

Abstract
A specialized insulin was recently found in the venom of a fish-hunting cone snail, Conus geographus. Here we show that many worm-hunting and snail-hunting cones also express venom insulins, and that this novel gene family has diversified explosively. Cone snails express a highly conserved insulin in their nerve ring; presumably this conventional signaling insulin is finely tuned to the Conus insulin receptor, which also evolves very slowly. By contrast, the venom insulins diverge rapidly, apparently in response to biotic interactions with prey and also possibly the cones’ own predators and competitors. Thus, the inwardly directed signaling insulins appear to experience predominantly purifying sele\ction to target an internal receptor that seldom changes, while the outwardly directed venom insulins frequently experience directional selection to target heterospecific insulin receptors in a changing mix of prey, predators and competitors. Prey insulin receptors may often be constrained in ways that prevent their evolutionary escape from targeted venom insulins, if amino-acid substitutions that result in escape also degrade the receptor’s signaling functions.
Keywords: venom, insulin gene family, diversification
Introduction
The fish-hunting geographer cone snail, Conus geographus, was recently shown to use a derived venom insulin, Con-Ins G1, in prey capture (Safavi-Hemami et al. 2015). The snails appear to release this “weaponized” insulin into the water to induce hypoglycemic shock in the fish on which they prey. Remarkably, Con-Ins G1 is more similar to fish insulins than to molluscan insulins expressed in endocrine cells, and it activates the fish insulin receptor, causing rapid depletion of blood glucose and impaired swimming behavior (Safavi-Hemami et al. 2015). Con-Ins G1 is the smallest functional insulin reported to date, and like other venom peptides in cone snails (but unlike any other characterized insulins), it carries several unusual post-translational modifications (γ-carboxylated glutamate residues and hydoxylated prolines). These unique features have probably evolved to maximize biological activity in the prey (Safavi-Hemami et al. 2015).
Insulins and related peptides (insulin-like peptides, insulin-like growth factors and relaxins) form a large superfamily of hormones that occurs throughout the animals (Shabanpoor et al. 2009). In mammals, insulin is produced and released by the endocrine β-cells of the pancreas where its primary role is the regulation of glucose homeostasis. Insulin consists of an A and B chain, cross-linked by two disulfide bonds and a third disulfide bridge within the A chain (Adams et al. 1969). In vertebrates, the structure and physiological role of insulin remains highly conserved (Ebberink et al. 1989; Blumenthal 2010). By contrast, invertebrate insulins are more variable and can serve more diverse functions, including regulation of haemolymph glucose levels, neuronal signaling, memory, reproduction and growth (Ebberink et al. 1989; Smit et al. 1998). In mollusks, insulins are produced in endocrine cells associated with the gastrointestinal tract and neuroendocrine cells of the central nervous system (Ebberink et al. 1989; Smit et al. 1998; Floyd et al. 1999).
Our recent finding that a fish-like insulin is used as a weapon for prey capture by the fish-hunting C. geographus provided the first example of insulin in a venom, and apparently the first example of insulin being used for a nefarious purpose outside of humans (Safavi-Hemami et al. 2015). The ancestors of fish-hunting cone snails preyed on worms, as do many extant cone lineages. A few lineages have evolved to feed on molluscs (Olivera et al. 2015). Here, we show that many but not all cone snail species express venom insulins (in some cases more than one), and that these unusual insulins evolve rapidly and episodically in a pattern that suggests adaptation to the physiologies of diverse prey species. Although highly diversified in their structures and amino-acid sequences, the venom insulins share a conserved signal sequence that differs from the one required to secrete the cone’s endocrine signaling insulin. All Conus species appear to express a signaling insulin in their nerve rings, and the amino-acid sequences of these conventional signaling insulins are highly conserved.
These findings suggest that early in the evolution of cone snails, a duplicated insulin gene was recruited for expression in the venom gland. As cones radiated and diversified ecologically, their venom insulins diverged in sequence and in expression levels, sometimes proliferating to form small families of venom insulins, but sometimes being lost.
Results
Venom Insulins are Widely Expressed in Diverse Cone Snail Lineages
Transcriptome analysis of venom glands from several cone-snail species representing major branches of the phylogeny led to the identification of a diverse set of insulins that share sequence similarity to the previously described C. geographus venom insulin (Safavi-Hemami et al. 2015). The inferred amino-acid sequences all include a conserved N-terminal signal sequence, while the region encoding proinsulin (the A and B chains and the C peptide) shows considerable variation (fig. 1). The average pairwise sequence identity is 84% for the signal peptides, followed by 43%, 60% and 57% for the B chains, C-peptides and A chains, respectively (fig. 1). Such close juxtaposition of conserved and hypervariable regions is also seen regularly in cone snail venom toxins (conotoxins) (Duda and Palumbi 1999; Olivera 2006). Several cone snail species were found to express more than one venom insulin, indicating that the gene family has expanded in some but not all cone-snail lineages. The largest number of distinct venom insulin mRNAs in a single species was seven (found in C. geographus). In all, we found 32 distinct sequences in the venom glands of 21 species.
Fig. 1.

Alignment of translated open reading frames of a diverse set of insulin sequences identified in the venom glands of various cone snail species. Sequence logo and approximate location of the insulin A and B chain and C-peptide regions are depicted above and below the alignment, respectively (generated in Geneious software, version 8.1.3). Average sequence identities (%) are provided for each region. Cysteines are highlighted in yellow; negatively and positively charged amino acids are shown in red and blue, respectively.
Of the 21 species examined, six including the fish-hunting cone snail C. bullatus did not express the venom insulin class at detectable levels in their venom glands (table 1). However, by mining the C. bullatus venom gland transcriptome we identified an insulin-like sequence that shared little sequence identity with members of the venom-insulin class, although it shared significant similarity to the insulin-like peptide precursor II from the marine gastropod Aplysia californica (32% sequence identity, 91% coverage, e-value: 2e−14). A similar sequence was also retrieved from the venom gland transcriptome of the worm-hunter C. virgo (96% sequence identity to the C. bullatus sequence). Members of the venom-insulin class generally do not show interspecific conservation as high as that seen for the C. virgo and C. bullatus insulin-like sequences (see supplementary fig. S1, Supplementary Material online, for sequence alignments). Furthermore, with between one and five reads per kilobase of transcript per million mapped reads (rpkm), expression levels were close to the lower limit of detection by RNAseq for venom gland transcripts, while reads for the venom insulin class were as abundant as 66,172 rpkm (table 2). This finding implied that there are at least two kinds of insulins in cone snails, one forming a major component of venom (the venom-insulin class), and the other expressed at low levels in endocrine or neuroendocrine cells (the signaling-insulin class).
