

Abstract
Hybrid seed failure represents an important postzygotic barrier to interbreeding among species of wild tomatoes (Solanum section Lycopersicon) and other flowering plants. We studied genome-wide changes associated with hybrid seed abortion in the closely related Solanum peruvianum and S. chilense where hybrid crosses yield high proportions of inviable seeds due to endosperm failure and arrested embryo development. Based on differences of seed size in reciprocal hybrid crosses and developmental evidence implicating endosperm failure, we hypothesized that perturbed genomic imprinting is involved in this strong postzygotic barrier. Consequently, we surveyed the transcriptomes of developing endosperms from intra- and inter-specific crosses using tissues isolated by laser-assisted microdissection. We implemented a novel approach to estimate parent-of-origin–specific expression using both homozygous and heterozygous nucleotide differences between parental individuals and identified candidate imprinted genes. Importantly, we uncovered systematic shifts of “normal” (intraspecific) maternal:paternal transcript proportions in hybrid endosperms; the average maternal proportion of gene expression increased in both crossing directions but was stronger with S. peruvianum in the maternal role. These genome-wide shifts almost entirely eliminated paternally expressed imprinted genes in S. peruvianum hybrid endosperm but also affected maternally expressed imprinted genes and all other assessed genes. These profound, systematic changes in parental expression proportions suggest that core processes of transcriptional regulation are functionally compromised in hybrid endosperm and contribute to hybrid seed failure.
Keywords: endosperm, seed failure, postzygotic isolation, genomic imprinting, Solanum
Introduction
Elucidating the evolutionary processes underlying the establishment of reproductive isolation between recently diverged lineages, as well as its molecular underpinnings, remains a fundamental problem in explaining the origins of biodiversity (Coyne and Orr 2004; Crespi and Nosil 2013). A somewhat neglected phenomenon in this regard is hybrid seed failure in flowering plants, albeit early work recognizing it as widespread and of significance for the potentially rapid establishment of postzygotic reproductive isolation (Cooper and Brink 1940; Brink and Cooper 1947; Haig and Westoby 1991; Lester and Kang 1998; Bushell et al. 2003). Following successful double fertilization, growth of the endosperm—an essential seed compartment in angiosperms—typically shows aberrant features in such crosses and eventually results in embryo and seed abortion; very recent work on Capsella (Rebernig et al. 2015) and Mimulus (Oneal et al. 2016) has provided fresh evidence for the importance of this type of postzygotic barrier in angiosperms.
Many empirical observations point to a decisive role for parental genome dosage and sensitivity to such dosage in the success or failure of particular crosses. For example, interploidy crosses typically have large effects on endosperm size (Cooper and Brink 1945; Lin 1984; Birchler 1993; Scott et al. 1998), and failure of crosses between different ploidy levels within species sometimes resembles failure in interspecific crosses of the same ploidy (Bushell et al. 2003; Gutierrez-Marcos et al. 2003). These concordant observations have fueled the hypothesis that parent-of-origin-dependent gene expression (genomic imprinting) might be causally involved in hybrid seed failure (Haig and Westoby 1991; Gutierrez-Marcos et al. 2003). Genomic imprinting is an epigenetic phenomenon known in angiosperms and mammals and refers to Allele-Specific Expression (ASE) that depends on whether the allele was inherited from the female or the male parent. In flowering plants, imprinting is most prevalent in the (normally) triploid endosperm and is critical for its proper development and thus for normal seed development (Raissig et al. 2011; Jiang and Köhler 2012; Gehring 2013).
In a seminal paper, Haig and Westoby (1991) interpreted the effects of between-species and interploidy crosses on seed development as reflecting genetic conflicts between maternally and paternally derived alleles over the allocation of resources from mother to offspring (also known as the “kinship theory” or “parental conflict theory” for the evolution of imprinting; Haig 2013; Pires and Grossniklaus 2014). Under this model, imprinting and the resulting levels of gene expression collectively secure successful seed development in the context of antagonistic parental forces. Normal seed development is therefore expected to be sensitive to changes in ploidy or any molecular divergence between parents that would affect genomic imprinting. However, the underlying cause of (hybrid) seed failure must not necessarily be sought in perturbed imprinting. More recently, alternative molecular mechanisms—that might well act in concert with perturbed imprinting—have been proposed to account for seed failure, such as small interfering (si) RNAs and the derepression of Transposable Elements (TEs) mediated by siRNAs (Castillo and Moyle 2012; Lu et al. 2012; Ng et al. 2012; Lafon-Placette and Köhler 2015).
With the advent of Next-Generation-Sequencing technologies, it has become possible to assess entire endosperm transcriptomes for evidence of genomic imprinting. Consequently, hundreds of candidate imprinted genes have been identified in Arabidopsis thaliana (Gehring et al. 2011; Hsieh et al. 2011; Wolff et al. 2011; Pignatta et al. 2014), rice (Luo et al. 2011; Rodrigues et al. 2013), and maize (Zhang et al. 2011; Waters et al. 2013). One of the major recent discoveries concerns evidence for allele-specific imprinting and, more generally, variation within species for imprinting status (Waters et al. 2013; Pignatta et al. 2014). These observations were mechanistically explained by transiently altered methylation patterns downstream of evolutionarily recent TE insertions (Pignatta et al. 2014), and imply that only a small minority of imprinted genes may be functionally important for normal endosperm and seed development. However, these recent endosperm RNA-Seq studies on plant model systems did not focus on reproductive isolation and the potential involvement of perturbed or dissimilar patterns of imprinting.
Previous investigations of the molecular signatures of hybrid seed failure are restricted to the genus Arabidopsis. Arguably the best-studied examples of interspecific hybrid seed failure at the same ploidy level involve A. thaliana × A. arenosa crosses (Josefsson et al. 2006; Walia et al. 2009; Burkart-Waco et al. 2013, 2015). This body of work documented biparental expression patterns of normally imprinted genes (MEDEA and PHERES1) in hybrid endosperm, as well as the reactivation of retrotransposons. Josefsson et al. (2006) established a causal link between perturbation of imprinting and the degree of interspecific seed abortion, equivalent to similar work on interploidy crosses in A. thaliana (Jullien and Berger 2010; Kradolfer et al. 2013). Moreover, these studies imply that this type of postzygotic barrier could be erected by evolutionary changes at very few genes. The most recent study of A. thaliana × A. arenosa crosses (Burkart-Waco et al. 2015) found evidence for a general shift toward higher maternal transcript proportions in hybrid endosperm and the concomitant mis-expression of paternally expressed imprinted genes. Because entire seeds were the source of RNA, however, this inference had to be restricted to the subset of genes expressed exclusively in the A. thaliana endosperm.
Wild tomatoes (Solanum section Lycopersicon) comprise close relatives of the cultivated tomato and exhibit variable levels of postzygotic isolation among pairs of taxa (Rick and Lamm 1955; Rick 1979, 1986; Peralta et al. 2008). In classical studies, C.M. Rick found very high proportions of hybrid seed failure in reciprocal crosses of Solanum peruvianum and S. chilense (Rick and Lamm 1955; Rick 1979, 1986), two closely related species with partly overlapping ranges in northern Chile and southwestern Peru. A few F1 hybrid seeds “escaped” abortion and after germination proved to be viable hybrid plants (Rick and Lamm 1955; Rick 1986), suggesting that the normal failure of such seeds can be attributed to disturbed endosperm–embryo interactions rather than early-acting incompatibilities between the two parental genomes in F1 embryos. This interpretation is strengthened by the success of F1 embryo culture in several interspecific crosses in the tomato clade, i.e., aborting embryos can be rescued by excising them from the seed and culturing them in vitro (Brink and Cooper 1947; Rick and Lamm 1955; Rick 1979).
Motivated by their evolutionarily interesting suite of biological properties, S. peruvianum and S. chilense have been the object of recent multilocus studies focusing on demography and speciation history, molecular evolution, and abiotic adaptation (Städler et al. 2005, 2008; Tellier et al. 2011; Böndel et al. 2015). Here, we provide evidence for genomic imprinting based on reciprocal crosses within both Solanum species, using endosperm tissue isolated by laser-assisted microdissection (LAM) as the source of RNA. Importantly, we characterized ASE in failing endosperm from the reciprocal hybrid crosses. While our work should not be considered a comprehensive study of imprinting in these taxa due to possible intraspecific variation in imprinting (Waters et al. 2013; Pignatta et al. 2014), it provides the first near-unbiased perspective on genome-wide changes in maternal:paternal transcript proportions in failing hybrid endosperm in flowering plants.
