

Abstract
Mutations in FGFR1 have recently been associated with Hartsfield syndrome, a clinically distinct syndromic form of holoprosencephaly (HPE) with ectrodactly, which frequently includes combinations of craniofacial, limb and brain abnormalities not typical for classical HPE. Unrelated clinical conditions generally without craniofacial or multi-system malformations include Kallmann syndrome and idiopathic hypogonadotropic hypogonadism. FGFR1 is a principal cause for these less severe diseases as well. Here we demonstrate that of the nine FGFR1 mutations recently detected in our screen of over 200 HPE probands by next generation sequencing, only five distinct mutations in the kinase domain behave as dominant-negative mutations in zebrafish over-expression assays. Three FGFR1 mutations seen in HPE probands behave identical to wild-type FGFR1 in rescue assays, including one apparent de novo variation. Interestingly, in one HPE family, a deleterious FGFR1 allele was transmitted from one parent and a loss-of-function allele in FGF8 from the other parent to both affected daughters. This family is one of the clearest examples to date of gene:gene synergistic interactions causing HPE in humans.
Introduction
FGF signaling plays multiple temporal and spatial roles in vertebrate development including neural and limb patterning, organ homeostasis and somatic cancer syndromes. There are 22 FGF ligands interacting with four classes of receptor tyrosine-kinases [FGFR1–4] that mediate tissue-specific responses in humans, and contributing to considerable molecular redundancy and complexity (1–4). In addition, according to the Online Mendelian Inheritance in Man (OMIM) there are at least six distinct clinical conditions associated with mutations in FGFR1 including craniosynostosis conditions, for example, Pfeiffer syndrome (5), Hartsfield syndrome (6–9), Kallmann syndrome (10–12) and also normosmic hypogonadotropic hypogonadism (nIHH) (9,13,14), among others. The molecular basis of the extensive phenotypic spectrum of FGFR1-associated mutations has not been fully explained. The more common FGFR1-associated diseases typically show an autosomal dominant mode of transmission. Craniosynostosis syndromes are considered gain-of-function dominant disorders with variable penetrance and expressivity. Both of the IHH conditions, Kallmann and nIHH, are frequently at the mild end of the clinical spectrum and are associated with FGFR1 mutations typified by gene deletion, frameshift and stop-codon mutations suggesting a general loss-of-function disease mechanism. The missense mutations in IHH or Kallmann syndrome can often be shown to display structural deficits, or aberrant surface expression, ligand binding or defective kinase activity consistent with the splice site, frameshift and nonsense mutations common to this group (Supplementary Material, Table S1). However, with the recent description of Hartsfield syndrome as both an autosomal dominant, and occasionally autosomal recessive condition (6–8), suggests an entirely different class of mutations in FGFR1 may be responsible for these phenotypic differences. Here we provide several lines of evidence supporting a dominant-negative, kinase-specific and frequently de novo class of mutations that help to explain the multi-system involvement in Hartsfield syndrome on a gene-product dosage mechanism versus haplo-insufficiency typical for the less-severe IHH conditions.
Holoprosencephaly (HPE) is the most common developmental malformation of the human forebrain and can present in neonates as only minor micro-signs, such as a single central incisor, over a nearly continuous spectrum to an undivided forebrain and single eye with a proboscis (15,16). Among cases with normal chromosomes and microarray, the clinical spectrum of HPE is still very wide and displays both genetic heterogeneity and variable expressivity similar to that described for IHH. Studies over the past two decades have identified four classical HPE genes that most clinical diagnostic centers test in routine practice: SHH, SIX3, ZIC2 and TGIF (16). Overall, nearly a dozen genes are implicated as likely causes for HPE. Additional genes are considered for diagnostic testing based on the clinical presentation or presence of additional findings. The deleterious coding region changes among these four classical HPE genes account for only 25% of our prospective case series. The overall presence of limb anomalies in HPE is rare and based on recent reports (6–8) should suggest Hartsfield syndrome and the pursuit of mutations in FGFR1.
HPE can be seen in all vertebrate species and is a reflection of a shared ancient highly conserved developmental gene regulatory scheme involving hedgehog and fgf signals (16–18). Consequently, extensive experimental manipulation in animal models, such as the mouse and zebrafish, have direct bearing on our understanding of the gene(s) participating in HPE pathogenesis in humans. Midline hedgehog signals from the prechordal plate are necessary to split the single embryonic eye field into left and right domains and allowing for binocular visual development and division of the forebrain into left and right hemispheres. In the vertebrate brain, several organizing centers direct the growth and regional specialization of adjacent neural tissue. Often these organizing centers interact such that regional neural specification simultaneously comes under the influence of multiple signaling centers (18). There are three principal organizing centers in the prosencephalon: the anterior neural ridge at the rostral edge of the neural plate secreting fgf ligands, the ventral midline prechordal plate secreting sonic hedgehog implicated in HPE and the dorsal signaling center which is a source of bmp and wnts. These centers in turn activate gene regulatory networks of transcription factors that establish distinct gene-expression territories. Rostral fgf signals activate the transcription factor foxg1 producing proliferative responses and suppression of apoptosis. Progressive reduction of fgf activity results in a small telencephalon and at extreme levels an HPE-like malformation (18,19). Fgf signals support the expression of shh, and vice versa. Fgf signals are required for the development of the olfactory bulbs (20) and gonadotropin secreting neurons (21), explaining the anosmia and infertility of Kallmann syndrome and IHH.