Table 1.
Venom Insulins are Widely But Not Ubiquitously Expressed in Cone Snails.
| Species | Subgenus | Prey | Venom insulin | Methodology |
|---|---|---|---|---|
| C. geographus | Gastridium | Fish | + | Illumina, 454 (Hu et al. 2012) |
| C. tulipa | Gastridium | Fish | + | RT-PCR (Safavi-Hemami et al. 2015) |
| C. bullatus | Textilia | Fish | − | Illumina |
| C. striatus | Pionoconus | Fish | − | Illumina |
| C. victoriae | Cylinder | Snail | + | 454 (Robinson et al. 2014, 2015) |
| C. textile | Cylinder | Snail | + | Illumina |
| C. marmoreus | Conus | Snail | + | Illumina |
| C. bandanus | Conus | Snail | + | RT-PCR |
| C. distans | Fraterconus | Worm | − | Illumina |
| C. planorbis | Strategoconus | Worm | + | Illumina |
| C. generalis | Strategoconus | Worm | − | Illumina |
| C. varius | Strategoconus | Worm | + | Illumina |
| C. tribblei | Splinoconus | Worm | + | Illumina |
| C. floridulus | Lividoconus | Worm | + | RT-PCR (Safavi-Hemami et al. 2015) |
| C. quercinus | Lividoconus | Worm | + | RT-PCR (Safavi-Hemami et al. 2015) |
| C. andremenezi | Turriconus | Worm | − | Illumina |
| C. praecellens | Turriconus | Worm | − | Illumina |
| C. tessulatus | Tesseliconus | Worm | + | Illumina |
| C. eburneus | Tesseliconus | Worm | + | Illumina |
| C. pulicarius | Puncticulis | Worm | + | Illumina |
| C. virgo | Virgiconus | Worm | + | Illumina |
Note.—Conventional signaling insulins were retrieved from all five species in which we looked for them (Conus geographus, C. striatus, C. bullatus, C. bandanus and C. virgo) and have very similar amino-acid sequences.
Table 2.
Expression Levels of Insulins in the Venom Glands of Several Conus Species.
| Prey | Species | rpkm |
|---|---|---|
| Venom insulins | ||
| Fish-hunter | Geographus 1 (fish-like) | 20,472 |
| Geographus 3 (fish-like) | 8,981 | |
| Geographus 2 (mollusk-like) | 6,249 | |
| Snail-hunter | Marmoreus | 66,172 |
| Textile | 5,773 | |
| Worm-hunter | Varius | 2,273 |
| Planorbis | 441 | |
| Tribblei | 2,478 | |
| Pulicarius | 117 | |
| Virgo | 170 | |
| Tessulatus | 71 | |
| Eburneus | 305 | |
| Nerve-ring insulins | ||
| Fish-hunter | Bullatus | 1 |
| Worm-hunter | Virgo | 5 |
Note.—Values are expressed as reads per million of mapped reads (rpkm).
Tissue-Specific Expression of Insulins in Conus
In other mollusks, signaling insulins are typically expressed in the nervous system (ganglia and nerve ring) and the epithelial lining of the digestive tract where they play roles in the regulation of haemolymph glucose levels, memory and learning (Ebberink et al. 1989; Smit et al. 1998). To establish whether the putative venom-insulin class was venom-gland specific and the signaling-insulin class was predominantly expressed in neuroendocrine cells, we carried out reverse transcription PCR (RT-PCR) and quantitative real-time PCR (qPCR) on venom glands and circumoesophageal nerve rings (containing ganglia) isolated from three cone-snail species belonging to diverse lineages (C. geographus, C. striatus and C. bandanus) (Puillandre et al. 2014). Venom insulins were amplified from the venom glands of C. geographus and C. bandanus but not C. striatus (supplementary fig. S2, Supplementary Material online). The lack of venom insulin expression in C. striatus is consistent with transcriptome data (see table 1). Venom insulins were absent from all three nervous tissues tested. In contrast, the putative signaling insulin class was expressed only in nerve rings and could not be detected in venom glands (supplementary fig. S2, Supplementary Material online). Identification of the signaling-insulin class in the venom-gland transcriptomes of C. bullatus and C. virgo (described earlier) is therefore most plausibly explained as tissue contamination from nerve cells surrounding the venom gland, consistent with very low read counts for these two sequences.
To further confirm tissue-specific expression of the venom and signaling insulin classes, we analyzed the transcriptomes of five tissue types isolated from C. geographus (venom gland, venom bulb, foot, oesophagus and nerve ring). Illumina reads were mapped to known C. geographus insulin sequences and normalized to the total number of reads obtained for each tissue (reads per million total reads [rpmr]). RNAseq analysis confirmed that the venom insulin class was highly expressed in the venom gland but absent from all other tissues tested with the exception of the low level expression observed in the venom bulb, a muscular organ located at the distal end of the venom gland (Safavi-Hemami et al. 2010) (fig. 2). Conversely, the signaling class was exclusively expressed in the oesophagus and nervous tissue, consistent with a role in insulin signaling (Ebberink et al. 1989; Smit et al. 1998).
Fig. 2.
