

Abstract
How do epigenetic modifications change across species and how do these modifications affect evolution? These are fundamental questions at the forefront of our evolutionary epigenomic understanding. Our previous work investigated human and chimpanzee brain methylomes, but it was limited by the lack of outgroup data which is critical for comparative (epi)genomic studies. Here, we compared whole genome DNA methylation maps from brains of humans, chimpanzees and also rhesus macaques (outgroup) to elucidate DNA methylation changes during human brain evolution. Moreover, we validated that our approach is highly robust by further examining 38 human-specific DMRs using targeted deep genomic and bisulfite sequencing in an independent panel of 37 individuals from five primate species. Our unbiased genome-scan identified human brain differentially methylated regions (DMRs), irrespective of their associations with annotated genes. Remarkably, over half of the newly identified DMRs locate in intergenic regions or gene bodies. Nevertheless, their regulatory potential is on par with those of promoter DMRs. An intriguing observation is that DMRs are enriched in active chromatin loops, suggesting human-specific evolutionary remodeling at a higher-order chromatin structure. These findings indicate that there is substantial reprogramming of epigenomic landscapes during human brain evolution involving noncoding regions.
Keywords: DNA methylation, human brain evolution, transcriptional divergence, differentially methylated regions, epigenomes
Introduction
Epigenetic changes such as DNA methylation and histone tail modifications leave distinctive marks on specific cell lineages, which otherwise harbor the same genetic material. Even though the functional significance of epigenetics in development (Gifford et al. 2013; Zhu et al. 2013), imprinting (Ferguson-Smith 2011; Hanna et al. 2016), and disease (Timp and Feinberg 2013; Ziller et al. 2013) is extremely well appreciated, whether and how epigenetic modifications are inherited across generations, and what heritable consequences may arise, remain as an unresolved and fundamental research area of epigenetics (Daxinger and Whitelaw 2012; Heard and Martienssen 2014). Even more uncertain is how epigenetic signals diverge in evolution, and what functional consequences, if any, may arise due to evolutionary epigenetic divergence.
In this paper, we investigate these pressing questions in the context of human brain evolution. Our brains are exceptionally large relative to body size compared to those of other animals (Jerison 1973), which occurred via a remarkably rapid expansion in recent evolutionary past (Kaas and Preuss 2013; Somel et al. 2013). Previous analyses have demonstrated that evolution has modified the human brain at almost every level of organization, including changes in long-distance corticocortical connectivity, histology, and local organization (Preuss 2011) as well as gene, protein, and metabolite expression (Brawand et al. 2011; Konopka et al. 2012; Liu et al. 2012). Intriguingly, increasing numbers of human brain studies are discovering the critical importance of epigenetic mechanisms in regulatory functions, development, and manifestations of neuropsychiatric diseases (Houston et al. 2013; Nikolova et al. 2014). We thus ask, do the human brain specific phenotypic transitions implicate changes at the epigenetic level? If so, how did the epigenetic modifications affect evolutionary innovations?
Recent studies have begun to elucidate epigenetic differences between brains of humans and nonhuman primates (Enard et al. 2004; Farcas et al. 2009; Shi et al. 2014; Shulha et al. 2012b; Zeng et al. 2012). In particular, we have previously compared the whole genome DNA methylation maps (referred to as “methylomes”) of human and chimpanzee brains (Zeng et al. 2012). However, our previous study focused on gene promoters, missing putative differentially methylated regions at un-annotated transcription start sites. Indeed, DNA methylation at gene bodies and gene deserts harbors strong regulatory potential (Hon et al. 2013; Huh et al. 2013; Mendizabal and Yi 2016). Zeng et al. (2012) also lacked data on outgroup species, limiting the inference on human-specific methylation changes. Importantly, to ascertain species-level epigenetic divergence with high confidence, it is critical to validate the observed epigenetic profiles in an independent dataset comprising a large number of brain samples, which were not available in previous studies in this domain. Addressing these concerns, here we present an unbiased identification and analysis of human brain specific DNA methylation changes. Our novel and robust research design allows us to infer lineage-specific epigenetic changes with high confidence. Moreover, using unbiased approaches, we illustrate the significance of epigenetic changes occurring in noncoding regions of the human genome. Comparative epigenomic analyses can offer significant insight into understanding evolution of human-specific regulatory processes.
Results
Whole-Genome Discovery of Differentially Methylated Regions in Human Brains
Our initial discovery data set consisted of whole genome bisulfite sequencing (WGBS) data from prefrontal cortices of three humans, three chimpanzees (Zeng et al. 2012), and two additional macaque methylomes from same brain region generated in this study (supplementary table S1, Supplementary Material online). We first identified differentially methylated regions (DMRs) between human and chimpanzee brain WGBS data sets using BSmooth (Hansen et al. 2012). BSmooth is specifically designed to utilize local averaging and information from biological replicates to identify DMRs from WGBS data (Hansen et al. 2012). We applied additional criteria, including a minimum DNA methylation difference of 0.3 between humans and chimpanzees. We used such a stringent criterion compared to other differential DNA methylation studies so that we can identify conservative candidate DMRs. In addition, using this criterion relieves concerns regarding differential cell composition (see below). We additionally imposed each DMR to harbor at least 10 mapped CpGs, to avoid effects of outlier CpGs.
The initial candidate DMRs following these procedures included 278 regions (supplementary table S2, Supplementary Material online for DMR coordinates). We used WGBS data from two rhesus macaques (Macaca mulatta) to estimate the polarity of human–chimpanzee differences (i.e., human-specific if DMR is present in human, but lacking in chimpanzees and macaques). Using this parsimony approach, we identified 85 human-specific and 102 chimpanzee-specific DMRs (refer “Material and Methods”). Interestingly, there was a significant excess of hypo-methylated DMRs in the human lineage (P = 0.005, χ2 test, supplementary table S3, Supplementary Material online). Nevertheless, given the current lack of knowledge on epigenetic evolutionary rates (refer “Discussion”), these results should be taken with caution.
Validation of DMRs via Expanded Sampling and Deep Sequencing
Patterns of DNA methylation at some genomic positions are variable among individuals within the same species. In addition, the average read depth for our discovery dataset based on whole-genome sequencing was low for some samples (ranged from 2 to 9, supplementary table S1, Supplementary Material online). To examine the consistency of the DMRs in the presence of these potential variabilities, we analyzed DNA methylation patterns of selected candidate DMRs from expanded samples using deep sequencing. Specifically, we performed targeted PCR and sequencing of bisulfite converted genomic DNA (average sequencing depth = 365X, supplementary table S4, Supplementary Material online) for a subset of DMRs (38 out of the 278) in the prefrontal cortex of a wider set of individuals and species: 23 humans, 2 chimpanzees, 1 Hoolock gibbon (H. leuconedys), 7 rhesus macaques, and 5 crab-eating macaques (M. fascicularis).
The validation set is biased toward human-specific DMRs because of sample availability (i.e., chimpanzee samples are not available in a large number). All validation DMRs showed significant human–chimpanzee differences (P < 0.05, one-sided Wilcoxon test), indicating that our discovery strategy successfully identified differentially methylated regions between the two species. Moreover, all but two validation DMRs showed consistent human brain specific DNA methylation in the extended comparison of human–chimpanzee–macaques. The majority of DMRs were human-specific even when we included two additional primate species (Hoolock gibbon and crab-eating macaques) (supplementary table S5, Supplementary Material online). In sum, human-specific methylation patterns of the majority of DMRs were consistent in a large number of individuals and in a larger panel of primates. The consistency between the discovery and validation sets is remarkable considering the age and sex differences found in the two sets (the discovery set consisted mostly of adult males, the validation set is composed of individuals of different ages and both genders, supplementary table S1, Supplementary Material online), which further support the idea that these DMRs represent true species-specific differences.
