled product in significant yields. Direct glycosylation with GlcNAc has also been reported by Jensen and co‐workers271 through the activation of thioglycosides 166 or pentenyl glycosides 167 by using various metal triflates (Table 7). However, when glycosylated with carbohydrate acceptors, the yields obtained were poorer than simple acceptors like 1‐octanol or (−)‐menthol. 2‐Deoxy‐2‐N‐sulfate glycosides 162 were also successfully utilized (Scheme 37) to give the desired β‐glycosylated products 163 in excellent yields272 on being protected with 2,2,2‐trichloroethyl (TCE) group, after which it could be easily deprotected by reacting Zn in the presence of ammonium chloride in methanol. A Yb(OTf)3‐mediated synthesis of N‐acetyl glucosamine derivatives273 has also been established by Crasto and Jones, which gave β‐glycosylated product in high yields. Direct synthesis of 1,2‐amino glycosides with p‐methoxybenzylidene group in the C‐2 position was achieved by Mensah and Nguyen by using nickel catalysts.274 Optimization of the reaction showed that Ni(4‐FPhCN)4(OTf)2 gave the best stereoselectivity with maximum effectiveness within a short reaction time.
Table 7.
A comparative study of metal‐catalyzed glycosylations with 2‐amino‐2‐deoxy donors with different acceptors.
| No | Donor | Acceptor | Product | Activation | Temp | Time | Yield [%] |
|---|---|---|---|---|---|---|---|
| 1 |
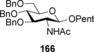 (2 equiv) (2 equiv) |
1‐octanol (1 equiv) |
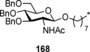
|
Sc(OTf)3, NIS | RT | 16 h | 88 |
| 2 | 167 (2 equiv) | 1‐octanol (1 equiv) | 168 | Cu(OTf)3, NIS | RT | 16 h | 84 |
| 3 | 167 (2 equiv) | 1‐octanol (1 equiv) | 168 | Zn(OTf)3, NIS | RT | 16 h | 88 |
| 4 | 167 (2 equiv) | (−)‐menthol (1 equiv) |
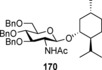
|
Cu(OTf)3, NIS | RT | 25 h | 76 |
| 5 | 167 (3 equiv) |
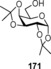 (1 equiv) (1 equiv) |
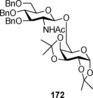
|
Cu(OTf)3, NIS | RT | 5 days | 37 |
| 6 | 167 (3 equiv) |
 (1 equiv) (1 equiv) |
|
Cu(OTf)3, NIS | RT | 2 days | 50 |
Scheme 37.
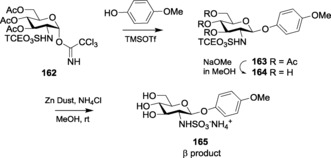
Use of 2‐deoxy −2‐N‐sulfate glycosides in glycosidations.
Thus, for amino sugars, the stereochemistry of the products largely depended on the directing properties of the protecting group in the C‐2 position. Modifications in the nature of the protecting group are still an active area of research.
3.8. Other Types of Glycosylation
3.8.1. Protecting‐Group‐Free Glycosylation
Until now, most of the glycosylating protocols described dealt with O‐protected glycosyl donors, aiding in the synthesis of varied glyconjugates and higher oligosaccharides. However, there has been much discontent about glycosylations performed with unprotected glycosyl donors with activated anomeric center. This is largely because accomplishing glycosylations without protecting the hydroxyl groups brings forth a number of advantages, the prime issue being the reduction in the protecting‐group manipulation steps of the complex oligosaccharide synthesis. It also opens the scope for iterative glycosylations, which lead to the continued coupling of the sugar fragments. Moreover, these hydroxyl glycosides also promises to overcome the issue of reduced reactivity with the acyl‐protected glycosides. The free hydroxyl groups also leave the opportunity for its ready use in various antibodies and therapeutics.
Despite all of the advantages, performing glycosylations with unprotected glycosyl donors present some major hurdles. Similar to 2‐deoxy glycosides, the absence of any directing group in the C‐2 position makes it highly difficult to control the stereochemistry of the formed glycosidic bond in the anomeric center. Moreover, the easy accessibility of more than one reactive hydroxyl groups requires the application of highly selective reagents capable of performing the coupling in the desired position. Hence, studies to optimize the reagents and the conditions to attain the best possible output are underway. Describing all glycosylation protocols developed in this field would require a complete detailed Review in its own right. The strategies of the past century have already been extensively reviewed.275 However, we aim here to describe the ones that show the most significant promise and can contribute to the challenges in performing regular glycosylations.
In an attempt to find a suitable robust donor capable of being activated in its unprotected state, Roy and Mukhopadhyay performed Fischer‐type glycosylation to establish the use of H2SO4–silica to couple unprotected free sugars with different alcohols.276 Interestingly, Pfaffe and Mahrwald showed that, when variously functionalized alcohols were reacted with free d‐ribose 175 catalyzed by 10 mol % of titanium‐(IV) tert‐butoxide and 50 mol % d‐mandelic acid at room temperature, only coupled ribose furanosides (Scheme 38) were obtained.277Addition of lithium bromide significantly enhanced the yield in acetonitrile medium. The same group in the following year introduced the concept of organo‐catalyzed glycosylation of unprotected and unactivated glycosides.278 Here, traces of PPh3 and CBr4 were used as the catalyst with LiClO4 as the essential additive. Surprisingly, this catalyst was successful in facilitating the formation of exclusive β‐products. Recently, Bhattacharrya and co‐workers also described the activation of both unprotected and unactivated glycosides by using 10 mol % of bismuth nitrate pentahydrate.279 The yields of the desired products were significantly increased when unprotected sugars were subjected to Fischer glycosylation by sulfamic acid.280 Sharma et al. reported the coupling of free unprotected and unactivated sugar molecules with alcohols on being mediated by NH4Cl at 90 °C.281 The yields of the products obtained were claimed to be higher and the process less time consuming compared to the standard glycosylation protocols using unprotected glycosyl donors in the presence of common catalysts like Amberlite IR‐120 (H+) resin, InCl3, In(OTf)3, Sc(OTf)3, and HCl.
Scheme 38.
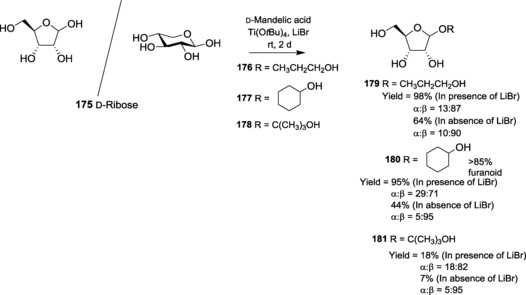
Reaction of d‐ribose with different alcohols.
The use of ionic liquids for performing glycosylations with unprotected glycosyl donors has also been explored. RTILs such as 1‐ethyl‐3‐methylimidazolium benzoate ([emIm][ba]) in the presence of Amberlite IR‐120 (H+) resin or p‐toluenesulfonic acid (TsOH) were used282 as promoters. These ionic liquids were reported to be successful in activating various unprotected sugar units for the desired oligosaccharide synthesis. Glycoconjugates derived from the glycosylations with this ionic liquid were also studied for their affinity towards lectins.283 Auǵe and Sizun reported the use of ionic liquids in Lewis‐acid‐catalyzed glycosylations in 2009. They first explored the catalytic efficiency of various metal salts like InCl3, In(OTf)3, Sc(OTf)3, and Yb(OTf)3 at different temperatures. The studies revealed that even a catalytic amount of Sc(OTf)3 provided the optimum yield at 80 °C on continuing the reaction for a longer period. Considering this and predicting the mechanism following the oxocarbenium ion intermediate, Auǵe and Sizun attributed the increase in yield and regioselectivity by the use of ionic liquids.284 A variety of ionic liquids, varying in their ionic part, were tested, among which 1‐butyl‐3‐methylimidazolium trifluoromethanesulfonate [bmim][OTf] gave encouraging results. Optimization of the reaction was, thus, accomplished with different unprotected sugars 182 and alcohol 183 catalyzed by Sc(OTf)3 in the presence of 1 mol % of [bmim][OTf] as the solvent (Scheme 39). The results of the reactions with octanol as the acceptor alcohol (Table 8) showed that the reagent is capable of generating the desired product in subtle α‐selectivity. Sc(OTf)3 as the catalyst has also been utilized for glycosylation of carbohydrates with different amino acids have also been accomplished in [bmim][OTf].285
Scheme 39.
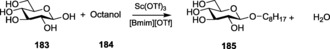
Reaction of unprotected sugars with alcohol.
Table 8.
Different yields obtained on the reaction of free sugars with octanol.
| Sugar | Temp [°C] | Time [h] | Yield [%] | α/β |
|---|---|---|---|---|
| d‐glucose | 80 | 24 | 74 | 75:25 |
| d‐galactose | 80 | 24 | 70 | 61:39 |
| d‐mannose | 80 | 24 | 88 | 100:0 |
| l‐fucose | 80 | 24 | 51 | 67:33 |
| N‐acetyl‐glucosamine | 110 | 24 | 60 | 79:21 |
Unprotected alkynyl glycosides were also explored for activation by Mamidyala and Finn,286 who tried to optimize the reaction conditions by using different solvent systems and employing variable temperatures catalyzed by AuIII salts. Detailed studies revealed the versatility of refluxing acetonitrile as the solvent to achieve the best possible yield. The same protocol was also verified by applying it for the synthesis of a trisaccharide. However, the stereoselectivity obtained in the reaction did not show much promise.
A photoinduced protocol was also employed by Toshima and co‐workers in 2013, who described a novel glycosylation protocol through photoinduced activation of unactivated deoxy thioglycosyl donors.119 Here, DDQ served as the SET reagent, whereas boronic acid was used as a temporary 1,3‐diol protection, leaving the primary hydroxyl group available for reaction to prevent the corresponding self‐coupling and the formation of anhydrosugars.
Nitz and co‐workers introduced different anomeric protecting groups that show promise to be activated in protecting‐group‐free conditions.287 N′‐Glycosyltoluenesulfonohydrazides (GSHs) (186) have also been used in the formation of various glycosyl O‐phosphates288 upon being activated by NBS in DMF. Glycosylation of these GSHs with non‐volatile alcohols were demonstrated to give moderate yields, but with poor stereoselectivity.50 However, the possibility of the formation of unprotected glycosyl chloride 187 from GSH was explored in situ (Scheme 40), which can further undergo coupling with alcohols at a faster rate, giving better yields. But, in both cases, poor stereoselectivity (β‐anomer predominating) limits the scope of such anomeric leaving groups.
Scheme 40.
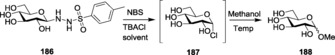
Glycosylation of unprotected N′‐glycosyltoluenesulfonohydrazides through the formation of glycosyl chlorides.
Further efforts are still being made in improving the stereoselectivity of glycosylation involving unprotected glycosyl donors. Atom economy and faster protocols show much promise towards expeditious oligosaccharide synthesis.
3.8.2. One‐Pot Glycosylation
Among the various progress achieved in oligosaccharide synthesis in the current century, the one‐pot glycosylation strategy has emerged as an important strategy. One‐pot glycosylations claim to be the shortest possible route for oligosaccharide synthesis. The strategies focus on an easy solution for issues related to purification and isolation of the intermediate products. However, one‐pot iterative glycosylations demand the use of strategically synthesized sugars. Such glycosylation strategies may be accomplished by employing either ‘armed–disarmed’ concept24 or chemoselective activation of the glycosyl donor. Thus, it becomes highly evident that one‐pot iterative glycosylations are primarily based on the reactivity difference of synthons at a particular reaction condition.
The present century has seen significant development in the domain of one‐pot glycosylations.289 In this Review, we report some of the most distinct illustrations that prove the versatility and novelty of such a glycosylation protocol and make it clear that one‐pot glycosylations are here to stay. In 2005, Demchenko and co‐workers utilized the reactivity difference of various thioimidates in convergent oligosaccharide synthesis.290 They illustrated the successful one‐pot synthesis of a tetrasaccharide, utilizing the reactivity difference of SBox, STaz, and SEt derivatives. Starting with an SBox 189 and a thioethyl glycoside 190, the first glycosylation was performed at room temperature in the presence of AgOTf as the catalyst, which selectively activated the SBox derivative to give the desired SEt disaccharide 191 exclusively. It was followed by the addition of another STaz acceptor 192 in the same reaction pot, and now the SEt group was activated by using the NIS/TfOH promoter system to afford the desired STaz trisaccharides 193 exclusively. Finally, the methyl glycoside acceptor 194 was added along with AgOTf to afford the required tetrasaccharide 195 (Scheme 41) through the activation of STaz. The final yield of the product obtained was quite significant, which highlighted the versatility of the iterative sequence. The advantage of one‐pot protocols over stepwise methodologies with leaving groups having different reactivities has also been illustrated by Cao and co‐workers291 in a recent report of synthesis of a branched penta‐mannoside.
Scheme 41.
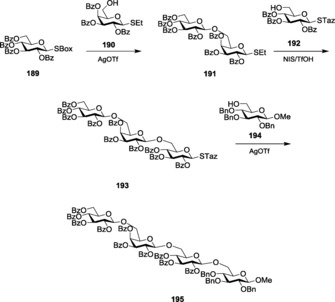
One‐pot glycosylation exploit reactivity differences of different thioglycosides donors.