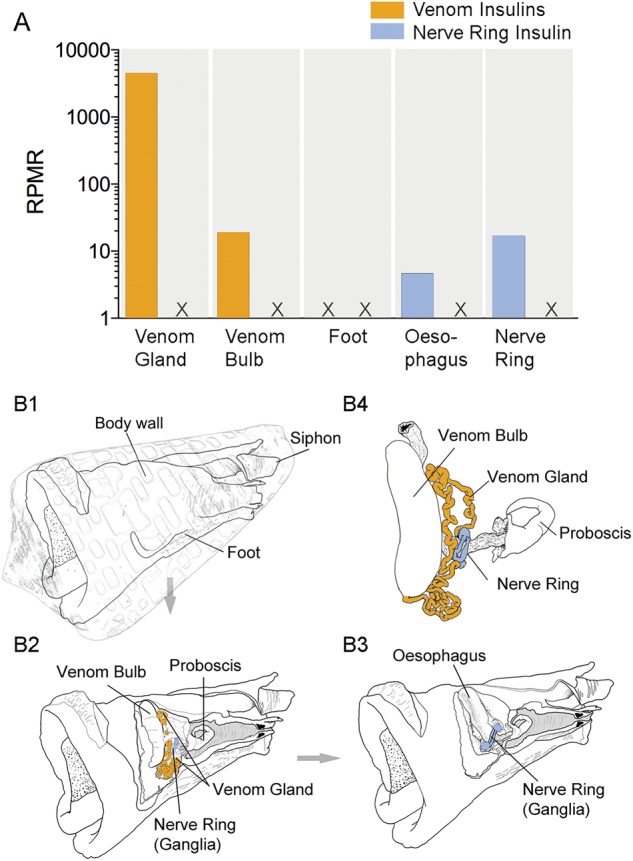
Tissue-specific expression of the venom and nerve ring insulin classes in different organs of Conus geographus. (A) Transcriptome analysis of five tissue types (venom gland, venom bulb, foot, oesophagus and nerve ring) demonstrated that venom insulins are exclusively expressed in the venom gland and venom bulb (orange bars) whereas expression of endogenous insulins is restricted to the nerve ring (including ganglia) and oesophagus (blue bars). Mapped reads were normalized to total reads obtained for each tissue and are expressed as reads per million total reads (rprm). (B) Schematic representation of the anatomy of a fixed specimen of C. tessulatus (anesthetized with isotonic magnesium chloride and preserved in 80% ethanol) showing equivalent tissues dissected and analyzed from C. geographus in this study. (B1–B3) Pictures were taken at different stages of dissections and converted into schematic drawings using a graphic tablet. Image editing was performed in Adobe Photoshop. (B1) Mantle removed. Position of the shell is shown for orientation. (B2) Proboscis cavity and body haemocoel dissected. Venom gland and nerve ring are depicted in orange and blue, respectively. (B3) Venom gland, radular diverticulum and salivary glands removed. (B4) Close-up of venom gland, venom bulb and nerve ring. In a hunting animal, a radula tooth would be positioned at the tip of the proboscis.
Contrasting Evolutionary Patterns and Rates in the Two Insulin Classes
Phylogenetic analysis of signaling and venom insulin sequences further established that the two groups form distinct gene classes and evolve with different tempos and modes (fig. 3). Signaling insulins group closely together on short branches and show consistently low dN/dS ratios (pairwise mean ω = 0.112, range 0.036–0.223, mean amino-acid identity = 96% for the signaling-insulin precursors of five species) (fig. 3 and supplementary table S1, Supplementary Material online), indicating that they are subject mainly to purifying selection. The A and B chains of the five sequences (i.e., the mature insulins) are nearly identical, with just one amino-acid substitution in the A chain of C. bandanus and one in the B chain of C. virgo (fig. 4).
Fig. 3.

Bayesian protein tree of nerve-ring and venom insulin sequences from diverse Conus species. Branches for venom insulins are colored to indicate prey preferences, with worm, snail and fish-hunting denoted by magenta, blue and yellow, respectively. Insulins from fish-hunting species are divided into mollusk- and fish-like sequences. Branch lengths are drawn proportional to estimated amino-acid change, highlighting the speed with which venom insulins have diverged, as compared with nerve-ring insulins. Tree was rooted with Aplysia californica (Californian sea hare) insulin as an outgroup. Multiple sequences from a given species are probably alleles in most cases, but clearly represent at least three paralogs in C. geographus. Posterior probabilities (Bayesian tree) and bootstrap values (ML tree) are provided for each branch. Codon deletions are depicted as dashes on branches and codon insertions are shown as boxes.
Fig. 4.

Alignments of A and B chains of venom and nerve-ring insulins from five species, showing strong conservation of the signaling insulins and divergence of the venom insulins. Cysteines are highlighted in yellow; positively and negatively charged amino acids are highlighted in blue and red, respectively.
By contrast, an alignment of the predicted A and B chains of venom insulins from four of these species plus C. varius reveals high levels of sequence divergence, even for alleles and/or paralogs within a species (fig. 4). There is also considerable variation in the lengths of the A and B chains, and in the numbers and placements of cysteine residues. Phylogenetic analysis of amino-acid substitutions in the venom insulins of 15 species yielded an overall branching pattern congruent with recently published phylogenies of the genus Conus (Puillandre et al. 2014), with distinct groups observed for worm-, snail- and fish-hunters. Within the worm-hunting group, the venom insulins also generally sort into clades that reflect the species’ phylogenetic relationships as determined by Puillandre and co-workers (fig. 3).
Venom insulins show spectacularly elevated dN/dS ratios, implying that their amino-acid sequences often have evolved under positive selection. For example, in pairwise comparisons among 17 representative sequences from the 15 species shown in figure 3, the mean dN/dS (ω) over 73 codons present in the proinsulins of all 17 sequences is 2.30, and 71% of the 136 pairwise estimates are >1.0 (supplementary table S2, Supplementary Material online). A tree-structured test for positive selection (comparing M8 to M7 in PAML) strongly favors a model in which 36% of the 73 codons evolve with an average ω of 3.09 (M8, lnL = −2053.87, 36 parameters), in comparison to the otherwise equivalent model where ω is constrained to be <1.0 at all sites (M7, lnL = −2082.08, 34 parameters, 2ΔlnL = 56.42, df = 2, P = 5.6e-13).
Branch-specific estimates of ω on the phylogeny are mostly >1.0 (fig. 5), but they are much more variable than the pairwise estimates, suggesting that adaptive evolution of the amino-acid sequences may have been highly episodic. This inference is supported by the much higher likelihood of a model with independent estimates of ω for each branch (lnL = −2085.94, 63 parameters) than of a model with one common value of ω (=1.23) for all branches (lnL = −2113.61, 33 parameters, 2ΔlnL = 55.34, df = 30, P = 0.003). Although this estimate of the overall ω for venom insulins suggests that dN may have exceeded dS on average, the 95% confidence interval (0.77–1.69) includes ω = 1.
Fig. 5.