Due to the chemical nature of the bisulfite conversion technique, DMRs could also potentially reflect underlying genetic polymorphisms instead of methylation levels (i.e., C-to-T polymorphisms not present in the reference genomes could be interpreted as unmethylated sites). To avoid confounding effects of SNPs, we identified variable positions within species by sequencing the validation DMR dataset at extremely high coverage (mean coverage > 1400X, supplementary table S4, Supplementary Material online) and analyzing the data following the Genome Analysis Toolkit (GATK) pipeline (McKenna et al. 2010). DMRs held the same methylation patterns after excluding polymorphic positions, indicating that they are not artifacts of genetic polymorphisms. Figure 1 illustrates one of those validated DMRs.
Fig. 1.

DMR identification. (A) Phylogenetic tree of the species analyzed in this study. Sample sizes are shown for whole-genome bisulfite and targeted bisulfite sequencing (N and n, respectively). (B) Example of a DMR located 19 kb upstream of the promoter of Alpha-2C adrenergic receptor (ADRA2C). Lines indicate smoothed methylation values from whole-genome bisulfite and each dot represents raw methylation values of each CpG site in the targeted validation dataset at high coverage. Genomic coordinates correspond to the hg19.
The prefrontal cortices of primates consist of different cell types, most notably glia and neurons, which may exhibit distinct epigenetic patterns at some loci. Furthermore, the ratios of neurons versus glial cells are divergent between primate brains (Herculano-Houzel 2014; Sherwood et al. 2006). However, our stringent cutoff value of methylation difference 0.3 makes the DMRs robust against cellular heterogeneity between human and chimpanzee brains. Specifically, given the mean glia/neuron ratios of 1.6 and 1.2 in humans and chimpanzees (Sherwood et al. 2006), even in the most extreme case of a region being completely divergent between the two cell types (e.g., 0 and 100% methylation in one cell type versus the other), the expected methylation difference between human and chimpanzee is only 8%. In addition, we analyzed neuron- and non-neuron (mostly glia) -specific DNA methylation markers from cell-sorted methylation datasets (Guintivano et al. 2013; Kozlenkov et al. 2014) and found no evidence of significant effect of cell type composition bias in our DMR (supplementary fig. S1 and table S6, Supplementary Material online).
Genomic Annotation of DMRs
Differentially methylated regions between human and chimpanzee brains identified in this study were on average 584 bp long (ranging between 67 and 2,015 bp). They were significantly CpG enriched compared to genomic background (average CpG O/E ratio of 0.65, >1.3-fold enrichment compared to control regions, P < 0.001 based on 1,000 bootstraps). We first sought to examine the genomic locations of DMRs according to the annotation of transcripts in the UCSC Genome Browser. Approximately half of DMRs (46%) were found at known promoters (fig. 2). Interestingly, slightly over half (54%) of DMRs were located outside annotated gene promoters, including many (30%) DMRs found in distal intergenic regions (>3kb away from known transcription start sites), and 21% of DMRs within gene bodies.
Fig. 2.
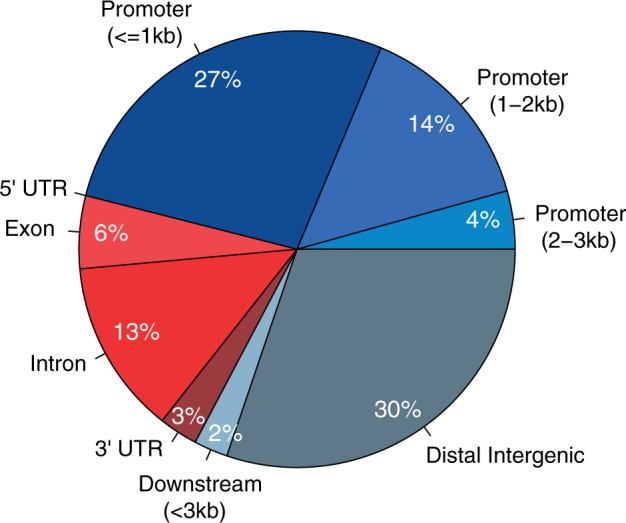
Genic annotation of DMRs. Annotation of DMRs with respect to known genes. Promoter region is divided into three according to distance to the TSS (<1 kb, 1–2 kb and 2–3 kb). Downstream region is considered up to 3 kb downstream of gene end, and distal intergenic is defined as >3 kb away from any gene.
To gain further insights into the functional role of DMRs, we examined enrichment of gene ontology (GO) and genome-wide association studies (GWAS) at DMRs and neighboring regions. Genes containing DMRs or closest to DMRs (supplementary table S2, Supplementary Material online) were significantly enriched for several GO developmental processes, including nervous system development, forebrain development, and embryonic morphogenesis (supplementary table S7, Supplementary Material online). In terms of GWAS, 146 DMR regions (DMR ± 50 kb) overlapped with GWAS signals. Among these, 24 DMR regions harbored variants associated with brain-related traits (supplementary table S7, Supplementary Material online), including those enriched in neurological disorders such as Asperger and Parkinson’s disease (P < 0.05, binomial test and FDR correction using HapMap SNPs as a null distribution see “Material and Methods” section and supplementary table S8, Supplementary Material online). Significantly enriched traits exclusively at human-specific DMRs (supplementary table S9, Supplementary Material online) included immune response, skeleton and tooth development, all with marked impacts on recent human evolution (Nielsen et al. 2007; Lachance and Tishkoff 2013). These patterns suggest that DMRs are located in genomic regions involved in brain development and other traits that are particularly relevant for our recent evolutionary history.
Coordinated Species-Specific Epigenetic Marks at DMRs
To understand the extent of coordinated evolutionary signatures of epigenetic modifications, we analyzed human-specific histone H3-trimethyl-lysine 4 (H3K4me3) modification data from prefrontal cortex neurons (Shulha et al. 2012b). DMRs are significantly enriched in regions harboring human-specific enrichment or depletion of H3K4me3 (51 DMRs overlap with human specific signatures of H3K4me3, 17-fold enrichment compared to control regions, P < 0.001, based upon 1,000 bootstraps). In concordance with the known antagonistic distribution of H3K4me3 and DNA methylation, human-specific hypo- and hyper-DNA methylation associated with significant H3K4me3 enrichment and depletion, respectively (P < 10−6, χ2 test, fig. 3). These observations illustrate coordinated epigenetic changes (of H3K4me3 and DNA methylation) during the evolution of human brains.
Fig. 3.

Coordinated epigenetic changes at DMRs (A) Example of human-hypomethylated DMR in human prefrontal cortex (hg19-based coordinates). (B) The region in panel A co-localizes with a human-specific H3K4me3 peak in cell sorted neurons. (C) Human-specific hyper-methylation of a DMR. (D) The region in panel C overlaps with a human-specific depletion of H3K4me3 histone mark, whereas chimpanzee and macaque show significant enrichment. Additional examples shown in supplementary figure S1.