Fine tuning of the donors has also facilitated synthesis of various convergent oligosaccharides. Polat and Wong demonstrated the successful one‐pot synthesis of the biologically relevant heparin‐like oligosaccharides292 by strategically synthesizing monosaccharide synthons. One‐pot glycosylations can also be performed by implementing the pre‐activation protocol. In 2008, Boons and co‐workers combined the reductive opening of the benzylidene acetals with sequential glycosylations in the course of synthesizing a trisaccharide.293 The most essential attribute of the whole process remains the selection of the proper reagent system. Likewise, after the first set of glycosylations with the trichloroacetimidate donor 196 and benzylidene protected acceptor 197 in the presence of TfOH, more TfOH and Et3SiH were added, following the reduction of the reaction temperature to −78 °C. The reagents were capable of selectively deprotecting the benzylidene group, which extended the scope to further coupling with another trichloroacetimidate donor 200 in the same reaction pot (Scheme 42). Desired trisaccharide 201 was subsequently formed in 63 % yield.
Scheme 42.
Synthesis of a trisaccharide through one‐pot glycosylation.
In a different sequence, using chemically different functionality in the anomeric center, Iadonisi and co‐workers modified the stepwise assembly of the biologically relevant antitumor PI‐88 pentasaccharide to a more robust and convenient one‐pot reaction strategy.138 The product was obtained by the use of mild Lewis acid catalysts like Yb(OTf)3 and Bi(OTf)3. Similarly, tumor‐associated carbohydrate antigen Globo‐H hexasaccharide was synthesized through the pre‐activation protocol294 in near‐stoichiometric yield. Various other expeditious oligosaccharide syntheses have been successfully achieved by using chemoselective one‐pot strategies.295 Ye and co‐workers introduced a new promoter system, benzenesulfinyl morpholine (BSM), to increase the efficiency of one‐pot glycosylations through pre‐activation method.296 For the synthesis of the trisaccharide 205, BSM was added for the complete activation of the thioglycoside 202 in the presence of Tf2O at −70 °C. Further addition of 203 after bringing the reaction temperature back to ambient led to the exclusive formation of disaccharide 204. Subsequent reduction of the reaction temperature to −70 °C and activation of the thioglycosides with BSM/Tf2O promoter system followed by addition of acceptor 205 provided trisaccharide 206 (Scheme 43) within 1 h in moderate yield.
Scheme 43.
Chemoselective one‐pot glycosylation of a trisaccharide.
In different activation methods, polymer‐assisted one‐pot synthesis of trisialic acid has been effectively achieved by Tanaka et al.297 Ionic liquids have also facilitated one‐pot syntheses. Galan et al. introduced the use of [bmim][OTf] as promoter in the presence of NIS for the region‐ and chemoselective glycosylation of glycosides with different reactivities.298
Despite the progress of one‐pot synthesis methods in the last two decades, there are still many limitations in its universal application. Studies and extensive investigations are being made to implement such processes in the expeditious synthesis of more natural oligosaccharides. However, this strategy shows immense promise, having the potential to emerge as a powerful synthetic tool in the domain of carbohydrate synthesis.
3.8.3. Solid‐Phase Synthesis
Solid‐phase oligosaccharide synthesis (SPOS) is an emerging field in carbohydrate chemistry that has attracted overwhelming interest in the present century. This modern method of carbohydrate synthesis claims to overcome various shortcomings of solution‐phase synthesis. Moreover, the need to perform purification at each step of a solution‐phase synthesis limits the applicability of the process, to a large extent, whereas SPOS involves a protocol that removes the excess reagents and other solution‐phase impurities by simple washing of the resin, thereby reducing the number of chromatographic steps involved. However, solid‐support‐based carbohydrate synthesis requires judicious planning and systematic implementation. The polymer support and the linker for its attachment to the sugar need to be planned very carefully so that it can withstand the conditions for the essential protecting‐group manipulations. However, at the same time, it should be labile so that it can be cleaved as and when required. Solid‐phase iterative strategies may be accomplished either by attaching the reducing or the non‐reducing end of the sugar to the solid polymeric support, leaving the growing end subjected to glycosylations. Attainment of stereospecific glycosylations in high yields through rationally designed strategies has made SPOS a widely used protocol. Taking cue from the first solid‐phase oligosaccharide synthesis by Fréchet and Schuerch299a in 1971 using Merrifield resin, rigorous attempts to develop smarter strategies have been observed in the later part of the last century.299b, 300 Various Reviews301 have revealed versatile and comprehensive overviews on SPOS. A study by Kanie et al. gives a broad comparative picture, as the group synthesized a wide range of fucosyl–galactosyl oligosaccharides in both the solution and solid phase.302
Monitoring the progress of reactions in SPOS is extremely essential. The crudest way is to take aliquots of the reacting resins and subject them to thin‐layer chromatography after cleaving them from the resins. Moreover, knowledge of the exact time required for the total completion of the reaction is crucial, as it has been observed that both a shorterned reaction time and exposure to excess time have been detrimental, increasing the amount of impurities in the product library. Thus, analysis of the on‐support conversions and yields becomes inevitable for the development of SPOS. 1H and 13C NMR spectroscopy has been explored in combination with other methods; but, it requires some time consuming and expensive protocols. Hence, in early 2001, Kihlberg and co‐workers implemented the use of gel‐phase 19F NMR spectroscopy303 by using fluorine‐labeled protecting groups in combination with fluorinated linkers. No countering signals were observed, owing to the absence of any fluorine group in the resins. Hence, monitoring solid‐phase oligosaccharide reactions by 19F NMR promised to provide much impact in the times to follow. By using the same protection protocols of fluorinated analogs, the same group utilized 19F NMR spectroscopy to monitor the solid‐phase synthesis of α‐Gal trisaccharide epitopes 207[ 304] after loading them with ArgoGel resin (Scheme 44). 13C NMR spectroscopy has also been found to monitor SPOS successfully, where the gated decoupling technique was implemented.305 Other methods for monitoring SPOS include single‐bead FTIR microspectroscopy, but it leads to significant overlap of spectra from the presence of multiple components including starting material, the involved resin, along with the formed product. To overcome this shortcoming, Yan and Yan introduced partial least squares (PLS), a chemometrics method for qualitative and quantitative analyses of samples.306 In this method, the removal or introduction of the organic functionalities in the ongoing reactions was displayed as positive or negative signals, respectively, thereby establishing itself as an effective and powerful analytical tool for SPOS.
Scheme 44.
Solid‐phase synthesis of α‐Gal trisaccharide epitope.
In another method, Manabe and Ito utilized a colorimetric method to establish real‐time monitoring of the on‐resin oligosaccharide synthesis by employing disperse red (C16H18N4O3) and (p‐nitrobenzyl)pyridine (PNBP) color tests.307 Owing to the complementary nature of the two tests, the progress of glycosylation reactions were successfully established by using these methods. The removal of the chloroacetyl group in the monosaccharide 209 was established by the positive red color in the disperse red test and negative result in the PNBP test. Subsequently, the disappearance of the hydroxyl groups in the glycosylation reaction was indicated by the negative disperse red and strongly positive PNBP tests (Table 9).
Table 9.
Colorimetric assessment of solid‐phase glycosylations.
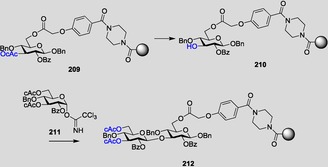
| Compound | PNBP test | Disperse red test | Remarks |
|---|---|---|---|
| 209 | positive | negative | positive PNBP test confirms presence of chloroacetyl group |
| 210 | negative | positive | positive disperse red test confirms presence of OH group |
| 212 | positive | negative | confirms of the presence of chloroacetyl group |
In SPOS, studies have shown that the presence of longer spacer connecting the synthons with the resin helps the coupling more than glycosides with a shorter spacer.308 Longer spacers help in bringing the sugar part more into the solution phase and provide more accelerated reactive platform.
Resin‐bound thioglycoside donors have successfully undergone glycosylation reactions with various acceptors under standard reaction conditions.309 Direct transfer of resin‐bound (JandaJel)310 thio glycosyl donors to complex aglycons was accomplished by Bennett and co‐workers.311 Optimization of reaction conditions implied benzenesulfinyl piperidine/triflic anhydride (BSP/Tf2O) as the best candidate of promoter in such a transformation. Glycosyl thioimidates have been successfully utilized for solid‐supported glycosylation strategies. In 2007, Demchenko and co‐workers performed glycosylations and, in one case, the solid‐bound acceptor was coupled with the imidate (SBox or STaz) donor and, in another case, the glycosyl thioimidate was bound to the resin followed by its glycosylation with the acceptor.164 Normal activation protocols using TMSOTf or AgOTf were employed for the glycosylations.
To establish a versatile SPOS strategy, Boons and co‐workers introduced polystryrene boronic acid (PSA)312 as the resin support that proved to be apt in its role. The ease of its preparation, the use of minimum amount of solvents for its loading, the cleavage, and its reusable nature made this resin widely used in oligosaccharide synthesis. When using PSA as the resin in oligosaccharide synthesis, protecting groups were selected, taking into consideration that the boronic ester linkage hydrolyzed in protic solvents. Hence, Fmoc protection was selected, as it is inert to protic solvents and could be cleaved by the treatment of triethylamine. Fmoc‐protected thioglycosides donor 213 was coupled with PSA‐immobilized acceptor 214 with the help of NIS/TMSOTf to obtain the resin‐bound disaccharide 215. The Fmoc group was then selectively cleaved to obtain the disaccharide acceptor 216, with the solid support intact. This PSA‐bound disaccharide was further coupled with thioglycoside donor 217 to obtain the final trisaccharide. Eventually, the resin could be cleanly cleaved by the application of an acetone–water mixture, obtaining trisaccharide 218 (Scheme 45), which can be further characterized by using spectroscopy. Capping reagents like benzoyl isocyanate have also been implemented, which aid in performing glycosylations by blocking the unconsumed acceptors and helping with cleaner SPOS.313 It helped in the purification of the formed glycosides, as it conveniently prevented the formation of unwanted sugar analogs with the unreacted counterparts.
Scheme 45.
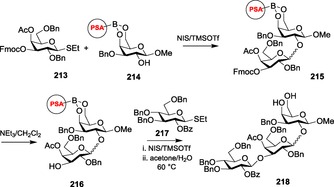
Glycosylations on polymer support.
Based on the convenient principles of solid‐phase polymer‐supported oligosaccharide synthesis, there have been examples of the synthesis of higher analogs of carbohydrates.314 Crich and Smith successfully applied a method for the synthesis of the unnatural β‐mannoside linkage.315 Schmidt and co‐workers also performed SPOS to synthesize a branched hexasaccharide, lacto‐N‐neohexaose derivative (found in human milk) by immobilization of linker‐loaded polystyrene resin.316 Glycosyl trichloroacetimidates were used as the efficient donors, whereas Fmoc and Lev protecting groups were exploited.
Thus, from where it was left in the previous century, SPOS has seen much progress in recent years. More vivid study on SPOS methods reveals widespread use of various glycoside donors like sulfoxides, thioglycosides, trichloroacetimidates, pentenyl glycoside, and so forth for effective glycosylation methodologies. However, solid‐phase synthesis still suffers certain limitations. These challenges open broader scopes of study to ascertain higher levels of stereochemistry, optimum, and suitable protecting‐group manipulations as well as the efficient use of the reagents in the synthesis of the solid‐supported glycosides. It is noteworthy that the present century promises to open new dimensions of solid‐phase oligosaccharide synthesis with its innovative and successful implementation in the introduction of a new domain involving automated oligosaccharide synthesis.
3.8.4. Automated Oligosaccharide Synthesis
Building on solid‐phase oligosaccharide synthesis, Seeberger et al. introduced the concept of automated oligosaccharide synthesis.317 Automated oligosaccharide synthesis is based on the same principle of SPOS, where the choice of the solid support and the linker plays a major role in the determination of the synthetic strategy. Moreover, protecting‐group manipulations should also be planned accordingly, with the inertness of the solid support and the linker with the glycosides. Considering the various aspects, Seeberger first utilized peptide synthesizer to develop an automated method for glycoside synthesis. Automated synthetic procedures enable the number various manual routine functions to be reduced by employing iterative synthesis comprising of selective deprotection, glycosylation, removal of the resin, and deprotection in a programmed reactor.317 The reactor was designed to perform conversions at variable temperatures subject to external programming, thus simplifying the synthesis of complex oligosaccharides. The widely used temporary protecting group suitable for such programmed reaction strategies, owing to their capability of selective cleavage, include Fmoc, Lev, and Nap, whereas the permanent protecting groups include benzyl, benzoyl, benzylidine, or azide.
With systematic synthetic strategy, Seeberger and co‐workers accomplished the first automated synthesis of two phytoalexin elicitors, a hexasaccharide 220 and a dodecasaccharide 219 (Figure 8).318 The widespread applicability of the glycosphingolipids, such as Globo‐H and Gb‐3, as probable antigens in a variety of cancers made them highly attractive biological targets. Automation of oligosaccharide synthesis has accomplished their synthesis in a linear fashion with glycosyl phosphates as the building blocks,319 facilitating the formation of the α‐galactosidic linkages immobilized on a solid support. LC–MS was successfully used to monitor reaction steps. Synthesis of the difficult β‐mannosidic bond was also achieved by using an automated protocol. Codée et al. demonstrated the β‐mannosylation by using carboxybenzyl glycosides320 on a solid support through automated synthetic cycles. Purification of the synthesized oligosaccharides through an automated strategy was also optimized by using the capping and tagging method of Seeberger and co‐workers.321 The use of any scavenger resin or proper filtration through a fluorinated silica gel has proved to be the best choice for such a process.
Figure 8.
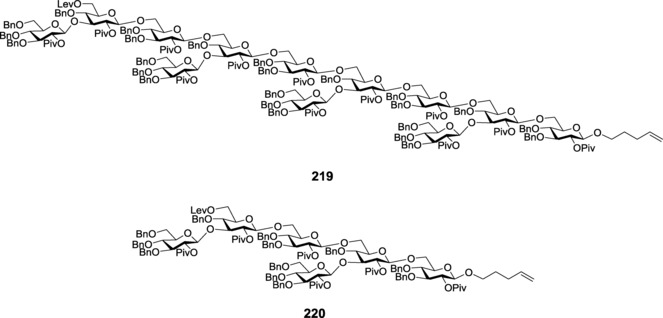
First oligosaccharides synthesized by using the automated protocol.