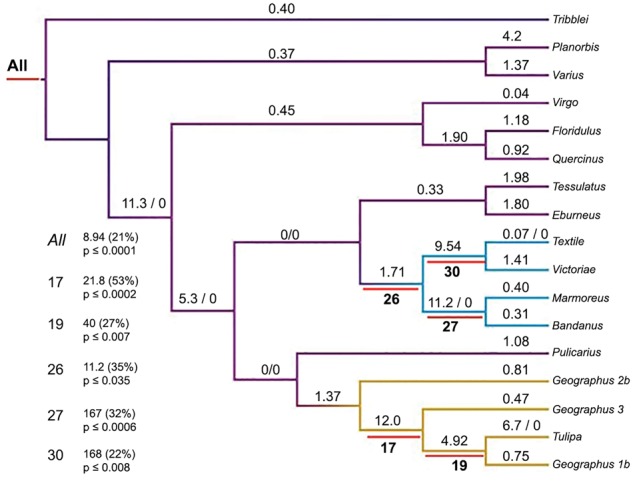
Estimated branch-specific dN/dS ratios (ω) for venom insulin sequences on a cladogram representing their evolutionary relationships. On branches with no estimated synonymous substitutions, the absolute numbers of estimated nonsynonymous (N) substitutions are shown, as N/S (i.e., N/0). Branches are colored to indicate prey preferences (worm-hunting, mollusk-hunting, fish-hunting) as in figure 3. Red bars (with branch numbers below the red bars) designate those considered as “foreground” branches in a priori tests for episodic positive selection conducted using the BUSTED method of Murrell et al. (2015). Statistics from those tests are shown to the left of the tree (branch number, ω3, percentage of branch-site combinations contributing to ω3, and P value from comparison to the null model). The topmost block (“All”) gives statistics for a test where all branches are classified as foreground, and where 21% of branch-site combinations are estimated to evolve with ω3 = 8.4.
Even within the hypervariable A and B chains, some amino-acid positions are highly conserved in some species (e.g., the cysteines that make disulfide bridges). This site-to-site rate heterogeneity is reflected visually in the sequence logo displayed along the top of figure 1, and is consistent with the result of the M8/M7 test described earlier. We used the method of Murrell et al. (2015) to test simultaneously for rate heterogeneity among sites and among branches in the tree, especially those branches following shifts from worm-hunting to mollusk-hunting, and from worm-hunting to fish-hunting. A model with all branches in the foreground estimated a very high rate of amino-acid substitution (ω3 = 8.94) at 21% of branch-site combinations, and yielded a significantly higher likelihood than the null model of no positive selection (P < 0.00001). Shifts from worm-hunting to mollusk-hunting and fish-hunting are also associated with strong signals of positive selection, on all five branches that we tested (fig. 5).
The three venom-insulin paralogs in C. geographus and C. tulipa also have evolved episodically. The paralog Geographus_2b has evolved relatively slowly since the speciation of C. geographus and the worm-hunting C. pulicarius, and it has retained its ancestral “snail-like” structure and chemical character (figs. 4 and 6). However, the duplicated copy ancestral to the “fish-like” venom insulins (Geographus_3, Geographus_1b and Tulipa; fig. 5) evolved very rapidly (ω = 12 over all sites, and ω3 = 21.8 at 53% of branch-site combinations) and on this branch it also acquired a dramatically different structure, with shortened A and B chains and a different pattern of cysteines (fig. 4). Then following a second duplication, one paralog hardly changed (Geographus_3) while the other (Geographus_1b/Tulipa) experienced a burst of amino-acid substitution (ω3 = 40 at 27% of branch-site combinations) and then slowed down in C. geographus following the speciation event giving rise to C. tulipa. However, in C. tulipa, the locus that is orthologous to Geographus_1b acquired six or seven more nonsynonymous substitutions (fig. 5), and C. tulipa apparently lost the other two paralogs, corresponding to Geographus_2b and 3, which presumably were still being expressed following C. tulipa’s separation from C. geographus.
Fig. 6.

Principal components analyses (PCA) of chemical characteristics of Conus venom and signaling insulins (A), and of venom insulins from cones and signaling insulins from putative prey species (B). Principal components were calculated from the correlation matrices of sequence characteristics using the prcomp package in R (R Core Team 2013). Ninety-five percent confidence ellipses for bivariate plots of the first two principal components were calculated using the data ellipse function in R. Black arrows point to snail-like insulin sequences from C. geographus. These two sequences and one sequence obtained from C. varius (gray arrow) were excluded from ellipse calculations. FH, fish-hunters (yellow); SH, snail-hunters (light blue); WH, worm-hunters (magenta); FP, fish prey (red); SP, snail prey (green); WP, worm prey (blue).
Venom Insulins Evolved from an Ancestral Endogenous Insulin Precursor
The insulin gene is remarkably well conserved across animals, and the insulins of modern vertebrates and invertebrates are believed to be orthologous (Chan and Steiner 2000). Canonical insulin genes contain three exons separated by two introns that are positioned upstream of the signal peptide region and within the C peptide region that separates the A and B chains (supplementary fig. S3, Supplementary Material online). A sequence corresponding to intron 2 (partial) and exon 3 (complete) of the venom insulin Con-Ins G1 was sequenced by Genome Walker PCR. Intron/Exon boundaries were shared with other insulin genes demonstrating that this venom insulin originated from a signaling insulin precursor. This partial sequence was blasted against sequences from a C. geographus fosmid DNA library, yielding a longer exact match in which intron 2 is at least 8,282 bp, which is longer than the intron 2 sequences described to date for other species (supplementary fig. S3, Supplementary Material online). Fosmid library mining identified a second homologous sequence that corresponds to the partial intron 2 and exon 3 of a paralogous C. geographus venom insulin (Con-Ins G2). The partial sequence of this intron was > 1,037 bp and the two introns share 68% identity (57% identity for exon 3). This finding confirms that several additional gene duplication events occurred in some lineages following the initial gene duplication that gave rise to the ancestral venom insulin gene.
Expression Levels of Venom Insulins Vary between Species and Correlate with Prey Preferences
Insulin expression varied significantly among different species, with highest expression observed in the venom gland of the snail-hunter C. marmoreus (66,172 rpkm) and the fish-hunter C. geographus (8,981–20,472 rpkm) and lowest expression in the worm-hunter C. tessulatus (71 rpkm) (table 2). In C. marmoreus, venom-insulin reads accounted for 2.6% of all RNAseq reads obtained from the venom gland. Similar values were obtained for C. geographus insulins (1.1% of all sequenced reads). Expression levels were also high in some worm-hunters (0.1% in C. tribblei and C. varius), but the average expression was lower in worm-hunters than in snail- and fish-hunters.