DMRs Cluster in Interacting Chromatin Loops
Recent analyses of three-dimensional configuration of eukaryotic genomes solidified the concept that the basic unit of genome organization is large (∼Mb scale), topologically associated domains (TADs) that form hierarchical structures (Dixon et al. 2012; Ea et al. 2015). At the sub-TAD level, interactions between distal sequences (such as between enhancers and promoters) are accomplished by chromatin loops (Dixon et al. 2012; Ea et al. 2015). Chromatin loops facilitate transcriptional regulation, by enabling enhancer–promoter interactions (Dixon et al. 2012; Ea et al. 2015; Whalen et al. 2016).
We hypothesized that some nearby DMRs may comprise chromatin loops that are potentially co-regulated. Indeed, DMRs were significantly clustered at megabase (Mb)-scale when compared to control regions with the similar CpG content, length and chromosomal distribution (supplementary fig. S3, Supplementary Material online). We further analyzed chromatin interaction maps compiled in the 4D Genome Database (Teng, et al. 2015). We found 19 significant interactions among 32 unique DMRs, as detected by Hi-C and ChiA-PET experiments (2.97-fold excess, P < 0.001, 1,000 bootstraps, supplementary table S10, Supplementary Material online). The majority of these interactions were found within the same DMR or between two adjacent DMRs, but one interacting loop included 15 consecutive DMRs on chromosome 2. Of note, a smaller scale interaction within this large loop was also captured by 3C assay of prefrontal cortex samples (Shulha et al. 2012b). Furthermore, the enrichment was largely driven by human hypo-methylated DMRs (1.22-fold enrichment, P = 0.05 vs. 0.45-fold of hyper-methylated regions, P = 0.1). Considering that DMRs are on average an order of magnitude shorter than chromatin loops (average 584 bp vs. 5,145 bp, for DMRs and chromatin loops respectively), we further investigated the degree of interactions of DMRs including flanking regions (±3 kb). We found that nearly half of DMRs (N = 132) were involved in a total of 227 significant chromatin interactions among themselves (odds ratio of 1.15, P = 0.024, supplementary table S2, Supplementary Material online) and 247 out of 278 DMRs (88.9%) showed at least one significant interaction with other non-DMR regions (1.07 fold enrichment, P = 0.007). Thus, DMRs appear to preferentially locate in regions participating in chromatin loops and therefore could potentially affect transcriptional regulation.
DMRs Mark Active Promoter/Enhancers with Enriched TF Binding Signatures
While evolutionary data on epigenomic marks are sparse, information on multiple epigenetic modifications from diverse cell types and developmental stages is available for humans. In order to understand the regulatory potential of DMRs, we explored the chromatin states of DMRs in 98 different tissues, inferred from ChIP-Seq experiments on six chromatin marks (H3K4me3, H3K4me1, H3K36me3, H3K27me3, H3K9me3, and H3K27ac) (Bernstein et al. 2010). We found that human-specific hypo-methylated DMRs were conspicuously marked by active regulatory chromatin states, mainly promoter and enhancer states (fig. 4). In contrast, human hyper-methylated regions harbored a large number of DMRs classified as quiescent chromatin states across tissue types (supplementary fig. S4, Supplementary Material online). Interestingly, the promoter and enhancer states of hypo-methylated DMRs were highest at brain samples, although not limited to this tissue type. In contrast, many DMRs currently annotated in gene deserts exhibit active promoter chromatin marks nearly exclusively in brain tissues (supplementary fig. S5, Supplementary Material online).
Fig. 4.

Chromatin states at human hypo-methylated DMRs. Heatmap of the fraction of DMRs exhibiting distinctive chromatin states (rows) in different tissues (columns). Each cell of the heatmap indicates the proportion of DMRs classified as that specific chromatin state in a given tissue type.
We further analyzed binding of specific transcription factors (TFs) using ChIP-Seq data of 161 transcription factors in 91 human cell lines from diverse tissues in the ENCODE project. This analysis revealed that DMRs were highly enriched in experimentally validated, functional transcription factor binding sites (TFBS). A majority of DMRs (84.5%) overlapped with TFBS (1.94 fold enrichment versus control regions, P < 0.001). Human-specific hypo-methylated DMRs were significantly more enriched with TFBS than hyper-methylated ones (P = 0.0015, χ2 test). Moreover, we found that four specific transcription factors were significantly more frequently bound in DMRs than expected, whereas bindings of 18 transcription factors were significantly depleted in DMRs (P < 0.05, fig. 5A). Among the enriched transcription factors we found NRF1, which associates with neurodegenerative diseases (Lee et al. 2011), and BRF1, implicated in neurodevelopmental processes (Borck et al. 2015). The experimentally supported transcription factor binding activity of DMRs and their promoter and enhancer-like epigenetic features advocate an active role of DMRs as regulatory hotspots in human brains.
Fig. 5.

Transcription factor binding sites at DMRs. (A) Transcription factors significantly enriched or depleted at DMRs. (B–C) Human-specific nucleotide change in position 3 of the binding site by transcription factor BHLHE4. This position is 1.1 kbp upstream of a human hypo-methylated DMR in the promoter of PSMB2 gene compared to chimpanzee and macaque. (D) Human-specific expression increase of PSMB2 gene observed in three different data sets.
DMRs Associate with Gene Expression Differences
We tested whether species-specific epigenetic changes and transcription factor binding potential of DMRs directly translated into gene expression differences. We analyzed three different expression datasets, generated via different methods (RNA-seq, SuperSAGE, and microarray) from prefrontal cortices of human, chimpanzee, and macaques (Brawand et al. 2011; Konopka et al. 2012; Liu et al. 2012). Specifically, we examined whether gene expression differences, if any, were consistent with the silencing effect of DNA methylation. Of note, the consistency across datasets was low, since only 66 out of 6,725 genes showed significant and consistent human-specific gene expression in all three datasets [in part due to low sample size, e.g., only 3 and 4 macaques were studied in Brawand et al. (2011) and Konopka et al. (2012), respectively]. Nonetheless, we found that 13 out of 61 human-specific DMRs with both methylation and expression information associated with significant human-specific expression patterns in at least one dataset (P < 0.05 in human-chimpanzee and human-macaque comparisons, Wilcoxon tests) including genes with key roles on neurological processes such as CSPG5, SPG7, and COBL (supplementary table S11, Supplementary Material online). We next tested whether the DMR-genes show an excess of human-specific expression compared to genes selected at random (1,000 randomizations), in each of the datasets independently as well as in the combined dataset. We observed a significant association between human-specific DMR-genes and human-specific expression patterns in one of the three datasets (the one with the largest sample sizes per species) as well as when all datasets were considered together (with 37 humans, 24 chimpanzee, and 38 macaques, supplementary table S12, Supplementary Material online). Therefore, despite the limitations of the available datasets, these results suggest that human-specific DMRs associate with gene expression changes of adjacent genes.
Signatures of Human-Specific Mutations at TFBS Motifs at DMRs
How do DMRs arise between species? One clue may be gained from recent studies demonstrating the effect of genetic variation on epigenetic divergence (Hernando-Herraez et al. 2015). Genetic changes can cause epigenetic changes is by generating or depleting functional transcription factor binding sites, since binding of transcription factors are known to reduce DNA methylation (e.g., Schübeler 2015). Consequently, we tested whether human-specific hypo-methylated DMRs associate with novel TFBS by identifying single nucleotide substitutions at functional TF motifs within or around DMRs (±3kb). We focused on experimentally validated TFBS sites (ChIP-Seq peaks within the ENCODE project), and fixed substitutions in the human lineage (i.e., <1% frequency in the 1,000 Genomes dataset). We further restricted to substitutions that generate a major TFBS (i.e., the position weight matrix of the human-specific nucleotide is at least 0.8).