Although many reactions have been accomplished with the peptide reactor turned carbohydrate reactor, Seeberger and co‐workers developed the first fully automated solid‐phase oligosaccharide synthesizer in 2012.322 The reactor not only allowed the iterative steps involved in the multistep synthesis, but is also equipped with various essential gadgets that facilitate the controlled syringe–pump driver reagent delivery and computerized temperature control ranging from −50 to 90 °C.
The choice of linker has always been the most significant aspect in planning the strategy for performing automated solid‐support‐based oligosaccharide synthesis. The limitations of their instability in different acidic and basic conditions prompted to develop photo‐labile linkers as a suitable choice in such automated synthesis. A photolabile nitrobenzyl‐ether‐based linker has been used to synthesize two chondroitin hexasaccharide fragments of glycosaminoglycans in a continuous flow reactor, implementing a carefully planned protecting group manipulations and glycosylation strategy.323 Seeberger's group carried out the synthesis of β‐(1,3)‐glucan dodecasaccharides by using both thio and phosphate glycosides as donors in the presence of photo‐labile linker immobilized on Merrifield's resin.324 Solid support 222 was used for the immobilization, which utilized three equivalents of glycosyl phosphate 221 in three repeats over 12 glycosylation cycles. TMSOTf was used as the Lewis acid promoter that could be conveniently added to the building blocks, 221 and the attacking acceptor glycoside through strategically designed syringe pumps in the automated flow reactor (Scheme 46). In between each glycosylation steps, the temporary Fmoc protecting group had to be selectively removed. After the successful completion of the glycosylation sequences, the cleavage of the solid support was also performed in the same reactor by photoradiation. The use of fully automated synthesizers has also been used for the synthesis of sialyl oligosaccharides,325 β‐mannuronic acid alginates,326 oligoarabinofuranosides and galactans,327 and hyaluronan oligosaccharides328 that offer broad scopes in conveniently evaluating their biological properties. Higher oligosaccahrides, such as a 30‐mer polysaccharide of mannose, were also synthesized by using an automated flow reactor where the purification was done by the catch‐release method.329 The yields obtained through this protocol were significant, with an average of 96 % in each step and the entire synthesis of the polysaccharide accomplished in less than a week.
Scheme 46.
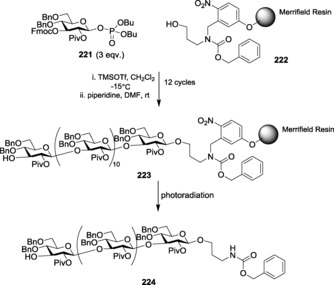
Oligosaccahride synthesis in automated flow reactor.
Nullifying the requirement of a sophisticated automated oligosaccharide synthesizer, methods of automated oligosaccharide synthesis in solution phase were also developed. Jaipuri and Pohl effectively synthesized linear and branched mannoside fragments based on solution‐phase automated iterative synthesis.330 The same group recently used the same protocol to synthesize β‐1,4‐mannuronate and β‐1,4‐mannan by using glycosyl trichloroacetimidates as the subsequent donors.331 Recently, Demchenko and co‐workers introduced the concept of HPLC‐based carbohydrate synthesis.332 They included a normal HPLC setup in order to facilitate all standard reaction steps essential in automated synthesis of a pentasaccharide 227 using repeated glycosylations with 225 and 226 as the synthons (Scheme 47). However, optimization of the various reaction conditions for the setup is under way. A suitable carbohydrate building block was rigorously optimized for performing glycosylations in an automated electrochemical assembly to synthesize a tetrasaccharide fragment from N,N,N‐trimethyl‐d‐glucosaminyl (TMG)–chitotriomycin.333 Thioglycosides have also been implemented in such an electrochemical assembly to synthesize poly‐β‐d‐(1‐6)‐N‐acetylglucosamine (PNAG),334 using six thioglycosides in a sequential one‐pot setup. Recently Seeberger's group performed a gold‐catalyzed solution‐phase oligosaccharide synthesis in a continuous flow reactor.200 Glycosyl ortho‐hexynylbenzoates were used as the donor for coupling with various sugar and non‐sugar derivatives catalyzed by PPh3AuOTf, furnishing the required glycosides/glycoconjugates in significant yields.
Scheme 47.
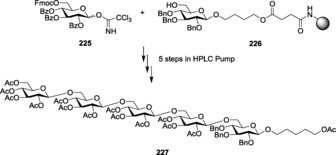
HPLC‐assisted synthesis of a pentasacchaide.
Automated glycoside synthesis in flow reactors is the latest addition in the chemistry of carbohydrate glycosylations based on the available instrumental facilities. This protocol claims to show much applicability in the following years by virtue of its ability to furnish complex higher analogs of polysaccharides in shorter time and in significant yields. However, the procedure still awaits further optimizations by solving the limitations it offers. The applicability of the process would facilitate interdisciplinary aid to the biologists in understanding the role of carbohydrates in the biological systems and in the development of carbohydrate‐derived antigens, thereby facilitating the advancement of the domain of therapeutics.
4. Conclusions and Outlook
Significant development in the area of chemical O‐glycosylation strategies is evident from the literature. These novel approaches offer a solid platform to deal with various complex oligosaccharides relevant for biological applications. The methods developed in the current century have helped us to get a better insight into the reactivity of glycosyl donors and acceptors. Novel promoter systems have enabled us to form glycosidic linkages with better stereoselectivity. Introduction of one‐pot iterative synthesis has minimized the labor required for complex oligosaccharide synthesis. With the development of solid‐phase synthesis and sophisticated automated glycosylation techniques, the future looks truly promising. However, the challenge of oligosaccharide synthesis is not yet over. Other than few ventures with protecting‐group‐free syntheses, chemical O‐glycosylations still suffer from the requirement of cumbersome protecting‐group manipulations that seriously defy the urge for atom economy. Moreover, the number of truly green reaction strategies is low in this domain. A sincere effort is needed to develop green and atom economic glycosylation strategies that can take us to a comparable domain of nature's enzymatic means of oligosaccharide preparation. We sincerely hope that the present survey of chemical O‐glycosylation strategies will help researchers to have a better understanding of the current scenario and pave the way for future developments in this important area.
Biographical Information
Rituparna Das was born in 1987 in India. She studied Chemistry at St. Xavier's College (Kolkata, India), where she obtained her B.Sc. in 2009. She then entered the Integrated Ph.D. at IISER Kolkata (India) and obtained her Ph.D. in 2015 under the supervision of Dr. Balaram Mukhopadhyay, working in the field of synthetic oligosaccharide chemistry. Currently, she is a Postdoctoral Fellow in the research group of Dr. Mukhopadhyay, working with glyco‐dendrimers focusing on multivalent carbohydrate–protein interactions.

Biographical Information
Balaram Mukhopadhyay obtained his Ph.D. in 2001 from the IACS, Jadavpur (India) under the supervision of Prof. Nirmolendu Roy, working in the field of synthetic carbohydrate chemistry. He then continued his Postdoctoral studies at the University of East Anglia (Norwich, UK; 2001–2005) with Prof. Robert A. Field. He started his independent career at CDRI, Lucknow (India) as Scientist‐C in 2005. In 2008, he moved to IISER Kolkata (India) as Assistant Professor and became Associate Professor in 2012. His research interests include the synthesis of oligosaccharides, glyco‐nanoparticles, and supramolecular chemistry of carbohydrates.

Acknowledgements
R.D. is thankful to IISER Kolkata for a Postdoctoral fellowship. The work is partially funded by the Science and Engineering Research Board (SERB), New Delhi through Grant No. SB/S1/OC‐48/2013 to B.M.
R. Das, B. Mukhopadhyay, ChemistryOpen 2016, 5, 401.
Contributor Information
Dr. Rituparna Das, Email: sugarnet73@hotmail.com
Dr. Balaram Mukhopadhyay, Email: ritu_iiser@yahoo.com
References
- 1.
- 1a. Stryer L., Biochemistry, W. H. Freeman, New York, 1988; [Google Scholar]
- 1b. Metzler D. E., Biochemistry-The Chemical Reactions of Living Cells, Academic, New York, 1977; [Google Scholar]
- 1c. Voet D., Voet J. G. in Biochemie (Eds.: A. Maelicke, W. Muller-Esterl), Wiley-VCH, Weinheim, 1994; [Google Scholar]
- 1d. Montreuil J., Adv. Carbohydr. Chem. Biochem. 1980, 37, 157–223. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Varki A., Glycobiology 1993, 3, 97–130; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2b. Varki A., Cummings R., Esko J., Freeze H., Hart G., Marth J., Essentials of Glycobiology, Cold Springer Harbor Laboratory Press, New York, 1999; [PubMed] [Google Scholar]
- 2c. Bertozzi G. E., Keissling L. L., Science 2001, 291, 2357–2364; [DOI] [PubMed] [Google Scholar]
- 2d. Sears P., Wong C. H., Science 2001, 291, 2344–2350. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Ritchie G. E., Mofatt B. E., Sim R. B., Morgan B. P., Dwek R. A., Rudd P. M., Chem. Rev. 2002, 102, 305–319; [DOI] [PubMed] [Google Scholar]
- 3b. Davis B. G., Chem. Rev. 2002, 102, 579–601; [DOI] [PubMed] [Google Scholar]
- 3c. OhtSubo K., Marth J. D., Cell 2006, 126, 855–867; [DOI] [PubMed] [Google Scholar]
- 3d. Bishop J. R., Schukz M., Esko J. D., Nature 2007, 446, 1030–1037; [DOI] [PubMed] [Google Scholar]
- 3e. Linhardt R. J., J. Med. Chem. 2003, 46, 2551–2564. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Kiessling L. L., Splain R. A., Annu. Rev. Biochem. 2010, 79, 619–653; [DOI] [PubMed] [Google Scholar]
- 4b. Boltje T., Buskas T., Boons G. J., Nat. Chem. 2009, 1, 611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Dwek R. A., Chem. Rev. 1996, 96, 683–720. [DOI] [PubMed] [Google Scholar]
- 6.Although we have now learned to synthesize oligosaccharides, it should be emphasized that each oligosaccharide synthesis remains an independent problem, whose resolution requires considerable systematic research and a good deal of know-how. There are no universal reaction condition for oligosaccharide synthesis’′–Hans Paulsen.
- 7.
- 7a. Toshima K., Tatsuta K., Chem. Rev. 1993, 93, 1503–1531; [Google Scholar]
- 7b. Bohé L., Crich D., C. R. Chim. C. R. Chimie 2011, 14, 3–16; [Google Scholar]
- 7c. Zhu X., Schmidt R. R., Angew. Chem. Int. Ed. 2009, 48, 1900–1934; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 1932–1967; [Google Scholar]
- 7d. Demchenko A. V., Synlett 2003, 9, 1225–1240; [Google Scholar]
- 7e. Jensen K. J., J. Chem. Soc. Perkin Trans. 1 2002, 2219–2233; [Google Scholar]
- 7f. Mulani S. K., Hung W. C., Ingle A. B., Shiau K. S., Tony Mong K.-K., Org. Biomol. Chem. 2014, 12, 1184–1197; [DOI] [PubMed] [Google Scholar]
- 7g. Davis B. G., J. Chem. Soc. Perkin Trans. 1 2000, 2137–2160. [Google Scholar]
- 8. Nicolaou K. C., Mitchell H. J., Angew. Chem. Int. Ed. 2001, 40, 1576–1624; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1624–1672. [Google Scholar]
- 9. Werz D. B., Ranzinger R., Herget S., Adibekian A., von der Lieth C. W., Seeberger P. H., ACS Chem. Biol. 2007, 2, 685–691. [DOI] [PubMed] [Google Scholar]
- 10.
- 10a. Seeberger P. H., Haase W. C., Chem. Rev. 2000, 100, 4349–4393; [DOI] [PubMed] [Google Scholar]
- 10b. Seeberger P. H., J. Carbohydr. Chem. 2002, 21, 613–643. [Google Scholar]
- 11.
- 11a. Hsu C. H., Hung S. C., Wu C. Y., Wong C. H., Angew. Chem. Int. Ed. 2011, 50, 11872–11923; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 12076–12129; [Google Scholar]
- 11b. Seeberger P. H., Chem. Commun. 2003, 1115–1121; [DOI] [PubMed] [Google Scholar]
- 11c. Seeberger P. H., Werz D. B., Nature 2007, 446, 1046–1051. [DOI] [PubMed] [Google Scholar]
- 12. Michael A., Am. Chem. J. 1879, 1, 305–312. [Google Scholar]
- 13. Fischer E., Ber. Dtsch. Chem. Ges. 1893, 26, 2400–2412. [Google Scholar]
- 14. Koenigs W., Knorr E., Ber. Dtsch. Chem. Ges. 1901, 34, 957–981. [Google Scholar]
- 15. Zémplén G., Gerecs A., Ber. Dtsch. Chem. Ges. 1930, 63, 2720–2729. [Google Scholar]
- 16. Helferich B., Wedermeyer K. F., Justus Liebigs Ann. Chem. 1949, 563, 139–145. [Google Scholar]
- 17. Bernstein S., Conrow R. B., J. Org. Chem. 1971, 36, 863–870. [DOI] [PubMed] [Google Scholar]
- 18. Lemieux R. U., Hendriks K. B., Stick R. V., James K., J. Am. Chem. Soc. 1975, 97, 4056–4062. [Google Scholar]
- 19. Ness R. K., Fletcher H. G., J. Am. Chem. Soc. 1956, 78, 4710–4714. [Google Scholar]
- 20.