Of the 22 members of the FGF ligand family, fgf8 stands out as a critical mediator of murine and human brain and limb patterning. It is expressed in key organizing centers of the mouse: the anterior neural ridge that patterns the rostral prosencephalon, the midbrain–hindbrain boundary and the apical ectodermal ridge of the limbs (3,4). An allelic series of decreasing fgf8 gene-product expression in the mouse clearly demonstrates that while fgf8 null/null mice are lethal the effective dose of fgf8 markedly affects the development, size and regional patterning of the brain and craniofacial structures (22,23). These murine models support the participation of FGF8 and FGFR1 in human diseases including isolated cleft-lip and palate (24), HPE (25), Hartsfield syndrome and the anosmia of Kallmann syndrome and the selective pituitary deficits of IHH. FGF8 mutations are an infrequent cause of IHH (25–27), whereas our previous investigation of HPE cases identified a single case with potential overlap between FGF8 mutations in HPE cohorts and IHH (25,26). Similarly, a single case of an autosomal recessive FGF8 mutation has been described as a rare cause of HPE (27). Taken together, these studies support a key role for FGF8 and FGFR1 in human development with phenotypes emerging based on the degree of impairment of FGF pathway signaling. We now provide a molecular mechanism of dominant-negative de novo kinase domain class of mutations in FGFR1 that can help to explain the severity of Hartsfield syndrome.
We recently conducted a targeted capture sequencing study in 206 HPE probands with a specific emphasis on key ligands and receptors of the hedgehog, bmp, wnt and fgf signaling pathways (3,4,18) in order to expand our knowledge of gene involvement and potential inter-pathway interactions forming the basis of HPE pathogenesis (Roessler et al., manuscript in preparation). Here we describe the clinical and molecular findings of all nine FGFR1 missense variations detected in our HPE cohort and their functional analysis in the zebrafish system. Although various bioinformatics programs proved useful in classification of variants into benign versus damaging groups, we show that functional testing in vivo is necessary to more fully characterize the biochemical consequences of these missense changes. Finally, we provide a case report of two sibs from a single family with an unusual functional interaction between FGFR1 and FGF8 supporting a model of occasional gene:gene interactions in HPE.
Results
Human HPE phenotypes
As detailed in Table 1, we detected nine different missense coding region changes in the FGFR1c reference isoform. Two of these changes, V102I and P772S are also detected among healthy controls described in public databases (see also Supplementary Material, Table S1). The clinical features of these three cases (two families) are typical for our HPE cohort and lack limb abnormalities (Fig. 1A–C). Of the remaining seven provisionally unique findings, three are confirmed to be de novo by molecular evaluation of the parental DNA: A152T, R627T (8) and D641N. A total of five mutations are observed in the kinase domain. Overall, we observe a clustering of the HPE-associated mutations within the kinase domain of the protein and strong genotype–phenotype correlations suggestive of typical Hartsfield syndrome combining brain, facial and limb findings in this subset of mutation carriers (Fig. 1D–J).
Table 1.
Clinical and molecular findings in patients with the FGFR1 variants
| Case | Amino acid | Mutation type | Domain | Co-morbid change | Relevant genetic findings (normal targeted capture results except as indicated) | Phenotype Craniofacial | Phenotype Brain | Phenotype Limb | Phenotype other |
|---|---|---|---|---|---|---|---|---|---|
| 1A | Pro772Ser | SNP [rs56234888] Presumed benign |
C-terminus | Arg257Trp SIX3 Presumed causative |
Normal chromosomes | Single central incisor | Microcephaly (10–25%), absent septum pellucidum, single azygous anterior cerebral artery | Normal hands and feet | Mild developmental delay, seizure disorder |
| 1B | Pro772Ser | SNP [rs56234888] Presumed benign |
C-terminus |
SIX3 frameshift Presumed causative |
Normal chromosomes, normal for microarray, SHH, ZIC2 and TGIF by Sanger | Microcephaly, cleft lip and palate, hypertelorism | Lobar HPE | Thin digits, nail hypoplasia | Developmental delay |
| 2 | Asp641Asn | De novo FGFR1 | Kinase NCBI ATP binding/active site |
None | None | Bilateral cleft lip and palate, dysplastic ears | Semi-lobar HPE, microcephaly, pituitary hypoplasia, GH neurosecretory dysfunction, neurogenic hypernatremia | Oligodactyly of both feet, syndactyly both hands | Developmental delay |
| 3 | Arg627Thr | De novo FGFR1a | Kinase NCBI ATP binding/active site |
None | None | Bilateral complete cleft lip and palate, right microtia, left malformed ear, undescended testes | Abnormal corpus callosum, central diabetes insipidus and growth hormone deficiency | Bilateral ectrodactly of both hands and feet | None |
| 4 | Asp623Glu | Singletonb | Kinase NCBI ATP binding/active site |
None | None | Not described | HPE | Split hand/split foot | None |
| 5 | Met535Lys | Singletonb | Kinase No known NCBI site |
None | None | Bilateral cleft lip and palate, single central incisor, missing columella, multiple hypodontia | Normal MRI | No reported limb anomalies | No seizures, normal psychomotor development, mild language delay |
| 6 | Gly487Asp | Singletonb | Kinase NCBI ATP binding/active site |
None | Normal chromosomes (550 band level) | Bilateral cleft lip and palate, high forehead, hypertelorism, | Lobar HPE, absence of septum pellucidum and anterior corpus callosum | Bilateral ectrodactyly of the feet | Developmental delay |
| 7 | Glu294Lys | Affected sibs Presumed synergy |
Ligand binding |
FGF8 indel Presumed synergy |
Normal chromosomes, microarray and normal results for SHH, ZIC2, TGIF and SIX3 | Both sisters with microcephaly (<2%) and diabetes insipidus | Two sisters with semi-lobar and lobar HPE, respectively | No reported limb anomalies | Global developmental delay, seizures |
| 8 | Ala152Thr |
De novo FGFR1 Presumed benign |
Linker domain |
De novo SIX3 Arg258Trp Presumed causative |
None | Mild microcephaly, hypotelorism, submucus cleft palate, single central incisor, choanal stenosis | Lobar HPE | Normal hands and feet | None |
| 9 | Val102Ile | SNP [rs55642501] Presumed benign |
First Ig domain | None | Normal chromosomes, microarray, SHH, ZIC2, SIX3 and TGIF | Mild microcephaly, midline cleft, hypotelorism, flat nose | No reported anatomical findings | No reported limb anomalies | None |
aAppears to be identical to the case described in Dhamija et al. (8).
bParents not available.
Figure 1.