We identified a total of 41 human substitutions within 51 TFBS motifs from this analysis (supplementary table S13, Supplementary Material online). One such mutation affects the E-box canonical binding sequence (enhancer-box, CANNTG) in the promoter of the PSMB2 gene, where a human-specific allele represents a putative de novo binding site for BHLHE40 (fig. 5B and C). The transcription factor BHLHE40 is a well-known circadian gene with strong cyclic expression in human brains that is deregulated in major depressive disorders. Consistent with the presence of the E-box motif and human-specific hypo-methylated DMR in the promoter of PSMB2, human brains have increased PSMB2 expression levels compared to chimpanzees and macaques (fig. 5D). Even though the motif was only predicted for BHLHE40, E-boxes are known to be utilized by a variety of transcription factors for regulation of gene expression in diverse tissues, including neurons (Massari and Murre 2000). Indeed, a large number of additional TFs (a total of 71) show binding to that specific region in ChIP-Seq experiments (supplementary table S14, Supplementary Material online). Notably, one of these TFs that bind to this region is FOXP2, a gene important for speech and language that regulates a suite of transcriptional networks in the human brain (Konopka et al. 2009). Another example of human-specific TFBS mutation with associated expression divergence is the stearoyl-CoA desaturase (SCD) gene (supplementary fig. S6, Supplementary Material online). Interestingly, mutations that generate functional transcription binding sites in DMRs are observed on average at a much lower rate compared to binding sites outside DMRs (nearly 6-fold less frequent, P < 0.02, 1,000 bootstraps), suggesting that such mutations do not occur frequently at regions of strong functional constraint.
Discussion
The human brain represents a challenging, yet extremely intriguing, system to study the intersection of evolution, epigenetics, and diseases. Human brains are characterized by dramatic evolutionary innovations such as increased brain size and cortical reorganization. Genetic and transcriptomic studies have revealed extensive changes in gene expression and gene-regulatory networks that could provide the basis for evolutionary specializations of human brain structure and function (Brawand et al. 2011; Konopka et al. 2012; Bauernfeind et al. 2015). Epigenetic modifications can cause such transcriptional changes, even in the absence of substantial sequence changes. In fact, epigenetics is known to be deeply involved in regulation of brain functions in humans (Akbarian and Huang 2009; Cheung et al. 2010; Zhu et al. 2013; Kozlenkov et al. 2014) as well as in several neuropsychiatric diseases (Shulha et al. 2012a; Jaffe et al. 2016).
To understand the role of epigenetic regulation on the evolution of human brains, we compared the methylomes from cortices of human and several nonhuman primates, in particular with chimpanzees. We combined genome-scans, nonparametric testing, and stringent cutoff criteria, and identified nearly 300 differentially methylated regions between human and chimpanzee cortices. To further confirm the observed patterns, we performed high quality targeted experiments on a validation panel, which included one of the largest number of brain specimens used for comparative epigenetic studies. Differential DNA methylation between human and chimpanzee brains was confirmed in all the validation DMRs tested, and most (>70%) were human-specific in the larger panel of all five species. These results provide a strong support for evolutionary divergence of DNA methylation. Our criterion of methylation difference > 0.3 is extremely stringent compared to the known examples of putatively functional methylation differences (Dias and Ressler 2014; Tobi et al. 2014; Jaffe et al. 2016). For example, we validate regions previously identified by targeted analyses (Farcas et al. 2009; Shi et al. 2014), although the observed fractional methylation differences fall below our criteria. Future studies with larger numbers of samples (specially for chimpanzees) and advanced statistical tools will likely identify many additional genomic regions of epigenetic divergence.
Using newly generated brain methylomes of rhesus macaques, we inferred evolutionary origins of DMRs following a simple parsimony rule. Rhesus macaques are often used as outgroups to assign evolutionary polarity to either the human or chimpanzee lineages in DNA sequence and gene expression comparisons (Uddin et al. 2004; Kim et al. 2006; Konopka et al. 2012; Rogers and Gibbs 2014). However, accurately quantifying how rapidly DNA methylation evolves and determining whether the use of rhesus macaques is adequate, require future research. Large-scale phylogenetic analyses of DNA methylation indicate some degree of conservation (Mendizabal et al. 2014; Keller et al. 2016). Indeed, when we test the difference between human and macaque DMRs, all but two putatively human-specific DMRs fit the expectation based upon parsimony. With few exceptions, data from crab-eating macaques (M. fascicularis), are largely consistent with those from rhesus macaques. These observations begin to provide information on the rates of epigenetic changes during evolution.
Even though many DMRs are found proximal to known TSS, the majority of DMRs locate outside promoters. This finding highlights the importance of employing unbiased whole-epigenome approaches, in contrast to those that interrogate predetermined regions such as gene promoters or classical CpG islands (Enard et al. 2004; Farcas et al. 2009; Zeng et al. 2012; Shi et al. 2014). Genomic and epigenomic analyses of DMRs in this study suggest that non-promoter DMRs have similar transcriptional potential as promoter DMRs, as judged by their epigenetic states, TF binding signatures, and association with gene expression (supplementary fig. S7, Supplementary Material online). In particular, many DMRs found in intergenic regions also exhibit chromatin features of active promoters, which interestingly show high brain specificity. Taken together, human brain-specific DNA methylation changes may contribute to regulation of transcription. Indeed, in light of the available expression datasets from the cortices of the three species, human-specific DMRs are significantly enriched for human-specific gene expression divergence at adjacent genes, in the direction that is expected according to the methylation patterns of the DMRs. Finally, in the human-specific DMRs, there was a significant excess of hypo-methylated DMRs. This could potentially be in line with increased gene expression levels in human brains (Caceres et al. 2003; Preuss et al. 2004), although some studies cast doubt on the generality of this pattern (Uddin et al. 2004; Babbitt et al. 2010).
Genes overlapping with DMRs and DMR-adjacent genes were enriched for several functional categories related to brain development. Moreover, many GWAS loci of neuropsychiatric diseases were over-represented in DMR regions, suggesting that some of these GWAS SNPs may affect epigenetic modifications. On the other hand, DMRs also associated with GWAS variants and GO categories not related with brain functions, but with other traits known to have played significant roles in human evolution. In fact, many DMRs showed chromatin features of promoters and enhancers in nonbrain tissues as well. This can be explained by the fact that DNA methylation patterns are often stable across tissues and developmental stages (Hon et al. 2013; Ziller et al. 2013; Zeng et al. 2014). For example, hypo-methylated embryonic enhancers can maintain their distinctive methylation profiles in adult tissues, even without direct enhancer activities in those tissues (Hon et al. 2013). Moreover, some of the functional ontology categories enriched in the current study overlap with those identified in previous comparative epigenomic studies, despite different methodologies and/or different tissues used (Pai et al. 2011; Hernando-Herraez et al. 2013; Hernando-Herraez et al. 2015). We propose that some of the DMRs are broadly used as regulatory regions in diverse tissues, or active regulatory elements in embryonic or fetal tissues that retained their epigenetic memories in adult brains.
