

Abstract
The synthesis of heteroglycoclusters (hGCs) is being subjected to rising interest, owing to their potential applications in glycobiology. In this paper, we report an efficient and straightforward convergent protocol based on orthogonal chemoselective ligations to prepare structurally well‐defined cyclopeptide‐based homo‐ and heterovalent glycoconjugates displaying 5‐N‐acetyl‐neuraminic acid (Neu5Ac), galactose (Gal), and/or N‐acetyl glucosamine (GlcNAc). We first used copper‐catalyzed azide–alkyne cycloaddition and/or thiol‐ene coupling to conjugate propargylated α‐sialic acid 3, β‐GlcNAc thiol 5, and β‐Gal thiol 6 onto cyclopeptide scaffolds 7–9 to prepare tetravalent homoglycoclusters (10–12) and hGCs (13–14) with 2:2 combinations of sugars. In addition, we have demonstrated that 1,2‐diethoxycyclobutene‐3,4‐dione can be used as a bivalent linker to prepare various octavalent hGCs (16, 19, and 20) in a controlled manner from these tetravalent structures.
Keywords: cyclopeptides, homoglycoclusters, heteroglycoclusters, multivalency, orthogonal ligations
1. Introduction
Multivalent interactions between carbohydrates and lectins play key roles in the initiation and control of numerous biological and pathological processes.1 For these reasons, a large panel of synthetic scaffolds2 decorated with clusters of identical sugar residues, namely homoglycoclusters, have been developed over the last decade and successfully used as antiadhesive agents, immunomodulators, or diagnostic tools.3 However, these compounds barely reflect the heterogeneity of the glycocalix, which limits their utility to decipher the most complex recognition processes and to develop more active and selective probes.4 Recently, a few reports highlighted the interest of heteroglycoclusters (hGCs), which display different sugar units, as they represent attractive tools to deepen the understanding of the influence of heterogeneous sugar display on biological systems.5 Unlike homoclusters, for which a number of potent synthetic methods are available, the assembly of structurally well‐defined hGCs remains challenging, even though a few procedures have been described so far. The most efficient methods rely on the use of orthogonal chemoselective ligations, which allow the controlled conjugation of sugar epitopes onto properly functionalized scaffolds. For example, our group has previously described the synthesis of tetravalent hGCs based on clickable cyclopeptide scaffolds decorated with two non‐identical sugar residues, either in 2:2 or 3:1 relative proportions, which were obtained in a stepwise protocol combining oxime ligation (OL) and CuI‐catalyzed azide–alkyne cycloaddition (CuAAC).6 Subsequently, we have demonstrated the orthogonality of both thiol‐ene and thiol‐chloroactetyl couplings (TEC and TCC, respectively) for the synthesis of hexavalent hGCs that display two different sugars, in a 4:2 relative proportion and in the two possible orientations on the scaffold.7 More recently, we have reported, for the first time, a multi‐click protocol involving four different conjugation methods: OL, TEC, CuAAC, and TCC.8 By using a proper reaction order, this approach gave access to unprecedented tetravalent hGCs, displaying four different sugar motifs in a sequential one‐pot assembly in excellent yields and with a single purification step.
With the aim to design a new series of cyclopeptide‐based glycoclusters, we synthesized diverse tetravalent homoglycoclusters and hGCs, and then octavalent structures from these building blocks were prepared by using a convergent protocol. Owing to their involvement in numerous biological and pathological recognition events, we focused our attention on 5‐N‐acetyl‐neuraminic acid (Neu5Ac), N‐acetyl glucosamine (GlcNAc), and galactose (Gal).9 In fact, we have introduced propargyl Neu5Ac on a cyclopeptide scaffold with four azide groups through CuAAC, whereas GlcNAc thiol units have been coupled on a scaffold displaying four allyloxycarbonyl (Alloc) groups, using TEC. Alternatively, a combination of both reactions have been used to generate tetravalent hGCs displaying Neu5Ac, Gal, and/or GlcNAc (2:2 combination). Furthermore, to obtain two series of octavalent hGCs (4–4 and 2:2–2:2 combinations), 1,2‐diethoxycyclobutene‐3,4‐dione, namely diethyl squarate (DS), was used as a linker to undergo selective couplings with the pending amine of those tetravalent scaffolds. The utilization of CuAAC, TEC, and DS coupling (DSC) in an orthogonal sequential protocol has not yet been reported.
2. Results and Discussion
2.1. Synthesis of Tetravalent Homo‐ and Heteroclusters 10–14
Three clickable carbohydrate building blocks, namely propargyl sialoside 4, GlcNAc thiol 5,10 and galactose thiol 6,10 have been prepared. The synthesis of propargyl sialoside 4 was adapted from the procedure described by Gan and Roy, who have reported the synthesis of the acetyated derivative of α‐sialoside propargyl 3 by using a Koenigs–Knorr glycosylation (Scheme 1).11
Scheme 1.
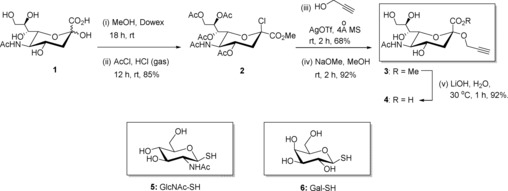
Synthesis of compound 4 and structures of 5 and 6.
Commercially available N‐acetyl neuraminic acid 1 was treated with dry MeOH in the presence of Dowex resin (H+), and then successively with acetyl chloride (saturated with HCl gas) at room temperature to give derivative 2 in good yield. Next, a glycosylation of derivative 2 with distilled propargyl alcohol in the presence of AgOTf and molecular sieves provided an anomeric mixture of propargyl sialoside, from which the alpha anomer 3 was obtained in 68 % yield after successive recrystallization in cold diethyl ether. The appearance of a characteristic triplet in the 1H NMR spectrum at 2.45 ppm (J=4.0 Hz), corresponding to the alkyne proton, and of a signal at 98.1 ppm in the 13C NMR spectrum for C‐2 confirmed the presence of the α‐linked propargyl functionality in compound 3. De‐O‐acetylation with NaOMe and methyl ester hydrolysis with LiOH afforded compound 4 in 85 % yield over two steps.