Results and Discussion
Phenotypic Asymmetry of Inviable Hybrid Seeds
Interspecific crosses between the two wild tomato accessions, one representing S. peruvianum and one S. chilense, respectively, resulted in almost complete seed failure, as expected from earlier studies (Rick and Lamm 1955; Rick 1979). Consequently, we recovered almost no viable seeds in interspecific crosses using the populations chosen for molecular analyses, in strong contrast to within-population crosses that yielded high proportions of viable seeds (fig. 1A). Importantly, the number of seeds per fruit was not significantly different between any of the six cross-type comparisons (Wilcoxon rank-sum tests, all P >0.05), emphasizing that there was no discernable post-mating, prezygotic interspecific barrier under the imposed noncompetitive pollination conditions (fig. 1A).
Fig. 1.
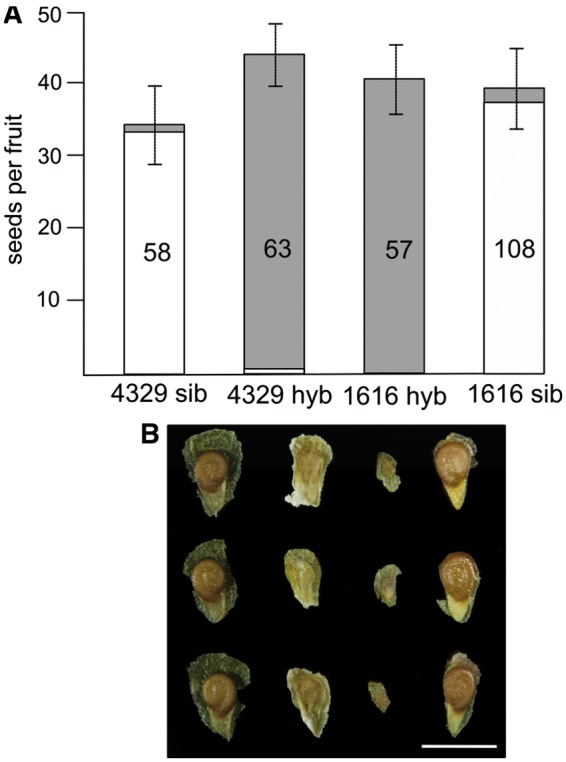
Comparisons of seed set and seed morphology for within-population vs. interspecific crosses. (A) From left to right, the bars quantify the average number of seeds per fruit in crosses among “sib” plants of LA4329 (S. chilense), hybrid crosses with LA1616 plants (S. peruvianum) as pollen donors, hybrid crosses with LA4329 plants as pollen donors, and among “sib” plants of LA1616. White bar proportions correspond to viable seeds, whereas gray proportions indicate shrivelled, empty seeds. Numbers within bars give the number of fruits analyzed per cross type, and error bars indicate standard deviation across fruits. (B) Representative seeds for each of the four cross types in (A), obtained 60 days after pollination. Note the coiled embryos in normal seeds from within-population crosses (4329sib and 1616sib) and the flat, inviable seeds (aborted embryos) from hybrid crosses, which are much smaller when LA4329 acts as pollen donor. Scale bar, 3 mm.
While almost exclusively yielding inviable seeds, the reciprocal interspecific crosses exhibited distinctly different seed size, with hybrid seeds being markedly smaller on S. peruvianum maternal plants (fig. 1B). Highly stable differences in average seed size have previously been documented for reciprocal interspecific and interploidy crosses (Scott et al. 1998; Lu et al. 2012; Willi 2013). Such differences have widely been attributed to parental genome conflict (Haig and Westoby 1991; Brandvain and Haig 2005), with relatively larger seeds exhibiting a “paternal excess” phenotype (thought to be due to enhanced nutrient-acquiring ability of the endosperm) in contrast to relatively smaller seeds exhibiting a “maternal excess” phenotype (thought to reflect balanced distribution of maternal resources amongst all seeds). Regardless whether interspecific differences in levels of parental conflict underlie the observed difference in hybrid seed size, the characteristic small-seed phenotype produced by the S. peruvianum × S. chilense cross (fig. 1B) may (or may not) be functionally linked to its larger increase in the maternal proportion of endosperm transcripts as revealed by our ASE analyses (see below).
Genomic Imprinting in the Endosperm
Two sets of reciprocal crosses were conducted within species to assess genomic imprinting in the “normal” endosperm of wild tomatoes. Deep sequencing of RNA obtained from endosperms isolated by LAM at 14 Days After Pollination (DAP) yielded a large number of sequencing reads (48–74 million across two replicate libraries) for each of the four genotypes. An average of 83.7% of the reads could be uniquely mapped to the gold-standard tomato reference genome (The Tomato Genome Consortium 2012; for details see supplementary table S1, Supplementary Material online). In contrast to A. thaliana, our focal plants are obligate outcrossers and both species harbor fairly high levels of nucleotide diversity (Städler et al. 2005, 2008; Tellier et al. 2011; Böndel et al. 2015); thus, we expected the majority of sequence differences between the parents to occur in a heterozygous state. To make use of information both from homo- and heterozygous parental differences, we implemented a novel approach to integrate differences between the parents of the type CC:AC (i.e., where one parent is a homozygote and the other a heterozygote carrying another nucleotide; for details see “Materials and Methods” section and supplementary fig. S1, Supplementary Material online). All sites with a minimum coverage of 10 reads were used to obtain a transcript-specific estimate of the maternal expression proportion. After filtering, ASE was successfully estimated for a total of 8,229 genes in S. peruvianum LA1616 and 2,560 genes in S. chilense LA4329, reflecting the higher number of parental sequence polymorphisms in the S. peruvianum cross (table 1).
Table 1.
Summary of Data Underlying the Estimation of Maternal Proportions in Endosperm-Expressed Genes in Three Reciprocal Cross Types.
| Peruvianum cross | Hybrid cross | Chilense cross | |
|---|---|---|---|
| Statistic | (1616A ↔ 1616J) | (1616A ↔ 4329B) | (4329B ↔ 4329K) |
| Endosperm-expressed genes with SNPs in reciprocal cross | 8,654 | 4,289 | 2,646 |
| Alternative homozygous sites | 6,408 | 9,225 | 1,263 |
| Heterozygous sites (e.g., CC:AC) | 41,610 | 13,821 | 7,421 |
| Mean no. of SNPs per gene | 5.55 | 5.37 | 3.28 |
| Informative genes after filtering | 8,229 | 4,111 | 2,560 |
| Candidate MEGs | 351 | (570) | 40 |
| Candidate PEGs | 172 | (6) | 70 |
| Median maternal proportion (all SNPs) | 0.646 ↔ 0.684 | 0.843 ↔ 0.701 | 0.630 ↔ 0.643 |
| Median maternal proportion (only homozygous SNPs) | 0.645 ↔ 0.696 | 0.847 ↔ 0.708 | 0.645 ↔ 0.645 |
| Median maternal proportion (only heterozygous SNPs) | 0.649 ↔ 0.683 | 0.843 ↔ 0.702 | 0.625 ↔ 0.642 |
Note.—Median maternal proportions were computed across all “relevant” genes per cross type and parental SNP category. Due to the global (asymmetric) misregulation of endosperm expression in the hybrid cross, numbers of candidate MEGs and PEGs for this cross are given in parentheses.
The endosperm has a genomic composition of 2:1 maternal:paternal haploid genomes, such that we expect the “normal” proportion of maternal expression to be approximately 0.67. Our empirical data broadly reflect these expectations, but in line with equivalent data from other plant studies (Waters et al. 2013; Pignatta et al. 2014), there was a lot of scatter in the distribution of maternal proportion estimates across individual transcripts (fig. 2A and supplementary fig. S2, Supplementary Material online). Candidate imprinted Maternally Expressed Genes (MEGs) exceed our imposed threshold of 0.833 maternal proportion in both cross directions, and candidate imprinted Paternally Expressed Genes (PEGs) are those with <0.333 maternal proportion in both cross directions. For S. peruvianum LA1616, we identified 351 candidate MEGs (fig. 2A, upper right) and 172 candidate PEGs (fig. 2A, lower left). The corresponding numbers for the less informative S. chilense LA4329 are 40 candidate MEGs and 70 candidate PEGs (table 1 and supplementary fig. S2, Supplementary Material online). Details regarding Single Nucleotide Polymorphisms (SNPs), maternal proportions, and functional annotation of candidate MEGs and PEGs identified in each species can be found in supplementary table S2, Supplementary Material online.