Putative Role of Venom Insulins in Cone Snails
The discovery of a venom insulin in the fish-hunting cone snail C. geographus showed that insulin is used by this species to induce hypoglycemic shock in the prey (Safavi-Hemami et al. 2011). Significant expansion of the venom insulin gene family, high dN/dS ratios, and high expression levels in the venom glands of diverse cone snail species strongly suggest that the use of insulins in prey capture (and/or defense and/or competition) is widespread in Conus. If the venom insulins principally target the insulin receptors of prey species, then they might be expected to resemble the native (signaling) insulins of those prey species. To test this prediction, we carried out principal components analyses (PCA) on a large suite of structural and chemical characteristics of the currently known venom and signaling insulins of cone snails, and the signaling insulins of potential prey species (fish, snails and worms); see supplementary table S3, Supplementary Material online, for a list of sequences used in these analyses. PCA transforms a large group of correlated variables into a smaller set of uncorrelated variables (the principal components) that are more easily interpretable and that account for most of the variability among the sequences. A similar method, factor analysis, has been used to distinguish among amino-acid sequences (Atchley and Fernandes 2005). The variables scored to characterize the insulins include length, charge, hydrophobicity, number and placement of cysteine residues, and other features of their chemical makeup (Materials and Methods). In analyses of the cone-snail insulins alone, the first two principal components clearly separate venom insulins from signaling insulins (fig. 6A). In addition, the venom insulins of fish-hunters are distinct from those of worm- and snail-hunters, except for two insulins previously identified in C. geographus as “snail-like” (black arrow) (Safavi-Hemami et al. 2015).
In analyses of venom insulins together with those of potential prey species, the venom insulins of fish-hunters group most closely with fish signaling insulins, especially on PC2 (fig. 6B and supplementary table S4, Supplementary Material online). A qualitatively similar pattern occurs for the venom insulins of mollusk-hunters; they differ significantly from the signaling insulins of mollusks, but nonetheless appear more similar to them than to the signaling insulins of worms and fish. These patterns are consistent with the hypothesis that venom insulins in these species mostly target the insulin receptors of their prey.
The venom insulins of worm-hunters do not group with worm signaling insulins, but instead with molluscan insulins (both venom and signaling). One potential explanation is that the venom insulins of worm-hunters are used mainly in defense against other mollusks rather than in predation on worms. Interestingly, worm signaling insulins appear to be more diverse than those of fish and snails (fig. 6B). Thus, the insulins of worms actually preyed on by Conus species could differ considerably from those used in this analysis, which were collected opportunistically from the public databases. Future functional studies of venom insulins isolated from worm- and snail-hunting cone snails should reveal their real targets.
Additional PCA restricted to the regions of the venom sequences subject to positive selection (A chain, B chain and C peptide) (supplementary fig. S6, Supplementary Material online) summarize the most influential chemical characteristics contributing to differences among the sequences for the three types of predatory snails.
Discussion
Several lines of evidence developed here show that diverse lineages of cone snails express two kinds of insulins. First, a conventional signaling insulin is expressed in the nerve ring of all Conus species analyzed. This insulin presumably regulates energy metabolism within the snail, and its amino-acid sequence is nearly identical in fish-hunting and nonfish-hunting species. Second, in several (but not all) species, one or more derived insulins are expressed in the venom gland. Some of these venom-insulin genes are transcribed at extremely high levels, and the encoded insulins are abundant components of the snail’s venom (as previously documented for the Con-Ins G1 insulin from C. geographus). The venom insulins differ dramatically from the signaling insulins and from each other.
In this and other respects, the venom insulins appear to function and to evolve like other components of Conus venoms. Like the well-known conotoxins that target neurotransmitter receptors, venom insulins appear to interact not with endogenous macromolecules but with molecular targets in other species (Duda and Palumbi 1999, 2000; Olivera 2006; Puillandre et al. 2010; Sunagar and Moran 2015). In addition, they evolve extremely rapidly like conotoxins (Puillandre et al. 2010; Chang and Duda 2012; Wu et al. 2013).
The ancestral prey of cone snails were polychaete worms. Some cone-snail lineages later shifted to mollusks and fish, and diversified extensively (Kohn 1956, 1966; Duda et al. 2001; Puillandre et al. 2014; Olivera et al. 2015). A venom insulin that efficiently targeted the insulin receptor of a fish would presumably differ from one that efficiently targeted the insulin receptor of a mollusk or a worm. Consistent with this expectation, the venom insulins of fish-hunting Conus tend to resemble the signaling insulins of fish (see fig. 6). Likewise, the venom insulins expressed in the mollusk-hunting clades tend to be relatively similar to gastropod insulins. This broad pattern is consistent with the hypothesis that at least many of these venom insulins are used in prey capture, and that as a consequence they are strongly selected to interact with the insulin receptors of prey that appear regularly in a cone’s diet.
The venom-insulin genes are clearly homologous to signaling-insulin genes, implying that they probably arose by duplication and divergence from an ancestral signaling insulin, early in the history of Conus. And apparently, in some lineages, subsequent duplications have given rise to multiple paralogs, as in C. geographus, where there are at least three distinct venom insulins. While two of these are fish-like in various chemical properties including the cysteine framework, the third differs considerably and more closely resembles molluscan insulins. One intriguing possibility is that this venom insulin may function mainly in defensive or competitive interactions with other gastropod molluscs, rather than in prey capture.
However, not all lineages that inherit one or more venom insulins seem to retain them. Among fish-hunting lineages, the subgenus Gastridium (which includes C. geographus and C. tulipa) does express venom insulins, but two other subgenera, Pionoconus (C. striatus) and Textilia (C. bullatus) do not appear to express a venom-insulin gene. Differing prey capture strategies may explain this difference. Conus geographus appears to release venom insulins into the water to make an entire school of small fish hypoglycemic, thereby enhancing the snail’s ability to engulf multiple fish. In contrast, fish-hunting species in the other subgenera capture fish by causing hyper-excitability of the nervous system and rapid onset of a tetanic paralysis. There may be no role for a venom insulin in this prey-capture strategy (Olivera et al. 2015). Conus tulipa is a close relative of C. geographus and presumably inherited the three or more paralogs found in that species, but it appears to express only one of them (fig. 5). Similarly, we found both mollusk-hunting and worm-hunting lineages that express a venom insulin, but two worm-hunting lineages that apparently do not. Additionally, several worm-hunting species exhibit low levels of venom insulin expression suggesting that insulins have been inherited but may have ceased to be important in these species.