With these compounds in hand, we next synthesized several cyclopeptide scaffolds by using solid‐phase peptide synthesis followed by cyclization of the linear peptide in solution. Two scaffolds displaying four azide (7)12 or allyloxycarbonyls (8) groups and one scaffold with a combination of two allyloxycarbonyl and azide groups 9 have thus been prepared (Scheme 2).
Scheme 2.
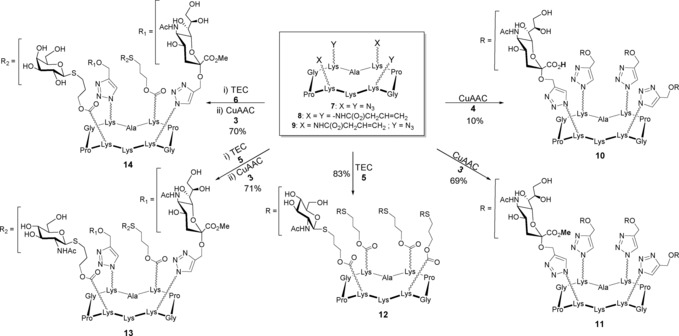
Synthesis of tetravalent homo‐ and heteroglycoclusters 10–14. Conditions for CuAAC: Cu micropowder, PBS pH 7.4, RT, 45 min; Conditions for TEC: 2,2‐dimethoxy‐2‐phenylacetophenone, DMF/H2O (2:1, v/v), hν (365 nm), RT, 45 min.
We first decided to perform the CuAAC reaction with the propargyl glycosides 4 and the scaffold 7 in the presence of copper micropowder in a mixture of isopropanol and sodium acetate buffer pH 7.4, as recently used in our group.13 Although the reaction was successful with other propargyl glycosides, no trace of the expected compound 10 was obtained by using propargyl sialoside 4 under these experimental conditions. We, thus, decided to perform the same reaction at room temperature in phosphate‐buffered saline (PBS) pH 7.4. The reaction was followed by analytical HPLC and UPLC–MS, and was found to be complete after 2 h, providing the tetravalent glycocluster 10 in a low yield (ca. 10 %) after preparative HPLC. Surprisingly, the utilization of a mixture of DMF and PBS (pH 7.4, 1:1, v/v) was found to be less efficient and led to product decomposition after 2 h. It should be mentioned that, unlike other carbohydrates, the preparation of sialylated conjugates is more frequently described with fully protected propargyl sialoside, except in a few cases.14 For example, Dondoni and co‐workers have reported the synthesis of calix[4]arene appended sialoclusters from acetylated propargyl thiosialoside and calixarene azides with good yields.15 For this reason, we hypothesized that the presence of the free carboxylic acid group in 4 might be the reason of these difficulties. To confirm this, we decided to perform the CuAAC reaction with the propargyl sialoside (3) protected as a methyl ester instead of 4. As expected, we were able to obtain the desired tetravalent glycocluster 11 in the presence of Cu micropowder in PBS (pH 7.4) in 69 % yield after RP–HPLC purification. Finally, tetravalent homoglycocluster 12 was prepared in 83 % yield by treating cyclopeptide 8 with GlcNAc thiol 5 in the presence of 2,2‐dimethoxy‐2‐phenylacetophenone (DMPA) under UV irradiation (λ max 365 nm), as reported previously.15, 16
Next, TEC and CuAAC were sequentially used to prepare the hetero‐tetravalent compounds 13 and 14, as the orthogonality of these two reactions has been demonstrated previously by Dondoni and co‐workers.15 In addition, we have determined the best reaction sequence is to use both reactions in a one‐pot fashion.8 Accordingly, we first performed the photoinduced (λ=365 nm) addition of GlcNAc thiol 5 (1.5 equiv per Alloc) on cyclopeptide 9 under UV irradiation in the presence of 2,2‐dimethoxy‐2‐phenylacetophenone (DMPA) as a radical initiator in water/DMF (2:1, v/v) for 45 min. The CuAAC ligation was subsequently carried out by adding to the crude reaction mixture propargyl sialoside 3 (1.6 equiv per azide) in the presence of Cu micropowder (4–5 equiv per azide) in PBS. This process gave the desired tetravalent glycoclusters 13 and 14 in 71 and 69 % overall yield after purification, respectively.
2.2. Synthesis of Octavalent hGC 16
A large number of artificial scaffolds that include carbohydrates,17 peptides,18 proteins,19 polymers,20 liposomes,21 dendrimers,22 nanostructures,23 or calyx[4]arenes24 have been exployed to conjugate multiple copies of sialoglycans, and the resulting glycoclusters have shown interesting binding efficiencies, particularly in the hemagglutination assay against influenza A viruses. In general, the strength of carbohydrate–protein interactions is closely dependent on the ligand structure. Many studies in this field have demonstrated that valency, linker structure, and scaffold geometry are also important parameters to consider in order to achieve high affinity. Our group has recently investigated the influence of these parameters in the binding efficiency between 4‐, 16‐ and 64‐valent series of glycocyclopeptides and diverse vegetal and bacterial lectins.25 Here, we extended the scope to generate the structural diversity of these compounds by preparing a new series of octavalent heteroclusters. For this, we focused our attention on DS, which is used in a variety of sequential amidation reactions by tuning the pH of the reaction mixture.26 In fact, this linker appears ideal to selectively conjugate the previous scaffolds 11–14 in two steps, which all contain a pending amine group. We first studied the reaction between compound 12 and DS in a mixture of Na2CO3 buffer (pH 8) and EtOH or DMF, as reported by Kunz and co‐workers.27 However, we found this procedure to be unsuccessful, presumably owing to solubility a problem. Instead, we determined that the treatment of 12 with DS (1.1 equiv) in the presence of N,N‐diisopropylethylamine (DIPEA) at pH 8.5 in DMF at room temperature led to the formation of mono‐amido ester 15 as the unique product after 4 h of reaction with 88 % yield (Scheme 3).
Scheme 3.
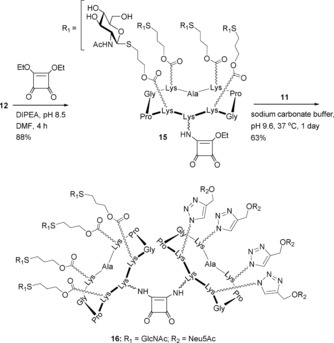
Synthesis of sialylated octavalent hGC 16.