Fig. 2.

Global patterns of parent-of-origin-specific maternal proportions in within-population vs. hybrid endosperm. (A) Endosperm maternal proportions for 8,229 genes in the reciprocal S. peruvianum crosses 1616A × 1616J (x axis) and 1616J × 1616A (y axis). Candidate MEGs have maternal proportions >0.833 in both directions of the cross (upper right sector), and candidate PEGs have maternal proportions <0.333 in both directions of the cross (lower left sector). Three classes of genes are distinguished by color: ASE information from only heterozygous SNPs (gray dots, n = 5,951), ASE information from only homozygous SNPs (orange dots, n = 383), and ASE information from both types of SNPs (green dots, n = 1,895). The 67 “complete” MEGs (mat. prop. >0.99) in the upper right corner cannot be adequately distinguished visually. (B) Endosperm maternal proportions for all 4,111 informative genes in the reciprocal hybrid crosses between 1616A (S. peruvianum) and 4329B (S. chilense). Note the marked shift toward higher maternal proportions in these hybrid endosperms, especially for 1616A in the maternal role (x axis, from median maternal proportion 0.646 in A to 0.843 in B).
Overall expression levels were lower for genes with evidence for ASE in one or both cross directions, compared to “normal” genes with biallelic expression (supplementary fig. S3, Supplementary Material online). However, median coverage per SNP was fairly high even for MEGs (median coverage MEGs, 29.4; PEGs, 59.2; all other genes, 79.3), the group of genes with lowest overall expression in both species, suggesting that our designations of candidate imprinted genes are unlikely to be driven by unusually low expression or “noisy” data. While speculative, systematically lower expression might be expected for genes experiencing epigenetically silenced parental copies, in addition to their perhaps attenuated expression as seed development proceeds in taxa with non-persisting endosperm.
Importantly, when analyzed separately using either homo- or heterozygous parental SNPs, the inferred maternal proportions within a given cross are very close to each other for all six cross directions (table 1). The use of both homo- and heterozygous parental SNPs implies that we can partition our ASE inferences among three “classes” of genes: (i) genes with exclusively homozygous SNPs, (ii) genes with exclusively heterozygous SNPs, and (iii) genes with both types of parental SNPs. The proportion of genes with ASE information from only heterozygous SNPs is highest in the within-species crosses (fig. 2A), while genes with information from both types of SNPs have larger representation in the hybrid cross (table 2). Invariably, our estimates of maternal proportions are very close to each other within a given cross, irrespective of the class of genes. Similarly, the proportions of candidate MEGs and PEGs identified among classes of genes broadly reflect their different proportions in the S. peruvianum, the S. chilense, and the hybrid cross, respectively (table 2). We thus conclude that our novel approach to incorporate information from heterozygous parental SNPs does not appear to bias our ASE inferences, which bodes well for future applications of this approach in other outcrossing, highly heterozygous plant species.
Table 2.
Partitioning of Total Evidence into Three Classes of Genes Based on Parental SNP Differences for the Three Reciprocal Crosses.
| Cross type and parameters | Homozygous SNPs only | Heterozygous SNPs only | Both types of SNPs |
|---|---|---|---|
| 1. S. peruvianum cross (n genes) | 383 | 5,951 | 1,895 |
| # candidate MEGs | 28 | 265 | 58 |
| # candidate PEGs | 8 | 136 | 28 |
| mat. prop. 1616A | 0.649 | 0.643 | 0.651 |
| mat. prop. 1616J | 0.702 | 0.682 | 0.687 |
| 2. S. chilense cross (n genes) | 278 | 2,078 | 204 |
| # candidate MEGs | 2 | 37 | 1 |
| # candidate PEGs | 7 | 57 | 6 |
| mat. prop. 4329B | 0.642 | 0.625 | 0.645 |
| mat. prop. 4329K | 0.644 | 0.639 | 0.655 |
| 3. Hybrid cross (n genes) | 995 | 1,265 | 1,851 |
| # candidate MEGs | 153 | 160 | 257 |
| # candidate PEGs | 2 | 4 | 0 |
| mat. prop. 1616A | 0.842 | 0.845 | 0.848 |
| mat. prop. 4329B | 0.706 | 0.688 | 0.708 |
Note.—“mat. prop.”, median maternal proportions for a given cross direction and class of genes.
Our current approach limits ASE inference to those endosperm-expressed genes that have at least some expression in flower buds. In other words, truly endosperm-specific genes are recalcitrant to our analyses because we do not have parental SNP information for them. Work on maize has found preferential expression of many MEGs in endosperm, whereas only 26% of PEGs showed this pattern (Waters et al. 2013). Comparable data in Arabidopsis appear to show that most endosperm-imprinted genes are also expressed in other stages of plant development (Pignatta et al. 2014). We consider this technical constraint to be modest, given our fairly high numbers of candidate imprinted genes and our main focus on the endosperm ASE differences between within-species and hybrid crosses. In any case, a future comprehensive assessment of imprinting in wild tomatoes would need to employ multiple reciprocal crosses within species, given the phenomenon of within-species variable imprinting status uncovered in maize and Arabidopsis.
Imprinted Genes’ Functional Roles and their Evolutionary Maintenance
Given our main focus on hybrid seed failure and its molecular correlates, here we highlight only novel aspects not predictable from previous large-scale endosperm RNA-Seq studies in plant model systems (Gehring et al. 2011; Hsieh et al. 2011; Luo et al. 2011; Wolff et al. 2011; Zhang et al. 2011; Rodrigues et al. 2013; Waters et al. 2013; Pignatta et al. 2014). Of note, a total of 35 genes potentially encoding subunits of Skp1–Cullin–F-box (SCF) protein complexes are among our candidate imprinted genes (9 MEGs and 26 PEGs; blue highlight in supplementary table S2, Supplementary Material online). Functioning of the SCF complex relies on proper coupling of its core proteins and cofactors. If genes coding for components of the SCF complex acquired imprinted expression for their role in modulating seed development as hypothesized by Dumbliauskas et al. (2011), the imprinted expression of such a high number of genes in wild tomatoes may have evolved under selection for coadaptation of gene expression. This scenario posits natural selection to favor the evolution of genomic imprinting because it facilitates closer coordination of coexpression of interacting gene products coded by different loci (Wolf 2013). Second, 30 nuclear-encoded chloroplast genes were found to be maternally expressed (green highlight in supplementary table S2, Supplementary Material online). Finding such a high number of nuclear-encoded genes whose protein products work in concert with chloroplast-encoded subunits as candidate MEGs is unprecedented in previous studies of imprinting in angiosperms (see references above). Interestingly, our results fit expectations under Wolf’s (2009) cytonuclear interactions model where nuclear-encoded organelle genes evolved to be maternally imprinted owing to coadaptation with organelle metabolism. Thirty of the 32 candidate imprinted nuclear-encoded chloroplast genes in wild tomatoes are MEGs, consistent with this model for imprinting due to cytonuclear epistasis of nuclear and chloroplast genomes (Wolf 2009).
Due to the much lower number of genes that could be assessed for ASE in the S. chilense cross, the seemingly low number of candidate imprinted genes that are shared among both species (23 PEGs and 13 MEGs) has to be evaluated carefully. When taking into account only those candidate genes that could be evaluated in both species, we find that between 19 and 32% of them are shared among species for the functional groups PEGs, MEGs, nuclear-encoded chloroplast genes, and SCF protein complex genes (table 3). Moreover, there is evidence that some of the “unshared” candidate genes, i.e., those that did not meet our strict thresholds in the other species, also show biased expression in the same direction, suggesting the possibility that we may have underestimated the true proportion of shared candidate genes (table 3). In any event, our data seem broadly consistent with published data from model systems suggesting a more limited conservation of imprinting in phylogenetically wide comparisons such as maize–rice and maize–Arabidopsis (Waters et al. 2013), and much higher levels of shared imprinting status among different genotypes of the same species (Waters et al. 2013; Pignatta et al. 2014).