- 20a. Hashimoto S., Sakamoto H., Honda T., Abe H., Nakamura S., Ikegami S., Tetrahedron Lett. 1997, 38, 8969–8972; [Google Scholar]
- 20b. Fraser-Reid B., Udodong U. E., Wu Z. F., Ottosson Z. H., Merritt J. R., Rao C. S., Roberts C., Madsen R., Synlett 1992, 927–942. [Google Scholar]
- 21. Roy R., Andersson F. O., Letellier M., Tetrahedron Lett. 1992, 33, 6053–6056. [Google Scholar]
- 22. Boons G. J., Isles S., J. Org. Chem. 1996, 61, 4262–4271. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Kanie O., lto Y., Ogawa T., Tetrahedron Lett. 1996, 37, 4551–4554; [Google Scholar]
- 23b. Ito Y., Ogawa T., J. Am. Chem. Soc. 1997, 119, 5562–5566. [Google Scholar]
- 24.
- 24a. Douglas N. L., Ley S. V., Lucking U., Warrier S. L., J. Chem. Soc. Perkin Trans. 1 1998, 51–65; [Google Scholar]
- 24b. Zhang Z., Ollmann I. R., Ye X. S., Wischnat R., Baasov T., Wong C.-H., J. Am. Chem. Soc. 1999, 121, 734–753. [Google Scholar]
- 25. Lemieux R. U., Pure Appl. Chem. 1971, 25, 527–548. [Google Scholar]
- 26. Edward J. T., Chem. Ind. 1955, 1102–1104. [Google Scholar]
- 27.
- 27a. Romers C., Altona C., Buys H. R., Havinga E., Top. Stereochem. 1969, 4, 39–97; [Google Scholar]
- 27b. Ardalan Z., Lucken E. A. C., Helv. Chim. Acta 1973, 56, 1715–1719. [Google Scholar]
- 28. Mo Y., Nat. Chem. 2010, 2, 666–671. [DOI] [PubMed] [Google Scholar]
- 29. Nukada T., Berces A., Whitfield D. M., Carbohydr. Res. 2002, 337, 765–774. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Boltje T. J., Kim J. H., Park J., Boons G.-J., Org. Lett. 2011, 13, 284–287; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30b. Walvoort M. T. C., Dinkelaar J., van der Bos L. J., Lodder G., Overkleeft H. S., Codeé J. D. C., van der Marel G. A., Carbohydr. Res. 2010, 345, 1252–1263; [DOI] [PubMed] [Google Scholar]
- 30c. Kumar R., Whitfield D. M., J. Org. Chem. 2012, 77, 3724–3739. [DOI] [PubMed] [Google Scholar]
- 31.
- 31a. Kim J.-H., Yang H., Park J., Boons G. J., J. Am. Chem. Soc. 2005, 127, 12090–12097; [DOI] [PubMed] [Google Scholar]
- 31b. Smoot J. T., Pornsuriyasak P., Demchenko A. V., Angew. Chem. Int. Ed. 2005, 44, 7123–7126; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 7285–7288. [Google Scholar]
- 32.
- 32a. Jensen H. H., Pederson C. M., Bols M., Chem. Eur. J. 2007, 13, 7576–7582; [DOI] [PubMed] [Google Scholar]
- 32b. Crich D., Vinogradova O., J. Org. Chem. 2006, 71, 8473–8480; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32c. Crich D., J. Org. Chem. 2011, 76, 9193–9209; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32d. Crich D., Acc. Chem. Res. 2010, 43, 1144–1153; [DOI] [PubMed] [Google Scholar]
- 32e. Ranade S. C., Demchenko A. V., J. Carbohydr. Chem. 2013, 32, 1–43; [Google Scholar]
- 32f. Mydock L. K., Demchenko A. V., Org. Biomol. Chem. 2010, 8, 497–510. [DOI] [PubMed] [Google Scholar]
- 33. Fischer E., Armstrong E. F., Ber. Dtsch. Chem. Ges. 1901, 34, 2885–2900. [Google Scholar]
- 34. Nitz M., Bundle D. R. in Glycoscience: Chemistry and Chemical Biology, Vol. 1 (Eds.: B. Fraser-Reid, K. Tatsuta, J. Thiem), Springer, Berlin, Heidelberg, New York, 2001, pp. 1497–1542. [Google Scholar]
- 35. Fischer E., Fischer H., Ber. Dtsch. Chem. Ges. 1910, 43, 2521–2536. [Google Scholar]
- 36. Koto S., Morishima N., Shichi S., Haigoh H., Hirooka M., Okamoto M., Higuchi T., Shimizu K., Hashimoto Y., Iriawa T., Kawasaki H., Takahashi Y., Yamazaki M., Mori Y., Kudo K., Ikegaki T., Suzuki S., Zen S., Bull. Chem. Soc. Jpn. 1992, 65, 3257–3274. [Google Scholar]
- 37. Fischer E., Ber. Dtsch. Chem. Ges. 1911, 44, 1898–1904. [Google Scholar]
- 38. Montero J. L., Winum J. Y., Leydet A., Kamal M., Pavia A. A., Roque J. P., Carbohydr. Res. 1997, 297, 175–180. [Google Scholar]
- 39. Hanessian S., Ponpipom M. M., Lavalle P., Carbohydr. Res. 1972, 24, 45. [Google Scholar]
- 40. Schmidt R. R., Rücker E., Tetrahedron Lett. 1980, 21, 1421–1424. [Google Scholar]
- 41. Yoon T. P., Ischay M. A., Du J., Nat. Chem. 2010, 2, 527–532. [DOI] [PubMed] [Google Scholar]
- 42. Tojino M., Hirose Y., Mizuno M., Tetrahedron Lett. 2013, 54, 7124–7126. [Google Scholar]
- 43. Yuan X., Cheng S., Shi Y., Xue W., Synthesis 2014, 46, 331–335. [Google Scholar]
- 44.
- 44a. Kröger L., Thiem J., J. Carbohydr. Chem. 2003, 22, 9–23; [Google Scholar]
- 44b. Watanabe Y., Shiozaki M., Carbohydr. Res. 2001, 335, 283–289. [DOI] [PubMed] [Google Scholar]
- 45. Beejmohun V., Grand E., Mesnard F., Fliniaux M. A., Kovensky J., Tetrahedron Lett. 2004, 45, 8745–8747. [Google Scholar]
- 46. Kumar V., Talisman I. J., Malhotra S. V., Eur. J. Org. Chem. 2010, 3377–3381. [Google Scholar]
- 47. Adinolfi M., Iadonisi A., Pezzella A., Ravida A., Synlett 2005, 1848–1852. [Google Scholar]
- 48.
- 48a. Turek D., Sundgren A., Lahmann M., Oscarson S., Org. Biomol. Chem. 2006, 4, 1236–1241; [DOI] [PubMed] [Google Scholar]
- 48b. Hou S., Kovac P., Carbohydr. Res. 2011, 346, 1394–1397; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48c. Ruttens B., Saksena R., Kovac P., Eur. J. Org. Chem. 2007, 4366–4375. [Google Scholar]
- 49.
- 49a. Jung K. H., Müller M., Schmidt R. R., Chem. Rev. 2000, 100, 4423–4442; [DOI] [PubMed] [Google Scholar]
- 49b. Iino K., Iwamoto S., Kasahara Y., Matsuo I., Tetrahedron Lett. 2012, 53, 4452–4456. [Google Scholar]
- 50. Williams R. J., Paul C. E., Nitz M., Carbohydr. Res. 2014, 386, 73–77. [DOI] [PubMed] [Google Scholar]
- 51. Gervay J., Nguyen T. N., Hadd M. J., Carbohydr. Res. 1997, 300, 119–125. [Google Scholar]
- 52.
- 52a. Chervin S. M., Abada P., Koreeda M., Org. Lett. 2000, 2, 369–372; [DOI] [PubMed] [Google Scholar]
- 52b. Adinolfi M., Iadonisi A., Ravida A., Schiattarella M., Tetrahedron Lett. 2003, 44, 7863–7866. [Google Scholar]
- 53. van Well R. M., Kartha K. P. R., Field R. A., J. Carbohydr. Chem. 2005, 24, 463–474. [Google Scholar]
- 54. Mukhopadhyay B., Kartha K. P. R., Russell D. A., Field R. A., J. Org. Chem. 2004, 69, 7758–7760. [DOI] [PubMed] [Google Scholar]
- 55. Bickley J., Cottrel J. A., Ferguson J. R., Field R. A., Harding J. R., Hughes D. L., Kartha K. P. R., Law J. L., Scheinmann F., Stachulski A. V., Chem. Commun. 2003, 1266–1267. [DOI] [PubMed] [Google Scholar]
- 56. Miquel N., Vignando S., Russo G., Lay L., Synlett 2004, 341–343. [Google Scholar]
- 57. Dabideen D. R., Gervay-Hague J., Org. Lett. 2004, 6, 973–975. [DOI] [PubMed] [Google Scholar]
- 58. El-Badry M. H., Gervay-Hague J., Tetrahedron Lett. 2005, 46, 6727–6728. [Google Scholar]
- 59. Lam S. N., Gervay-Hague J., Org. Lett. 2002, 4, 2039–2042. [DOI] [PubMed] [Google Scholar]
- 60.
- 60a. Kobashi Y., Mukaiyama T., Chem. Lett. 2004, 33, 874–875; [Google Scholar]
- 60b. Gemma E., Lahmann M., Oscarson S., Carbohydr. Res. 2005, 340, 2558–2562. [DOI] [PubMed] [Google Scholar]
- 61. Lam S. N., Gervay-Hague J., J. Org. Chem. 2005, 70, 8772–8779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.
- 62a. Baldoni L., Marino C., Carbohydr. Res. 2013, 374, 75–81; [DOI] [PubMed] [Google Scholar]
- 62b. Baldoni L., Marino C., J. Org. Chem. 2009, 74, 1994–2003; [DOI] [PubMed] [Google Scholar]
- 62c. Gu X., Chen L., Wang X., Liu X., You Q., Xi W., Gao L., Chen G., Chen Y.-L., Xiong B., Shen J., J. Org. Chem. 2014, 79, 1100–1110; [DOI] [PubMed] [Google Scholar]
- 62d. Adinolfi M., Iadonisi A., Pezzella A., Ravidà A., Synlett 2005, 12, 1848–1852. [Google Scholar]
- 63.
- 63a. Morales-Serna J. A., Diaz Y., Mattheu I., Castillon S., Eur. J. Org. Chem. 2009, 3849–3852; [Google Scholar]
- 63b. Hsieh H.-W., Schombs M. W., Gervay-Hague J., J. Org. Chem. 2014, 79, 1736–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Hsieh H.-W., Davis R. A., Hoch J. A., Gervay-Hague J., Chem. Eur. J. 2014, 20, 6444–6454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Ying L., Gervay-Hague J., Carbohydr. Res. 2003, 338, 835–841. [DOI] [PubMed] [Google Scholar]
- 66. Perrie J. A., Harding J. R., King C., Sinnott D., Stachulski A. V., Org. Lett. 2003, 5, 4545–4548. [DOI] [PubMed] [Google Scholar]
- 67. Murakami T., Sato Y., Shibakami M., Carbohydr. Res. 2008, 343, 1297–1308. [DOI] [PubMed] [Google Scholar]
- 68. Mukaiyama T., Murai Y., Shoda S., Chem. Lett. 1981, 431–432. [Google Scholar]
- 69.
- 69a. Yokoyama H., Carbohydr. Res. 2000, 327, 5–14; [DOI] [PubMed] [Google Scholar]
- 69b. Toshima K., Carbohydr. Res. 2000, 327, 15–26. [DOI] [PubMed] [Google Scholar]
- 70. Huang K. T., Winssinger N., Eur. J. Org. Chem. 2007, 1887–1890. [Google Scholar]
- 71.
- 71a. López J. C., Bernal-Albert P., Uriel C., Valverde S., Gómez A. M., J. Org. Chem. 2007, 72, 10268–10271; [DOI] [PubMed] [Google Scholar]
- 71b. Tsegay S., Williams R. J., Williams S. J., Carbohydr. Res. 2012, 357, 16–22; [DOI] [PubMed] [Google Scholar]
- 71c. Kunigami M., Hara S., Carbohydr. Res. 2015, 417, 78–80; [DOI] [PubMed] [Google Scholar]
- 71d. Fraser-Reid B., Lopez J. C., Bernal-Albert P., Gomez A. M., Uriel C., Ventura J., Can. J. Chem. 2013, 91, 51–65. [Google Scholar]
- 72.
- 72a. Ohtsuka I., Ako T., Kato R., Daikoku S., Koroghi S., Kanemitsu T., Kanie O., Carbohydr. Res. 2006, 341, 1476–1487; [DOI] [PubMed] [Google Scholar]
- 72b. Suzuki K., Ito Y., Kanie O., Carbohydr. Res. 2012, 359, 81–91. [DOI] [PubMed] [Google Scholar]
- 73.
- 73a. Francesconi O., Nativi C., Gabrielli G., Gentili M., Palchetti M., Bonora B., Roelens S., Chem. Eur. J. 2013, 19, 11742–11752; [DOI] [PubMed] [Google Scholar]
- 73b. Hojo H., Tanaka H., Hagiwara M., Asahina Y., Ueki A., Katayama H., Nakahara Y., Yoneshige A., Matsuda J., Ito Y., Nakahara Y., J. Org. Chem. 2012, 77, 9437–9446. [DOI] [PubMed] [Google Scholar]
- 74.
- 74a. Nicolaou K. C., Caulfield T. J., Kataoka H., Carbohydr. Res. 1990, 202, 177–191; [DOI] [PubMed] [Google Scholar]
- 74b. Kondo T., Tomoo T., Abe H., Isobe M., Goto T., Chem. Lett. 1996, 337–338. [Google Scholar]
- 75.