We next performed the second amidation reaction to afford the octavalent compound 16 by using a slight excess of glycocluster 11 (1.5 equiv) in sodium phosphate buffer (pH 9.5) at 37 °C. This reaction was found to be slow and low yielding; moreover, the partial hydrolysis of methyl esters of Neu5Ac moieties was also observed by UPLC analysis. To avoid this problem, we decided to use sodium carbonate buffer pH 9.5 instead of sodium phosphate buffer to perform the second amidation and concomitant full deprotection of the methyl esters. Under these conditions, compound 16 was obtained in a 63 % yield after semi‐preparative HPLC purification, along with the formation of compound 10 in a 31 % yield as a side product.
With the aim to prepare Neu5Ac‐containing heteroclusters with different geometrical distributions to 16, we synthesized compounds 19 and 20, displaying Neu5Ac with GlcNAc and Gal residues, respectively, at alternating positions on the cyclopeptide scaffold, following the protocol described above (Scheme 4).
Scheme 4.
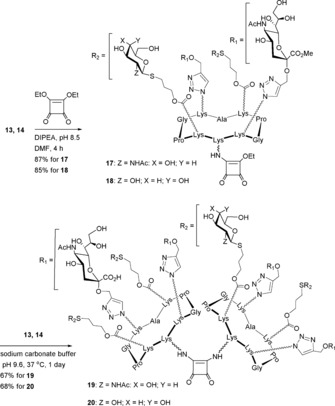
Synthesis of octavalent heteroclusters 19 and 20.
Compound 17 was first prepared in an 87 % yield from hGC 13 (1 equiv) and 1,2‐diethoxycyclobutene‐3,4‐dione (1.1 equiv) in DMF at room temperature. After purification with semi‐preparative HPLC, the addition of a slight excess of compound 13 (1.5 equiv) in the presence of sodium carbonate buffer at 37 °C afforded compound 19 in 67 % yield. Similar treatment of compound 14 under identical conditions afforded the desired heterocluster 20 in a 68 % yield.
3. Conclusions
An efficient and straightforward protocol for the controlled synthesis of multivalent homo‐ and hGCs has been developed. This convergent protocol is based on the use of three different chemoselective ligations that are CuAAC, TEC, and squaric acid diester as the central core. We first demonstrated that tetravalent homo‐ and heteroglycocluster precursors can be synthesized in high yield by using Cu‐micropowder‐catalyzed azide–alkyne cycloadditon, thiol‐ene coupling reaction, or both, using propargyl sialoside 3 and GlcNAc thiol 5, or galactoside 6 and cyclopeptides 7, 8, and 9, respectively. During the process, an alternative method to synthesize propargyl sialoside 4 through silver‐triflate‐mediated Koenigs–Knorr glycosylation was also developed in a good yield. Furthermore, we also described that squaric acid diester represents a simple and versatile linker to connect two amine‐terminated glycoclusters through two consecutive coupling reactions at basic pH. The overall protocol gave access to octavalent hGCs in good yields and purity. This synthetic approach together with our previous reports in this field represent versatile synthetic tools to develop original series of structurally well‐defined homo‐ and heteroglycodendrimers with promising biological properties. In particular, we are currently exploring the best chemoselective ligation sequences to prepare multi‐antigenic vaccines composed of tumor‐associated carbohydrate and peptide antigens.
Experimental Section
General Procedures
All chemical reagents were purchased from Aldrich (Saint Quentin Fallavier, France) or Acros (Noisy‐Le‐Grand, France). All protected amino acids and Fmoc‐Gly‐Sasrin® resin were obtained from Advanced ChemTech Europe (Brussels, Belgium). For peptides and glycopeptides, analytical RP‐HPLC was performed on a Waters system equipped with a Waters 2695 separations module and a Waters 2487 Dual Absorbance UV/Vis Detector. Analysis was carried out at 1.0 mL min−1 (EC 125/3 nucleosil 300–5 C18) with UV monitoring at 214 and 250 nm by using a linear A–B gradient (buffer A: 0.09 % CF3CO2H in water; buffer B: 0.09 % CF3CO2H in 90 % acetonitrile). Purifications were carried out at 22.0 mL min−1 (VP 250/21 nucleosil 100–7 C18) with UV monitoring at 214 and 250 nm by using a linear A–B gradient. For carbohydrate derivatives, moisture‐sensitive reactions were performed under an argon atmosphere by using oven‐dried glassware and reactions was monitored by using TLC with silica gel 60 F254 precoated plates (Merck). Spots were inspected by UV light and visualized by charring with 10 % H2SO4 in EtOH for carbohydrates. Silica gel 60 (0.063–0.2 mm or 70–230 mesh, Merck) was used for column chromatography. 1H and 13C NMR spectra were recorded on Bruker Avance 400 MHz or Brucker Avance III 500 MHz spectrometers and chemical shifts (δ) were reported in parts per million (ppm). Spectra were referenced to the residual proton solvent peaks relative to the signal of CDCl3 (δ=7.26 and 77.0 ppm for 1H and 13C) and D2O (4.79 ppm for 1H), assignments were performed by using GCOSY and GHMQC experiments. Standard abbreviations s, d, t, dd, br s,m refer to singlet, doublet, triplet, doublet of doublet, broad singlet, multiplet, respectively. The anomeric configuration was established from J 1,2 coupling constants. HRMS and ESI‐MS spectra of peptides and glycopeptides were measured on an Esquire 3000 spectrometer from Bruker or on an Acquity UPLC/MS system from Waters equipped with an SQ Detector 2. MALDI‐TOF experiments were performed on a AutoFlex I Bruker after sample pretreatment in an OligoR3 microcolumn (Applied Biosystems, USA) using 2,5‐dihydroxybenzoic acid matrix.