Table 3.
Proportions of Shared Imprinted Genes among Both Species, and Evidence for Near-Biased Expression of Non-Candidate Genes in Either Species.
| Group of genes | # PEGs/MEGs shared among both species | ASE data for both species (n genes) | % PEGs/MEGs shared | Median mat. prop. of peru candidate genes in chil cross (both directions, n genes) | Median mat. prop. of chil candidate genes in peru cross (both directions, n genes) |
|---|---|---|---|---|---|
| Candidate PEGs (n = 219) | 23 | 72 | 31.9 | 0.059 (n = 34) | 0.298 (n = 61) |
| Candidate MEGs (n = 378) | 13 | 68 | 19.1 | 0.834 (n = 45) | 0.829 (n = 36) |
| Nuclear-encoded cp genes (30 MEGs, 2 PEGs) | 4 | 13 | 30.8 | 0.883 (n = 10) | 0.965 (n = 7) |
| SCF protein complex genes (9 MEGs, 26 PEGs) | 2 | 9 | 22.2 | – | – |
Note.—Median maternal proportions are not shown for the last row due to the co-occurrence of candidate MEGs and PEGs.
Genome-Wide Increase of Maternal Transcript Proportions in Hybrid Endosperms
The reciprocal hybrid cross yielded 4,111 transcripts that could be assessed for their maternal:paternal expression proportions (table 1). Intriguingly, overall maternal transcript proportions were elevated in both directions of the hybrid cross (fig. 2B). However, this trend was more pronounced for hybrid seeds developing on the S. peruvianum 1616A plant, exhibiting an increase in median maternal proportion from 0.646 in the within-population cross to 0.843 in the hybrid cross. The shift in median maternal transcript proportion on the S. chilense 4329B plant was more modest, from 0.630 in the within-population cross to 0.701 in the hybrid cross (table 1).
These systematic shifts toward higher maternal transcript proportions in hybrid endosperms led us to assess whether expression-level differences might also be biased toward the respective seed parent, as has recently been found in rosette leaf tissue of inter-population hybrids in A. lyrata (Videvall et al. 2016). Briefly, for genes found to be differentially expressed between the two species, we compared hybrid endosperm expression levels with those of normal (within-population) endosperms from the same mothers. Rather than “copying” the maternal biases in hybrid ASE, endosperm expression levels in both hybrid cross directions were closer to those of the S. peruvianum within-population expression distribution (supplementary fig. S4, Supplementary Material online). While the molecular underpinnings and a more extensive analysis of these patterns are beyond the scope of this article, they may signal a preponderance of S. peruvianum expression dominance in both parental roles.
To explore the scale and direction of hybrid endosperm ASE changes at the gene level, we assessed maternal proportions in normal and hybrid endosperms for all genes with parental sequence differences in both within-population and hybrid crosses (S. peruvianum: 3,647 genes; S. chilense: 1,856 genes). Maternal proportions in normal endosperms are compared to those in hybrid endosperms from the same seed parents in fig. 3 (S. peruvianum) and supplementary fig. S5 (S. chilense), Supplementary Material online. The marked shift of the large cluster of “normal” genes toward higher maternal proportions in hybrid endosperms is perhaps the most obvious feature, particularly in S. peruvianum (fig. 3B). In addition, those candidate PEGs in S. peruvianum that could be assessed in the hybrid cross (n = 73), with few exceptions, show marked expression from the maternal genome, rendering them non-PEGs in the hybrid background. While the general pattern in hybrid endosperms shows elevated maternal transcript proportions, candidate MEGs generally do not show an average increase in maternal proportions but tend to exhibit a slight decrease of maternal transcript proportions in hybrid endosperms (fig. 3B and supplementary fig. S5B, Supplementary Material online).
Fig. 3.

Changes in maternal proportions for S. peruvianum genes with ASE information from both within-species and the independent hybrid crosses. (A) Endosperm maternal proportions for 3,647 genes in the reciprocal S. peruvianum crosses 1616A × 1616J (x axis) and 1616J × 1616A (y axis); the same cross as in fig. 2A, but restricted to those genes with independent ASE information from the hybrid cross. Red and blue dots mark candidate PEGs and MEGs, respectively, and all other genes are marked as open circles. (B) Comparison of endosperm maternal proportions for the same 3,647 genes for 1616A in the within-species cross maternal role (x axis; same data as in A) vs. 1616A in the hybrid cross maternal role (y axis, 1616A × 4329B). Red and blue dots identify (within-species) candidate PEGs and MEGs, respectively. Note the marked shift of the “normal” genes (0.333< mat. prop. <0.833) toward higher mean maternal proportions in the hybrid cross, and the particularly large shift of many PEGs.
Assessed quantitatively at the “candidate-group” level, the median upward shift in maternal proportions is considerably smaller for candidate PEGs in S. chilense hybrid endosperm (Δ PEGs = 0.276) than in S. peruvianum hybrid endosperm (Δ PEGs = 0.532). The corresponding shifts for candidate imprinted MEGs are toward slightly lower maternal proportions in hybrid endosperms (S. chilense, median Δ MEGs = –0.036; S. peruvianum, median Δ MEGs = –0.018). These results suggest that, at PEG loci, maternal alleles that are normally repressed become de-repressed in hybrid tomato endosperm. However, as this perturbation of normal transcriptional regulation apparently is not confined to imprinted genes but affects the entire spectrum of maternal expression proportions, we conclude that the regulatory machinery of transcription appears to be fundamentally compromised in hybrid endosperm.
Perturbation of Candidate MEGs and PEGs in Hybrid Endosperms
We further quantified changes in the parental transcript proportions of candidate imprinted genes, separately for both species. As a consequence (or more precisely, an epiphenomenon) of the genome-wide shift toward higher hybrid maternal transcript proportions, the paternal expression bias of many candidate PEGs was eliminated in hybrid endosperm (supplementary figs. S6A, C, Supplementary Material online). Furthermore, this shift toward higher maternal proportions in candidate PEGs is larger in the S. peruvianum LA1616 data (median Δ = 0.532) than in the S. chilense LA4329 data (median Δ = 0.276), consistent with the global trend of stronger maternal allelic bias in the hybrid endosperm of S. peruvianum (table 1; fig. 3B). About 47% (18/38) of the S. chilense candidate PEGs remain PEGs in the hybrid cross with 1616A as the male parent (supplementary fig. S6C [shift along the x axis], Supplementary Material online). In keeping with the generally smaller shifts in the “high” range of maternal proportion, most candidate MEGs remain MEGs in hybrid endosperm of both species (86% in S. peruvianum [124/145] and 62% in S. chilense [13/21]; supplementary figs. S6B, D, Supplementary Material online).
Finally, we evaluated differences in maternal transcript proportions between within-population and hybrid endosperms for the smaller group of genes with evidence for being imprinted in both Solanum species (so-called “shared” imprinted genes; fig. 4). The average shift for candidate PEGs is less severe for S. chilense in the maternal role in this set of genes, but the overall pattern mirrors that of the larger set of species-specific candidate PEGs (supplementary fig. S6, Supplementary Material online), in particular the sweeping disruption of paternal expression bias in S. peruvianum. Likewise, the shared MEGs show rather slight departures from their corresponding estimates in intra-population endosperms, with most of them retaining their MEG status in hybrid endosperms of both species (fig. 4). This pattern is consistent with that in the larger set of species-specific candidate MEGs (supplementary fig. S6, Supplementary Material online).
Fig. 4.