- 75a. Mukaiyama T., Jona H., Takeuchi K., Chem. Lett. 2000, 696–697; [Google Scholar]
- 75b. Jona H., Mandai H., Chavasiri W., Takeuchi K., Mukaiyama T., Bull. Chem. Soc. Jpn. 2002, 75, 291–309; [Google Scholar]
- 75c. Jona H., Takeuchi K., Muaiyama T., Chem. Lett. 2000, 1278–1279. [Google Scholar]
- 76.
- 76a. Yang Y., Yu B., Tetrahedron 2014, 70, 1023–1046. [Google Scholar]
- 77. Kim H. M., Kim I. J., Danishefsky S. J., J. Am. Chem. Soc. 2001, 123, 35–48. [DOI] [PubMed] [Google Scholar]
- 78. Nicolaou K. C., Seitz S. P., Papahatjis D. P., J. Am. Chem. Soc. 1983, 105, 2430–2434. [Google Scholar]
- 79. Pozsgay V., Jennings H. J., J. Org. Chem. 1987, 52, 4635–4637. [Google Scholar]
- 80. Sasaki M., Tachibana K., Nakanishi H., Tetrahedron Lett. 1991, 32, 6873–6876. [Google Scholar]
- 81. Kihlberg J. O., Leigh D. A., Bundle D. R., J. Org. Chem. 1990, 55, 2860–2863. [Google Scholar]
- 82.
- 82a. Veeneman G. H., van Leeuwen S. H., van Boom J. H., Tetrahedron Lett. 1990, 31, 1331–1334; [Google Scholar]
- 82b. Konradsson P., Mootoo D. R., McDevitt R. E., Fraser-Reid B., J. Chem. Soc. Chem. Commun. 1990, 270–272; [Google Scholar]
- 82c. Konradsson P., Udodong U. E., Fraser-Reid B., Tetrahedron Lett. 1990, 31, 4313–4316. [Google Scholar]
- 83. Veeneman G. H., van Boom J. H., Tetrahedron Lett. 1990, 31, 275–278. [Google Scholar]
- 84. Veeneman G. H., van Leeuwen S. H., Zuurmond H., van Boom J. H., J. Carbohydr. Chem. 1990, 9, 783–796. [Google Scholar]
- 85.
- 85a. Kartha K. P. R., Cura P., Aloui M., Readman S. K., Rutherford T. J., Field R. A., Tetrahedron: Asymmetry 2000, 11, 581–593; [Google Scholar]
- 85b. Cura P., Aloui M., Kartha K. P. R., Field R. A., Synlett 2000, 1279–1280. [Google Scholar]
- 86. Fukase K., Hasuoka A., Kinoshita I., Kusumoto S., Tetrahedron Lett. 1992, 33, 7165–7168. [Google Scholar]
- 87. Xiong D.-C., Zhang L. H., Ye X. S., Adv. Synth. Catal. 2008, 350, 1696–1700. [Google Scholar]
- 88. Peng P., Ye X. S., Org. Biomol. Chem. 2011, 9, 616–622. [DOI] [PubMed] [Google Scholar]
- 89. Kaeothip S., Jagodige P., Yasomanee J. P., Demchenko A. V., J. Org. Chem. 2012, 77, 291–299. [DOI] [PubMed] [Google Scholar]
- 90. Crich D., Smith M., Org. Lett. 2000, 2, 4067–4069. [DOI] [PubMed] [Google Scholar]
- 91. Crich D., Smith M., J. Am. Chem. Soc. 2001, 123, 9015–9020. [DOI] [PubMed] [Google Scholar]
- 92. Codée J. D. C., Litjens R. E. J. N., Heeten R., Overkleeft H. S., van Boom J. H., van der Marel G. A., Org. Lett. 2003, 5, 1519–1522. [DOI] [PubMed] [Google Scholar]
- 93. Codée J. D. C., van den Bos L. J., Litjens R. E. J. N., Overkleeft H. S., van Boeckel C. A., van Boom J. H., van der Marel G. A., Tetrahedron 2004, 60, 1057–1064. [Google Scholar]
- 94. Wang C., Wang H., Huang H., Zhang L. H., Ye X. S., Synlett 2006, 2846–2850. [Google Scholar]
- 95. Tatai J., Fügedi P., Org. Lett. 2007, 9, 4647–4650. [DOI] [PubMed] [Google Scholar]
- 96. Durón S. G., Polat T., Wong C. H., Org. Lett. 2004, 6, 839–841. [DOI] [PubMed] [Google Scholar]
- 97. Maity S. K., Basu N., Ghosh R., Carbohydr. Res. 2012, 354, 40–48. [DOI] [PubMed] [Google Scholar]
- 98. Basu N., Maity S. K., Ghosh R., RSC Adv. 2012, 2, 12661–12664. [Google Scholar]
- 99. Takeuchi K., Tamura T., Jona H., Mukaiyama T., Chem. Lett. 2000, 29, 692–693. [Google Scholar]
- 100. Jona H., Takeuchi K., Saitoh T., Mukaiyama T., Chem. Lett. 2000, 29, 1178–1179. [Google Scholar]
- 101. Takeuchi T., Tamura T., Mukaiyama T., Chem. Lett. 2000, 29, 124–125. [Google Scholar]
- 102. Chung S.-K., Park K. H., Tetrahedron Lett. 2001, 42, 4005–4007. [Google Scholar]
- 103. Hashihayata T., Ikegai K., Takeuchi K., Jona H., Mukaiyama T., Bull. Chem. Soc. Jpn. 2003, 76, 1829. [Google Scholar]
- 104. Qin Z. H., Li H., Cai M. S., Li Z. J., Carbohydr. Res. 2002, 337, 31–36. [DOI] [PubMed] [Google Scholar]
- 105. Mukherjee S., Mukhopadhyay B., Synlett 2010, 19, 2853–2856. [Google Scholar]
- 106.
- 106a. Budhadev D., Mukhopadhyay B., Carbohydr. Res. 2014, 384, 51–55; [DOI] [PubMed] [Google Scholar]
- 106b. Budhadev D., Mukhopadhyay B., Carbohydr. Res. 2014, 394, 26–31. [DOI] [PubMed] [Google Scholar]
- 107. Mukhopadhyay B., Collet B., Field R. A., Tetrahedron Lett. 2005, 46, 5923–5925. [Google Scholar]
- 108.
- 108a. Roy B., Pramanik K., Mukhopadhyay B., Glycoconjugate J. 2008, 25, 157–166; [DOI] [PubMed] [Google Scholar]
- 108b. Dasgupta S., Pramanik K., Mukhopadhyay B., Tetrahedron 2007, 63, 12310–12316; [Google Scholar]
- 108c. Mandal S., Mukhopadhyay B., Tetrahedron 2007, 63, 11363–11370; [Google Scholar]
- 108d. Dasgupta S., Mukhopadhyay B., Eur. J. Org. Chem. 2008, 5770–5777; [Google Scholar]
- 108e. Verma P. R., Mukhopadhyay B., Carbohydr. Res. 2010, 345, 432–436; [DOI] [PubMed] [Google Scholar]
- 108f. Roy B., Field R. A., Mukhopadhyay B., Carbohydr. Res. 2009, 344, 2311–2316; [DOI] [PubMed] [Google Scholar]
- 108g. Verma P., Mukhopadhyay B., Carbohydr. Res. 2009, 344, 2554–2558. [DOI] [PubMed] [Google Scholar]
- 109.
- 109a. Das R., Mukhopadhyay B., Carbohydr. Res. 2013, 376, 1–6; [DOI] [PubMed] [Google Scholar]
- 109b. Verma P. R., Mukhopadhyay B., RSC Adv. 2013, 3, 201–207; [Google Scholar]
- 109c. Pal K. B., Mukhopadhyay B., Carbohydr. Res. 2013, 379, 26–29. [DOI] [PubMed] [Google Scholar]
- 110. Yao C. H., Lee J. C., Tetrahedron 2014, 70, 6757–6762. [Google Scholar]
- 111. Périon R., Lemée L., Ferriéres V., Duval R., Plusquellec D., Carbohydr. Res. 2003, 338, 2779–2792. [DOI] [PubMed] [Google Scholar]
- 112. Valerio S., Iadonisi A., Adinolfi M., Ravidà A., J. Org. Chem. 2007, 72, 6097–6106. [DOI] [PubMed] [Google Scholar]
- 113. Goswami M., Ellern A., Pohl N. L. B., Angew. Chem. Int. Ed. 2013, 52, 8441–8445; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 8599–8603. [Google Scholar]
- 114. Vibhute A. M., Dhaka A., Athiyarath V., Sureshan K. M., Chem. Sci. 2016, 7, 4259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Mao R.-Z., Xiong D.-C., Guo F., Li Q., Duan J., Ye X.-S., Org. Chem. Front. 2016, 3, 737. [Google Scholar]
- 116. Wever W. J., Cinelli M. A., Bowers A. A., Org. Lett. 2013, 15, 30–33. [DOI] [PubMed] [Google Scholar]
- 117. Spell M. L., Deveaux K., Bresnahan C. G., Bernard B. L., Sheffield W., Kumar R., Ragains J. R., Angew. Chem. Int. Ed. 2016, 55, 6515–6519; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 6625–6629. [Google Scholar]
- 118. Mao R.-Z., Guo F., Xiong D. C., Li Q., Duan J., Ye X. S., Org. Lett. 2015, 17, 5606–5609. [DOI] [PubMed] [Google Scholar]
- 119. Nakanishi M., Takahashi D., Toshima K., Org. Biomol. Chem. 2013, 11, 5079–5082. [DOI] [PubMed] [Google Scholar]
- 120. Chu A.-H. A., Minciunescu A., Montanari V., Kumar K., Bennett C. S., Org. Lett. 2014, 16, 1780–1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Tanaka N., Ohnishi F., Uchihata D., Toriib S., Nokamia J., Tetrahedron Lett. 2007, 48, 7383–7387. [Google Scholar]
- 122. Schmidt R. R., Michel J., Angew. Chem. Int. Ed. Engl. 1980, 19, 731–732; [Google Scholar]; Angew. Chem. 1980, 92, 763–764. [Google Scholar]
- 123. Schaubach R., Hemberger J., Kinzy W., Liebigs Ann. Chem. 1991, 607–614. [Google Scholar]
- 124. Zimmermann P., Bommer B., Bär T., Schmidt R. R., J. Carbohydr. Chem. 1988, 7, 435. [Google Scholar]
- 125. Wegmann B., Schmidt R. R., J. Carbohydr. Chem. 1987, 6, 357–375. [Google Scholar]
- 126. Dobarro-Rodriguez A., Trumtel M., Wessel H. P., J. Carbohydr. Chem. 1992, 11, 255–263. [Google Scholar]
- 127. Urban F. J., Moore B. S., Breitenbach R., Tetrahedron Lett. 1990, 31, 4421–4424. [Google Scholar]
- 128. Douglas S. P., Whitfield D. M., Krepinsky J. J., J. Carbohydr. Chem. 1993, 12, 131–136. [Google Scholar]
- 129. Adinolfi M., Barone G., Iadonisi A., Schiattarella M., Synlett 2002, 0269–0270. [Google Scholar]
- 130. Lian G., Gao Q., Lin F., Carbohydr. Res. 2008, 343, 2992–2996. [DOI] [PubMed] [Google Scholar]
- 131. Li M., Han X., Yu B., J. Org. Chem. 2003, 68, 6842–6845. [DOI] [PubMed] [Google Scholar]
- 132. Yang W., Sun J., Yang Z., Han W., Zhang W. D., Yu B., Tetrahedron Lett. 2012, 53, 2773–2776. [Google Scholar]
- 133. Li Y., Mo H., Lian G., Yu B., Carbohydr. Res. 2012, 363, 14–22. [DOI] [PubMed] [Google Scholar]
- 134. Mattson A. L., Michel A. K., Cloninger M. J., Carbohydr. Res. 2012, 347, 142–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Adinolfi M., Barone G., Guariniello L., Iadonisi A., Tetrahedron Lett. 2000, 41, 9005–9008. [Google Scholar]
- 136. Adinolfi M., Barone G., Iadonisi A., Mangoni L., Schiattarella M., Tetrahedron Lett. 2001, 42, 5967–5969. [Google Scholar]
- 137.
- 137a. Adinolfi M., Iadonisi A., Ravidà A., Synlett 2006, 4, 583–586; [Google Scholar]
- 137b. Adinolfi M., Barone G., Iadonisi A., Schiattarella M., Tetrahedron Lett. 2002, 43, 5573–5577. [Google Scholar]
- 138. Valerio S., Pastore A., Adinolfi M., Iadonisi A., J. Org. Chem. 2008, 73, 4496–4503. [DOI] [PubMed] [Google Scholar]
- 139. Adinolfi M., Iadonisi A., Ravidà A., Valerio S., Tetrahedron Lett. 2006, 47, 2595–2599. [Google Scholar]
- 140. Du Y., Wei G., Cheng S., Huaa Y., Linhardt R. J., Tetrahedron Lett. 2006, 47, 307–310. [Google Scholar]
- 141. Ludek O. R., Gu W., Gildersleeve C. J., Carbohydr. Res. 2010, 345, 2074–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Adinolfi M., Barone G., Iadonisi A., Schiattarella M., Org. Lett. 2003, 5, 987–989. [DOI] [PubMed] [Google Scholar]
- 143. Tian Q., Zhang S., Yu Q., He M. B., Yang J. S., Tetrahedron 2007, 63, 2142–2147. [Google Scholar]
- 144. Tanaka S., Takashina M., Tokimoto H., Fujimoto Y., Tanaka K., Fukase K., Synlett 2005, 2325–2328. [Google Scholar]
- 145. Goetze S., Fitzner R., Kunz H., Synlett 2009, 3346–3348. [Google Scholar]
- 146. Roy R., Palanivel A. K., Mallick A., Vankar Y. D., Eur. J. Org. Chem. 2015, 4000–4005. [Google Scholar]
- 147. Peng P., Schmidt R. R., J. Am. Chem. Soc. 2015, 137, 12653–12659. [DOI] [PubMed] [Google Scholar]
- 148. Yang J., Cooper-Vanosdell C., Mensah E. A., Nguyen H. M., J. Org. Chem. 2008, 73, 794–800. [DOI] [PubMed] [Google Scholar]
- 149. Zandanel C., Dehuyser L., Wagner A., Baati R., Tetrahedron 2010, 66, 3365–3369. [Google Scholar]
- 150. Iwata R., Uda K., Takahashi D., Toshima K., Chem. Commun. 2014, 50, 10695. [DOI] [PubMed] [Google Scholar]
- 151. Jayakanthan K., Vankar Y. D., Carbohydr. Res. 2005, 340, 2688–2692. [DOI] [PubMed] [Google Scholar]
- 152. Shirahata T., Matsuo J.-I., Teruya S., Hirata N., Kurimoto T., Akimoto N., Sunazuka T., Kaji E., Omura S., Carbohydr. Res. 2010, 345, 740–749. [DOI] [PubMed] [Google Scholar]
- 153.