Synthesis of Propargyl Sialoside (4)
Methyl (5‐Acetamido‐4,7,8,9‐tetra‐O‐acetyl‐2‐chloro‐2,3,5‐trideoxy‐d‐glycereo‐β‐d‐galacto‐2‐nonulopyranosid)onate (2)
Dowex‐50WX2 resin (1 g, H+ form) was added to a solution of N‐acetyl neuraminic acid 1 (2 g, 6.5 mmol) in dry MeOH (100 mL) at room temperature under an argon atmosphere. The reaction mixture was stirred for 18 h and the resin was filtered through a Celite pad, washed with dry methanol (10 mL), and the filtrate was evaporated under vacuum to give a white solid. The crude reaction mixture was used for next reaction without further purification. Acetyl chloride (50 mL) was added to the crude reaction mixture at room temperature under an HCl (gas) environment. The white suspension gradually became pale pink and the reaction mixture was stirred continuously for 12 h. The solvents were removed in vacuo, co‐evaporated with toluene (2×50 mL), and the residue was purified by flash chromatography (SiO2) to afford 2 (2.81 g, 85 %) as a white solid. R f=0.33 (MeOH/CH2Cl2=24:1). The NMR data is consistent with literature.28
Methyl (Prop‐2‐ynyl 5‐acetamido‐3,5‐dideoxy‐d‐glycero‐α‐d‐galacto‐2‐nonulopyranosid)onate (3)
A mixture of 2 (2.81 g, 5.5 mmol), AgOTf (2.1 g, 8.2 mmol) and powdered 4 Å molecular sieves (4 g) in freshly distilled propargyl alcohol (60 mL) was stirred at room temperature under an Ar atmosphere. The reaction mixture was protected from light and stirred for 2 h in the dark. The crude mixture was filtered through a Celite pad, washed with CH2Cl2 (2×50 mL), and the filtrate was concentrated under vacuum to dryness. The residue was dissolved in CH2Cl2 (500 mL), washed with water (2×200 mL), brine (100 mL), and aqueous Na2S2O3 (10 % m/v). The combined organics were dried over MgSO4, filtered, and the solvent removed in vacuo. The crude was purified by using flash chromatography (SiO2) and crystallized from diethyl ether at −4 °C to give the peracetylated precursor of 3 (1.98 g, 68 %) as a colorless solid. R f=0.30 (MeOH/CH2Cl2=1:24); (400 MHz, CDCl3): δ H=5.45–5.41 (1 H, m, H‐8), 5.33 (1 H, dd, J=1.4, 8.0 Hz, H‐7), 5.18 (1 H, d, J=8.0 Hz, NHAc), 4.93–4.86 (1 H, m, H‐4), 4.43 (1 H, dd, J=4.0, 12.0 Hz, CH2aC≡CH), 4.31 (1 H, dd, J=4.0, 12.0 Hz, H‐9′), 4.19 (1 H, dd, J=4.0, 16.0 Hz, CH2bC≡CH), 4.13–4.09 (3 H, m, H‐9, H‐5, H‐6), 3.84 (3 H, s, OCH3), 2.66 (1 H, dd, J=4.0, 12.0 Hz, H‐3a), 2.45 (1 H, t, J=4.0 Hz, CH2≡CH), 2.17–2.05 (4×3 H, 4 s, 4×OCOCH3), 1.90 (3 H, s, NHCOCH3), 1.85 ppm (1 H, m, H‐3b); (100 MHz, CDCl3): δ C=170.9–170.0 (4×OCOCH3 and CONH), 167.7 (C‐1), 98.1 (C‐2), 78.9 (C≡CH), 74.4 (C≡CH), 72.6 (C‐6), 68.8 (C‐4), 68.2 (C‐8), 67.1 (C‐7), 62.3 (C‐9), 52.8 (OCH3), 52.7 (OCH2≡), 49.3 (C‐5), 37.9 (C‐3), 23.1 (NHCOCH3), 21.0, 20.8, 20.7, 20.5 ppm (4 x OCOCH3); HRMS (ESI+‐TOF) m/z: calcd for C23H31NO13Na [M+Na]+: 552.1693; found 552.1698.
NaOMe/MeOH (200 μL, pH 9) was added to a solution of the protected precursor of 3 (0.65 g, 1.23 mmol) in MeOH (20 mL), and the reaction mixture was stirred at room temperature for 3 h. Amberlite IR‐120 resin (H+) was added to neutralize the reaction and the solvent was removed under vacuum. The obtained residue was washed with Et2O (2×20 mL) and dried to give 3 (0.41 g, 92 %) as a white powder. HRMS (ESI+‐TOF) m/z: calcd for C15H23NO9Na [M+Na]+: 384.1271; found 384.1267.
Prop‐2‐ynyl 5‐acetamido‐3,5‐dideoxy‐d‐glycero‐α‐d‐galacto‐2‐onulopyranose (4)
LiOH (62.0 mg, 2.59 mmol) was added to a solution of 3 (0.30 g, 0.86 mmol) in water (15 mL), and the reaction stirred at room temperature for 1 h. The reaction mixture was neutralized with amberlite IR‐120 resin (H+) and filtered; the aqueous solution was washed with Et2O (2×20 mL) and freeze‐dried to give 4 (275 mg, 92 %) as a flocculent powder. (400 MHz, D2O): δ H=4.25 (2 H, ddd, J=1.6, 8.0 20.8 Hz, H‐8 NHAc), 3.84–3.72 (3 H, m, H‐2, H‐7, H‐4), 3.64–3.51 (7 H, m, H‐5, H‐6a,b, H‐9a,b, −CH2≡), 2.68 (1 H, dd, J=4.0, 12.0 Hz, H‐3a), 1.97 (3 H, s, Me), 1.61 ppm (1 H, t, J=12 Hz, H‐3b); (100 MHz, D2O): δ C=174.6 (C=O), 173.9 (C‐1), 95.3 (C‐2), 70.6 (C‐3), 70.5 (C‐7), 68.3 (C‐4), 66.4 (C‐6), 63.1 (C‐5), 52.0 (Me), 48.9 (−OCH2≡), 38.6 (C‐3), 21.9 ppm (Me); HRMS (ESI+‐TOF) m/z: calcd for C14H21NO9 [M]+: 347.1216; found 347.1208.