Phenotypes of individuals with FGFR1 mutations. (A) Female at 7 months of age (Patient 7 in Table 1) with lobar HPE; facial features short bulbous nose, long philtrum, tented upper lip and low-set ears; (B) Female at 23 months of age [sister of (A)] with semi-lobar HPE. She has a flat nasal bridge, short bulbous nose, anteverted nares, long philtrum and tented upper lip, additionally large low-set ears are present; (C) Three-year-old female with lobar HPE (Patient 8 in Table 1), from this photo, hypotelorism and single central incisor are apparent, her extremity exam was normal; (D–F) 11-year-old female with semi-lobar HPE (Patient 2 in Table 1), note cleft lip repair, dysplastic ears, syndactyly of fingers 3 and 4 bilaterally and split feet; (G–J) Male with lobar HPE at 7 months of age (Patient 6 in Table 1), facial features include hypertelorism, cleft lip/palate; hands have tapering of fingers, left third digit camptodactyly and bilateral fifth digit clinodactyly; note bilateral split foot and the absence of second digit phalanges bilaterally on X-ray.
Analysis of FGFR1 missense variants by zebrafish morphant rescue assays
Many, but not all, novel mutations are pathogenic. Therefore, to experimentally evaluate the effects of all nine of these missense changes we established a series of functional tests exploiting the zebrafish system. Given the strikingly conserved gene regulatory networks across vertebrate species, very often the synthetically engineered human variants are bioactive when introduced into a model organism, such as the zebrafish or mouse. Our first approach was a morpholino (MO)-based rescue experiment involving the injection of human FGFR1 synthetic transcripts and measuring the relative biological activity of each variant on developing zebrafish embryos. This method has previously been widely used to evaluate the biological effects of variants using co-injection of human orthologous mRNA in zebrafish. We confirmed that the published combination of zebrafish fgfr1a and fgfr1b MO mimics the established loss of FGF signaling with a shortened body trunk and tail, and that these morphant phenotypes can be rescued with zebrafish fgfr1 mRNA (28) (data not shown). Therefore, we proceeded to optimize this assay by empirically determining the most effective dose for morphant rescue using the wild-type (wt) human FGFR1. We found our ideal assay conditions to be 25 pg of human wt FGFR1 RNA and a combination of 8 ng of fgfr1a MO plus 4 ng of fgfr1b MO based on the observed trunk and tail size recovery (Fig. 2B and C). We could independently demonstrate that 25 pg of wt FGFR1 mRNA alone (without MOs) has no phenotypic consequences similar to our gfp control mRNA-injected embryos (see below) (Fig. 3B). No microscopically distinct phenotype has been detected up to 100 pg of FGFR1 mRNA (data not shown).
Figure 2.

Functional evaluation of FGFR1 missense mutations by mRNA rescue of morphant phenotypes. (A) Schematic drawing of the 822 residue wt FGFR1 protein and its functional domains. Ig, immune globulin-like (green); TM, transmembrane (blue); PTK, phospho tyrosine kinase domain (red). (B) Lateral view of the injection phenotype (Types I–III) observed at 24 h post-fertilization (hpf). Three different outcomes of injected embryos are observed after injection with MO alone, or the co-injection of MO and FGFR1 construct-derived mRNA. Type I represents a normal phenotype for gfp, wt and rescued embryo, Type II is typical for the fgfr1a + 1b morphant. Type III is a severe phenotype with a missing tail which is exclusively observed in co-injection of fgfr1a + 1b MO plus FGFR1 D641N mRNA. The arrow indicates the developing tail. (C) Injected embryos are scored based on phenotypic severity and presented as a percentage of each observed Type. Quantified embryos for this study have been described in Supplementary Material, Table S3.
Figure 3.

Characterization of the over-expression microinjection consequence of all nine FGFR1 missense variants at 24 hpf. (A) Lateral view of the live injection phenotype and in situ hybridization with a blood cell marker hbbe1.1 gene. Two distinct types are observed; Type I with normal development and Type II with reduced trunk size and enlarged blood cells. An asterisk in Type II embryos identifies the blood island. (B) Quantification of mRNA-injected embryos. Among nine variants, six have typical Type II phenotype from FGFR1 E294K to FGFR1 D641N while other variants are similar to wt and induce no phenotype. (C and E) In situ hybridization of mRNA-injected FGFR1 with chd at 8 hpf. Mild reduction of chd expression is observed (C) and quantified (E). (D and F) In situ hybridization of FGFR1 mRNA-injected embryos with dusp6. Mildly reduced dusp6 expression levels are found (D) and quantified (F). Only six variants show a suppression of the FGF signaling pathway with various degrees of activity. D, dorsal; V, ventral. Quantified embryos for this study are described in Supplementary Material, Table S3.
As detailed in Figure 2C, the zebrafish morphant Type II phenotype is efficiently rescued by microinjection of wt and variants V102I, A152T and P772S synthetic mRNAs. In contrast, FGFR1 E294K, G487D, M535K, D623E, R627T and D641N failed to rescue. The rescue activity of the E294K allele in the extra-cellular ligand-binding region of IgIIIc was clearly abnormal and was detected in two sisters affected with HPE with normal limbs and a co-morbid FGF8 mutation (discussed subsequently) (Table 1 and Fig. 4D). Interestingly, all of the remaining five variants in the protein tyrosine kinase domain failed to rescue the morphant phenotype. At first glance, this could be interpreted as either normal activity of common SNP variants or loss-of-function in the patient-specific mutations. However, we observed that in the case of the FGFR1 D641N variant, our MO rescue injection shows an unexpected dramatic increase of severity at 24 hpf with loss of posterior structures (Fig. 2B Type III). This result indicated that a simple loss-of-function mechanism might not be the best explanation.
Figure 4.