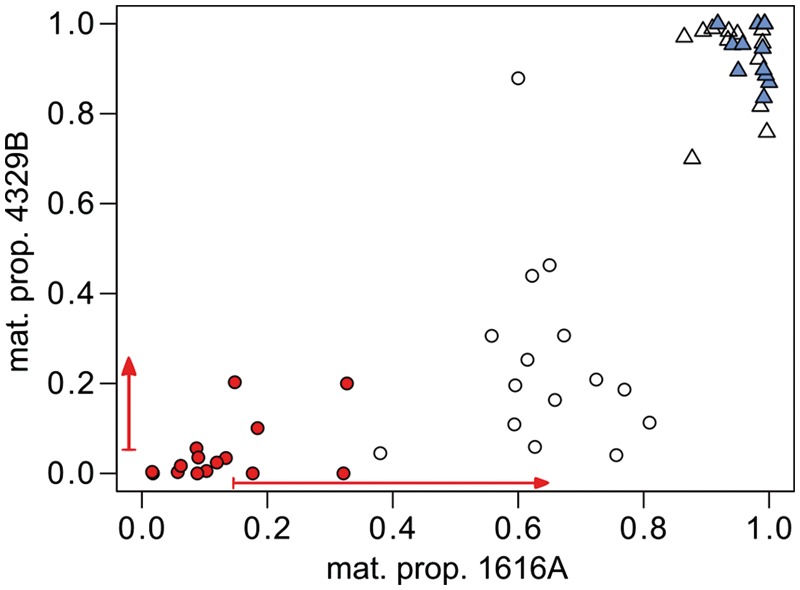
Shift in maternal proportion between within-population and hybrid endosperm for 15 candidate PEGs and 12 candidate MEGs conserved between S. peruvianum (LA1616) and S. chilense (LA4329). For within-population crosses, PEGs are indicated as red dots and MEGs as blue triangles, and their respective maternal proportion in hybrid endosperm is shown with open symbols. The average shift for PEGs (red arrows along both axes) is less pronounced for S. chilense in the maternal role.
Recent work on hybridization between A. thaliana and A. arenosa has uncovered similar perturbations in maternal transcript proportions for specific genes, with several previously known PEGs exhibiting normal-to-high expression from the maternal allele in hybrid seeds, among other changes (Burkart-Waco et al. 2015). These similarities may reflect shared responses to hybridization in both Brassicaceae and Solanaceae, possibly due to equivalent molecular mechanisms involved in the establishment of imprinted expression as well as its misregulation upon interspecific hybridization. Burkart-Waco et al. (2015) argued that Polycomb Repressive Complex 2 may be responsible for this behavior, due to its known role in regulating some imprinted loci in Arabidopsis (Köhler et al. 2005; Jullien et al. 2006). Another transcriptional regulatory mechanism in the developing seed is RNA-directed DNA methylation (RdDM), which regulates imprinting at specific loci expressed in the endosperm and is mediated by siRNAs (Vu et al. 2013). While RdDM has been shown to regulate imprinting at a handful of Arabidopsis loci, we consider this mechanism improbable to account for the transcriptome-wide trend we have uncovered; such a scenario would imply that all genes undergoing shifts in maternal expression in hybrid endosperm have TEs in their respective genomic neighborhoods that are targeted by RdDM. Alternatively, the RdDM pathway may also regulate non-TE targets in the endosperm as it was found in young Arabidopsis embryos, where the lack of maternal components of the RdDM pathway leads to de-repression of paternal alleles at many loci (Autran et al. 2011). Given the general, genome-wide shift of maternal transcript proportions, however, it is more likely that general functions in transcription are affected. We speculate that the composition of multimeric complexes involved in transcription and its regulation is optimized for its respective genome. In hybrid endosperms, more of these subunits are encoded by the maternal genome, leading to the formation of a higher proportion of multimeric complexes that are composed of mostly maternal isoforms, thus transcribing the maternal genome more efficiently.
Conclusions
Our interspecific crosses have uncovered unprecedented, systematic shifts of maternal transcript proportions in (failing) hybrid endosperms of two wild tomato species. Related work on Arabidopsis was constrained by targeting known candidate imprinted genes, particularly PEGs (Burkart-Waco et al. 2015; Wolff et al. 2015), and could not assess the breadth of transcriptional changes because RNA was isolated from entire seeds in these studies. The shifts in maternal transcript proportions documented here clearly affect a majority of endosperm-expressed genes and are neither restricted to imprinted genes, nor solely PEGs or MEGs. Nevertheless, our data reveal that the average shift toward higher maternal proportion in candidate PEGs is sufficient to eliminate their “normal” paternal expression bias in the hybrid yielding very small seeds (S. peruvianum as female parent). In light of recent evidence from both Arabidopsis (Wolff et al. 2015) and Mimulus (Garner et al. 2016), it is thus plausible that misregulation of some of these PEGs might contribute to the hybrid seed failure phenotype. These intriguing patterns notwithstanding, which of the assessed changed expression properties (if any) in hybrid endosperm contribute to interspecific seed failure in these wild tomatoes cannot be determined with the data at hand. Future studies will encompass more crosses with greater numbers of parental nucleotide differences, evaluating expression-level changes, and work with a more mechanistic focus to investigate the molecular basis of the genome-wide shift in maternal transcript proportions.
Materials and Methods
Plant Material, Crossings, and Seed Evaluation
All seeds were obtained from the C.M. Rick Tomato Genetics Resource Center at U.C. Davis (http://tgrc.ucdavis.edu, last accessed 16 June 2016). Representing Solanum peruvianum, we used seeds from accession LA1616 (Lima, Peru), and representing S. chilense, we used seeds from accession LA4329 (Antofagasta, Chile). Both accessions are strictly self-incompatible, eliminating the need to emasculate flowers before applying pollen from a different plant. Several plants per accession were grown under standard, insect-free greenhouse conditions. Freshly opened flowers were used both as sources and recipients of pollen from other plants, either from the same accession (i.e., within-LA1616 and within-LA4329 crosses) or among accessions (i.e., heterospecific crosses). Pollinations were performed by manually collecting pollen from the paternal plant and immediately transferring it to the stigmas of the maternal plant. Stigmas were completely covered with pollen to secure enough seed production. All hand-pollinated flowers were individually marked and ripe fruits were collected 60 DAP. All resulting seeds (viable and non-viable) were counted and seed viability was determined by the presence of a fully developed embryo that had reached a coiled stage, irrespective of seed size. Statistical analyses and plotting were performed using the R software, version 3.2.1 (R Development Core Team 2014).
Crossing Design for Endosperm Transcriptome Sequencing
For the molecular component of this study, we used four of the plants from among the larger cohort, referred to as 1616A, 1616J, 4329B, and 4329K. We analyzed three different parental combinations: the within-species S. peruvianum case with plants 1616A and 1616J as parents, the within-species S. chilense case with plants 4329B and 4329K as parents, and the between-species case with plants 1616A and 4329B as parents. For each of these four parental plants, transcriptomes were obtained by sequencing RNA from flower buds. Young flower bud tissue was collected in liquid nitrogen and RNA extracted with the RNAeasy mini RNA isolation kit (Qiagen), according to the manufacturer’s instructions. Libraries were prepared with the Illumina TrueSeq RNA Sample Preparation Kit v2 following the manufacturer’s instructions and were sequenced on the Illumina HiSeq2000 platform, generating 150-bp paired-end reads.
Using transcriptome data as the source of parental SNP calls implies the potential for mis-scoring genotypes, such as under cis-regulated differences in allelic expression levels in heterozygous plants. We have systematically explored the expected consequences of mis-scoring a parental true heterozygous position as a homozygote under the range of possible expression behavior in the endosperm (i.e., “normal” 2m:1p expectation in the endosperm with no allelic bias, or assuming ASE like in the parental flower buds). Under the latter endosperm expression pattern, our inference pipeline (see below) would uniformly return expectations of 2m:1p for all three possible cross combinations involving “apparent” (i.e., wrongly scored) homozygotes. If endosperm expression were normal and not biased like in parental bud tissue, we would expect to see evidence of plant-specific expression, i.e., biased expression from one plant in both cross directions (supplementary table S3, Supplementary Material online). We thus conclude that potentially mis-scoring parental genotypes due to allelic biases in flower bud expression level is not expected to generate false positive candidate imprinted genes under any realistic circumstances.