Our study highlights the synergistic nature of epigenetic changes during evolution. DMRs extensively overlap with human-specific signatures of H3K4me3 modifications identified in a previous study (Shulha et al. 2012b). Moreover, many of the DMRs cluster locally and exhibit evidence of physical interactions by participating in chromatin loops, indicating that human brain-specific epigenetic changes affect large-scale spatial organization of three-dimensional interactions among chromosomal regions (Shulha et al. 2012b). Notably, a strong signature of human-specific hypo-methylation overlaps with a human-specific H3K4me3 peak in the second intron of DDP10 gene (supplementary fig. S1, Supplementary Material online), which also harbors the TSS of a human expressed sequence tag (BF979467). DPP10 encodes a dipeptidyl peptidase-related protein involved in neuronal excitability that plays roles in brain development. Genetic variation in DPP10 is associated with autism, bipolar disorder, and schizophrenia (Djurovic et al. 2010). DMRs found in the vicinity of the DPP10 locus participate in interacting domains of chromatin loops in a fetal lung derived cell line (Jin et al. 2013), as well as in human prefrontal cortex samples (Shulha et al. 2012b). These observations indicate that hotspots of human-specific epigenetic modification may exist in this region.
We identified several fixed nucleotide substitutions in the human lineage that associate with human-specific epigenetic and gene expression divergence. The patterns of nucleotide substitution, DNA methylation changes, transcription factor binding, and gene expression profiles of these mutations are all in accord with the well-established functional roles of different components of gene expression regulation. Comparative epigenetic studies such as the current investigation can provide critical information to prioritize candidate regulatory changes for necessary experimental validation, ultimately to elucidate the functional roles of human-specific sequence changes.
Materials and Methods
Whole Genome Bisulfite Sequencing Method
Briefly, genomic DNA from macaque prefrontal cortices was extracted following the Qiagen DNeasy Blood & Tissue Kit. In total, 300–500 ng of genomic DNA were then fragmented by sonication, end repaired and ligated to custom-synthesized methylated Illumina PE adapters (Eurofins MWG Operon, Huntsville, AL) according to manufacturer’s (Illumina, San Diego, CA) instructions for gDNA library construction. Adaptor-ligated libraries were subjected to two successive treatments of sodium bisulfite conversion using the EpiTect Bisulfite kit (Qiagen, Valencia, CA) as outlined in the manufacturer’s instructions. Five to ten nanograms of bisulfite-converted libraries was PCR amplified with the following condition: 2.5 U of ExTaq DNA polymerase (Takara), 5 µl of 10X Extaq reaction buffer, 25 µM dNTPs, 1 µl of 10 µM PE PCR primer 1.0, and 1 µl of 10 µM PE PCR Primer 2.0 in a 50 µl PCR reaction. The thermocyling was as follows: 95 °C 3 min, then 10 cycles of 95 °C 30 s, 65 °C 30 s, and 72 °C 60 s. The enriched libraries were purified twice with SPRI method using 0.8x v/v AMPure beads (Beckman Coulter, Inc. Brea CA), and sequenced using an Illumina HiSeq2000 at the Vincent J. Coates Genomic Sequencing Laboratory at the University of California, Berkeley that was supported by NIH S10 Instrumentation Grants S10RR029668 and S10RR027303. WGBS data from human and chimpanzee prefrontal cortices were downloaded from GEO (GSE37202).
Mapping and DMR Identification
Raw files were processed by FastQC for quality control. Quality and adapter trimming was performed using TrimGalore (Babraham Institute). Reads were mapped to the respective reference genomes (hg19, pantro4, and rheMac3 for human, chimpanzee, and macaque, respectively) using Bismark (Krueger and Andrews 2011) and BsMap (Xi and Li 2009). We chose to remove all reads with identical start and ends, considering them as duplicates. This strategy is likely to be conservative, given the uncertainty on the best practice to deal with such reads (Balzer et al. 2013). After de-duplication, we calculated fractional methylation levels at individual CpGs (Lister et al. 2009).
We identified differentially methylated regions (DMRs) between human and chimpanzee brain methylomes using BSmooth (Hansen et al. 2012). We used quantile cutoff of 1% of the t-statistics using only CpGs supported by at least two reads in two or more individuals per species. We additionally filtered the detected DMRs to select those with at least 10 CpGs per DMR, and a minimum difference of fractional methylation level of 0.3 between the two species. DMRs identified from the two mappers were combined.
DMR Polarization
We used the macaque methylome data to polarize the DMRs. A DMR was classified as human-specific when the methylation difference between the human and macaque was greater than the difference between chimpanzee and macaque. Using this simple parsimony approach, 100 DMRs could be assigned as human-specific, and 127 DMRs as chimpanzee-specific. The remaining 51 DMRs did not have matching CpGs mapped thus remained unclassified. We then imposed additional criteria to make our inference more conservative. Specifically, the difference between human and macaque should be greater than 0.15, which led to 85 human-specific and 102 chimpanzee-specific DMRs. A stricter threshold of 0.30 was also applied, which resulted in 62 human-specific, 72 chimp-specific, and 144 unknown DMRs (supplementary table S2, Supplementary Material online).
Targeted Validation Analyses
We performed genomic and bisulfite sequencing for 38 DMRs on the prefrontal cortices of 23 humans, 2 chimpanzees, 1 gibbon, 7 rhesus macaques, and 5 crab eating macaques using a MiSeq machine (supplementary table S4, Supplementary Material online). Frozen tissues were obtained from the prefrontal cortices of the individuals, all of which had no known neuronal diseases or history of drug abuse. For the human subjects, informed written consent was obtained from the relatives of the human subjects prior to sample collection or analysis. The internal review board of Kunming Institute of Zoology, Chinese Academy of Sciences, approved all protocols of this study.
Genomic DNA was extracted according to the DNeasy Blood&Tissue Kit (QIAGEN) protocol. We used the EpiTect Bisulfite Kits (Qiagen, Valencia, CA) to conduct bisulfite conversions of genomic DNA following the manufacturer's instructions. Sodium bisulfite converts unmethylated cytosine to uracil, which is then PCR amplified as thymidine and methylated cytosine remain cytosine. PCR primers were designed using Methyl Primer Express 1.0 (ABI). To detect potential SNPs, we also performed the PCR to the untreated genomic DNA. PCR primers were designed using Primer Premier 5. The primer information is available under request.
The targeted bisulfite PCR products from single reactions were first quantified before proceeding with sequencing. For quality control purposes, we also performed qPCR. Following the Nextera XT DNA Sample Preparation protocol, we used 1 ng of total DNA (0.2ng/µl) per sample to prepare MiSeq libraries. Genomic libraries of individuals used for DMR validation were also generated and subjected to MiSeq sequencing. All of the MiSeq sequencing was performed in the Kunming Institute of Zoology sequencing core.
Mapping and Analyses of Validation Dataset
For validation, we performed bisulfite sequencing on 38 DMRs using a MiSeq machine. One region failed to be amplified in all individuals. After quality control, reads were mapped using Bismark (Krueger and Andrews 2011) to the following reference genomes: hg19, pantro4, rheMac3, macFas5, and nomLeu3 for human, chimpanzee, rhesus Macaque, crab eating macaque, and gibbon, respectively.
We also sequenced the corresponding genomic regions of the specific individuals analyzed to identify any potential SNPs that may affect our inference. Genome sequence reads were aligned using bwa (Li and Durbin 2009). Duplicate reads were removed using Picard (http://picard.sourceforge.net). We then identified SNPs using GATK v3.4 (McKenna et al. 2010) and using standard hard filtering parameters (QD < 2.0 ‖ FS > 60.0 ‖ MQ < 40.0 ‖ MQRankSum < −12.5 ‖ ReadPosRankSum < −8.0).