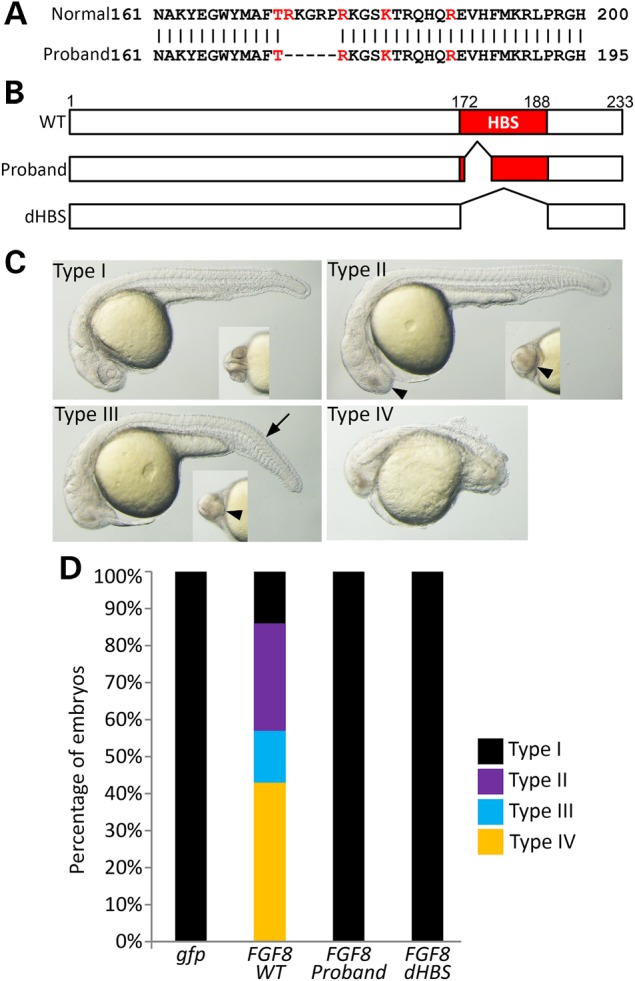
Disruption or elimination of the heparin binding site (HBS) domain abolishes FGF8 bioactivity. (A) Amino acid alignment of wild-type (wt) and the patient-specific 15 bp deletion region of FGF8. Red characters indicate that conserved five residues around HBS domain (glycine box). The missing five amino acid residues are indicated by ‘–’. (B) Injection constructs. Two different deletions of the HBS region have been generated by PCR. The first faithfully represents the 15 bp deletion from the proband, while the other represents the full deletion of the HBS as a control. (C) Lateral view of the injection phenotype of FGF8 mRNA at 24 h after fertilization (hpf). Small inner images of each Type are visualized by a ventral view of the forebrain. Decreased size of the eye (arrowhead) and short trunk (arrow) are the major phenotypic findings. Type I with normal development, Type II with only reduced eye size, Type III with reduced eye and short trunk, Type IV with reduced eye and extremely short trunk size. (D) Bioactivity measurements were made by counting embryos based on their phenotypes (Type I–IV) and calculating their percentage. Quantified embryos for this study have been described in Supplementary Material, Table S3.
Over-expression analysis of FGFR1 variants
To better understand the differences among the rescue assay findings, we began a series of over-expression experiments. We again microinjected construct-derived mRNA for the wt and all nine variants (in the absence of MOs) and assessed the consequences. The results show that FGFR1 V102I, A152T and P772S variants are again indistinguishable from wt control mRNA-injected embryos. Combining the findings from both assays, we conclude that despite being de novo, the A152T change is effectively benign. Note that this patient has a de novo SIX3 mutation that can readily explain the HPE findings (Table 1).
The other six variants previously shown to fail to rescue fgfr1a and fgfr1b morphants have an additional microscopically visible defect with reduced mesoderm-derived structures such as trunk and tail (Fig. 3A and B) and enlarged endoderm-derived blood cell islands (Fig. 3A, marker hbbe1.1). The fact that these variants not only fail to rescue morphants, but also trigger dominant effects on the trunk and tail support a dominant-negative mechanism of action of these mutations. Note that the dose of 25 pg wt FGFR1 mRNA was chosen as the lowest dose of mRNA that is insufficient to result in a measurable morphological phenotype (compare Fig. 3A and B; Type I with Type II), but is able to fully rescue the morphant phenotype (Fig. 2C).
To more fully characterize the variants causing a morphological phenotypic abnormality at a molecular level, we performed in situ hybridization at gastrulation stages using zebrafish chd (Fig. 3C and E) and dusp6 (Fig. 3D and F). The expression level of chd showed that E294K and all five kinase domain mutations have a Type II phenotype with suppression of chd expression at 8 hpf. The dusp6 expression pattern showed a more variable effect between different constructs. This well-known fgf downstream signaling marker dual specificity phosphatase (dusp6) was not significantly changed by the E294K variant at the dose analyzed. However, when taken together the same six mutations are consistently dominant-negative mutations based on morphological criteria and the analysis of three independent markers (hbb1.1, chd and dusp6).
Structural analysis of Hartsfield-associated missense mutations
We can now establish a strong relationship between the location of key residues in the ATP binding site/active site of the protein tyrosine-kinase domain PTKc_FGFR1 [NCBI cd05098] and those affected by mutation in Hartsfield syndrome. Previous reports of Hartsfield syndrome implicate FGFR1 residues G490R, D623Y (8), N628K and C725Y. Those described for IHH/SHSF invoke FGFR1 kinase domain residues G485R, E670A, L712P and possibly V688L (Supplementary Material, Table S1). To this list we can now add G487D, M535K, D623Y (8), R627T and D641N. Note the clustering of these mutations (ATP binding/active site). Residues 484–492 are implicated in ATP binding and the active site for kinase function, as are residues 623, 627 and 641. This analysis indicates that certain residues within the PTKc_FGFR1 are susceptible to having dominant effects and that other kinase missense changes seen in some Kallmann syndrome patients affect residues leading to more typical loss-of-function changes.
Analysis of the effects on FGF8 function of the 15 bp deletion in affected sibs with a co-morbid deleterious FGFR1 allele
One of our families evaluated in the next generation sequencing study included two sisters who each inherited the functionally abnormal FGFR1 mutation E294K from their father (see above) and an in-frame 15 bp indel of unknown significance in the last exon of FGF8 from their mother (Supplementary Material, Fig. S1). Bioinformatic programs generally are not helpful in determining the consequences of this type of indel mutation, nevertheless, all FGF8 isoforms are predicted to be affected (29). Therefore, we set out to establish a biosensor assay of FGF8 activity in zebrafish.