General Procedure for Solid‐Phase Peptide Synthesis
Assembly of the protected linear peptide was performed manually by employing a solid‐phase peptide synthesis (SPPS) protocol using the Fmoc/tBu strategy and the Fmoc‐Gly‐SasrinTM resin. Coupling reactions were performed by using, relative to the resin loading, 2 equivalents of N‐Fmoc‐protected amino acid activated in situ with PyBOP (2 equiv) and DIPEA (5 equiv) in DMF (10 mL g−1 resin) for 30 min. The coupling reaction was checked by using the TNBS test with a solution of 1 % trinitrobenzenesulfonic acid in DMF. N‐Fmoc protecting groups were removed upon treatment with piperidine–DMF (1:4, 10 mL g−1 resin) for 10 min. The process was repeated three times and the resin was further washed five times with DMF (10 mL g−1 resin) for 2 min. The peptide was released from the resin by treating ten times with a cleavage solution of TFA/CH2Cl2 (1:99). The combined cleavage fractions were concentrated under reduced pressure; ice‐cold Et2O was added to induce precipitation and the linear peptide was obtained as a white powder after filtration and used without any further purification.
General Procedure for Peptide Cyclization
All linear peptides were dissolved in CH2Cl2 (0.5 mm) and the pH of the solution was adjusted to 8.5–9 by addition of DIPEA. PyBOP (1.2 equiv) was added and the reaction mixture was stirred at room temperature for 1 h. The solvent was removed under reduced pressure and precipitation from diethyl ether afforded desired Boc‐protected cyclic peptides as white solids. Boc‐protected cyclic peptides were treated with a solution of TFA/CH2Cl2 (10 mL, 3:2, v/v) for 1 h; the crude reaction mixture was concentrated to afford ‐NH2 terminated cyclic peptides 7–9.
Compound 8
The linear peptide sequence A (0.49 mmol) (see Scheme 1 in the Supporting Information) was synthesized following the general procedure for SPPS, then cyclized, and finally the Boc group was cleaved. Yield: 90 % (over three steps, 569 mg) after purification by preparative RP‐HPLC and lyophilization; RP‐HPLC: Rt=7.6 min (C18, λ=214 nm 5–100 % B in 15 min); HRMS (ESI+‐TOF) m/z: calcd for C63H102N15O18 [M+H]+:1356.7527, found: 1356.7614
Compound 9
The linear peptide sequence B (0.40 mmol) (see Scheme 2 in the Supporting Information) was synthesized following the general procedure for SPPS, then cyclized, and finally the Boc group was cleaved. Yield: 88 % (over three steps, 569 mg) after purification by preparative RP‐HPLC and lyophilization; RP‐HPLC: Rt=7.6 min (C18, λ=214 nm 5–100 % B in 15 min); HRMS (ESI+‐TOF) m/z: calcd for C55H90N19O14 [M+H]+: 1240.6915, found 1240.6980.
General Procedure for CuAAC Ligation
A solution of cyclopeptide and propargyl sialoside (1.5–1.8 equiv/N3) in PBS buffer (10 mm, pH 7.4) was degassed at room temperature under an Ar atmosphere for 15 min. Copper micropowder (4–5 equiv/N3) was added to the reaction mixture under a nitrogen atmosphere and the solution was vigorously stirred at room temperature. After 45 min, analytical HPLC indicated complete coupling. ChelexTM resin was added to remove excess copper and the solution was filtered, concentrated, and purified by using RP‐HPLC to afford pure compound as a white powder after lyophilization.
General Procedure for TEC Ligation
To a solution of cyclopeptide (1–5 mm) and thiol glycoside (1.5–1.8 equiv/allyl) in a mixture DMF/H2O (2:1), 2,2‐dimethoxy‐2‐phenylacetophenobe (DMPA, 2–3 mg) was added and the solution was irradiated at 365 nm for 45 min while stirring. The reaction was directly purified by using semi‐preparative RP‐HPLC to afford pure compound as white foam after lyophilization.
General Procedure for DSC Ligation
Procedure A: mono‐Amido Ester Formation
To a solution of amino‐containing cyclopeptide (1–5 mm) and 1,2‐diethoxycyclobutene‐3,4‐dione (1.1 equiv) in DMF, DIPEA was added at room temperature to reach pH 8.5. After 4 h of stirring, analytical RP‐HPLC showed mono‐amide product formation, the crude mixture was then purified by using semi‐preparative RP‐HPLC to afford the desired compound as a white foam after lyophilization.
Procedure B: bis‐Amido Ester Formation
To a solution of squaric acid mono‐amide compound (1–5 mm) in sodium carbonate buffer (pH 9.5), amino‐containing cyclopeptide (1.1–1.5 equiv) was added at room temperature. The reaction mixture was incubated at 37 °C and the evolution was monitored by using analytical RP‐HPLC. After 1 day, the crude reaction mixture was purified by using semi‐preparative RP‐HPLC to give the desired compound as a white foam after lyophilization.
Compound 11
Compound 11 was obtained by coupling 7 (10.0 mg, 8.9 μmol) with 3 (19.3 mg, 53.4 μmol), following the general procedure for CuAAC ligation, in 69 % yield (15.8 mg) as a white foam after lyophilization. RP‐HPLC: Rt=8.6 min (C18, λ=214 nm 0–40 % B in 15 min); ESI+‐MS m/z: calcd for C107H170N27O46 [M+H]+: 2570.6, found: 2569.9; calcd for C107H171N27O46 [M+2 H]2+: 1285.8, found: 1285.7.
Compound 12
Compound 12 was obtained by coupling 8 (11.0 mg, 8.5 μmol) with 5 10 (12.1 mg, 51.0 μmol), following the general procedure for TEC ligation, in 83 % yield (16.3 mg) as a white foam after lyophilization. RP‐HPLC: Rt=9.4 min (C18, λ=214 nm 0–40 % B in 15 min); ESI+‐MS m/z: calcd for C95H162N19O38S4 [M+H]+: 2306.7, found: 2306.5; calcd for C95H163N19O38S4 [M+2 H]2+: 1153.8, found: 1153.8.