Functional Annotation and Epigenetic Features of DMRs
To test the significance of different genomic features of DMRs, we selected 1,000 control regions of same length and chromosome location as the DMRs, and containing at least 10 CpGs (the same criteria we used for DMR discovery). P-values were computed by counting the number of simulations showing values as extreme as the observed one. Enrichments were computed as the ratio between the observed value and the mean of the control regions.
We used ChIPSeeker (Yu et al. 2015) to annotate DMRs to genes using hg19 KnowGene table from UCSC. GO analyses were performed using GoStat Rpackage (Falcon and Gentleman 2007) and using 13,455 human–chimpanzee orthologous genes (The Chimpanzee Sequencing and Analysis Consortium 2005). GWAS enrichment analyses were performed with traseR package (Chen and Qin 2015). Specifically, we considered 44,078 SNP-trait associations from the Association Results Browser (http://www.ncbi.nlm.nih.gov/projects/gapplusprev/sgap_plus.htm, last accessed 24 August 2016) that combines associations from both dbGaP and NHGRI GWAS Catalog. We also considered linked SNPs (LD >0.8 and located within 100 kb of GWAS SNPs in European populations from Thousand Genomes, hg19). Including linked variants, we have 90,700 SNP-trait associations and 78,247 unique trait-associated SNP and 573 unique traits. For enrichment testing, we considered background SNPs from HapMap CEU population (HapMap phases I + II + III, 4,029,840 variants excluding those on Y-chromosome). Specifically, a contingency table was created using counts of the number of trait-associated SNPs found within DMR and non-DMR regions, and compared those numbers with the counts for HapMap SNPs that fall in DMR vs. non-DMR regions. Significance was tested using binomial test and FDR correction was applied for multiple testing. We tested each of the original 573 traits independently, as well as grouped into 26 categories (supplementary table S8, Supplementary Material online). The rationale for joining traits into related categories was to gain statistical power as well as to account for biases due to heterogeneity on reported trait names across GWAS studies (i.e., the independent entries in the database alcohol drinking, alcoholism, or drinking behavior were considered within Addictive Behavior category, supplementary table S8, Supplementary Material online).
We identified the overlap between our DMR dataset and the human-specific H3K4me3 peaks (Shulha et al. 2012b). For plotting purposes, we re-mapped raw reads to hg19, pantro4, and rheMac3 genomes. We analyzed the chromatin interactions (those with Confidence Score 1 ≤ 0.05) from the 4DGenome Database (Teng et al. 2015). We analyzed data derived from ENCODE ChIP-Seq experiments (UCSC wgEncodeRegTfbsClusteredV3 table for peaks and factorbookMotifCanonical for motifs).
We explored the 18 chromatin state-model maps from the Roadmap Epigenomics dataset (Roadmap Epigenomics Consortium et al. 2015). We joined several categories related to similar states, such as active Tss (TssA, TssFlnk, TssFlnkU, and TssFlnkD), enhancers (EnhG1, EnhG2, EnhA1, EnhA2, and EnhWk), transcription (Tx and TxWk), and repressed PolyComb (ReprPC and ReprPCWk), yielding a total of nine final states. We followed the tissue classification in the original dataset. In addition we combined related tissues to the following categories: “fetal” (IMR90 fetal lung fibroblasts Cell Line, Fetal Adrenal Gland, Fetal Brain Male, Fetal Brain Female, Fetal Heart, Fetal Intestine Large, Fetal Intestine Small, Fetal Kidney, Fetal Lung, Fetal Muscle Trunk, Fetal Muscle Leg, Fetal Stomach, and Fetal Thymus) and “stem-cell” (H1 Derived Mesenchymal Stem Cells, Adipose Derived Mesenchymal Stem Cell Cultured Cells, Bone Marrow Derived Cultured Mesenchymal Stem Cells, Primary hematopoietic stem cells, Primary hematopoietic stem cells short term culture, Primary hematopoietic stem cells G-CSF-mobilized Female, and Primary hematopoietic stem cells G-CSF-mobilized Male).
We further analyzed binding of specific transcription factors (TFs) using ChIP-Seq data of 161 transcription factors in 91 human cell lines from diverse tissues in the ENCODE project (Gerstein et al. 2012). For human-specific mutations potentially generating novel TFBS within DMRs we excluded DMRs that were chimp-specific. Then we searched for positions within and around DMRs (3 kb) where human reference genomes showed a different allele compared to that in macaque and chimpanzee genomes (which need to be identical). We only considered positions within TFBS motifs where the human nucleotide represented at least 80% frequency within the motif. Finally, we excluded any positions that were polymorphic within humans (i.e., non-reference alleles with >1% frequency in worldwide populations in the 1000Genome Dataset (The 1000 Genomes Project Consortium, 2015).
Analyses of Gene Expression
We focused on human-specific DMRs and tested if humans showed significant gene expression values (Wilcoxon test, P < 0.05) in both human–chimpanzee and human–macaque comparisons (assuming repressive effect of DNA methylation on expression, i.e., human hypo-methylated DMR, increased expression in humans). To test for significance at genome-wide level, we randomly selected the same number of genes as in the DMR gene sets from the total genes in a given dataset (while maintaining the observed numbers of hypo- and hyper-methylation), and examined whether they show increase or decrease of gene expression. We repeated this process 1,000 times and counted the number of genes exhibiting human-specific expression values with P <0.05. We tested each dataset independently as well as using a combined data set (using standardized z-scores and the subset of genes overlapping among the three datasets).
Supplementary Material
Supplementary figures S1 to S7 and tables S1 to S14 are available at Molecular Biology and Evolution online (http://www.mbe.oxfordjournals.org/).
Acknowledgment
This work was supported by the National Science Foundation (SBE-131719 to SVY), the National Institutes of Health (1R01MH103517-01A1 to SVY, GK and TMP), the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB13010000 to BS), the National Natural Science Foundation of China (NSFC 31130051 to BS and NSFC to LS), the Youth Innovation Promotion Association of the Chinese Academy of Sciences (to LS), the Research Personnel Improvement Program by the Department of Education, Language Policy, and Culture by the Basque Government (POS_2013_1_130 to IM); and the Yerkes National Primate Center Base Grant (ORIP/OD P51OD011132).