- 153a. Carbohydrates in Chemistry and Biology, Vol. 1 (Eds.: R. R. Schmidt, K.-H. Jung, B. Ernst, G. W. Hart, P. Sinaÿ), Wiley-VCH, Weinheim, 2000, pp. 5–59; [Google Scholar]
- 153b. Schmelzer U., Zhang Z., Schmidt R. R., J. Carbohydr. Chem. 2007, 26, 223–238. [Google Scholar]
- 154. Ranade S. C., Hasty S. C., Demchenko A. V., J. Carbohydr. Chem. 2013, 32, 360–379. [Google Scholar]
- 155. Kamat M. N., Meo C. D., Demchenko A. V., J. Org. Chem. 2007, 72, 6947–6955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Kamat M. N., Rath N. P., Demchenko A. V., J. Org. Chem. 2007, 72, 6938–6946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Demchenko A. V., Malysheva N. N., Meo C. D., Org. Lett. 2003, 5, 455–458. [DOI] [PubMed] [Google Scholar]
- 158.
- 158a. Kamat M. N., Demchenko A. V., Org. Lett. 2005, 7, 3215–3218; [DOI] [PubMed] [Google Scholar]
- 158b. Crich D., Li M., Org. Lett. 2007, 9, 4115–4118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Kaeothip S., Pornsuriyasak P., Rath N. P., Demchenko A. V., Org. Lett. 2009, 11, 799–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Demchenko A. V., Kamat M. N., De Meo C., Synlett 2003, 9, 1287–1290. [Google Scholar]
- 161. Bongat A. F. G., Kamat M. N., Demchenko A. V., J. Org. Chem. 2007, 72, 1480–1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.
- 162a. Hasty S. J., Kleine M. A., Demchenko A. V., Angew. Chem. Int. Ed. 2011, 50, 4197–4201; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 4283–4287; [Google Scholar]
- 162b. Demchenko A. V., Pornsuriyasak P., De Meo C., Malysheva N. N., Angew. Chem. Int. Ed. 2004, 43, 3069–3072; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3131–3134. [Google Scholar]
- 163. Kaeothip S., Demchenko A. V., J. Org. Chem. 2011, 76, 7388–7398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Parlato M. C., Kamat M. N., Wang H., Stine K. J., Demchenko A. V., J. Org. Chem. 2008, 73, 1716–1725. [DOI] [PubMed] [Google Scholar]
- 165. Pornsuriyasak P., Demchenko A. V., Chem. Eur. J. 2006, 12, 6630–6646. [DOI] [PubMed] [Google Scholar]
- 166. Kaeothip S., Pornsuriyasak P., Demchenko A. V., Tetrahedron Lett. 2008, 49, 1542–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Ranade S. C., Kaeothip S., Demchenko A. V., Org. Lett. 2010, 12, 5628–5631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Ranade S. N., Demchenko A. V., Carbohydr. Res. 2015, 403, 115–122. [DOI] [PubMed] [Google Scholar]
- 169. Mukaiyama T., Nakatsuka T., Shoda S.-i., Chem. Lett. 1979, 8, 487. [Google Scholar]
- 170. Hasty S. J., Demchenko A. V., Chem. Heterocycl. Compd. 2012, 48, 220–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Padungros P., Alberch L., Wei A., J. Org. Chem. 2014, 79, 2611–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172. Lee J.-C., Pan G.-R., Kulkarni S. S., Luo S.-Y., Liao C.-C., Hung S.-C., Tetrahedron Lett. 2006, 47, 1621–1624. [Google Scholar]
- 173. Luo S.-Y., Tripathi A., Manuel M., Zulueta L., Hung S.-C., Carbohydr. Res. 2012, 352, 197–201. [DOI] [PubMed] [Google Scholar]
- 174. Fortin M., Kaplan J., Pham K., Kirk S., Andrade R. B., Org. Lett. 2009, 11, 3594–3597. [DOI] [PubMed] [Google Scholar]
- 175.
- 175a. Jayaprakash K. N., Fraser-Reid B., Carbohydr. Res. 2007, 342, 490–498; [DOI] [PubMed] [Google Scholar]
- 175b. Jayaprakash K. N., Radhakrishnan K. V., Fraser-Reid B., Tetrahedron Lett. 2002, 43, 6953–6955. [Google Scholar]
- 176.
- 176a. Fraser-Reid B., Grimme S., Piacenza M., Mach M., Schlueter U., Chem. Eur. J. 2003, 9, 4687–4692; [DOI] [PubMed] [Google Scholar]
- 176b. Jayaprakash K. N., Fraser-Reid B., Org. Lett. 2004, 6, 4211–4214; [DOI] [PubMed] [Google Scholar]
- 176c. Jayaprakash K. N., Fraser-Reid B., Synlett 2004, 301–305. [Google Scholar]
- 177. López J. C., Agocs A., Uriel C., Gómeza A. M., Fraser-Reid B., Chem. Commun. 2005, 5088–5090. [DOI] [PubMed] [Google Scholar]
- 178. Anilkumar G., Nair L. G., Fraser-Reid B., Org. Lett. 2000, 2, 2587–2589. [DOI] [PubMed] [Google Scholar]
- 179. Mathew F., Jayaprakash K. N., Fraser-Reid B., Mathew J., Scicinski J., Tetrahedron Lett. 2003, 44, 9051–9054. [Google Scholar]
- 180. Uriel C., Ventura J., Gómez A. M., López J. C., Fraser-Reid B., Eur. J. Org. Chem. 2012, 3122–3131. [DOI] [PubMed] [Google Scholar]
- 181. Hotha S., Kashyap S., J. Am. Chem. Soc. 2006, 128, 9620–9621. [DOI] [PubMed] [Google Scholar]
- 182. Kayastha A. K., Hotha S., Tetrahedron Lett. 2010, 51, 5269–5272. [Google Scholar]
- 183. Kayastha A. K., Hotha S., Chem. Commun. 2012, 48, 7161–7163. [DOI] [PubMed] [Google Scholar]
- 184. Vidadala S. R., Gayatri G., Sastry N., Hotha S., Chem. Commun. 2011, 47, 9906–9908. [DOI] [PubMed] [Google Scholar]
- 185. Adhikari S., Baryal K. N., Zhu D., Xiaohua L., Zhu J., ACS Catal. 2013, 3, 57–60. [Google Scholar]
- 186. Chen X., Shen D., Wang Q., Yang Y., Yu B., Chem. Commun. 2015, 51, 13957–13960. [DOI] [PubMed] [Google Scholar]
- 187.
- 187a. Thadke S. A., Mishra B., Hotha S., J. Org. Chem. 2014, 79, 7358–7371; [DOI] [PubMed] [Google Scholar]
- 187b. Thadke S. A., Mishra B., Hotha S., Org. Lett. 2013, 15, 2466–2469. [DOI] [PubMed] [Google Scholar]
- 188. Bound D. J., Bettadaiah B. K., Srinivas P., Synth. Commun. 2014, 44, 2565–2576. [Google Scholar]
- 189. Sureshkumar G., Hotha S., Tetrahedron Lett. 2007, 48, 6564–6568. [Google Scholar]
- 190. Sureshkumar G., Hotha S., Chem. Commun. 2008, 4282–4284. [DOI] [PubMed] [Google Scholar]
- 191. Li Y., Yang Y., Yu B., Tetrahedron Lett. 2008, 49, 3604–3608. [Google Scholar]
- 192. Tang Y., Li J., Zhu Y., Li Y., Yu B., J. Am. Chem. Soc. 2013, 135, 18396–18405. [DOI] [PubMed] [Google Scholar]
- 193.
- 193a. Liao J., Sun J., Niu Y., Yu B., Tetrahedron Lett. 2011, 52, 3075–3078; [Google Scholar]
- 193b. Li Y., Sun J., Yu B., Org. Lett. 2011, 13, 5508–5511; [DOI] [PubMed] [Google Scholar]
- 193c. Li Y., Yang X., Liu Y., Zhu C., Yang Y., Yu B., Chem. Eur. J. 2010, 16, 1871–1882; [DOI] [PubMed] [Google Scholar]
- 193d. Ma Y., Li Z., Shi H., Zhang J., Yu J., J. Org. Chem. 2011, 76, 9748–9756; [DOI] [PubMed] [Google Scholar]
- 193e. Li Y., Yang W., Ma Y., Sun J., Shan L., Zhang W. D., Yu B., Synlett 2011, 915–918. [Google Scholar]
- 194.
- 194a. Sun P., Wang P., Zhang Y., Zhang X., Wang C., Liu S., Lu J., Li M., J. Org. Chem. 2015, 80, 4164–4175; [DOI] [PubMed] [Google Scholar]
- 194b. Zhu Y., Yu B., Chem. Eur. J. 2015, 21, 8771–8780. [DOI] [PubMed] [Google Scholar]
- 195. Yang Y., Li Y., Yu B., Tetrahedron Lett. 2010, 51, 1504–1507. [Google Scholar]
- 196. Zhu Y., Yu B., Angew. Chem. Int. Ed. 2011, 50, 8329–8332; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 8479–8482. [Google Scholar]
- 197. Dutta S., Sarkar S., Gupta S. J., Sen A. K., Tetrahedron Lett. 2013, 54, 865–870. [Google Scholar]
- 198. Imagawa H., Kinoshita A., Fukuyama T., Yamamoto H., Nishizawa M., Tetrahedron Lett. 2006, 47, 4729–4731. [Google Scholar]
- 199. Zhu D., Cao X., Yu B., Org. Chem. Front. 2015, 2, 360–365. [Google Scholar]
- 200. Matthies S., McQuade D. T., Seeberger P. H., Org. Lett. 2015, 17, 3670–673. [DOI] [PubMed] [Google Scholar]
- 201. Yang F., Wang Q., Yu B., Tetrahedron Lett. 2012, 53, 5231–5234. [Google Scholar]
- 202. Koppolu S. R., Niddana R., Balamurugan R., Org. Biomol. Chem. 2015, 13, 5094–5097. [DOI] [PubMed] [Google Scholar]
- 203. Ferrier R. J., Overend W. G., Ryan A. E., J. Chem. Soc. C 1962, 3667–3670. [Google Scholar]
- 204. Ferrier R. J., in O. A. Zubkov Organic Reactions; L. E. Overman (Ed.); John Wiley & Sons, Inc.: New York, 2003. ; 62, 569–736. [Google Scholar]
- 205. Rafiee E., Tangestaninejad S., Habibi M. H., Mirkhani V., Bioorg. Med. Chem. Lett. 2004, 14, 3611–3614. [DOI] [PubMed] [Google Scholar]
- 206. Bettadaiah B. K., Srinivas P., Tetrahedron Lett. 2003, 44, 7257–7259. [Google Scholar]
- 207. Masson C., Soto J., Bessodes M., Synlett 2000, 1281–1282. [Google Scholar]
- 208. Zhang G., Liu Q., Shi L., Wang J., Tetrahedron 2008, 64, 339–344. [Google Scholar]
- 209. Chen P., Wang S., Tetrahedron 2012, 68, 5356–5362. [Google Scholar]
- 210. Gorityala B. K., Lorpitthaya R., Bai Y., Liu X. W., Tetrahedron 2009, 65, 5844–5848. [Google Scholar]
- 211. Ding F., William R., Gorityala B. K., Ma J., Wang S., Liu X. W., Tetrahedron Lett. 2010, 51, 3146–3148. [Google Scholar]
- 212. Narasimha G., Srinivas B., Krishna P. R., Kashyap S., Synlett 2014, 523–526. [Google Scholar]
- 213. Kim H., Men H., Lee C., J. Am. Chem. Soc. 2004, 126, 1336–1337. [DOI] [PubMed] [Google Scholar]
- 214. Schuff B. P., Mercer G. J., Nguyen H. M., Org. Lett. 2007, 9, 3173–3176. [DOI] [PubMed] [Google Scholar]
- 215.
- 215a. Ji L., Xiang S.-H., Leng W.-L., Hoang K. L. M., Liu X.-W., Org. Lett. 2015, 17, 1357–1360; [DOI] [PubMed] [Google Scholar]
- 215b. Reddy Y. S., Lahiri R., Vankar Y. D., Eur. J. Org. Chem. 2012, 4751–4761. [Google Scholar]
- 216.
- 216a. Xiang S., He J., Tan Y. J., Liu X.-W., J. Org. Chem. 2014, 79, 11473–11482; [DOI] [PubMed] [Google Scholar]
- 216b. Xiang S., He J., Ma J., Liu X.-W., Chem. Commun. 2014, 50, 4222–4224. [DOI] [PubMed] [Google Scholar]
- 217. Sherry B. D., Loy R. N., Toste F. D., J. Am. Chem. Soc. 2004, 126, 4510–4511. [DOI] [PubMed] [Google Scholar]
- 218. Balamurugan R., Koppolu S. R., Tetrahedron 2009, 65, 8139–8142. [Google Scholar]
- 219. Srinivas B., Reddy T. R., Radha Krishna P., Kashyap S., Synlett 2014, 1325–1330. [Google Scholar]
- 220.