As shown in Figure 4A and B, the 15 bp deletion of FGF8 is part of the heparin-binding site (HBS) (glycine box) that is critical for the binding of the ligand FGF to its receptor FGFR (30,31). To evaluate whether this partial deletion of the HBS is damaging to FGF8 function during zebrafish development, we first confirmed the presence of the indel in the probands by Sanger sequencing and generated the same deletion in an FGF8 expression construct. As a control, we generated a deletion construct that eliminated the entire heparin-binding sequence (dHBS) (Fig. 4B). The biological activities of FGF8 mutant constructs were tested in zebrafish with various doses from 0.2 pg (Fig. 4C Type II) to 20 pg (all dead). In this study, we injected 0.4 pg of mRNA with a control gfp mRNA to observe significant phenotypic difference between wt and mutant. Wild-type human FGF8 mRNA-injected embryos show a strong activity during early embryonic development with a mildly reduced eye and reduced size of the body and trunk (Fig. 4C and D). These effects have been previously described as a dorsalizing effect in zebrafish (32). Interestingly, both the 15 bp deletion and the deletion of the entire HBS eliminated the biological activity of the modified constructs. This result suggests that partial or complete loss of the HBS can strongly interfere with ligand and/or receptor dimerization required for tyrosine-kinase downstream signaling. Furthermore, the combination of the dominant acting FGFR1 mutation from the father and the loss-of-function mutation from the mother almost certainly act synergistically in the two affected sisters.
Discussion
FGF signaling plays an important role in brain and mesoderm formation during early development (2), and at later stages the specification of the limb and digits (4). Recently, Hartsfield syndrome has been described as a unique association of HPE and ectrodactyly caused by mutations in FGFR1, a molecule well known to be active in both developmental fields. Our study clearly extends these initial findings, with the addition of new cases, and identifies a novel mechanism of action of critical residues in the kinase domain. These mutations are distinguished by the inability to cooperate with the normal FGFR1 allele, and also interfere with normal signaling activity in vivo. The dominant-negative activity of a selected subset of these mutations can account for the more severe phenotypic findings of Hartsfield syndrome.
The signaling of receptor tyrosine kinase requires the formation of a ligand dimer with receptor dimers prior to the activation step by trans-phosphorylation and triggering of downstream activation cascades. We propose that receptor subunits incapable of binding ATP create dimers frozen in the inactive stage that fail to proceed with trans-phosphorylation and activation. Such complexes would comprise only a distinct subset of mutation types, as seen here. Basic researchers have previously generated dominant-negative FGFR1 receptors in the frog (33) that recapitulate many of the findings displayed by our Hartsfield mutations in zebrafish. Far less commonly, rare instances of two structurally abnormal homozygous FGFR1 extracellular mutations may phenocopy the kinase-domain mutations. In these cases, no normal FGFR1 molecules are present.
Over the past decade, multiple investigators into the genetic causes of infertility have cataloged nearly 50 distinct mutations in FGFR1 associated with Kallmann syndrome and IHH (Supplementary Material, Table S1). These mutations occur both extra-cellularly and intra-cellularly. Furthermore, simple microdeletions of FGFR1 can cause IHH. Similarly, microdeletions of FGF8 are implicated as a rare cause of HPE (34) (Supplementary Material, Table S2). Elegant functional studies in several laboratories have consistently described ex vivo loss-of-function characteristics to most of these disease-associated changes (35,36). In this study, we demonstrate that zebrafish provides yet another useful tool to evaluate the bioactivity of human FGFR1 variants in a living host. The assays are robust and reproducible. As demonstrated by this study, both loss-of-function and gain-of-function assays are equally informative. Similarly, assays for FGF8 are highly sensitive and readily quantifiable. The utility of zebrafish is particularly informative with developmental genes shared among all vertebrates. Rather than attempting to model a human disease in all of its phenotypic aspects, we instead use zebrafish as a biosensor system to compare wt and mutant forms of ligands or receptors linked to human disorders. A final advantage of zebrafish models is the ability to test non-coding enhancers or other regulatory units surrounding developmental genes (37) in order to better understand gene-regulatory networks and pathway interactions.
As newer sequencing platforms have become increasingly available and cost-effective, genotype findings in human patients have increased exponentially. HPE and IHH-related conditions are both extremely genetically heterogeneous and display unexplained incomplete penetrance and variable expressivity. Instances of potential gene:gene interactions, consistent with an oligogenetic model to explain this variability, have been proposed in both contexts (38, Roessler et al., manuscript in preparation). Detailed examination of Patients 1A and 1B (each with the benign P772S variant) have SIX3 mutations that more convincingly explain the HPE phenotype. Similarly, Patient 8 with the de novo benign A152T variant also have a de novo SIX3 mutation that is likely causative.
Here we describe one of the most convincing examples from our patient collection of a genetic synergy between two experimentally proven mutations: E294K in FGFR1 and an in-frame 15 bp deletion in the last exon of FGF8. While we are actively seeking such interactions in our cohort, we caution that these appear to be the exception rather than the rule. Similar FGFR1 and FGF8 interactions have been described for Kallmann syndrome, however, the incidence of co-occurrence is uncommon and many of the proposed FGF8 variants may need to be re-classified as rare variants of uncertain significance unless functional studies are performed (Supplementary Material, Table S2). An extremely helpful feature now readily available to all investigators is the high depth control data set established by in exome aggregation consortium (http://exac.broadinstitute.org/). Bone fide HPE, Hartsfield and Kallmann syndrome mutations are frequently unique and absent in these public control databases.
While many aspects of genotype:phenotype correlations attributed to disease causing mutations in FGFR1 remain unresolved, we assert that the type of mutation is a key to our understanding of the phenotypic consequences. Gain-of-function mutations help explain the craniosynostosis malformations, loss-of-function mutations help explain the anosmia and infertility of IHH, and now dominant-negative mutations begin to explain the craniofacial, brain and limb anomalies characteristic of Hartsfield syndrome.