References
- Akbarian S, Huang HS. 2009. Epigenetic regulation in human brain-focus on histone lysine methylation. Biol Psychiatry. 65:198–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbitt CC, Fedrigo O, Pfefferle AD, Boyle AP, Horvath JE, Furey TS, Wray GA. 2010. Both noncoding and protein-coding RNAs contribute to gene expression evolution in the primate brain. Genome Biol Evol. 2:67–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzer S, Malde K, Grohme MA, Jonassen I. 2013. Filtering duplicate reads from 454 pyrosequencing data. Bioinformatics 29:830–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauernfeind AL, Reyzer ML, Caprioli RM, Ely JJ, Babbitt CC, Wray GA, Hof PR, Sherwood CC. 2015. High spatial resolution proteomic comparison of the brain in humans and chimpanzees. J Comp Neurol. 523:2043–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BE, Stamatoyannopoulos JA, Costello JF, Ren B, Milosavljevic A, Meissner A, Kellis M, Marra MA, Beaudet AL, Ecker JR, et al. 2010. The NIH Roadmap Epigenomics Mapping Consortium. Nat Biotechnol. 28:1045–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borck G, Hog F, Dentici ML, Tan PL, Sowada N, Medeira A, Gueneau L, Thiele H, Kousi M, Lepri F, et al. 2015. BRF1 mutations alter RNA polymerase III-dependent transcription and cause neurodevelopmental anomalies. Genome Res. 25:155–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brawand D, Soumillon M, Necsulea A, Julien P, Csardi G, Harrigan P, Weier M, Liechti A, Aximu-Petri A, Kircher M, et al. 2011. The evolution of gene expression levels in mammalian organs. Nature 478:343–348. [DOI] [PubMed] [Google Scholar]
- Caceres M, Lachuer J, Zapala MA, Redmond JC, Kudo L, Geschwind DH, Lockhart DJ, Preuss TM, Barlow C. 2003. Elevated gene expression levels distinguish human from non-human primate brains. Proc Natl Acad Sci U S A. 100:13030–13035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Qin ZS. 2015. traseR: an R package for performing trait-associated SNP enrichment analysis in genomic intervals. Bioinformatics 15:1214–1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung I, Shulha HP, Jiang Y, Matevossian A, Wang J, Weng Z, Akbarian S. 2010. Developmental regulation and individual differences of neuronal H3K4me3 epigenomes in the prefrontal cortex. Proc Natl Acad Sci U S A. 107:8824–8829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daxinger L, Whitelaw E. 2012. Understanding transgenerational epigenetic inheritance via the gametes in mammals. Nat Rev Genet. 13:153–162. [DOI] [PubMed] [Google Scholar]
- Dias BG, Ressler KJ. 2014. Parental olfactory experience influences behavior and neural structure in subsequent generations. Nat Neurosci. 17:89–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon JR, Selvaraj S, Yue F, Kim A, Li Y, Shen Y, Hu M, Liu JS, Ren B. 2012. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 485:376–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djurovic S, Gustafsson O, Mattingsdal M, Athanasiu L, Bjella T, Tesli M, Agartz I, Lorentzen S, Melle I, Morken G, Andreassen OA. 2010. A genome-wide association study of bipolar disorder in Norwegian individuals, followed by replication in Icelandic sample. J Affect Disord. 126:312–316. [DOI] [PubMed] [Google Scholar]
- Ea V, Baudement M-O, Lesne A, Forné T. 2015. Contribution of topological domains and loop formation to 3D chromatin organization. Gene 6:734–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enard W, Fassbender A, Model F, Adorjan P, Pääbo S, Olek A. 2004. Differences in DNA methylation patterns between humans and chimpanzees. Curr Biol. 14:R148–R149. [PubMed] [Google Scholar]
- Falcon S, Gentleman R. 2007. Using GOstats to test gene lists for GO term association. Bioinformatics 23:257–258. [DOI] [PubMed] [Google Scholar]
- Farcas R, Schneider E, Frauenknecht K, Kondova I, Bontrop R, Bohl J, Navarro B, Metzler M, Zischler H, Zechner U, et al. 2009. Differences in DNA methylation patterns and expression of the CCRK gene in human and nonhuman primate cortices. Mol Biol Evol. 26:1379–1389. [DOI] [PubMed] [Google Scholar]
- Ferguson-Smith AC. 2011. Genomic imprinting: the emergence of an epigenetic paradigm. Nat Rev Genet. 12:565–575. [DOI] [PubMed] [Google Scholar]
- Gerstein MB, Kundaje A, Hariharan M, Landt SG, Yan KK, Cheng C, Mu XJ, Khurana E, Rozowsky J, Alexander R, et al. 2012. Architecture of the human regulatory network derived from ENCODE data. Nature 489:91–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gifford CA, Ziller MJ, Gu H, Trapnell C, Donaghey J, Tsankov A, Shalek AK, Kelley DR, Shishkin AA, Issner R, et al. 2013. Transcriptional and epigenetic dynamics during specification of human embryonic stem cells. Cell 153:1149–1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guintivano J, Aryee MJ, Kaminsky ZA. 2013. A cell epigenotype specific model for the correction of brain cellular heterogeneity bias and its application to age, brain region and major depression. Epigenetics 8:290–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanna CW, Peñaherrera MS, Saadeh H, Andrews S, McFadden DE, Kelsey G, Robinson WP. 2016. Pervasive polymorphic imprinted methylation in the human placenta. Genome Res. 26:756–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, Irizarry RA. 2012. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol. 13:R83.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heard E, Martienssen RA. 2014. Transgenerational epigenetic inheritance: myths and mechanisms. Cell 157:95–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herculano-Houzel S. 2014. The glia/neuron ratio: how it varies uniformly across brain structures and species and what that means for brain physiology and evolution. Glia 62:1377–1391. [DOI] [PubMed] [Google Scholar]
- Hernando-Herraez I, Heyn H, Fernandez-Callejo M, Vidal E, Fernandez-Bellon H, Prado-Martinez J, Sharp AJ, Esteller M, Marques-Bonet T. 2015. The interplay between DNA methylation and sequence divergence in recent human evolution. Nucleic Acids Res. 43:8204–8214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernando-Herraez I, Prado-Martinez J, Garg P, Fernandez-Callejo M, Heyn H, Hvilsom C, Navarro A, Esteller M, Sharp AJ, Marques-Bonet T. 2013. Dynamics of DNA methylation in recent human and great Ape evolution. PLoS Genet. 9:e1003763.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. 2013. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 45:1198–1206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houston I, Peter CJ, Mitchell A, Straubhaar J, Rogaev E, Akbarian S. 2013. Epigenetics in the human brain. Neuropsychopharmacology 38:183–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huh I, Zeng J, Park T, Yi SV. 2013. DNA methylation and transcriptional noise. Epigenetics Chromatin 6:9.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaffe AE, Gao Y, Deep-Soboslay A, Tao R, Hyde TM, Weinberger DR, Kleinman JE. 2016. Mapping DNA methylation across development, genotype and schizophrenia in the human frontal cortex. Nat Neurosci. 19:40–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jerison HJ. 1973. Evolution of the Brain and Intelligence. New York: Academic Press. [Google Scholar]
- Jin F, Li Y, Dixon JR, Selvaraj S, Ye Z, Lee AY, Yen C-A, Schmitt AD, Espinoza CA, Ren B. 2013. A high-resolution map of the three-dimensional chromatin interactome in human cells. Nature 503:290–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaas JH, Preuss TM. 2013. Human Brain Evolution In: Squire L, Berg D, Bloom FE, du Lac S, Ghosh A, Spitzer NC, editors. Fundamental neuroscience. 4th ed San Diego (CA): Academic Press; p. 901–918. [Google Scholar]