We chose to base our exploratory work on genomic imprinting on intra-population crosses (at the possible cost of having lower power to distinguish maternal from paternal reads due to fewer SNPs between parents) to minimize the incidence of failed seed development observed in many intraspecific, inter-population crosses (our unpublished data). Several months before the beginning of the experiment, the four focal plants were transferred from the greenhouse to climate-controlled chambers. The conditions in the climate chambers were 12 hours of light (18 Klux) at 22 °C with 50% relative humidity and 12 h of darkness (0 Klux) at 18 °C with 60% relative humidity. For each of the three cross types, reciprocal hand pollinations were performed and developing fruits were collected on each plant for each cross type. Based on previous observations on seed development in Solanum (Beamish 1955; Dnyansagar and Cooper 1960; Pacini and Sarfatti 1978; Briggs 1993) and our histological analyses of seed development (unpublished), we chose an early globular embryo stage to collect the material for library preparation. We collected fruits 14 DAP, always in late afternoon. This time point was chosen because it was early enough to distinguish the developing embryo from the surrounding endosperm tissue, while the latter was large enough to extract RNA in the quantities needed for library preparation. For each plant and cross type, two separate RNA libraries were prepared from laser-captured endosperm tissue, for a total of 12 endosperm libraries.
Laser Microdissection, RNA Extraction, and Library Preparation
For analysis of the endosperm transcriptomes, the collected fruits were immediately placed in a fixation solution (9:1 ethanol:acetic acid). All solutions were maintained cold at <4 °C from fixation to transfer of the samples into the embedding machine (see below). The samples were swiftly transferred to a refrigerator and remained in the fixing solution for a minimum of 24 h and a maximum of 48 h. During this fixing step, the samples were submitted to a vacuum for at least 30 min to allow infiltration of the fixative. The samples were then transferred to a cold (<4 °C) solution of 90% ethanol and shortly thereafter placed on a Leica embedding machine for paraffin infiltration (settings as in Florez-Rueda et al. 2016). Prior to LAM, 8-µm sections were prepared from the samples embedded in paraffin blocks with a RM2145 Leica microtome (Leica Microsystems GmbH, Wetzlar, Germany). The sections were mounted on nuclease-free membrane metal-frame slides (MicroDissect GmbH, Wetzlar, Germany) using water. Slides were dried on a heating table at 42 °C for a maximum of 2 h. The samples were deparaffinized in xylol at room temperature in two 15-min washes.
To isolate endosperm tissue for RNA sequencing we followed the protocol described in Florez-Rueda et al. (2016). In brief, LAM was performed using a CellCut Plus device (MMI Molecular Machines & Industries AG, Glattbrugg, Switzerland), carefully separating the endosperm from the embryo and surrounding sporophytic (i.e., maternal) seed coat tissues. Endosperm tissue was collected using MMI isolation caps and RNA extraction was performed immediately or within 24 h; in the latter case, the caps were stored at –80 °C prior to extraction. RNA was extracted using the Applied Biosystems® Arcturus®PicoPure® RNA Isolation Kit (ref. KIT0204) according to the manufacturer’s instructions. The quality and quantity of total RNA was assessed with Agilent Bioanalyzer Pico Chips. RNA that showed clear ribosomal peaks was used for library preparation. We used a minimum of 82 ng RNA for preparing each library, an amount reached by pooling separate extracts. For each library, 200–700 sections of endosperm were isolated, representing several fruits and at least two independent pollination events. Sequencing libraries were prepared with the Illumina TruSeq RNA Sample Preparation Kits v2 following the manufacturer’s instructions.
Sequencing, Read Mapping, and Gene Expression Analyses
The 12 endosperm-derived libraries were sequenced on three lanes of the Illumina HiSeq2000 platform at the Functional Genomics Center Zurich (www.fgcz.ch, last accessed 22 June 2016), generating 100-bp single-end reads. The quality of each library was assessed using the FastQC program (www.bioinformatics.babraham.ac.uk/projects/fastqc, last accessed 15 May 2014). Adapters were removed from the reads with the Cutadapt program (Martin 2011) and quality filtering was performed with the ConDeTri program (Smeds and Künstner 2011) using a minimum quality threshold of 20. The tuxedo pipeline encompassing TopHat, Cuffdiff, and Cufflinks (Trapnell et al. 2012) was used for the assembly of reads, mapping to the reference genome, and tests of differential expression. RNA-Seq quality-filtered reads for each library were mapped using TopHat version 1.4.0 (Trapnell et al. 2010). Mapping was done against the International Tomato Annotation Group (ITAG) Release 2.3 of the tomato reference genome sequence (The Tomato Genome Consortium 2012) on the SL2.40 genome build which includes the mitochondrial and chloroplast genome, downloaded via the SOL Genomics Network ftp site (ftp://ftp.sgn.cornell.edu/genomes/, last accessed 15 December 2015). A maximum of six mismatches was allowed between reads and the reference, and reads that mapped to more than one position in the genome were discarded. Furthermore, transcripts mapping to the organelle genomes were excluded for all analyses. For estimates of gene expression and tests of differential gene expression, two replicate libraries were used per cross direction. Specifically, we were interested in the expression behavior of genes in the hybrid crosses that were differentially expressed between 1616A (S. peruvianum) and 4329B (S. chilense) in their respective within-species crosses (i.e., changes in expression level with identical seed parents but different pollen parents). Cuffdiff uses a test statistic assessing the significance of the observed changes in Fragments Per Kilobase per Million mapped reads (FPKM), and the P-value of this test statistic is then corrected for false discovery rate (FDR) (Trapnell et al. 2013). Differentially expressed genes met our requirements of at least two-fold expression differences and >1 FPKM expression value. Pairwise comparisons were performed using Wilcoxon rank sum tests with Bonferroni P-value adjustments in R (R Development Core Team 2014).
SNP Calling and ASE Analyses
The mpileup command of SAMtools (Li et al. 2009) was used to call variant sites in the flower bud and endosperm transcriptomes. Popoolation2 (Kofler et al. 2011) was subsequently used to recover the allelic counts of major and minor alleles at each site, using a minimum read mapping quality threshold of 20. Using the allelic counts, we estimated the maternal:paternal transcript proportions separately for each SNP. Our approach is explained below and was implemented in Python 2.7 (scripts are available at https://github.com/anaflo/tomato, last accessed December 29, 2015).
Variant sites between the parental plants were recovered using the flower bud transcriptome sequencing. To make use of as many genotypic differences between the parents as possible, we developed a novel approach integrating information from both heterozygous and homozygous SNPs (table 1 and supplementary fig. S1, Supplementary Material online). The variant sites that can provide information for quantifying ASE are (1) the homozygote cases, that is, reciprocally different homozygous parental genotypes, and (2) the heterozygote cases, i.e., sites at which one parent is homozygous for a given base and the other parent is heterozygous.
Using our custom Python program, we calculated the proportion of the minor allele relative to the major allele for each polymorphic site and determined homozygous and heterozygous SNPs. First, sites with minor allelic proportion <5% were considered as homozygotes in order to avoid biases due to sequencing errors observed in next-generation sequencing. Only reciprocal homozygotes (i.e., different nucleotides between the parental individuals) were kept for the analyses. Then, heterozygote sites were defined using a conservative minor allelic proportion >40% in the heterozygous parent (supplementary fig. S1A, Supplementary Material online). For the heterozygous genotypes, the bases were categorized as “discriminant” or “fixed”. The “fixed” base corresponds to the base identity of the homozygous parent, whereas a “discriminant” base refers to the other base observed in the heterozygous parent. It is the discriminant base that allows the estimation of the endosperm ASE in the heterozygote cases (see below).
In the endosperm data, maternal proportion of overall transcription was calculated based on the parental identity of the alternative bases at each site, i.e., whether a given base in the endosperm data was of maternal or paternal origin (supplementary fig. S1B, Supplementary Material online). We note that each of the three reciprocal crosses has independent variant sites dependent on each parental genotype. The two replicated libraries per cross direction were pooled and only sites covered by a minimum of 10 reads were kept for analyses. For reciprocal homozygous sites in the parents, maternal proportion was calculated as the proportion of reads with the maternal base compared to the total number of reads for that site (supplementary fig. S1B, Supplementary Material online). For the heterozygous SNPs, we inferred the maternal proportion based on the discriminant base counts, as follows. For paternally heterozygous sites, maternal proportion (mat. prop.) was estimated by subtracting twice the observed proportion of the discriminant base (freq. discr.) from unity, i.e., mat. prop. = 1 – (2 * freq. discr.). Conversely, for maternally heterozygous sites, maternal proportion was estimated by doubling of the observed discriminant base frequency, i.e., mat. prop. = 2 * freq. discr. (supplementary fig. S1B, Supplementary Material online). This rationale assumes “fair” segregation of alleles at meiosis. Estimates of maternal proportions were constrained to fall within the range 0–1. For genes with multiple informative sites in a given cross, per-gene estimates of maternal proportions were calculated using a weighted average of the independent per-base estimates within genes, implying more weight for more highly covered polymorphic sites within a given gene (supplementary fig. S1B, Supplementary Material online).