Material and Methods
Patient population and mutation screening
Clinically affected patients, with an emphasis on parent–child trios, ascertained world-wide have been prospectively collected over two decades under an NIH research protocol on brain research into HPE and related malformations. A similar research protocol is active in Brazil and all patients provided informed consent for molecular studies supervised by these respective institutional review boards. A collection of 206 HPE patients representative of the entire HPE clinical spectrum were screened by targeted capture next-generation sequencing (NIH Intramural Sequencing Center, NHGRI, NIH) for ligands and receptors of developmental genes, including FGFR1 and FGF8 (Roessler et al., manuscript in preparation). Their primary physicians provided additional clinical information as articulated in Table 1 allowing for genotype–phenotype comparisons.
Human cDNA constructs
Wild-type versions of both of human FGF8 (NM_033164.3) and FGFR1 (NM_023110.2) full-length cDNA clones were purchased (Creative Biogene) and subcloned into the mammalian expression vector pCS2+. Human FGF8 deletion constructs were generated by PCR allowing for the removal of patient-associated 15 bp indel, or the entire heparin-sulfate-binding region (dHBS). All human FGFR1 missense variant constructs were generated using the QuikChange Site-Directed Mutagenesis kit (Agilent Technologies) according to the manufacturer's instructions. The sequence integrity of all expression constructs was confirmed by Sanger sequencing.
Injection of MO and transcribed mRNAs into zebrafish embryos
The effects of Zebrafish fgfr1a and fgfr1b morphants has been previously studied (28) and, therefore, we ordered the identical MOs (Gene-Tools LLC). The MOs sequences for this study are: the fgfr1a translation-blocking MO (ATG MO), 5′-GCAGCAGCGTGGT CTTCATTATCAT-3′ and the fgfr1b splicing MO (SP MO), 5′- CAAGAGAGCGCAT GCTGCTTACGTA-3′. The amount of MO used in this study was 8 ng for fgfr1a MO and 4 ng for fgfr1b MO, and 10 ng of a standard control MO. Both zebrafish fgfr1a and fgfr1b MO sites are not present in human FGFR1 mRNA. To generate mRNA, wild-type (wt) and mutant forms of FGF8 and FGFR1 cDNAs were linearized by digestion with the restriction enzyme NotI, purified and subsequently transcribed with mMESSAGE mMACHINE SP6 Transcription Kit (Life Technologies). For the FGF8 RNA over-expression experiment, we used 0.4 pg of mRNA for all experiments, and for the FGFR1 mRNA injection studies were used 25 pg of mRNA for MO rescue and characterization of injected embryo phenotypes. All injection experiments were performed at one-cell stage embryos. Microinjected FGFR1 transcripts remained detectable by RT–PCR throughout the developmental stages investigated. All experiments were performed at least twice to assure that the effects were reproducible.
In situ hybridization experiments
Developmentally staged zebrafish (8 hpf and 24 hpf) embryos were fixed with 4% paraformaldehyde (Sigma) during an overnight incubation at 4°C. Whole-mount in situ hybridization was performed using the conventional alkaline-phosphatase-based single-color method (39). We used a gene hemoglobin beta embryonic-1.1 (hbbe1.1; previously known as beta-globin) (40) at 24 h, and chordin (chd; also known as chordino) (41) and dual specificity phosphatase 6 (dusp6; previously known as mkp3) (42) at 8 hpf.
Imaging studies
3% Methylcellulose (Sigma) was used as embedding solution for imaging live embryos and in situ stained embryos were cleared through a glycerol series and photographed using ZEISS AxioCam HRC Camera and AxioVision SE64 software.
Web Resources
Accession numbers and URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.ncbi.nlm.nih.gov/Omim/, (clinical cross-reference).
GeneBank, http://www.ncbi.nlm.nih.gov/GeneBank/ (for human gene annotation).
ECR Browser, http://ecrbrowser.dcode.org/, (selection of non-coding conserved regions).
University of California Santa Cruz Bioinformatics Site, http://genome.ucsc.edu/, (gene structure and annotation).
PolyPhen2, http://genetics.bwh.harvard.edu/pph2/bgi.shtml/, (used as definition of ref protein and ref transcript).
1000 genomes browser, http://browser.1000genomes.org/index.html/, (used for Polyphen and SIFT annotation and transcript and protein cross-reference).
PROVEAN, JCVI, http://provean.jcvi.org/, (PROVEAN and SIFT scores for functional annotation)
dbSNP, http://www.ncbi.nlm.nih.gov/projects/SNP/snp_ref.cgi/,(variant characterization).
Exome Variant Server, http://evs.gs.washington.edu/EVS/, (EVS, variant characterization).
Exome Aggregation Consortium, http://exac.broadinstitute.org/, (ExAC, variant frequency determination).
Supplementary Material
Funding
The NIH Sequencing Center (NISC) provided technical expertise and financial support through a Flagship award. This work was supported in part by the Division of Intramural Support, NHGRI, National Institutes of Health, USA.
Supplementary Material
Acknowledgements
We would like to thank the patients that participated in these studies and the continuing support of clinicians from around the world.
Conflict of Interest statement. None declared.