- Keller TE, Han P, Yi SV. 2016. Evolutionary transition of promoter and gene body DNA methylation across invertebrate-vertebrate boundary. Mol Biol Evol. 33:1019–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SH, Elango N, Warden C, Vigoda E, Yi SV. 2006. Heterogeneous genomic molecular clocks in primates. PLoS Genet. 2:e163.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka G, Bomar JM, Winden K, Coppola G, Jonsson ZO, Gao F, Peng S, Preuss TM, Wohlschlegel JA, Geschwind DH. 2009. Human-specific transcriptional regulation of CNS development genes by FOXP2. Nature 462:213–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopka G, Friedrich T, Davis-Turak J, Winden K, Oldham MC, Gao F, Chen L, Wang GZ, Luo R, Preuss TM, Geschwind DH. 2012. Human-specific transcriptional networks in the brain. Neuron 75:601–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kozlenkov A, Roussos P, Timashpolsky A, Barbu M, Rudchenko S, Bibikova M, Klotzle B, Byne W, Lyddon R, Di Narzo AF, et al. 2014. Differences in DNA methylation between human neuronal and glial cells are concentrated in enhancers and non-CpG sites. Nucleic Acids Res. 42:109–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krueger F, Andrews SR. 2011. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 27:1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lachance J, Tishkoff SA. 2013. Population Genomics of Human Adaptation. Annu Rev Ecol Evol Syst. 44:123–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CS, Lee C, Hu T, Nguyen JM, Zhang J, Martin MV, Vawter MP, Huang EJ, Chan JY. 2011. Loss of nuclear factor E2-related factor 1 in the brain leads to dysregulation of proteasome gene expression and neurodegeneration. Proc Natl Acad Sci U S A. 108:8408–8413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462:315–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Somel M, Tang L, Yan Z, Jiang X, Guo S, Yuan Y, He L, Oleksiak A, Zhang Y, et al. 2012. Extension of cortical synaptic development distinguishes humans from chimpanzees and macaques. Genome Res. 22:611–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massari ME, Murre C. 2000. Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol Cell Biol. 20:429–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna A, Hanna M, Banks E, Sivachenko A, Cibulskis K, Kernytsky A, Garimella K, Altshuler D, Gabriel S, Daly M, DePristo MA. 2010. The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20:1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, Keller TE, Zeng J, Yi SV. 2014. Epigenetics and evolution. Integr Comp Biol. 54:31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendizabal I, Yi SV. 2016. Whole-genome bisulfite sequencing maps from multiple human tissues reveal novel CpG islands associated with tissue-specific regulation. Hum Mol Genet. 25:69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen R, Hellmann I, Hubisz MJ, Bustamante C, Clark AG. 2007. Recent and ongoing selection in the human genome. Nat Rev Genet. 8:857–868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nikolova YS, Koenen KC, Galea S, Wang C-M, Seney ML, Sibille E, Williamson DE, Hariri AR. 2014. Beyond genotype: serotonin transporter epigenetic modification predicts human brain function. Nat Neurosci. 17:1153–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pai AA, Bell JT, Marioni JC, Pritchard JK, Gilad Y. 2011. A genome-wide study of DNA methylation patterns and gene expression levels in multiple human and chimpanzee tissues. PLoS Genet. 7:e1001316.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss TM. 2011. The human brain: rewired and running hot. Ann N Y Acad Sci. 1225 Suppl 1:E182–E191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preuss TM, Caceres M, Oldham MC, Geschwind DH. 2004. Human brain evolution: insights from microarrays. Nat Rev Genet. 5:850–860. [DOI] [PubMed] [Google Scholar]
- Roadmap Epigenomics Consortium, Kundaje A, Meuleman W, Ernst J, Bilenky M, Yen A, Heravi-Moussavi A, Kheradpour P, Zhang Z, Wang J, et al. 2015. Integrative analysis of 111 reference human epigenomes. Nature 518:317–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers J, Gibbs RA. 2014. Comparative primate genomics: emerging patterns of genome content and dynamics. Nat Rev Genet. 15:347–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D. 2015. Function and information content of DNA methylation. Nature 517:321–326. [DOI] [PubMed] [Google Scholar]
- Sherwood CC, Stimpson CD, Raghanti MA, Wildman DE, Uddin M, Grossman LI, Goodman M, Redmond JC, Bonar CJ, Erwin JM, Hof PR. 2006. Evolution of increased glia-neuron ratios in the human frontal cortex. Proc Natl Acad Sci U S A. 103:13606–13611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Lin Q, Su B. 2014. Human-specific hypomethylation of CENPJ, a key brain size regulator. Mol Biol Evol. 31:594–604. [DOI] [PubMed] [Google Scholar]
- Shulha HP, Cheung I, Whittle C, Wang J, Virgil D, Lin CL, Guo Y, Lessard A, Akbarian S, Weng Z. 2012a. Epigenetic signatures of autism: trimethylated H3K4 landscapes in prefrontal neurons. Arch Gen Psychiatry 69:314–324. [DOI] [PubMed] [Google Scholar]
- Shulha HP, Crisci JL, Reshetov D, Tushir JS, Cheung I, Bharadwaj R, Chou HJ, Houston IB, Peter CJ, Mitchell AC, et al. 2012b. Human-specific histone methylation signatures at transcription start sites in prefrontal neurons. PLoS Biol. 10:e1001427.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somel M, Liu X, Khaitovich P. 2013. Human brain evolution: transcripts, metabolites and their regulators. Nat Rev Neurosci. 14:112–127. [DOI] [PubMed] [Google Scholar]
- Teng L, He B, Wang J, Tan K. 2015. 4DGenome: a comprehensive database of chromatin interactions. Bioinformatics 31:2560–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The 1000 Genomes Project Consortium 2015. A global reference for human genetic variation. Nature 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Chimpanzee Sequencing and Analysis Consortium 2005. Initial sequence of the chimpanzee genome and comparison with the human genome. Nature 437:69–87. [DOI] [PubMed] [Google Scholar]
- Timp W, Feinberg AP. 2013. Cancer as a dysregulated epigenome allowing cellular growth advantage at the expense of the host. Nat Rev Cancer 13:497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tobi EW, Goeman JJ, Monajemi R, Gu H, Putter H, Zhang Y, Slieker RC, Stok AP, Thijssen PE, Müller F, et al. 2014. DNA methylation signatures link prenatal famine exposure to growth and metabolism. Nat Commun. 5:5592.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin M, Wildman DE, Liu G, Xu W, Johnson RM, Hof PR, Kapatos G, Grossman LI, Goodman M. 2004. Sister grouping of chimpanzees and humans as revealed by genome-wide phylogenetic analysis of brain gene expression profiles. Proc Natl Acad Sci U S A. 101:2957–2962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whalen S, Truty RM, Pollard KS. 2016. Enhancer-promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet. 48:488–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi Y, Li W. 2009. BSMAP: whole genome bisulfite sequence MAPping program. BMC Bioinformatics 10:232.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G, Wang L-G, He Q-Y. 2015. ChIPseeker: an R/Bioconductor package for ChIP peak annotation, comparison and visualization. Bioinformatics 31:2382–2383. [DOI] [PubMed] [Google Scholar]
- Zeng J, Konopka G, Hunt BG, Preuss TM, Geschwind D, Yi SV. 2012. Divergent whole-genome methylation maps of human and chimpanzee brains reveal epigenetic basis of human regulatory evolution. Am J Hum Genet. 91:455–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng J, Nagrajan HK, Yi SV. 2014. Fundamental diversity of human CpG islands at multiple biological levels. Epigenetics 9:483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Adli M, Zou JY, Verstappen G, Coyne M, Zhang X, Durham T, Miri M, Deshpande V, De Jager PL, et al. 2013. Genome-wide chromatin state transitions associated with developmental and environmental cues. Cell 152:642–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller MJ, Gu H, Muller F, Donaghey J, Tsai LTY, Kohlbacher O, De Jager PL, Rosen ED, Bennett DA, Bernstein BE, et al. 2013. Charting a dynamic DNA methylation landscape of the human genome. Nature 500:477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.