- 220a. Srinivas B., Narasimha G., Krishna P. R., Kashyap S., Synthesis 2014, 1191–1196; [Google Scholar]
- 220b. Chen P., Lin L., Tetrahedron 2013, 69, 10045–10051; [Google Scholar]
- 220c. Battina S. K., Reddy T. R., Krishna P. R., Kashyap S., Tetrahedron Lett. 2015, 56, 1798–1800; [Google Scholar]
- 220d. Srinivas B., Reddy T. R., Kashyap S., Carbohydr. Res. 2015, 406, 86–92. [DOI] [PubMed] [Google Scholar]
- 221. Hotha S., Tripathi A., Tetrahedron Lett. 2005, 46, 4555–4558. [Google Scholar]
- 222. Yadav J. S., Reddy B. V. S., Murthy C. V. S. R., Kumar G. M., Synlett 2000, 1450–1451. [Google Scholar]
- 223. Chen P., Li S., Tetrahedron Lett. 2014, 55, 5813–5816. [Google Scholar]
- 224. Chen P., Li S., Tetrahedron 2013, 69, 4524–4531. [Google Scholar]
- 225. Xue S., He L., Han K.-Z., Zheng X.-Q., Guo Q.-X., Carbohydr. Res. 2005, 340, 303–307. [DOI] [PubMed] [Google Scholar]
- 226. Smitha G., Reddy C. S., Synthesis 2004, 834–836. [Google Scholar]
- 227. Yadav J. S., Subba Reddy B. V., Reddy J. S. S., J. Chem. Soc. Perkin Trans. 1 2002, 2390–2394. [Google Scholar]
- 228. Suryakiran N., Reddy S. M., Srinivasulu M., Venkateswarlu Y., Synth. Commun. 2008, 38, 170–176. [Google Scholar]
- 229. Procopio A., Dalpozzo R., Nino A. D., Maiuolo L., Nardi M., Oliverioa M., Russoa B., Carbohydr. Res. 2007, 342, 2125–2131. [DOI] [PubMed] [Google Scholar]
- 230.
- 230a. Stevanović D., Pejović A., Damljanović I., Vukićević M., Bogdanović G. A., Vukićević R. D., Tetrahedron Lett. 2012, 53, 6257–6260; [Google Scholar]
- 230b. Stevanovic D., Pejovic A., Damljanovic I., Minic A., Bogdanovic G. A., Vukicevic M., Radulovic N. S., Vukicevic R. D., Carbohydr. Res. 2015, 407, 111–121. [DOI] [PubMed] [Google Scholar]
- 231. Williams D. B. G., Simelanea S. B., Kinfea H. H., Org. Biomol. Chem. 2012, 10, 5636. [DOI] [PubMed] [Google Scholar]
- 232. Chen Y.-B., Wang S. I., Lin Z.-P., Lin C.-H., Hsieh M. T., Lin H.-C., Tetrahedron 2015, 71, 350–358. [Google Scholar]
- 233.
- 233a. Boga S. B., Balasubramanian K. K., Arkivoc 2004. (viii) 87–102; [Google Scholar]
- 233b. Yadav J. S., Reddy B. V. S., Raman J. V., Niranjan N., Kumar S. K., Kunwar A. C., Tetrahedron Lett. 2002, 43, 2095–2098; [Google Scholar]
- 233c. Das S. K., Reddy K. A., Abbineni C., Roy J., Rao K. V. L. N., Sachwani R. H., Iqbal J., Tetrahedron Lett. 2003, 44, 4507–4509; [Google Scholar]
- 233d. Das S. K., Reddy K. A., Roy J., Synlett 2003, 11, 1607–1610; [Google Scholar]
- 233e. Mukherjee D., Yousuf S. K., Taneja S. C., Tetrahedron Lett. 2008, 49, 4944–4948. [Google Scholar]
- 234. Nagaraj P., Ramesh N. G., Tetrahedron Lett. 2009, 50, 3970–3973. [Google Scholar]
- 235. Swamy N. R., Venkateswarlu Y., Synthesis 2002, 5, 598–600. [Google Scholar]
- 236. Babu J. L., Khare J. A., Vankar Y. D., Molecules 2005, 10, 884–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.
- 237a. Freitas J. C. R., de Freitas J. R., Menezes P. H., J. Braz. Chem. Soc. 2010, 21, 2169–2172; [Google Scholar]
- 237b. Freitas J. C. R., Couto T. R., Paulino A. A. S., de Freitas Filho J. R., Malvestiti I., Oliveira R. A., Menezes P. H., Tetrahedron 2012, 68, 10611–10620. [Google Scholar]
- 238. De K., Legros L., Crousse B., Bonnet-Delpon D., Tetrahedron 2008, 64, 10497–10500. [Google Scholar]
- 239.
- 239a. de Oliveira R. N., de Freitas Filho J. R., Srivastava R. M., Tetrahedron Lett. 2002, 43, 2141–2143; [Google Scholar]
- 239b. Shanmugasundaram B., Bose A. K., Balasubramanian K. K., Tetrahedron Lett. 2002, 43, 6795–6798. [Google Scholar]
- 240. Oba M., Ueno Y., Kitani S., Hayakawa T., Takahashi T., Suzuki T., Sato M., Ikeda K., Heterocycles 2014, 89, 69–81. [Google Scholar]
- 241.
- 241a. Agarwal A., Rani S., Vankar Y. D., J. Org. Chem. 2004, 69, 6137–6140; [DOI] [PubMed] [Google Scholar]
- 241b. Misra A. K., Tiwari P., Agnihotri G., Synthesis 2005, 2, 260–266; [Google Scholar]
- 241c. Tiwari P., Agnihotri G., Misra A. K., Carbohydr. Res. 2005, 340, 749–752. [DOI] [PubMed] [Google Scholar]
- 242. Chen P., Wang S., Tetrahedron 2013, 69, 583–588. [Google Scholar]
- 243. Kinfe H. H., Mebrahtu F. M., Sithole K., Carbohydr. Res. 2011, 346, 2528–2532. [DOI] [PubMed] [Google Scholar]
- 244. Zhang J., Zhanga B., Zhou J., Chena H., Li J., Yang G., Wang Z., Tang J., J. Carbohydr. Chem. 2013, 32, 380–391. [Google Scholar]
- 245. Gorityala B. K., Cai S., Ma J., Liu X.-W., Bioorg. Med. Chem. Lett. 2009, 19, 3093–3095. [DOI] [PubMed] [Google Scholar]
- 246. Yang X., Li N., Scientific World Journal 2014, 2014, 307895, 1–6.24574881 [Google Scholar]
- 247. Khan A. T., Basha R. S., Lal M., Arkivoc 2013. (ii) 201–212. [Google Scholar]
- 248. Bodipati N., Palla S. R., Komera V., Peddinti R. K., Tetrahedron Lett. 2014, 55, 6878–6881. [Google Scholar]
- 249. Vieira A. S., Fiorante P. F., Hough T. L. S., Ferreira F. P., Lüdtke D. S., Stefani H. A., Org. Lett. 2008, 10, 5215–5218. [DOI] [PubMed] [Google Scholar]
- 250. Di Bussolo V., Caselli M., Romano M. R., Pineschi M., Crotti P., J. Org. Chem. 2004, 69, 7383–7386. [DOI] [PubMed] [Google Scholar]
- 251.
- 251a. Di Bussolo V., Caselli M., Romano M. R., Pineschi M., Crotti P., J. Org. Chem. 2004, 69, 8702–8708; [DOI] [PubMed] [Google Scholar]
- 251b. Di Bussolo V., Frau I., Favero L., Uccello-Barretta G., Balzano F., Crotti P., Tetrahedron 2015, 71, 6276–6284. [Google Scholar]
- 252. Di Bussolo V., Caselli M., Pineschi M., Crotti P., Org. Lett. 2002, 4, 3695–3698. [DOI] [PubMed] [Google Scholar]
- 253. Di Bussolo V., Romano M. R., Pineschi M., Crotti P., Org. Lett. 2005, 7, 1299–1302. [DOI] [PubMed] [Google Scholar]
- 254. Di Bussolo V., Caselli M., Pineschi M., Crotti P., Org. Lett. 2003, 5, 2173–2176. [DOI] [PubMed] [Google Scholar]
- 255.
- 255a. Marzabadi C. H., Franck R. W., Tetrahedron 2000, 56, 8385–8417; [Google Scholar]
- 255b. De Lederkremer R. M., Marino C., Adv. Carbohydr. Chem. Biochem. 2007, 61, 143–216; [DOI] [PubMed] [Google Scholar]
- 255c. Hou D., Lowary T. L., Carbohydr. Res. 2009, 344, 1911–1940; [DOI] [PubMed] [Google Scholar]
- 255d. Borovika A., Nagorny P., J. Carbohydr. Chem. 2012, 31, 255–283. [Google Scholar]
- 256. Lear M. J., Yoshimura F., Hirama M., Angew. Chem. Int. Ed. 2001, 40, 946–949; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 972–975. [Google Scholar]
- 257. Lu Y. S., Li Q., Zhang L. H., Ye X. S., Org. Lett. 2008, 10, 3445–3448. [DOI] [PubMed] [Google Scholar]
- 258. Issa J. P., Bennett C. S., J. Am. Chem. Soc. 2014, 136, 5740–5744. [DOI] [PubMed] [Google Scholar]
- 259. Kaneko M., Herzon S. B., Org. Lett. 2014, 16, 2776–2779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260. Jaunzems J., Sourkouni-Argirusi G., Jesberger M., Kirschning A., Tetrahedron Lett. 2003, 44, 637–639. [Google Scholar]
- 261. Tomono S., Kusumi S., Takahashi D., Toshima K., Tetrahedron Lett. 2011, 52, 2399–2403. [Google Scholar]
- 262. Ruei J.-H., Venukumar P., Ingle A. B., Tony Mong K. K., Chem. Commun. 2015, 51, 5394–5397. [DOI] [PubMed] [Google Scholar]
- 263. Park J., Boltje T. J., Boons G. J., Org. Lett. 2008, 10, 4367–4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.
- 264a. Petitou M., van Boeckel C. A. A., Angew. Chem. Int. Ed. 2004, 43, 3118; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 3180; [Google Scholar]
- 264b. Danishefsky S. J., Allen J. R., Angew. Chem. Int. Ed. 2000, 39, 836; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2000, 112, 882. [Google Scholar]
- 265.
- 265a. Banoub J., Boullanger P., Lafont D., Chem. Rev. 1992, 92, 1167–1195; [Google Scholar]
- 265b. Debenham J., Rodebaugh R., Fraser-Reid B., Liebigs Ann. Recl. 1997, 791–802. [Google Scholar]
- 266.
- 266a. Mandal S., Das R., Mukhopadhyay B., Tetrahedron: Asymmetry 2011, 22, 1108–1113; [Google Scholar]
- 266b. Mitra A., Mukhopadhyay B., Synthesis 2015, 3061–3066; [Google Scholar]
- 266c. Sarkar V., Mukhopadhyay B., Carbohydr. Res. 2015, 406, 65–70; [DOI] [PubMed] [Google Scholar]
- 266d. Misra A. K., Ding Y., Lowe J. B., Hindsgaul O., Bioorg. Med. Chem. Lett. 2000, 10, 1505–1509; [DOI] [PubMed] [Google Scholar]
- 266e. Chang S.-S., Lin C.-C., Li Y.-K., Tony Mong K.-K., Carbohydr. Res. 2009, 344, 432–438. [DOI] [PubMed] [Google Scholar]
- 267. Yang L., Ye X. Y., Carbohydr. Res. 2010, 345, 1713–1721. [DOI] [PubMed] [Google Scholar]
- 268. Zulueta M. M. L., Lin S.-Y., Lin Y.-T., Huang C.-J., Wang C.-C., Ku C.-C., Shi Z., Chyan C.-L., Irene D., Lim L.-H., Tsai T.-I., Hu Y. P., Arco S. D., Wong C.-H., Hung S.-C., J. Am. Chem. Soc. 2012, 134, 8988–8995. [DOI] [PubMed] [Google Scholar]
- 269. Wei G., Lv X., Du Y., Carbohydr. Res. 2008, 343, 3096–3099. [DOI] [PubMed] [Google Scholar]
- 270. Kajimoto T., Morimoto K., Ogawa R., Dohi T., Kita Y., Eur. J. Org. Chem. 2015, 2138–2142. [Google Scholar]
- 271. Krag J., Christiansen M. S., Petersen J. G., Jensen H. H., Carbohydr. Res. 2010, 345, 872–879. [DOI] [PubMed] [Google Scholar]
- 272. Chen J., Yu B., Tetrahedron Lett. 2008, 49, 1682–1685. [Google Scholar]
- 273. Crasto C. F., Jones G. B., Tetrahedron Lett. 2004, 45, 4891–4894. [Google Scholar]
- 274.