Selection should strongly favor resistance to Conus venoms. Do prey insulin receptors ever acquire amino-acid substitutions that allow them to escape their predator’s venom insulin? And if so, do venom insulins then acquire compensating substitutions that restore binding to recently escaped receptors, and so on, in the dynamic often referred to as “Red Queen” coevolution (Van Valen 1973)? A prey species that modified its insulin receptor to escape a venom insulin might suffer large costs arising from interference with its own insulin signaling pathways, which would then need to be readjusted in other ways. For the resulting benefits to be large enough to offset such costs, the prey species would probably need to experience a particular Conus species as a major, persistent source of mortality. Dietary breadth varies greatly among cone-snail lineages, from highly specialized feeding to omnivory (Duda et al. 2001). Conus imperialis feeds exclusively on fire worms (Nybakken 1970) and might therefore present a good system in which to look for effects of predation on the molecular adaptation of venom targets in prey. Whether specialized Conus species loom large in the fears of their prey, and actively coevolve with those prey, are questions about which we currently know little.
Materials and Methods
Transcriptome Analysis
Assembled and annotated venom-gland transcriptomes of cone snails belonging to 13 different subgenera, including ones that prey on worms (12 species), mollusks (3 species) and fish (3 species), were mined for transcripts encoding insulin-like peptides (see table 1 for species and platforms used in this analysis). Briefly, total RNA was isolated from venom glands using TRIzol Reagent (Life Technologies) or the RNeasy kit (Qiagen) following the manufacturers’ instructions. RNA integrity, quantity and purity were determined on a 2100 Bioanalyzer (Agilent Technologies). cDNA libraries were prepared and sequenced on an Illumina HiSeq 2000 instrument (Sanger/Illumina 1.9 reads, 101- or 125-bp paired-end). Adapter clipping and quality trimming of raw reads were performed using fqtrim software (version 0.9.4, http://ccb.jhu.edu/software/fqtrim/) and PRINSEQ (version 0.20.4; Schmieder and Edwards 2011), by calculating the complexity using the DUST method with a maximum allowed score of 7. Poly-A/T tails longer than 3 bases were trimmed, and sequences shorter than 70 bps and those containing >5% ambiguous bases (Ns) were discarded. De novo transcriptome assembly was performed using Trinity version 2.0.5 (Grabherr et al. 2011) with a kmer size for building De Bruijn Graphs of 32, a minimum percent identity for two paths to be merged of 99, a maximum allowed difference between combined path sequences of 1, a maximum internal gap length allowed for combining paths of 3 and a minimum output contig length of 75. Assembled transcripts were annotated using BLASTx (ncbi-blast-2.2.28+; Altschul et al. 1990) against the UniProt database (2015), and insulin transcripts extracted based on annotation. To check fidelity of assembled insulin transcripts, raw reads were mapped back using the map-to-reference tool in Geneious version 8.1.3 (Kearse et al. 2012) and manually examined.
To determine tissue-specific insulin expression, several different tissues were dissected from C. geographus (foot, venom bulb, oesophagus, nerve ring and venom gland) and total RNA extracted using Direct-zol RNA MiniPrep Plus (Zymo Research) following the manufacturer’s instructions. RNA integrity, quantity and purity were determined on a 2100 Bioanalyzer (Agilent Technologies). Libraries were prepared using the TruSeq Stranded mRNA Sample Prep with poly(A) selection (Illumina) and sequenced on a HiSeq instrument (Illumina, 125-bp paired-end). Reads were mapped onto the open reading frame of venom and nerve ring insulin sequences using Geneious (version 8.1.3; Kearse et al. 2012) and normalized to the total number of reads obtained for each RNAseq data set.
Expression Levels
Read counts for transcripts were determined by mapping RNAseq data sets onto contigs using Bowtie2 software with default settings (Langmead and Salzberg 2012). Expression levels were obtained by calculating the number of reads per kilobase of transcript per million total mapped reads (rpkm). Average rpkm values were taken for allelic variants because high sequence similarities do not allow for unambiguous assignment of reads. Read counts could not be determined for sequences obtained by PCR and for insulin from C. victoriae because the transcriptome library was normalized prior to sequencing (Robinson et al. 2014).
Comparative RT-PCR and qPCR
Venom glands and nerve rings were dissected from C. geographus, C. striatus and C. bandanus and total RNA extracted using the Ambion Purelink RNA Mini Kit according to the manufacturer’s instructions, with on-column DNase treatment. cDNA was prepared using SmartScribe reverse transcriptase (Clontech) with a 1:1 mixture of random hexamer and oligo-dT primers. RT-PCR was performed using the Clontech Advantage 2 PCR kit (30 cycles using an annealing temperature of 51°C). Venom insulin primers were designed against known C. geographus sequences (Safavi-Hemami et al. 2015) and nerve ring insulin primers were designed based on sequences obtained from the venom gland transcriptomes of C. bullatus and C. virgo (see supplementary table S5, Supplementary Material online, for all primers used). PCR amplicons were gel purified (Qiagen gel extraction kit), ligated into the pGEM-T Easy vector (Promega) and transformed into competent NB 10-beta Escherichia coli (New England Biolabs). Plasmid DNA was isolated from overnight cultures using the Qiagen plasmid mini kit and sequenced at the University of Utah Sequencing and Genomics core facility using Sanger sequencing. Accession numbers are provided in supplementary table S3, Supplementary Material online. qPCR was performed using the SsoFast EvaGreen Supermix with 20 ng cDNA template for nerve ring and venom gland on a CFX96 instrument (Bio-Rad) and analyzed in CFX Manager software (Bio-Rad). The TMX1 and mitochondrial NADH dehydrogenase gene were used for normalization.