Compound 13
Following the procedure for TEC ligation, compound 9 (10.0 mg, 8.3 μmol) was reacted with 5 10 (5.9 mg, 25.0 μmol) in the presence of 2,2‐dimethoxy‐2‐phenylacetophenone (DMPA, 2.6 mg, 10.1 μmol). After 45 min, the crude reaction mixture was concentrated under reduced pressure, the residue was dissolved in PBS buffer (1.0 mL, pH 7.4, 10 mm), and then 3 (10.8 mg, 29.9 μmol) was added to this solution. The coupling was performed by following the general procedure for CuAAC ligation. After crude mixture purification through semi‐preparative RP‐HPLC, compound 13 was obtained in 71 % yield (14.4 mg) over two steps as a white foam after lyophilization. RP‐HPLC: Rt=8.9 min (C18, λ=214 nm 0–40 % B in 15 min); ESI+‐MS m/z: calcd for C101H166N23O42S2 [M+H]+: 2348.7, found: 2348.4; calcd for C101H167N23O42S2 [M+2 H]2+: 1219.9, found: 1219.7; calcd for C101H168N23O42S2 [M+3 H]3+: 813.6, found: 813.4.
Compound 14
Compound 14 was obtained in 70 % yield (13.7 mg) from 9 (10.0 mg, 8.3 μmol), 6 10 (4.9 mg, 24.9 μmol) and 3 (9.6 mg, 26.6 μmol), following the same strategy used for compound 13. RP‐HPLC: Rt=8.4 min (C18, λ=214 nm 0–40 % B in 15 min); ESI+‐MS m/z: calcd for C97H160N21O42S2 [M+H]+: 2356.6, found: 2356.2; calcd for C97H161N21O42S2 [M+2H]2+: 1178.8, found: 1178.5; calcd for C97H162N21O42S2 [M+3 H]3+: 786.2, found: 786.1.
Compound 15
Compound 15 was obtained by coupling 12 (12.0 mg, 5.2 μmol) with 3,4‐diethoxy‐3‐cyclobutene‐1,2‐dione (850 μL, 5.7 μmol), following procedure A for the DSC coupling, in 88 % yield (11.1 mg) as a white foam after lyophilization. RP‐HPLC: Rt=8.0 min (C18, λ=214 nm 0–60 % B in 15 min); ESI+‐MS m/z: calcd for C101H166N19O41S4 [M+H]+: 2430.8, found: 2429.9, calcd for C101H167N19O41S4 [M+2 H]2+: 1215.9, found: 1215.8; calcd for C101H168N19O41S4 [M+3 H]3+: 810.9, found: 810.7.
Compound 16
Compound 16 was obtained by coupling compound 15 (3.9 mg, 1.6 μmol) with compound 11 (6.2 mg, 2.4 μmol), following procedure A for the DSC coupling, in 63 % yield (5.0 mg) as a white foam after lyophilization. Compound 10 (1.5 mg, 0.5 μmol, 31 %) was also obtained as by‐product. RP‐HPLC: Rt=7.2 min (C18, λ=214 nm 0–60 % B in 15 min); ESI−‐MS m/z: calcd for C202H318N46O86S4 [M−2H]2−: 2447.6, found: 2447.6; calcd for C202H317N46O86S4 [M−3H]3−: 1631.4, found: 1631.5.
Compound 17
Compound 17 was obtained by coupling 13 (4.5 mg, 1.8 μmol) with 3,4‐diethoxy‐3‐cyclobutene‐1,2‐dione (300 μL, 2.0 μmol), following procedure A for the DSC coupling, in 87 % yield (4.0 mg) as a white foam after lyophilization. RP‐HPLC: Rt=7.9 min (C18, λ=214 nm 0–60 % B in 15 min); ESI+‐MS m/z: calcd for C107H170N23O45S2 [M+H]+: 2562.8, found: 2562.3, calcd for C107H171N23O45S2 [M+2 H]2+: 1281.9, found: 1281.7.
Compound 18
Compound 18 was obtained by coupling 14 (6.0 mg, 2.6 μmol) with 3,4‐diethoxy‐3‐cyclobutene‐1,2‐dione (410 μL, 2.8 μmol), following procedure A for the DSC coupling, in 85 % yield (5.5 mg) as a white foam after lyophilization. RP‐HPLC: Rt=7.6 min (C18, λ=214 nm 0–60 % B in 15 min); ESI+‐MS m/z: calcd for C103H164N21O45S2 [M+H]+: 2480.6, found: 2480.5, calcd for C103H165N21O45S2 [M+2 H]2+: 1240.8, found: 1240.6, calcd for C103H166N21O45S2 [M+3 H]3+: 827.6, found: 827.4
Compound 19
Compound 19 was obtained by coupling compound 17 (4.1 mg, 1.6 μmol) with compound 13 (5.6 mg, 2.3 μmol), following procedure A for the DSC coupling, in 67 % yield (5.2 mg) as a white foam after lyophilization. RP‐HPLC: Rt=7.2 min (C18, λ=214 nm 0–60 % B in 15 min); ESI−‐MS m/z: calcd for C202H318N46O86S4 [M−2 H]2−: 2447.6, found: 2446.6; calcd for C202H317N46O86S4 [M−3 H]3−: 1631.4, found: 1631.1.
Compound 20
Compound 20 was obtained by coupling compound 18 (4.0 mg, 1.6 μmol) with compound 14 (5.9 mg, 2.5 μmol), following procedure A for the DSC coupling, in 68 % yield (5.1 mg) as a white foam after lyophilization. RP‐HPLC: Rt=7.3 min (C18, λ=214 nm 0–60 % B in 15 min); ESI−‐MS m/z: calcd for C194H306N42O86S4 [M−2 H]2−: 2365.5, found: 2364.7; calcd for C194H305N42O86S4 [M−3 H]3−: 1576.7, found: 1576.3.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The authors acknowledge support from the “Communauté d′agglomération Grenoble‐Alpes Métropole” (Nanobio program), ICMG FR 2607, LabEx ARCANE (ANR‐11‐LABX‐0003‐01), the french Agence Nationale de la Recherche (ANR‐12‐JS07‐0001‐01 “VacSyn”) and the European Research Council Consolidator Grant “LEGO” (647938) to O.R.
G. C. Daskhan, C. Pifferi, O. Renaudet, ChemistryOpen 2016, 5, 477.
This article is part of the Virtual Issue “Carbohydrates in the 21st Century: Synthesis and Applications”
References
- 1.