References
- 1.Ornitz D.M., Itoh N. (2015) The fibroblast growth factor signaling pathway. Wiley Interdiscip. Rev. Dev. Biol., 4, 215–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mason I. (2007) Initiation to endpoint: multiple roles of fibroblast growth factors in neural development. Nat. Rev. Neurosci., 8, 583–596. [DOI] [PubMed] [Google Scholar]
- 3.Hébert J.M., Fischell G. (2008) The genetics of early telencephalon patterning: some assembly required. Nat. Rev. Neurosci., 9, 678–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li C., Xu X., Nelson D.K., Williams T., Kuehn M., Deng C.X. (2005) FGFR1 function at the earliest stages of limb development plays and indispensible role in subsequent autopod development. Development, 132, 4755–4764. [DOI] [PubMed] [Google Scholar]
- 5.Muenke M., Schell U., Hehr A., Robin N., Losken H.A., Schinzel P., Pulleyn L.J., Rutland P., Reardon W., Malcolm S. et al. (1994) A common mutation in the fibroblast growth factor receptor 1 gene in Pfeiffer syndrome. Nat. Genet., 8, 269–274. [DOI] [PubMed] [Google Scholar]
- 6.Hartsfield J.K., Bixler D., DeMeyer W.E. (1984) Syndrome identification case report 119. Hypertelorism associated with holoprosencephaly and ectrodactyly. Clin. Dysmorphol., 2, 27–31. [Google Scholar]
- 7.Simonis N., Migeotte I., Lambert N., Perrazolo C., de Silva D.C., Dimitrov B., Heinrichs C., Jannsens S., Kerr B., Mortier G. et al. (2013) FGFR1 mutations cause Hartsfield syndrome, the unique associate of holoprosencephaly and ectrodactyly. J. Med. Genet., 50, 585–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dhamija R., Kirmani S., Wang X., Ferber M.J., Wieben E.D., Lazaridis K.N., Babovic-Vuksanovic D. (2014) Novel de novo heterozygous mutation in two siblings with Hartsfield syndrome: a case of gonadal mosaicism. Am. J. Med. Genet., 164A, 2356–2359. [DOI] [PubMed] [Google Scholar]
- 9.Villanueva C., Jacobson-Dickman E., Xu C., Manouvrier S., Dwyer A.A., Sykiotis G.P., Beenken A., Liu Y., Tommiska J., Hu Y. et al. (2015) Congenital hypogonadism with split hand/foot malformation: a clinical entity with high frequency of FGFR1 mutations. Genet. Med., 17, 651–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Dodé C., Levilliers J., Dupont J.M., De Paepe A., Le Dû N., Soussi-Yanicostas N., Coimbra R.S., Delmaghani S., Complain-Nouaille S., Baverel F. et al. (2003) Loss-of-function mutations in FGFR1 cause autosomal dominant Kallmann syndrome. Nat. Genet., 33, 463–465. [DOI] [PubMed] [Google Scholar]
- 11.Pitteloud N., Meysing A., Quinton R., Acierno J.S. Jr, Dwyer A.A., Plummer A., Fliers E., Boepple P., Hayes F., Seminara S. et al. (2004) Mutations in FGFR1 cause Kallmann syndrome with a wide spectrum of reproductive phenotypes. Mol. Cell. Endocrinol., 254–255, 60–69. [DOI] [PubMed] [Google Scholar]
- 12.Dodé C., Fouveaut C., Mortier F., Jannsens S., Bertherat J., Mahoudeau J., Kottler M.L., Chabrolle C., Gancel A., Françios I. et al. (2006) Novel FGFR1 Sequence variants in Kallmann syndrome, and genetic evidence that FGFR1c isoform is required in olfactory bulb and palate morphogenesis. Hum. Mutat., 28, 97–98. [DOI] [PubMed] [Google Scholar]
- 13.Tommiska J., Känsäkaski J., Christiansen P., Jørgensen N., Lawaetz J.G., Juul A. (2014) Genetics of congenital hypogonadotropic hypogonadism in Denmark. Eur. J. Med. Genet., 57, 345–348. [DOI] [PubMed] [Google Scholar]
- 14.Hero M., Laitinen E.M., Varimo T., Vaaralahti K., Tommiska J., Ravio T. (2015) Childhood growth of females with Kallmann syndrome and FGFR1 mutations. Clin. Endo., 82, 122–126. [DOI] [PubMed] [Google Scholar]
- 15.Muenke M., Beachy P.A. (2001) Holoprosencephaly. In Scriver C.R., Beaudet A.L., Valle D., Sly W.S. (eds), The Metabolic and Molecular Bases of Inherited Disease, 8th edn, Vol. 4 McGraw-Hill, New York, NY, pp. 6203–6230. [Google Scholar]
- 16.Roessler E., Muenke M. (2010) The molecular genetics of holoprosencephaly. Am. J. Med. Genet., 154C, 52–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Deng C.X., Wynshaw-Boris A., Shen M.M., Daugherty C., Ornitz D.M., Leder P. (1994) Murine FGFR-1 is required for early post-implantation growth and axial organization. Genes. Dev., 8, 3045–3057. [DOI] [PubMed] [Google Scholar]
- 18.Storm E.E., Garel S., Borello U., Hebert J.M., Martinez S., McConnell S.K., Martin G.R., Rubenstein J.L.R. (2006) Dose-dependent functions of fgf8 in regulating telencephalic patterning centers. Development, 133, 1831–1844. [DOI] [PubMed] [Google Scholar]
- 19.Paek H., Gutin G., Hébert J.M. (2009) FGF signaling is strictly required to maintain early telencephalic cell survival. Development, 136, 2457–2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hébert J.M., Lin M., Partanen J., Rossant J., McConnell S.K. (2003) FGF signaling through FGFR1 is required for olfactory bulb morphogenesis. Development, 130, 1101–1111. [DOI] [PubMed] [Google Scholar]
- 21.Chung W.C.J., Moyle S.S., Tsai P.S. (2008) Fibroblast growth factor 8 signaling through fibroblast growth factor receptor 1 is required for the emergence of gonadotropic-releasing hormone neurons. Endocrinology, 149, 4497–5003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Crossley P.H., Martin G.R. (1995) The mouse Fgf8 gene encodes a family of polypeptides and is expressed in regions that direct outgrowth and patterning in the developing embryo. Development, 121, 439–451. [DOI] [PubMed] [Google Scholar]