Phylogenetic Analysis
Multiple protein sequence alignment of nerve-ring and venom insulin sequences was performed using MAFFT v7.017 with the slow, iterative refinement setting (FFT-NS-i) (Katoh et al. 2005). Signaling insulin from the sea hare Aplysia californica was used as an outgroup (GenBank: NP_001191503). Alignment gaps were removed prior to phylogenetic analysis. A bayesian tree was estimated by MrBayes 3.2.2 (Huelsenbeck and Ronquist 2001) with two runs each of four Markov chains sampling every 200 generations. The likelihood score stabilized after 1,100,000 generations. The consensus tree was calculated after omitting the first 25% of the samples as burn-in. Bootstrap branch support values were obtained for an equivalent ML tree estimated by PhyML (Guindon and Gascuel 2003) (WAG substitution model, 4 rate categories, 100 bootstraps).
Selection Analysis
For selection analysis, one venom insulin-coding sequence was randomly selected from each of the 15 species with the exception of C. geographus for which one sequence was randomly selected from each of the three clearly distinct paralogous loci. The purpose of this sampling scheme was to ensure that selection analysis was carried out on different loci (fig. 3), not on alleles. The program codeml from PAML 4.7 (Yang 2007) was used to estimate synonymous and nonsynonymous substitutions under the F3x4 codon frequency model, over all codon positions without gaps in an alignment of the proinsulin sequences (i.e., the A and B chains plus C peptide, omitting the highly conserved signal peptide). Analyses were conducted under several different models of sequence evolution: (1) pairwise comparisons of the 17 sequences; (2) a beta distribution of dN/dS ratios (0 < ω < 1) over sites (M7); (3) a beta distribution over some sites, with a single higher value of ω at others (M8); (4) a single value of ω for all branches (M0); and (5) independent estimates of ω for all branches in the phylogeny (M1) (Yang et al. 2000). Similar analyses were carried out for an alignment of the five nerve-ring insulin sequences. Conventional likelihood-ratio tests were used to compare models that form nested pairs.
Using the same venom-insulin alignment, we carried out branch-site statistical tests for episodic selection in HyPhy (Kosakovsky Pond et al. 2005) using the BUSTED algorithm (Murrell et al. (2015). BUSTED is an extension of the Nielsen and Yang (1998) episodic model that averages ω values across sites within the foreground (the branches included in an a priori specified hypothesis of positive selection) and within the background (all other branches). The averaging tends to wash out the effect of localized selection pressures. In contrast, BUSTED models site-wise variable rates for each branch of the tree to more sensitively detect local episodic selection. The test accounts for stochastically variable rates across sites and across branches by estimating three categories of selection, under the alternative constraints that ω1 ≤ ω2 ≤ 1 ≤ ω3, for the foreground and the background. The constraints for the null hypothesis of no episodic positive selection are ω1 ≤ ω2 ≤ ω3 ≤ 1.00. The proportions of branch-site combinations in each category of ω are estimated. We tested a model with all branches in the foreground (unrestricted positive selection), and five different a priori models motivated by the phylogenetically inferred shifts in prey preferences.
Statistical Sequence Comparison
Principal components analysis was performed on two sets of amino-acid sequences: (1) venom and nerve-ring (signaling) insulins from cone snails, and (2) venom insulins from cone snails and signaling insulins in potential prey species (see supplementary table S3, Supplementary Material online, for all sequences used in this analysis). The list of variables used in the PCA analyses (sequence length, number of cysteine residues, molecular weight, isoelectric point, and percentage of positive amino acids) is given in supplementary table S4, Supplementary Material online. Variables were determined for intact insulin precursors, predicted A and B chains, and distinct regions within the A and B chains. These regions included cysteine loops and the N- and C-terminal tails following the first and last cysteine of each chain. This is illustrated in supplementary figure S5, Supplementary Material online. Principal components were calculated using the prcomp package in R (R Core Team 2013). Values for a given sequence feature were centered to zero and scaled to have unit variance prior to analysis. The two components with the best resolving power were chosen to generate plots.
Supplementary Material
Supplementary tables S1–S5 and figures S1–S3, S5 and S6 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank Kevin Chase for help with the PCA analysis, and the staff of the sequencing and genomics core at the University of Utah for fast and excellent DNA sequencing and for helpful advice. This work was supported in part by National Institutes of Health Grants GM 48677 (to B.M.O.) and GM 099939 (to M.Y.), and by an International Outgoing Fellowship Grant from the European Commission (CONBIOS 330486, to H.S.-H.).
References
- Adams MJ, Blundell TL, Dodson EJ, Dodson GG, Vijayan M, Baker EN, Harding MM, Hodkin DC, Rimmer B, Sheat S. 1969. Structure of rhombohedral 2 zinc insulin crystals. Nature 224:491–495. [Google Scholar]
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. [DOI] [PubMed] [Google Scholar]
- Atchley WR, Fernandes AD. 2005. Sequence signatures and the probabilistic identification of proteins in the Myc-Max-Mad network. Proc Natl Acad Sci U S A. 102:6401–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blumenthal S. 2010. From insulin and insulin-like activity to the insulin superfamily of growth-promoting peptides: a 20th-century odyssey. Perspect Biol Med. 53:491–508. [DOI] [PubMed] [Google Scholar]
- Chan SJ, Steiner DF. 2000. Insulin through the ages: phylogeny of a growth promoting and metabolic regulatory hormone. Am Zool. 40:213–222. [Google Scholar]
- Chang D, Duda TFJ. 2012. Extensive and continuous duplication facilitates rapid evolution and diversification of gene families. Mol Biol Evol. 29:2019–2029. [DOI] [PubMed] [Google Scholar]
- Consortium U. 2015. UniProt: a hub for protein information. Nucleic Acids Res. 43:D204–D212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duda TF, Jr., Kohn AJ, Palumbi S. 2001. Origins of diverse feeding ecologies within Conus, a genus of venomous marine gastropods. Biol J Linnean Soc. 73:391–409. [Google Scholar]
- Duda TF, Palumbi SR. 2000. Evolutionary diversification of multigene families: allelic selection of toxins in predatory cone snails. Mol Biol Evol. 17:1286–1293. [DOI] [PubMed] [Google Scholar]