- 274a. Mensah E. A., Nguyen H. M., J. Am. Chem. Soc. 2009, 131, 8778; [DOI] [PubMed] [Google Scholar]
- 274b. Mensah E. A., Yu F., Nguyen H. M., J. Am. Chem. Soc. 2010, 132, 14288–14302. [DOI] [PubMed] [Google Scholar]
- 275. Hanessian S., Chem. Rev. 2000, 100, 4443–4463. [DOI] [PubMed] [Google Scholar]
- 276. Roy B., Mukhopadhyay B., Tetrahedron Lett. 2007, 48, 3783–3787. [Google Scholar]
- 277. Pfaffe M., Mahrwald R., Org. Lett. 2012, 14, 792–795. [DOI] [PubMed] [Google Scholar]
- 278. Schmalisch S., Mahrwald R., Org. Lett. 2013, 15, 5854–5857. [DOI] [PubMed] [Google Scholar]
- 279. Polanki I. K., Kurma S. H., Bhattacharya A. K., J. Carbohydr. Chem. 2015, 34, 196–205. [Google Scholar]
- 280. Guchhait G., Misra A. K., Catal. Commun. 2011, 14, 52–57. [Google Scholar]
- 281. Sharma D. K., Lambu M. R., Sidiq T., Khajuria A., Tripathi A. K., Yousuf S. K., Mukherjee D., RSC Adv. 2013, 3, 11450–11455. [Google Scholar]
- 282. Park T.-J., Weiwer M., Yuan X., Baytas S. N., Munoz E. M., Murugesana S., Linhardt R. J., Carbohydr. Res. 2007, 342, 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283. Mũnoz F. J., Andŕe S., Gabius H.-J., Sinisterra J. V., Herńaiz M. J., Linhardt R. J., Green Chem. 2009, 11, 373–379. [Google Scholar]
- 284. Auǵe J., Sizun G., Green Chem. 2009, 11, 1179–1183. [Google Scholar]
- 285. Monasson O., Sizun-Thomé G., Lubin-Germain N., Uziel J., Augé J., Carbohydr. Res. 2012, 352, 202–205. [DOI] [PubMed] [Google Scholar]
- 286. Mamidyala S. K., Finn M. G., J. Org. Chem. 2009, 74, 8417–8420. [DOI] [PubMed] [Google Scholar]
- 287.
- 287a. Gudmundsdottir A. V., Nitz M., Org. Lett. 2008, 10, 3461–3463; [DOI] [PubMed] [Google Scholar]
- 287b. Dasgupta S., Nitz M., J. Org. Chem. 2011, 76, 1918–1921. [DOI] [PubMed] [Google Scholar]
- 288. Edgar L. J. G., Dasgupta S., Nitz M., Org. Lett. 2012, 14, 4226–4229. [DOI] [PubMed] [Google Scholar]
- 289.
- 289a. Wang Y., Ye X.-S., Zhang L.-H., Org. Biomol. Chem. 2007, 5, 2189–2200; [DOI] [PubMed] [Google Scholar]
- 289b. Zulueta M. L. L., Janreddy D., Hung S.-C., Isr. J. Chem. 2015, 55, 347–359; [Google Scholar]
- 289c. Codée J. D. C., Litjens R., van den Bos L. J., Overkleeft H. S., van der Marel G. A., Chem. Soc. Rev. 2005, 34, 769–782; [DOI] [PubMed] [Google Scholar]
- 289d. Koeller K. M., Wong C. H., Chem. Rev. 2000, 100, 4465–4493; [DOI] [PubMed] [Google Scholar]
- 289e. Huang T.-Y., Zulueta M. M. L., Hung S.-C., Org. Biomol. Chem. 2014, 12, 376–382. [DOI] [PubMed] [Google Scholar]
- 290. Pornsuriyasak P., Demchenko A. V., Tetrahedron: Asymmetry 2005, 16, 433–439. [Google Scholar]
- 291. Zhang Y., Chen C., Jin L., Tan H., Wang F., Cao H., Carbohydr. Res. 2015, 401, 109–114. [DOI] [PubMed] [Google Scholar]
- 292. Polat T., Wong C.-H., J. Am. Chem. Soc. 2007, 129, 12795–12800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293. Vohra Y., Vasan M., Venot A., Boons G. J., Org. Lett. 2008, 10, 3247–3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294. Wang Z., Zhou L., El-Boubbou K., Ye X.-S., Huang X., J. Org. Chem. 2007, 72, 6409–6420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.
- 295a. Dinkelaar J., Gold H., Overkleeft H. S., Codée J. D. C., van der Marel G. A., J. Org. Chem. 2009, 74, 4208–4216; [DOI] [PubMed] [Google Scholar]
- 295b. Ohtsuka I., Hada N., Atsumi T., Kakiuchi N., Tetrahedron 2013, 69, 1470–1475; [Google Scholar]
- 295c. Sarkar S., Dutta S., Das G., Sen A. K., Tetrahedron 2011, 67, 4118–4122; [Google Scholar]
- 295d. Bhattacharyya S., Magnusson B. G., Wellmar U., Nilsson U. J., J. Chem. Soc. Perkin Trans. 1 2001, 886–890; [Google Scholar]
- 295e. Pastore A., Adinolfi M., Iadonisi A., Valerio A., Eur. J. Org. Chem. 2010, 711–718; [Google Scholar]
- 295f. Manabe S., Ishii K., Ito Y., J. Am. Chem. Soc. 2006, 128, 10666–10667; [DOI] [PubMed] [Google Scholar]
- 295g. Deng L.-M., Liu X., Liang X.-Y., Yang J.-S., J. Org. Chem. 2012, 77, 3025–3037. [DOI] [PubMed] [Google Scholar]
- 296. Wang C., Wang H., Huang X., Zhang L.-H., Ye X.-S., Synlett 2006, 2846–2850. [Google Scholar]
- 297. Tanaka H., Tateno Y., Nishiura Y., Takahashi T., Org. Lett. 2008, 10, 5597–5600. [DOI] [PubMed] [Google Scholar]
- 298. Galan M. C., Tran A. T., Whitaker S., Chem. Commun. 2010, 46, 2106–2108. [DOI] [PubMed] [Google Scholar]
- 299.
- 299a. Fréchet J. M. J., Schuerch C., J. Am. Chem. Soc. 1971, 93, 492–496; [Google Scholar]
- 299b. Yan L., Taylor C. M., R. Goodnow, Jr. , Kahne D., J. Am. Chem. Soc. 1994, 116, 6953–6954. [Google Scholar]
- 300.
- 300a. Kononov L. O., Ito Y., Ogawa T., Tetrahedron Lett. 1997, 38, 1599–1602; [Google Scholar]
- 300b. Doi T., Sugiki M., Yamada H., Takahasi T., Tetrahedron Lett. 1999, 40, 2141–2144; [Google Scholar]
- 300c. Nukada T., Berces A., Whitfield D. M., J. Org. Chem. 1999, 64, 9030–9045. [Google Scholar]
- 301.
- 301a. Fréchet J. M. in Polymer-supported Reactions in Organic Synthesis (Eds.: P. Hodge, D. Sherrington), Wiley, Chichester, 1980, 407–434; [Google Scholar]
- 301b. Ito Y., Manabe S., Curr. Opin. Chem. Biol. 1998, 2, 701–708; [DOI] [PubMed] [Google Scholar]
- 301c. Seeberger P. H., Haase W.-C., Chem. Rev. 2000, 100, 4349–4393; [DOI] [PubMed] [Google Scholar]
- 301d. Bennett C. S., Org. Biomol. Chem. 2014, 12, 1686–1698. [DOI] [PubMed] [Google Scholar]
- 302. Kanie O., Ohtsuka I., Ako T., Daikoku S., Kanie Y., Kato R., Angew. Chem. Int. Ed. 2006, 45, 3851–3854; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 3935–3938. [Google Scholar]
- 303. Mogemark M., Elofsson M., Kihlberg J., Org. Lett. 2001, 3, 1463–1466. [DOI] [PubMed] [Google Scholar]
- 304. Mogemark M., Elofsson M., Kihlberg J., J. Org. Chem. 2003, 68, 7281–7288. [DOI] [PubMed] [Google Scholar]
- 305. Kanemitsu T., Wong C. H., Kanie O., J. Am. Chem. Soc. 2002, 124, 3591–3599. [DOI] [PubMed] [Google Scholar]
- 306. Yan B., Yan H., J. Comb. Chem. 2001, 3, 78–84. [DOI] [PubMed] [Google Scholar]
- 307. Manabe S., Ito Y., J. Am. Chem. Soc. 2002, 124, 12638–12639. [DOI] [PubMed] [Google Scholar]
- 308. Ganesh N. V., Fujikawa K., Tan Y. H., Nigudkar S. S., Stine K. J., Demchenko A. V., J. Org. Chem. 2013, 78, 6849–6857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309. Ferguson J., Marzabadi C. M., Tetrahedron Lett. 2003, 44, 3573–3577. [Google Scholar]
- 310. Ako T., Daikoku S., Ohtsuka I., Kato R., Kanie O., Chem. Asian J. 2006, 1, 798–813. [DOI] [PubMed] [Google Scholar]
- 311. Nguyen S. H., Trotta A. H., Cao J., Straub T. J., Bennett C. S., Org. Biomol. Chem. 2012, 10, 2373–2376. [DOI] [PubMed] [Google Scholar]
- 312.
- 312a. Belogi G., Zhu T., Boons G.-J., Tetrahedron Lett. 2000, 41, 6965–6968; [Google Scholar]
- 312b. Belogi G., Zhu T., Boons G.-J., Tetrahedron Lett. 2000, 41, 6969–6972. [Google Scholar]
- 313.
- 313a. Wu X., Schmidt R. R., J. Org. Chem. 2004, 69, 1853–1857; [DOI] [PubMed] [Google Scholar]
- 313b. Wu J., Guo Z., J. Org. Chem. 2006, 71, 7067–7070. [DOI] [PubMed] [Google Scholar]
- 314. Guedes N., Czechura P., Echeverria B., Ruiz A., Michelena O., Martin-Lomas M., Reichardt N.-C., J. Org. Chem. 2013, 78, 6911–6934. [DOI] [PubMed] [Google Scholar]
- 315. Crich D., Smith M., J. Am. Chem. Soc. 2002, 124, 8867–8869. [DOI] [PubMed] [Google Scholar]
- 316. Roussel F., Mohamed Takhi M., Schmidt R. R., J. Org. Chem. 2001, 66, 8540–8548. [DOI] [PubMed] [Google Scholar]
- 317.
- 317a. Seeberger P. H., Werz D. B., Nat. Rev. Drug Discovery Nat. Rev. 2005, 4, 751–763; [DOI] [PubMed] [Google Scholar]
- 317b. Seeberger P. H., Acc. Chem. Res. 2015, 48, 1450–1463. [DOI] [PubMed] [Google Scholar]
- 318. Plante O. J., Palmacci E. R., Seeberger P. H., Science 2001, 291, 1523–1527. [DOI] [PubMed] [Google Scholar]
- 319. Werz D. B., Castagner B., Seeberger P. H., J. Am. Chem. Soc. 2007, 129, 2770–2771. [DOI] [PubMed] [Google Scholar]
- 320. Codée J. D. C., Kröck L., Castagner B., Seeberger P. H., Chem. Eur. J. 2008, 14, 3987–3994. [DOI] [PubMed] [Google Scholar]
- 321. Palmacci E. R., Hewitt M. C., Seeberger P. H., Angew. Chem. Int. Ed. 2001, 40, 4433–4437; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 4565–4569. [Google Scholar]
- 322. Kröck L., Esposito D., Castagner B., Wang C.-C., Bindschädler P., Seeberger P. H., Chem. Sci. 2012, 3, 1617–1622. [Google Scholar]
- 323. Eller E., Collot M., Yin J., Hahm H. S., Seeberger P. H., Angew. Chem. Int. Ed. 2013, 52, 5858–5861; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5970–5973. [Google Scholar]
- 324. Weishaupt M. W., Matthies S., Seeberger P. H., Chem. Eur. J. 2013, 19, 12497–12503. [DOI] [PubMed] [Google Scholar]
- 325. Esposito D., Hurevich M., Castagner B., Wang C.-C., Seeberger P. H., Beilstein J. Org. Chem. 2012, 8, 1601–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326. Walvoort M. T. C., van den Elst H., Plante O. J., Kröck L., Seeberger P. H., Overkleeft H. S., van der Marel G. A., Codée J. D. C., Angew. Chem. Int. Ed. 2012, 51, 4393–4396; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 4469–4472. [Google Scholar]
- 327.
- 327a. Kandasamy J., Hurevich M., Seeberger P. H., Chem. Commun. 2013, 49, 4453–4455; [DOI] [PubMed] [Google Scholar]
- 327b. Bartetzko M. P., Schuhmacher F., Hahm H. S., Seeberger P. H., Pfrengle F., Org. Lett. 2015, 17, 4344–4347; [DOI] [PubMed] [Google Scholar]
- 327c. Schmidt D., Schuhmacher F., Geissner A., Seeberger P. H., Pfrengle F., Chem. Eur. J. 2015, 21, 5709–5713. [DOI] [PubMed] [Google Scholar]
- 328. Walvoort M. T. C., Volbeda A. G., Reintjens N. R. M., van den Elst H., Plante O. J., Overkleeft H. S., van der Marel G. A., Codée J. D. C., Org. Lett. 2012, 14, 3776–3779. [DOI] [PubMed] [Google Scholar]
- 329. Calin O., Eller S., Seeberger P. H., Angew. Chem. Int. Ed. 2013, 52, 5862–5865; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 5974–5977. [Google Scholar]
- 330. Jaipuri F. A., Pohl N. L. B., Org. Biomol. Chem. 2008, 6, 2686–2691. [DOI] [PubMed] [Google Scholar]
- 331. Tang S.-L., Pohl N. L. B., Org. Lett. 2015, 17, 2642–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332. Ganesh N. V., Fujikawa K., Tan Y. H., Stine K. J., Demchenko A. V., Org. Lett. 2012, 14, 3036–3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333. Nokami T., Isoda Y., Sasaki N., Takaiso A., Hayase S., Itoh T., Hayashi R., Shimizu A., Yoshida J.-I., Org. Lett. 2015, 17, 1525–1528. [DOI] [PubMed] [Google Scholar]
- 334. Nokami T., Hayashi R., Saigusa Y., Shimizu A., Liu C.-Y., Mong K.-K. T., Yoshida J.-I., Org. Lett. 2013, 15, 4520–4523. [DOI] [PubMed] [Google Scholar]