Once a per-gene value was obtained for each direction of the cross, we used thresholds for maternal proportions in reciprocal crosses to call a given gene as potentially imprinted and consider moderately and strongly imprinted genes, as follows: moderate MEGs > 0.833, strong MEGs > 0.917, moderate PEGs < 0.333, and strong PEGs < 0.167 maternal proportion (supplementary fig. S1B, Supplementary Material online). These thresholds are reciprocally symmetric and consistent with the expectation of a 0.667 maternal proportion of gene expression in the triploid endosperm (2m:1p). Our “moderately” and “strongly” imprinted genes reflect greater than 2-fold and 4-fold deviations, respectively, from the expected maternal:paternal proportions. Furthermore, genes considered as candidate MEGs and PEGs exhibited significant departures from the expected 2m:1p ratio, as assessed by χ2 tests with False Discovery Rate corrections. While the above thresholds largely determine whether or not a given gene is considered as candidate imprinted, they have no bearing on the major finding of this study, i.e., the systematic shifts of maternal:paternal transcript proportions in hybrid endosperms (figs. 2–4 and supplementary figs. S5 and S6, Supplementary Material online).
Supplementary Material
Supplementary tables S1–S3 and figures S1–S6 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgments
We thank Maja Frei for meticulous plant care, Niklaus Zemp for advice on statistics, Mathias Scharmann for help with the Python script and photography for fig. 1B, and the Genetic Diversity Center Zurich (GDC) for use of its facilities. We also thank the C.M. Rick Tomato Genetics Resource Center at U.C. Davis for generously supplying seed samples, and four anonymous reviewers whose comments led to improvements in the final version of this paper. This work was supported by the Swiss National Science Foundation (31003A_130702 to T.S.), the “Staatssekretariat für Bildung und Forschung” (COST action FA0903 to A.S. and U.G.), funds from the University of Zurich (U.G.), and an ETH Research Grant (ETH-40 13-2 to T.S. and A.W.). The RNA-Seq reads have been deposited at the NCBI Sequence Read Archive under accession no. SRX1850236.
References
- Autran D, Baroux C, Raissig MT, Lenormand T, Wittig M, Grob S, Steimer A, Barann M, Klostermeier UC, Leblanc O, et al. 2011. Maternal epigenetic pathways control parental contributions to Arabidopsis early embryogenesis. Cell 145:707–719. [DOI] [PubMed] [Google Scholar]
- Beamish KI. 1955. Seed failure following hybridization between the hexaploid Solanum demissum and four diploid Solanum species. Am J Bot. 42:297–304. [Google Scholar]
- Birchler JA. 1993. Dosage analysis of maize endosperm development. Annu Rev Genet. 27:181–204. [DOI] [PubMed] [Google Scholar]
- Böndel KB, Lainer H, Nosenko T, Mboup M, Tellier A, Stephan W. 2015. North–south colonization associated with local adaptation of the wild tomato species Solanum chilense. Mol Biol Evol. 32:2932–2943. [DOI] [PubMed] [Google Scholar]
- Brandvain Y, Haig D. 2005. Divergent mating systems and parental conflict as a barrier to hybridization in flowering plants. Am Nat. 166:330–338. [DOI] [PubMed] [Google Scholar]
- Briggs CL. 1993. Endosperm development in Solanum nigrum L. Formation of the zone of separation and secretion. Ann Bot. 72:303–313. [Google Scholar]
- Brink RA, Cooper DC. 1947. The endosperm in seed development. Bot Rev. 13:423–541. [Google Scholar]
- Burkart-Waco D, Ngo K, Dilkes B, Josefsson C, Comai L. 2013. Early disruption of maternal-zygotic interaction and activation of defense-like responses in Arabidopsis interspecific crosses. Plant Cell 25:2037–2055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkart-Waco D, Ngo K, Lieberman M, Comai L. 2015. Perturbation of parentally biased gene expression during interspecific hybridization. PLoS ONE 10:e0117293.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushell C, Spielman M, Scott RJ. 2003. The basis of natural and artificial postzygotic hybridization barriers in Arabidopsis species. Plant Cell 15:1430–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castillo DM, Moyle LC. 2012. Evolutionary implications of mechanistic models of TE-mediated hybrid incompatibility. Intl J Evol Biol. 2012: article ID 698198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC, Brink RA. 1940. Somatoplastic sterility as a cause of seed failure after interspecific hybridization. Genetics 25:593–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper DC, Brink RA. 1945. Seed collapse following matings between diploid and tetraploid races of Lycopersicon pimpinellifolium. Genetics 30:376–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyne JA, Orr HA. 2004. Speciation. Sunderland (MA: ): Sinauer Associates. [Google Scholar]
- Crespi B, Nosil P. 2013. Conflictual speciation: species formation via genomic conflict. Trends Ecol Evol. 28:48–57. [DOI] [PubMed] [Google Scholar]
- Dnyansagar VR, Cooper DC. 1960. Development of the seed of Solanum phureja. Am J Bot. 47:176–186. [Google Scholar]
- Dumbliauskas E, Lechner E, Jaciubek M, Berr A, Pazhouhandeh M, Alioua M, Cognat V, Brukhin V, Koncz C, Grossniklaus U, et al. 2011. The Arabidopsis CUL4-DDB1 complex interacts with MSI1 and is required to maintain MEDEA parental imprinting. EMBO J. 30:731–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Florez-Rueda AM, Grossniklaus U, Schmidt A. 2016. Laser-assisted microdissection (LAM) as a tool for transcriptional profiling of individual cell types. J Vis Exp. 111:e53916.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garner AG, Kenney AM, Fishman L, Sweigart AL. 2016. Genetic loci with parent-of-origin effects cause hybrid seed lethality in crosses between Mimulus species. New Phytol. 211:319–331. [DOI] [PubMed] [Google Scholar]
- Gehring M. 2013. Genomic imprinting: insights from plants. Annu Rev Genet. 47:187–208. [DOI] [PubMed] [Google Scholar]
- Gehring M, Missirian V, Henikoff S. 2011. Genomic analysis of parent-of-origin allelic expression in Arabidopsis thaliana seeds. PLoS ONE 6:e23687.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez-Marcos JF, Pennington PD, Costa LM, Dickinson HG. 2003. Imprinting in the endosperm: a possible role in preventing wide hybridization. Phil Trans R Soc Lond B. 358:1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haig D. 2013. Kin conflict in seed development: an interdependent but fractious collective. Annu Rev Cell Dev Biol. 29:189–211. [DOI] [PubMed] [Google Scholar]
- Haig D, Westoby M. 1991. Genomic imprinting in endosperm: its effect on seed development in crosses between species, and between different ploidies of the same species, and its implications for the evolution of apomixis. Phil Trans R Soc Lond B. 333:1–13. [Google Scholar]
- Hsieh T-F, Shin J, Uzawa R, Silva P, Cohen S, Bauer MJ, Hashimoto M, Kirkbride RC, Harada JJ, Zilberman D, et al. 2011. Regulation of imprinted gene expression in Arabidopsis endosperm. Proc Natl Acad Sci U S A. 108:1755–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Köhler C. 2012. Evolution, function, and regulation of genomic imprinting in plant seed development. J Exp Bot. 63:4713–4722. [DOI] [PubMed] [Google Scholar]
- Josefsson C, Dilkes B, Comai L. 2006. Parent-dependent loss of gene silencing during interspecific hybridization. Curr Biol. 16:1322–1328. [DOI] [PubMed] [Google Scholar]
- Jullien PE, Katz A, Oliva M, Ohad N, Berger F. 2006. Polycomb group complexes self-regulate imprinting of the Polycomb group gene MEDEA in Arabidopsis. Curr Biol. 16:486–492. [DOI] [PubMed] [Google Scholar]
- Jullien PE, Berger F. 2010. Parental genome dosage imbalance deregulates imprinting in Arabidopsis. PLoS Genet. 6:e1000885.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kofler R, Pandey RV, Schlötterer C. 2011. PoPoolation2: identifying differentiation between populations using sequencing of pooled DNA samples (Pool-Seq). Bioinformatics 27:3435–3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhler C, Page DR, Gagliardini V, Grossniklaus U. 2005. The Arabidopsis thaliana MEDEA Polycomb group protein controls expression of PHERES1 by parental imprinting. Nat Genet. 37:28–30. [DOI] [PubMed] [Google Scholar]