- Duda TF, Palumbi SR. 1999. Molecular genetics of ecological diversification: duplication and rapid evolution of toxin genes of the venomous gastropod Conus. Proc Natl Acad Sci U S A. 96:6820–6823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebberink RHM, Smit AB, Van Minnen J. 1989. The insulin family: evolution of structure and function in vertebrates and invertebrates. Biol Bull. 177:176–182. [Google Scholar]
- Floyd PD, Li L, Rubakhin SS, Sweedler JV, Horn CC, Kupfermann I, Alexeeva VY, Ellis TA, Dembrow NC, Weiss KR, et al. 1999. Insulin prohormone processing, distribution, and relation to metabolism in Aplysia californica. J Neurosci. 19:7732–7741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. 2011. Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat Biotechnol. 29:644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guindon S, Gascuel O. 2003. A simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst Biol. 52:696–704. [DOI] [PubMed] [Google Scholar]
- Hu H, Bandyopadhyay PK, Olivera BM, Yandell M. 2012. Elucidation of the molecular envenomation strategy of the cone snail Conus geographus through transcriptome sequencing of its venom duct. BMC Genomics 13:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huelsenbeck JP, Ronquist F. 2001. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 17:754–755. [DOI] [PubMed] [Google Scholar]
- Katoh K, Kuma K, Toh H, Miyata T. 2005. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 20:511–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearse M, Moir R, Wilson A, Stones-Havas S, Cheung M, Sturrock S, Buxton S, Cooper A, Markowitz S, Duran C, et al. 2012. Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28:1647–1649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AJ. 1956. Piscivorous gastropods of the genus Conus. Proc Natl Acad Sci U S A. 42:168–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohn AJ. 1966. Food specialization in Conus in Hawaii and California. Ecology 47:1041–1043. [Google Scholar]
- Kosakovsky Pond SL, Frost SD, Muse SV. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676–679. [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrell B, Weaver S, Smith MD, Wertheim JO, Murrell S, Aylward A, Eren K, Pollner T, Martin DP, Smith DM, et al. 2015. Gene-wide identification of episodic selection. Mol Biol Evol 32:1365–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Yang Z. 1998. Likelihood models for detecting positively selected amino acid sites and applications to the HIV-1 envelope gene. Genetics 148:929–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nybakken J. 1970. Correlation of radula tooth structure and food habits of three vermivorous species of Conus. Veliger 12:316–318. [Google Scholar]
- Olivera BM. 2006. Conus peptides: biodiversity-based discovery and exogenomics. J Biol Chem. 281:31173–31177. [DOI] [PubMed] [Google Scholar]
- Olivera BM, Seger J, Horvath MP, Fedosov AE. 2015. Prey-capture strategies of fish-hunting cone snails: behavior, neurobiology and evolution. Brain Behav Evol. 86:58–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puillandre N, Bouchet P, Duda TF, Jr., Kauferstein S, Kohn AJ, Olivera BM, Watkins M, Meyer C. 2014. Molecular phylogeny and evolution of the cone snails (Gastropoda, Conoidea). Mol Phylogenet Evol. 78:290–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puillandre N, Watkins M, Olivera BM. 2010. Evolution of Conus peptide genes: duplication and positive selection in the A-superfamily. J Mol Evol. 70:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team. 2013. R: A language and environment for statistical computing. Vienna (Austria): R Foundation for Statistical Computing. Available from: http://www.R-project.org/. [Google Scholar]
- Robinson SD, Li Q, Bandyopadhyay PK, Gajewiak J, Yandell M, Papenfuss AT, Purcell AW, Norton RS, Safavi-Hemami H. 2015. Hormone-like peptides in the venoms of marine cone snails. Gen Comp Endocrinol. Advance Access published August 25, 2015, doi: 10.1016/j.ygcen.2015.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson SD, Safavi-Hemami H, McIntosh LD, Purcell AW, Norton RS, Papenfuss AT. 2014. Diversity of conotoxin gene superfamilies in the venomous snail, Conus victoriae. PLoS One 9:e87648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavi-Hemami H, Gajewiak J, Karanth S, Robinson SD, Ueberheide B, Douglass AD, Schlegel A, Imperial JS, Watkins M, Bandyopadhyay PK, et al. 2015. Specialized insulin is used for chemical warfare by fish-hunting cone snails. Proc Natl Acad Sci U S A. 112:1743–1748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safavi-Hemami H, Siero WA, Gorasia DG, Young ND, MacMillan D, Williamson NA, Purcell AW. 2011. Specialisation of the venom gland proteome in predatory cone snails reveals functional diversification of the conotoxin biosynthetic pathway. J Proteome Res. 10:3904–3919. [DOI] [PubMed] [Google Scholar]
- Safavi-Hemami H, Young ND, Williamson NA, Purcell AW. 2010. Proteomic interrogation of venom delivery in marine cone snails – novel insights into the role of the venom bulb. J Proteome Res. 9:5610–5619. [DOI] [PubMed] [Google Scholar]
- Schmieder R, Edwards R. 2011. Quality control and preprocessing of metagenomic datasets. Bioinformatics 27:863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabanpoor F, Separovic F, Wade JD. 2009. The human insulin superfamily of polypeptide hormones. Vitam Horm. 80:1–31. [DOI] [PubMed] [Google Scholar]
- Smit AB, van Kesteren RE, Li KW, Van Minnen J, Spijker S, Van Heerikhuizen H, Geraerts WP. 1998. Towards understanding the role of insulin in the brain: lessons from insulin-related signaling systems in the invertebrate brain. Prog Neurobiol. 54:35–54. [DOI] [PubMed] [Google Scholar]
- Sunagar K, Moran Y. 2015. The rise and fall of an evolutionary innovation: contrasting strategies of venom evolution in ancient and young animals. PLoS Genet. 11:e1005596.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Valen L. 1973. A new evolutionary law. Evol. Theory 1:1–30. [Google Scholar]
- Wu Y, Wang L, Zhou M, You Y, Zhu X, Qiang Y, Qin M, Luo S, Ren Z, Xu A. 2013. Molecular evolution and diversity of Conus peptide toxins, as revealed by gene structure and intron sequence analyses. PLoS One 8:e82495.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z. 2007. PAML 4: phylogenetic analysis by maximum likelihood. Mol Biol Evol. 24:1586–1591. [DOI] [PubMed] [Google Scholar]
- Yang Z, Nielsen R, Goldman N, Pedersen AM. 2000. Codon-substitution models for heterogeneous selection pressure at amino acid sites. Genetics 155:431–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.