- 23.Meyers E.N., Lewandoski M., Martin G.R. (1998) An Fgf8 mutant allelic series generated by Cre- and Flp mediated recombination. Nat. Genet., 18, 136–141. [DOI] [PubMed] [Google Scholar]
- 24.Riley B.M., Mansilla M.A., Ma J., Daack-Hirsch S., Maher B.S., Raffensperger L.M., Russo E.T., Vieira A.R., Dodé C., Mohammadi M. et al. (2006) Impaired FGF signaling contributes to cleft lip and palate. Proc. Natl Acad. Sci. USA, 104, 4512–4517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Arauz R.F., Solomon B.D., Pineda-Alvarez D.E., Wendland J., Groupman A.L., Parsons J.A., Roessler E., Muenke M. (2010) A hypomorphic allele in the FGF8 gene contributes to holoprosencephaly and is allelic to gonadotropin-releasing hormone deficiency in humans. Mol. Syndrom., 1, 59–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Falardeau J., Chung W.C.J., Beenken A., Raivio T., Plummer L., Sidis Y., Jacobson-Dickman E.E., Eliseenkova E.V., Ma J., Dwyer A. et al. (2008) Decreased FGF8 signaling causes deficiency of gonadotropin-releasing hormone in humans and mice. J. Clin. Invest., 118, 2822–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.McCabe M.J., Gaston-Massuet C., Tziaferi V., Gregory L.C., Alatzoglou K.S., Signore M., Puelles E., Gerrelli D.I., Farooqi S., Raza J. et al. (2011) Novel FGF8 mutations associated with holoprosencephaly, craniofacial defects, and hypothalamo-pituitary dysfunction. J. Clin. Endocrinol. Met., 96, E1709–E1718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rohner N., Bercsényi M., Orbán L., Kolanczyk M.E., Linke D., Brand M., Nüsslein-Volhard C., Harris M.P. (2009) Duplication of fgfr1 permits Fgf signaling to serve as a target for selection during domestication. Curr. Biol., 19, 1642–1647. [DOI] [PubMed] [Google Scholar]
- 29.Olsen S.K., Li J.Y.H., Bromleigh C., Eliseenkova A.V., Ibrahimi O.A., Lao Z., Zhang F., Linhardt R.J., Joyner A.L., Mohammadi M. (2006) Structural basis by which alternative splicing modulates the organizer activity of FGF8 in the brain. Genes. Dev., 20, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu X., Komiya H., Chirino A., Faham S., Fox G.M., Arakawa T., Hsu B.T., Rees D.C. (1991) Three-dimensional structures of acidic and basic fibroblast growth factors. Science, 251, 90–93. [DOI] [PubMed] [Google Scholar]
- 31.Spivak-Kroizman T., Lemmon M.A., Dikic I., Ladbury J.E., Pinchasi D., Huang J., Jaye M., Crumley G., Schlessinger J., Lax I. (1994) Heparin-induced oligomerization of FGF molecules is responsible for FGF receptor dimerization, activation, and cell proliferation. Cell, 79, 1015–1024. [DOI] [PubMed] [Google Scholar]
- 32.Fürthauer M., Thisse C., Thisse B. (1997) A role for FGF-8 in the dorsoventral patterning of the zebrafish gastrula. Development, 124, 4253–4264. [DOI] [PubMed] [Google Scholar]
- 33.Amaya E., Musci T.J., Kirshner M.W. (1991) Expression of a dominant negative mutant of the FGF receptor disrupts mesoderm formation in Xenopus embryos. Cell, 66, 257–270. [DOI] [PubMed] [Google Scholar]
- 34.Peltekova I.T., Hurteau-Millar J., Armour C.M. (2014) Novel interstitial deletion of 10q24.3–25.1 associated with multiple congenital anomalies including lobar holoprosencephaly, cleft lip and palate, and hypoplastic kidneys. Am. J. Med. Genet. Part A., 164A, 3132–3136. [DOI] [PubMed] [Google Scholar]
- 35.Pitteloud N., Quinton R., Pearce S., Ravio T., Acierno J., Dwyer A., Plummer L., Hughes V., Seminara S., Cheng Y.Z. et al. (2007) Digenic mutations account for variable phenotypes in idiopathic hypogonadotropic hypogonadism. J. Clin. Invest., 117, 457–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Raivio T., Sidis Y., Plummer L., Chen H., Ma J., Mukherjee A., Jacobson-Dickman Q.R., Vliet V., Lavoie H., Van Hughes V.A. et al. (2009) Impaired fibroblast growth factor receptor 1 signaling as a cause of normosmic idiopathic hypogonadotropic hypogonadism. J. Clin. Endocrinol. Metab., 94, 4380–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Marinić M., Aktas T., Ruf S., Spitz F. (2013) An integrated holo-enhancer unit defines tissue and gene specificity of the FGF8 regulatory landscape. Dev. Cell., 24, 530–542. [DOI] [PubMed] [Google Scholar]
- 38.Sykiotisa G.P., Plummer L., Hughes V.A., Au M., Durrania S., Nayak Y.S., Dwyer A.A., Quinton R., Hall J.E., Gusella J.F. et al. (2010) Oligogenic basis of isolated gonadotropin-releasing hormone deficiency. Proc. Natl Acad. Sci. USA, 107, 15140–15144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kudoh T., Tsang M., Hukriede N.A., Chen X., Dedekian M., Clarke C.J., Kiang A., Schultz S., Epstein J.A., Toyama R. et al. (2001) A gene expression screeing in zebrafish embryogenesis. Genome Res., 11, 1979–1987. [DOI] [PubMed] [Google Scholar]
- 40.Brownlie A., Hersey C., Oates A.C., Paw B.H., Falick A.M., Witkowska H.E., Flint J., Higgs D., Jessen J., Bahary N. et al. (2003) Characterization of embryonic globin genes of the zebrafish. Dev. Biol., 255, 48–61. [DOI] [PubMed] [Google Scholar]
- 41.Miller-Bertoglio V.E., Fisher S., Sánchez A., Mullins M.C., Halpern M.E. (1997) Differential regulation of chordin expression domains in mutant zebrafish. Dev. Biol., 192, 537–550. [DOI] [PubMed] [Google Scholar]
- 42.Tsang M., Maegawa S., Kiang A., Habas R., Weinberg E., Dawid I.B. (2004) A role for mkp3 in axial patterning of the zebrafish embryo. Development, 131, 2769–2779. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.