- Kradolfer D, Wolff P, Jiang H, Siretskiy A, Köhler C. 2013. An imprinted gene underlies postzygotic reproductive isolation in Arabidopsis thaliana. Dev Cell. 26:525–535. [DOI] [PubMed] [Google Scholar]
- Lafon-Placette C, Köhler C. 2015. Epigenetic mechanisms of postzygotic reproductive isolation in plants. Curr Opin Plant Biol. 23:39–44. [DOI] [PubMed] [Google Scholar]
- Lester RN, Kang JH. 1998. Embryo and endosperm function and failure in Solanum species and hybrids. Ann Bot. 82:445–453. [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 genome project data processing subgroup. 2009. The sequence alignment/map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin BY. 1984. Ploidy barrier to endosperm development in maize. Genetics 107:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Zhang C, Baulcombe DC, Chen ZJ. 2012. Maternal siRNAs as regulators of parental genome imbalance and gene expression in endosperm of Arabidopsis seeds. Proc Natl Acad Sci U S A. 109:5529–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M, Taylor JM, Spriggs A, Zhang H, Wu X, Russell S, Singh M, Koltunow A. 2011. A genome-wide survey of imprinted genes in rice seeds reveals imprinting primarily occurs in the endosperm. PLoS Genet. 7:e1002125.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. 2011. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.J 17:10–12. [Google Scholar]
- Ng DW-K, Lu J, Chen ZJ. 2012. Big roles for small RNAs in polyploidy, hybrid vigor, and hybrid incompatibility. Curr Opin Plant Biol. 15:154–161. [DOI] [PubMed] [Google Scholar]
- Oneal E, Willis JH, Franks RG. 2016. Disruption of endosperm development is a major cause of hybrid seed inviability between Mimulus guttatus and Mimulus nudatus. New Phytol. 210:1107–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacini E, Sarfatti G. 1978. Reproductive calendar of Lycopersicon peruvianum Mill. Bull Soc Bot France. 125:295–299. [Google Scholar]
- Peralta IE, Spooner DM, Knapp S. 2008. Taxonomy of wild tomatoes and their relatives (Solanum sect. Lycopersicoides, sect. Juglandifolia, sect. Lycopersicon; Solanaceae). Syst Bot Monogr. 84:1–186. [Google Scholar]
- Pignatta D, Erdmann RM, Scheer E, Picard CL, Bell GW, Gehring M. 2014. Natural epigenetic polymorphisms lead to intraspecific variation in Arabidopsis gene imprinting. eLife 3:e03198.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pires ND, Grossniklaus U. 2014. Different yet similar: evolution of imprinting in flowering plants and mammals. F1000Prime Rep. 6:63.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raissig MT, Baroux C, Grossniklaus U. 2011. Regulation and flexibility of genomic imprinting during seed development. Plant Cell 23:16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebernig CA, Lafon-Placette C, Hatorangan MR, Slotte T, Köhler C. 2015. Non-reciprocal interspecies hybridization barriers in the Capsella genus are established in the endosperm. PLoS Genet 11:e1005295.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team. 2014. R: a language and environment for statistical computing. Vienna (Austria: ): R Foundation for Statistical Computing; [cited 16 June, 2016]. Available from: http://www.R–project.org/. [Google Scholar]
- Rick CM. 1979. Biosystematic studies in Lycopersicon and closely related species of Solanum In: Hawkes JG, Lester RN, Skelding AD, editors. The biology and taxonomy of the Solanaceae. New York: Academic Press; p. 667–678. [Google Scholar]
- Rick CM. 1986. Reproductive isolation in the Lycopersicon peruvianum complex In: D’Arcy WG, editor. Solanaceae—biology and systematics. New York: Columbia University Press; p. 477–495. [Google Scholar]
- Rick CM, Lamm R. 1955. Biosystematic studies on the status of Lycopersicon chilense. Am J Bot. 42:663–675. [Google Scholar]
- Rodrigues JA, Ruan R, Nishimura T, Sharma MK, Sharma R, Ronald PC, Fischer RL, Zilberman D. 2013. Imprinted expression of genes and small RNA is associated with localized hypomethylation of the maternal genome in rice endosperm. Proc Natl Acad Sci U S A. 110:7934–7939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RJ, Spielman M, Bailey J, Dickinson HG. 1998. Parent-of-origin effects on seed development in Arabidopsis thaliana. Development 125:3329–3341. [DOI] [PubMed] [Google Scholar]
- Smeds L, Künstner A. 2011. ConDeTri – A content dependent read trimmer for Illumina data. PLoS One 6:e26314.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Städler T, Roselius K, Stephan W. 2005. Genealogical footprints of speciation processes in wild tomatoes: demography and evidence for historical gene flow. Evolution 59:1268–1279. [PubMed] [Google Scholar]
- Städler T, Arunyawat U, Stephan W. 2008. Population genetics of speciation in two closely related wild tomatoes (Solanum section Lycopersicon). Genetics 178:339–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tellier A, Laurent SJY, Lainer H, Pavlidis P, Stephan W. 2011. Inference of seed bank parameters in two wild tomato species using ecological and genetic data. Proc Natl Acad Sci U S A. 108:17052–17057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Tomato Genome Consortium 2012. The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485:635–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 28:511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Roberts A, Goff L, Pertea G, Kim D, Kelley DR, Pimentel H, Salzberg SL, Rinn JL, Pachter L. 2012. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat Protocols. 7:562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Hendrickson DG, Sauvageau M, Goff L, Rinn JL, Pachter L. 2013. Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat Biotechnol. 31:46–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Videvall E, Sletvold N, Hagenblad J, Ågren J, Hansson B. 2016. Strong maternal effects on gene expression in Arabidopsis lyrata hybrids. Mol Biol Evol. 33:984–994. [DOI] [PubMed] [Google Scholar]
- Vu TM, Nakamura M, Calarco JP, Susaki D, Lim PQ, Kinoshita T, Higashiyama T, Martienssen RA, Berger F. 2013. RNA-directed DNA methylation regulates parental genomic imprinting at several loci in Arabidopsis. Development 140:2953–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walia H, Josefsson C, Dilkes B, Kirkbride R, Harada J, Comai L. 2009. Dosage-dependent deregulation of an AGAMOUS-LIKE gene cluster contributes to interspecific incompatibility. Curr Biol. 19:1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waters AJ, Bilinski P, Eichten SR, Vaughn MW, Ross-Ibarra J, Gehring M, Springer NM. 2013. Comprehensive analysis of imprinted genes in maize reveals allelic variation for imprinting and limited conservation with other species. Proc Natl Acad Sci U S A. 110:19639–19644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willi Y. 2013. The battle of the sexes over seed size: support for both kinship genomic imprinting and interlocus contest evolution. Am Nat. 181:787–798. [DOI] [PubMed] [Google Scholar]
- Wolf JB. 2009. Cytonuclear interactions can favor the evolution of genomic imprinting. Evolution 63:1364–1371. [DOI] [PubMed] [Google Scholar]
- Wolf JB. 2013. Evolution of genomic imprinting as a coordinator of coadapted gene expression. Proc Natl Acad Sci U S A. 110:5085–5090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff P, Weinhofer I, Seguin J, Roszak P, Beisel C, Donoghue MTA, Spillane C, Nordborg M, Rehmsmeier M, Köhler C. 2011. High-resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLoS Genet 7:e1002126.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolff P, Jiang H, Wang G, Santos-González J, Köhler C. 2015. Paternally expressed imprinted genes establish postzygotic hybridization barriers in Arabidopsis thaliana. eLife 4:e10074.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Zhao H, Xie S, Chen J, Xu Y, Wang K, Zhao H, Guan H, Hu X, Jiao Y, et al. 2011. Extensive, clustered parental imprinting of protein-coding and noncoding RNAs in developing maize endosperm. Proc Natl Acad Sci U S A. 108:20042–20047. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.