- 1a. Varki A., Cummings R. D., Esko J. D., Freeze H. H., Stanley P., Bertozzi C. R., Hart G. W., Etzler M. E., Essential of Glycobiology, 2nd ed., Cold Spring Harbour, New York, 2009; [PubMed] [Google Scholar]
- 1b. Gabius H.-J., Siebert H.-C., Andre S., Jimenez-Barbero J., Rüdiger H., ChemBioChem 2004, 5, 740. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Martínez A., Mellet C. O., Garcia Fernandez J. M., Chem. Soc. Rev. 2013, 42, 4746; [DOI] [PubMed] [Google Scholar]
- 2b. Sansone F., Casnati A., Chem. Soc. Rev. 2013, 42, 4623; [DOI] [PubMed] [Google Scholar]
- 2c. Hartmann M., Lindhorst T. K., Eur. J. Org. Chem. 2011, 3583; [Google Scholar]
- 2d. Galan M. C., Dumy P., Renaudet O., Chem. Soc. Rev. 2013, 42, 4599; [DOI] [PubMed] [Google Scholar]
- 2e. Renaudet O., Mini-Rev. Org. Chem. 2008, 5, 274; [Google Scholar]
- 2f. Daskhan G. C., Berthet N., Thomas B., Fiore M., Renaudet O., Carbohydr. Res. 2015, 405, 13; [DOI] [PubMed] [Google Scholar]
- 2g. Reymond J.-L., Bergmann M., Dabre T., Chem. Soc. Rev. 2013, 42, 4814; [DOI] [PubMed] [Google Scholar]
- 2h. Chen Y., Star A., Vidal S., Chem. Soc. Rev. 2013, 42, 4532; [DOI] [PubMed] [Google Scholar]
- 2i. Marradi M., Chiodo F., Garcia I., Penadés S., Chem. Soc. Rev. 2013, 42, 4728; [DOI] [PubMed] [Google Scholar]
- 2j. Spinelli N., Defrancq E., Morvan F., Chem. Soc. Rev. 2013, 42, 4557; [DOI] [PubMed] [Google Scholar]
- 2k. Chabre Y. M., Roy R., Chem. Soc. Rev. 2013, 42, 4657; [DOI] [PubMed] [Google Scholar]
- 2l. Ladmiral V., Melia E., Haddleton D. M., Eur. Polym. J. 2004, 40, 431. [Google Scholar]
- 3.
- 3a. Synthesis and biological applications of glycoconjugates (Eds.: O. Renaudet, N. Spinelli), Bentham Science Publishers Ltd., U. A. E., 2011; [Google Scholar]
- 3b. Chabre Y. M., Roy R., Adv. Carbohydr. Chem. Biochem. 2010, 63, 165; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3c. Bernardi A., Jiménez-Barbero J., Casnati A., De Castro C., Darbre T., Fieschi F., Finne J., Funken H., Jaeger K.-E., Lahmann M., Lindhorst T. K., Marradi M., Messner P., Molinaro A., Murphy P., Nativi C., Oscarson S., Penadés S., Peri F., Pieters R. J., Renaudet O., Reymond J.-L., Richichi B., Rojo J., Sansone F., Schäffer C., Turnbull W. B., Velasco-Torrijos T., Vidal S., Vincent S., Wennekes T., Zuilhof H., Imberty A., Chem. Soc. Rev. 2013, 42, 4709; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3d. Branson T. R., Turnbull W. B., Chem. Soc. Rev. 2013, 42, 4613; [DOI] [PubMed] [Google Scholar]
- 3e. Peri F., Chem. Soc. Rev. 2013, 42, 4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Déniaud D., Julienne K., Gouin S. G., Org. Biomol. Chem. 2011, 9, 966; [DOI] [PubMed] [Google Scholar]
- 4b. Pieters R. J., Org. Biomol. Chem. 2009, 7, 2013. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Müller C., Despras G., Lindhorst T. K., Chem. Soc. Rev. 2016, 45, 3275; [DOI] [PubMed] [Google Scholar]
- 5b. Vincent S. P., Buffet K., Nierengarten I., Imberty A., Nierengarten J.-F., Chem. Eur. J. 2016, 22, 88; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5c. Ponader D., Maffre P., Aretz J., Pussak D., Ninnemann N. M., Schmidt S., Seeberger P. H., Rademacher C., Nienhaus G. U., Hartmann L., J. Am. Chem. Soc. 2014, 136, 2008; [DOI] [PubMed] [Google Scholar]
- 5d. Jiménez Blanco J. L., Ortiz Mellet C., García Fernández J. M., Chem. Soc. Rev. 2013, 42, 4518; [DOI] [PubMed] [Google Scholar]
- 5e. Gómez-García M., Benito J. M., Butera A. P., Ortiz Mellet C., García Fernández J. M., Jiménez Blanco J. L., J. Org. Chem. 2012, 77, 1273. [DOI] [PubMed] [Google Scholar]
- 6. Thomas B., Fiore M., Bossu I., Dumy P., Renaudet O., Beilstein J. Org. Chem. 2012, 8, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fiore M., Daskhan G. C., Thomas B., Renaudet O., Beilstein J. Org. Chem. 2014, 10, 1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Thomas B., Fiore M., Daskhan G. C., Spinelli N., Renaudet O., Chem. Commun. 2015, 51, 5436. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Yarema K. J., Bertozzi C. R., Curr. Opin. Chem. Biol. 1998, 2, 49; [DOI] [PubMed] [Google Scholar]
- 9b. Lanctot P. M., Gage F. H., Varki A., Curr. Opin. Chem. Biol. 2007, 11, 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gamblin D. P., Garnier P., van Kasteren S., Oldham N. J., Fairbanks A. J., Davis B. G., Angew. Chem. Int. Ed. 2004, 43, 828; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2004, 116, 846. [Google Scholar]
- 11. Gan Z., Roy R., Can. J. Chem. 2002, 80, 908. [Google Scholar]
- 12. Thomas B., Pifferi C., Daskhan G. C., Fiore M., Berthet N., Renaudet O., Org. Biomol. Chem. 2015, 13, 11529. [DOI] [PubMed] [Google Scholar]
- 13. Bossu I., Berthet N., Dumy P., Renaudet O., J. Carbohydr. Chem. 2011, 30, 458. [Google Scholar]
- 14. Caraballo R., Saleeb M., Bauer J., Liaci A. M., Chandra N., Storm R. J., Frängsmyr L., Qian W., Stehle T., Arnberg N., Elofsson O., Org. Biomol. Chem. 2015, 13, 9194. [DOI] [PubMed] [Google Scholar]
- 15. Fiore M., Chambery A., Marra A., Dondoni A., Org. Biomol. Chem. 2009, 7, 3910. [DOI] [PubMed] [Google Scholar]
- 16. Fiore M., Berthet N., Marra A., Gillon E., Dumy P., Dondoni A., Imberty A., Renaudet O., Org. Biomol. Chem. 2013, 11, 7113. [DOI] [PubMed] [Google Scholar]
- 17. Makimura Y., Watanabe S., Suzuki T., Suzuki Y., Ishida H., Kiso M., Katayama T., Kumagai H., Yamamoto K., Carbohydr. Res. 2006, 341, 1803. [DOI] [PubMed] [Google Scholar]
- 18.
- 18a. Ogata M., Murata T., Murakami K., Suzuki T., Hidari K. I. P. J., Suzuki Y., Usui T., Bioorg. Med. Chem. 2007, 15, 1383; [DOI] [PubMed] [Google Scholar]
- 18b. Kamitakahara H., Suzuki T., Nishigori N., Suzuki Y., Kanie O., Wong C.-H., Angew. Chem. Int. Ed. 1998, 37, 1524; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 1607. [Google Scholar]
- 19.
- 19a. Roy R., Laferriére C. A., J. Chem. Soc. Chem. Commun. 1990, 1709; [Google Scholar]
- 19b. Roy R., Laferriére C. A., Gamian A., Jennings H. J., J. Carbohydr. Chem. J. Carbohydr. Res. 1987, 6, 161. [Google Scholar]
- 20.
- 20a. Roy R., Laferriére C. A., Carbohydr. Res. 1988, 177, c1; [DOI] [PubMed] [Google Scholar]
- 20b. Roy R., Andersson F. O., Harms G., Kelm S., Schauer R., Angew. Chem. Int. Ed. Engl. 1992, 31, 1478; [Google Scholar]; Angew. Chem. 1992, 104, 1551; [Google Scholar]
- 20c. Lees W. J., Spaltenstein A., Kingery-Wood J. E., Whitesides G. M., J. Med. Chem. 1994, 37, 3419; [DOI] [PubMed] [Google Scholar]
- 20d. Sigal G. B., Mammen M., Dahmann G., Whitesides G. M., J. Am. Chem. Soc. 1996, 118, 3789; [Google Scholar]
- 20e. Reuter J. D., Myc A., Hayes M. M., Gan Z., Roy R., Qin D., R, Yin , Piehler L. T., Esfand R., Tomalia D. A., Baker J. R., Bioconjugate Chem. 1999, 10, 271; [DOI] [PubMed] [Google Scholar]
- 20f. Yang Z.-Q., Puffer E. B., Pontrello J. K., Kiessling L. L., Carbohydr. Res. 2002, 337, 1605; [DOI] [PubMed] [Google Scholar]
- 20g. Gambaryan A. S., Boravleva E. Y., Matrosovich T. Y., Matrosovich M. N., Klenk H.-D., Moiseeva E. V., Tuzikov A. B., Chinarev A. A., Pazynina G. V., Bovin N. V., Antiviral Res. 2005, 68, 116. [DOI] [PubMed] [Google Scholar]
- 21.
- 21a. Kingery-Wood J. E., Williams K. W., Sigal G. B., Whitesides G. M., J. Am. Chem. Soc. 1992, 114, 7303; [Google Scholar]
- 21b. Spevak W., Nagy J. O., Charych D. H., Schaefer M. E., Gilbert J. H., Bednarski M. D., J. Am. Chem. Soc. 1993, 115, 1146; [Google Scholar]
- 21c. Guo C.-T., Sun X.-L., Kanie O., Shortridge K. F., Suzuki T., Miyamoto D., Hidari K. I. P. J., Wong C.-H., Suzuki Y., Glycobiology 2002, 12, 183. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Roy R., Zanini D., Meunier S. J., Romanowska A., J. Chem. Soc. Chem. Commun. 1993, 1869; [Google Scholar]
- 22b. Roy R., Zanini D., J. Org. Chem. 1996, 61, 7348; [DOI] [PubMed] [Google Scholar]
- 22c. Llinares M., Roy R., Chem. Commun. 1997, 2119; [Google Scholar]
- 22d. Meunier S. J., Wu Q., Wang S.-N., Roy R., Can. J. Chem. 1997, 75, 1472; [Google Scholar]
- 22e. Sakamoto J.-I., Koyama T., Miyamoto D., Yingsakmongkon S., Hidari K. I. P. J., Jampangern W., Suzuki T., Suzuki Y., Esumi Y., Hatano K., Terunuma D., Matsuoka K., Bioorg. Med. Chem. Lett. 2007, 17, 717. [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Tuzikov A. B., Chinarev A. A., Gambaryan A. S., Oleinkov V. A., Kinov D. V., Matsko N. B., Kadykov V. A., Ermishov M. A., Demin I. V., Demin V. V., Rye P. D., Bovin N. V., ChemBioChem 2003, 4, 147; [DOI] [PubMed] [Google Scholar]
- 23b. Bovin N. V., Tuzikov A. B., Gambaryan A. S., Glycoconjugate J. 2004, 21, 471. [DOI] [PubMed] [Google Scholar]
- 24. Marra A., Moni L., Pazzi D., Corallini A., Bridi D., Dondoni A., Org. Biomol. Chem. 2008, 6, 1396. [DOI] [PubMed] [Google Scholar]
- 25. Thomas B., Berthet N., Dumy P., Renaudet O., Chem. Commun. 2013, 49, 10796. [DOI] [PubMed] [Google Scholar]
- 26. Wurm F. R., Klok H.-A., Chem. Soc. Rev. 2013, 42, 8220. [DOI] [PubMed] [Google Scholar]
- 27. Kaiser A., Gaidzik N., Westerlind U., Kowalczyk D., Hobel A., Schmitt E., Kunz H., Angew. Chem. Int. Ed. 2009, 48, 7551; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 7688. [Google Scholar]
- 28. Wolf S., Warnecke S., Ehrit J., Gerardy-Schahn R., Meier C., ChemBioChem 2012, 13, 2605. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary