

Abstract
Elevated sweat chloride levels, failure to thrive (FTT), and lung disease are characteristic features of cystic fibrosis (CF, OMIM #219700). Here we describe variants in CA12 encoding carbonic anhydrase XII in two pedigrees exhibiting CF-like phenotypes. Exome sequencing of a white American adult diagnosed with CF due to elevated sweat chloride, recurrent hyponatremia, infantile FTT and lung disease identified deleterious variants in each CA12 gene: c.908-1 G>A in a splice acceptor and a novel frameshift insertion c.859_860insACCT. In an unrelated consanguineous Omani family, two children with elevated sweat chloride, infantile FTT, and recurrent hyponatremia were homozygous for a novel missense variant (p.His121Gln). Deleterious CFTR variants were absent in both pedigrees. CA XII protein was localized apically in human bronchiolar epithelia and basolaterally in the reabsorptive duct of human sweat glands. Respiratory epithelial cell RNA from the adult proband revealed only aberrant CA12 transcripts and in vitro analysis showed greatly reduced CA XII protein. Studies of ion transport across respiratory epithelial cells in vivo and in culture revealed intact CFTR-mediated chloride transport in the adult proband. CA XII protein bearing either p.His121Gln or a previously identified p.Glu143Lys missense variant localized to the basolateral membranes of polarized Madin-Darby canine kidney (MDCK) cells, but enzyme activity was severely diminished when assayed at physiologic concentrations of extracellular chloride. Our findings indicate that loss of CA XII function should be considered in individuals without CFTR mutations who exhibit CF-like features in the sweat gland and lung.
Introduction
Persistently elevated sweat chloride concentration caused by loss of function mutations in the cystic fibrosis transmembrane conductance regulator (CFTR) gene is the diagnostic hallmark of cystic fibrosis (CF). Individuals with features of CF who do not carry any disease-causing CFTR alleles have been reported. These patients were phenotypically indistinguishable from CF patients carrying two known CF-causing mutations (1). Some individuals presenting a milder, atypical CF were found to carry variants that altered the function of subunits that form the epithelial sodium channel (ENaC) (2,3).
CA12, the gene encoding carbonic anhydrase (CA) XII, has been implicated as a cause of elevated sweat chloride concentration, failure to thrive in infancy, and recurrent hyponatremia in two consanguineous Bedouin kindreds (4,5). The same missense variant was identified in both pedigrees. However, the variant caused only a modest reduction (∼30%) in enzymatic activity (5), which was unexpected as autosomal recessive disorders are generally associated with severe loss of function variants. The authors speculated that the minimal reduction in CA XII function produced a phenotype limited to the sweat gland (OMIM #143860) (5,6).
In this study, we report the discovery and analysis of loss of function variants in CA12 that associate with elevated sweat chloride concentrations in two unrelated pedigrees. An adult proband in one pedigree also displayed pulmonary features that overlap with CF; namely recurrent pulmonary exacerbations, Pseudomonas in sputum cultures, and mild but distinct bronchiectasis upon high resolution chest CT scanning. These findings indicate that loss of CA XII activity is uncompensated in certain epithelia and that CA XII may play a key role in the function of the pulmonary airways as well as the sweat gland.
Results
Identification of CA12 variants segregating in two unrelated pedigrees
The proband in pedigree A (II:1, Fig. 1A) presented with failure to thrive at 2.5 months of age and sweat chloride concentrations ranging from 82 to 88 mEq/l. She was diagnosed with cystic fibrosis (CF) and prescribed pancreatic enzymes to improve growth. At 7 months of age she had an episode of hyponatremic dehydration requiring hospitalization (plasma sodium 120 mm/l upon admission). Spirometry from ages 7–9 years indicate three episodes of airway obstruction, with forced expiratory volumes (FEV1) and forced expiratory flows (FEF25-75%) falling below 80%. At age nine, repeat sweat chloride testing revealed elevated levels (range = 112–116 mEq/l) and serum IRT levels were within normal range, resulting in the discontinuation of pancreatic enzymes. Nasal potential difference (NPD) testing performed at this time reported aberrant chloride transport consistent with CF. It should be noted that NPD testing can have considerable technical variability and was standardized after this test was administered to proband A. Available clinical records between the ages of 19–22 revealed a persistent cough and cultures of bacteria common to CF patients, including Pseudomonas aeruginosa in the throat (age 19), Stenotrophomonas maltophilia in the throat (age 20), and Pseudomonas fluorescens in sputum (age 22). The proband's pulmonary exacerbations were often treated with a regiment consistent with her CF diagnosis, including bronchodilators, antibiotics, and steroids. At age 25, NPD testing repeated at the same clinical facility was not consistent with CF (response to low chloride and isoproterenol: −24 mV on right and −16 mV on left). The proband continued to be seen regularly at an accredited CF care center and reported compliance with daily respiratory treatments including aerosolized albuterol, acetylcysteine, hypertonic saline, and chest physiotherapy. High resolution chest CT scanning revealed mild bronchiectasis without scarring, inflammation, or mucus plugging (Fig. 2). Assessment of airway dilatation was confirmed by two adult CF pulmonologists and an additional interpretation by a radiologist who was masked to the clinical status of proband A. The proband has also been treated by a dermatologist for axillary hyperhidrosis. Exome sequencing was performed on the proband, her unaffected sister, and both parents. Average depth of coverage was 87X, and 93.2% of the targeted regions were covered at a depth ≥10X. No deleterious variants were found in CFTR or the three genes (SCNN1A, SCNN1B, and SCNN1G) that encode the epithelial sodium channel (ENaC). Loss of ENaC function can cause pseudohypoaldosteronism, a secondary and rare cause of elevated sweat chloride concentration (2,7). Two variants within CA12 were discovered in trans (compound heterozygosity) in the proband (II:1): a variant inherited from her father (I:1) in the canonical splice acceptor site of exon 10, c.908-1 G>A (rs148438059, chr15:63,619,433, ClinVar accession# SVC000255965) and an insertion variant of four nucleotides inherited from her mother (I:2), c.859_860insACCT (chr15:63,631,029-63,631,030, ClinVar accession# SVC000255963) in exon 8. The splice acceptor variant is found in heterozygosity in 53 individuals in the Exome Aggregation Consortium (ExAC) variant browser (8) with a global MAF of 0.00471%. It is the most common predicted deleterious CA12 variant found in ExAC. The proband's insertion mutation was not found in ExAC. CA12 variants were confirmed in all family members via Sanger sequencing.
Figure 1.

Segregation of putative deleterious CA12 variants in two unrelated families. Filled shapes indicate status as affected and arrows indicate the proband in each family. The ages indicate the age of the individual at the time of exome sequencing. (A) Pedigree A: A white American family in which the proband exhibits consistently elevated sweat chloride concentration and bronchiectasis. The proband carries a splice acceptor variant c.908-1 G>A and an insertion frameshift variant c.859_860insACCT. (B) Pedigree B: An Omani family with first-cousin parents as indicated by the double horizontal line. The proband and affected sister exhibit elevated sweat chloride concentrations and have experienced multiple episodes of hyponatremic dehydration. Only the proband and affected sister are homozygous for c.363 C>A (p.His121Gln).
Figure 2.

Axial plane high resolution CT images of proband A. Examples of enlargement of the airways (tram-tracking and signet rings) are indicated by arrows. This bronchiectasis is seen in the absence of mucus plugging, scar tissue, or surrounding inflammation. It is unknown as to whether this mild pulmonary phenotype would be more exacerbated had the proband not been undergoing daily preventative lung therapies (aerosolized albuterol, hypertonic saline, chest physiotherapy, etc.) due to her original CF diagnosis.
In a second unrelated family, a six year old Omani boy (proband B, II:3, Fig. 1B) presented with a history of hyponatremic dehydration, elevated sweat chloride, and bilateral hyperkeratosis of the heels. Hyponatremic dehydration was alleviated with administration of Pedialyte and unrestricted access to dietary salt. Four sweat chloride measurements ranged from 90 to 110 mEq/l. Pulmonary function tests and fecal elastase measurements were within the normal ranges, ruling out chronic pulmonary and exocrine pancreatic insufficiency associated with CF. Aldosterone measurements excluded pseudohypoaldosteronism. Clinical diagnostic sequencing of the coding and intron flanking regions of CFTR and SCNN1A encoding the α subunit of ENaC in proband B did not detect sequence variations predicted to be deleterious. An 11 year old sister (II:1) of proband B initially considered to be asymptomatic was discovered to have a sweat chloride of 130 mEq/l. At a two year follow up, this sister was found to have developed a phenotype concordant with that of proband B, reporting episodes of hyponatremic dehydration as well as mild bilateral hyperkeratosis of the heels. Hyperkeratosis of the heels was not observed in proband A or the previously reported patients (6) and could be due to unrelated deleterious recessive alleles that may be present in this consanguineous pedigree. Unaffected siblings in pedigree B had sweat chloride measurements within the normal range for the referring laboratory (<50 mEq/l, personal communication Jozef Hertecant). No evidence of respiratory disease was reported in either patient; however, we were unable to obtain high resolution chest CT scans. Proband B was reported to have a normal chest X-ray at age 11 and his affected sister had no pulmonary testing of any kind. CFTR, SCNN1B, and SCNN1G were excluded via genetic linkage analysis of all individuals in pedigree B with the assumption of recessive inheritance. Exome sequencing was conducted on proband B, his affected sister, and all unaffected siblings in pedigree B. The average depth of coverage was 34X, and 62.5% of the targeted regions were covered at a depth ≥8X. A previously unreported variant c.363 C>A (chr15:63,637,742, ClinVar accession# SVC000255964) in exon 4 of CA12 was found in homozygosity only in the two affected individuals (Fig. 1B). It is predicted to cause a substitution of His at codon 121 with Gln (p.His121Gln). Segregation of the CA12 c.363 C>A variant in an autosomal recessive inheritance pattern was confirmed in all family members in pedigree B via Sanger sequencing.
CA XII is localized to the basolateral membrane of ductal epithelia in sweat gland and apical membrane in airway epithelia
To ascertain whether CA XII was expressed in the organs affected in the two probands, namely the sweat gland and the airways, immunohistochemistry (IHC) of normal skin and lung sections was performed. IHC of whole skin tissue showed robust sweat gland expression of CA XII in the basolateral compartment of the reabsorptive ductal cells (Fig. 3B and C). Basolateral CA XII staining in the reabsorptive sweat duct was distinguishable from apically localized CA II, a ubiquitously expressed cytosolic carbonic anhydrase (Fig. 3D and E). To determine if CA XII is expressed in the airways, IHC of lung was performed and showed robust apical localization of CA XII in bronchiolar epithelia (Fig. 3F and G). Of note, IHC of CA XII in the lung was performed using the same anti-CA XII antibody (ProteinTech #15180-1-AP) that was utilized for IHC of the sweat gland. The varying subcellular localization of CA XII (basolateral within the sweat gland; apical within bronchioles), may indicate alternative roles for this protein in reabsorptive or secretory epithelial membranes.
Figure 3.

Immunohistochemical staining of CA XII and CA II in human skin and lung. (A) Diagram depicting longitudinal view of sweat gland components with red line indicating a hypothetical plane used to generate slices for the micrographs shown in (B)–(E). (B) Two populations of sweat gland resorptive ducts (‘RD’) and secretory coils (‘SC’) immunostained for CA XII and counter-stained with hematoxylin and eosin. The resorptive ducts (n = 9 different cross-sections captured in this panel) show positive staining for CA XII in a two cell thick layer of cuboidal epithelia cells. The myoepithelial cells surrounding the secretory coils (n = 3 different cross-sections captured in this panel) show light staining of CA XII that may be non-specific. Magnification of this micrograph is 100×. (C) Enlargement of CA XII positive staining of the basolateral membrane in resorptive ductal cells from (B). (D) Positive staining of apically localized control protein CA II in sweat ducts and secretory coils. Magnification of this micrograph is 100×. (E) Enlargement of resorptive ducts from (E) showing apical localization of CA II. (F) CA XII positive staining of the luminal edge of a bronchiole cross-section. Magnification of this micrograph is 100×. (G) Positive staining of apically localized CA XII in the terminal bar of bronchial epithelia. Magnification of this micrograph is 400×.
The c.908-1 G>A and c.859_860insACCT variants found in proband A generate aberrant RNA transcripts
The two variants identified in proband A were predicted to affect RNA processing. To evaluate this supposition, respiratory epithelia from the inferior nasal turbinate was obtained for RNA and functional studies. CA XII is expressed in nasal epithelial cells (Supplementary Material, Table S1) and respiratory epithelia of the nasal turbinates have been used as a proxy for respiratory epithelia of the airways (9). The CA12 gene is composed of 11 exons that constitute its primary mRNA transcript (Fig. 4A). Alternative splicing of CA12 has been observed in native and cancerous tissues by both RT-PCR and RNA sequencing (10). The most common alternative isoform of CA12 (CCDS# 10186) removes exon 9, a small exon composed of 33 bp, allowing the downstream transcript to retain the same reading frame. A review of publicly available RNA-sequencing splicing data from the Human Protein Atlas (11) reveals this isoform is predominately expressed in brain, and select other tissues (Supplementary Material, Table S1). Two additional rare isoforms of CA12 result from skipping of exons 9 and 10, or exon 10 only. These alternative CA12 transcripts are of very low abundance in nasal and bronchial epithelial cells compared to the full-length transcript with 11 exons. PCR of cDNA derived from nasal epithelial cell RNA of proband A generated DNA products of 1069, 980 and 947 bp. Each product was gel purified and subject to Sanger sequencing. The 1069 bp product corresponded to full-length CA12 transcript bearing the insertion c.859_860insACCT. This variant introduces a frameshift that is predicted to lead to the incorporation of 49 novel residues following codon 287 and a premature termination codon (PTC) in exon 11 (predicted size 336 residues; Fig. 4B). Despite the presence of a PTC, the transcript was stable due to the location of the PTC in the last exon of CA12, thereby allowing the transcript to evade nonsense mediated RNA decay (12). The splice site variant found in proband A, c.908-1 G>A, was predicted to cause misplicing of CA12 exon 10 as it alters an invariant nucleotide of the canonical 3′ splice acceptor site. Indeed, the 980 bp amplicon was CA12 transcript missing exon 10 and the 947 bp product was an alternatively spliced CA12 transcript missing exons 9 and 10 (Fig. 4C). Loss of exon 10 was predicted to result in a frameshift beginning at codon 302 and translational read-through of the native termination codon. A novel termination codon in the 3′ UTR occurs at amino acid position 413. The resulting protein is predicted to be composed of the first 302 amino acids of CA XII followed by 111 novel residues, 89 of which are translated from the 3′ UTR. Skipping of exons 9 and 10 would add the same 111 novel residues but the frameshift would start at codon 291 (predicted size 402 residues). Finally, amplification from exon 8 to exon 10 and Sanger sequencing verified that all transcripts bearing exons 9 and 10 contained the c.859_860insACCT insertion (data not shown). In summary, all CA12 mRNA transcripts in the nasal epithelial RNA of proband A were abnormal and each was predicted to generate aberrant CA XII protein.
Figure 4.

Effect of CA12 variants upon RNA processing in nasal epithelial cells from proband A. (A) Exon and intron structure of CA12 with locations of proband A variants identified by NGS. Rectangles represent exons and the lines through the center of the rectangles represent the genomic axis. Variants are indicated with triangles and HGVS cDNA names. The gray rectangle indicating exon 9 is spliced out in an alternative CA12 transcript whose function and tissue distribution is unknown. (B) (Left) Electropherogram of Sanger sequencing of cDNA reverse transcribed from RNA extracted from proband A cultured nasal epithelial. Sequencing detected a heterozygous insertion variant c.859_860insACCT on a transcript retaining alternatively spliced exon 9. A second transcript detected by sequencing does not bear the insertion, retains exon 9, and is consistent with transcript lacking exon 10 as a result of the in trans variant c.908-1 G>A. Presence of this second transcript in this sequencing reaction is likely due to imperfect isolation of the similarly sized transcripts by gel purification as transcript bearing the insertion is only 89 bp longer than transcripts lacking exon 10 only. (Right) Gene models depict RNA processing of the insertion variant and the predicted gene product. The insertion variant causes a frameshift starting in exon 8, a premature termination codon in exon 11, and was predicted to generate a misfolded protein targeted by ERAD. Hashed rectangles indicate an altered exonic reading frame. (C) (Left) Electropherogram of Sanger sequencing detecting transcript missing exons 9 and 10, and a second transcript missing exon 10 only, due to heterozygous splice acceptor variant c.908-1 G>A. Imperfect isolation of transcripts in this reaction is due to alternatively spliced exon 9 which is only 33 bp long. (Right) Gene models depict the two processed RNAs and protein products lacking exon 10 observed by RT-PCR: one with exon 9 alternatively spliced out and one retaining exon 9. This variant is predicted to cause skipping of exon 10, a frameshift resulting in read-through of the native stop codon, translation of 267 nucleotides from the 3′ UTR, and a misfolded protein targeted by ERAD. Hashed rectangles indicate an altered exonic reading frame and filled rectangles indicate translation of the 3′ UTR.
CA12 variants found in proband A generate unstable CA XII protein
Expression vectors with CA12 cDNA modified to correspond to each of the three transcripts observed in the nasal epithelial cells of proband A, the missense variant p.His121Gln (c.363 C>A) observed in proband B, and a previously described Bedouin missense variant p.Glu143Lys (c.427 G>A) were transfected into HEK293 cells and lysates were subjected to analysis by Western blot. Wild-type (WT) CA XII was present in both unglycosylated (39 kDa) and fully glycosylated (43 kDa) forms (13) (Fig. 5, lane 2). CA XII protein was severely reduced in the lysate of cells transfected with CA12 expression vectors bearing the insertion variant c.859_860insACCT (7.2–13.3% of WT) and only a single band of the predicted mass of the unglycosylated protein (37.9 kDa) was observed (Fig. 5, lane 4). CA XII lacking residues encoded by exon 10 and exons 9 and 10 were barely visible (Fig. 5, lanes 5, and 6). CA XII bearing the missense variants p.His121Gln, and p.Glu143Lys generated protein of a molecular mass comparable to WT and unglycosylated and glycosylated forms were observed (Fig. 5, lanes 7 and 8). CA XII with p.Glu143Lys had a higher fraction of unglycosylated protein, suggesting a possible effect of the amino acid substitution on processing and post-translation modifications. These results indicate that each of the changes in amino acid composition due to the CA12 variants found in proband A cause substantial instability in CA XII.
Figure 5.

Expression of transiently transfected wild-type and mutant CA XII protein in HEK 293 cells. Western blot of cell lysates extracted from HEK 293 cells (top) following transfection with CA XII expression vectors. Probing with anti-CA XII antibody (Novus) shows unglycosylated (39 kDa) and glycosylated (43 kDa) protein generated from transfections with WT CA12 (lane 2), c.363 C>A (lane 7) and c.427 G>A (lane 8). The insertion variant in proband A (c.859_860insACCT) produced a faint band of approximately 38 kDa while CA XII cDNA missing exon 10 and exons 9 and 10 sequence generated only faint bands. The negative control in the first lane is a mock transfection. The third lane has no lysate. GAPDH loading control (bottom) shows loading of cell lysates.
Nasal respiratory epithelial cells from proband A demonstrate CFTR-mediated chloride transport
Cultured epithelial cells from proband A and controls were mounted in Ussing chambers for short circuit current measurements. To increase the driving force for chloride secretion through CFTR, the apical membrane was hyperpolarized by administration of amiloride that inhibits sodium current conducted by epithelial sodium channels. To specifically examine chloride currents mediated by CFTR, calcium-activated chloride channels were inhibited by DIDS (4, 4′-diisothiocyanoto-stilbene -2, 2′-disulfonic acid). Application of DIDS did not result in a significant change in current in any sample. CFTR was activated by elevating cellular levels of cAMP with forskolin and 3-isobutyl-1-methylxanthine (IBMX). Upon treatment with forskolin and IBMX (‘F+I’), the change in CFTR-mediated chloride transport in nasal epithelia from proband A (9.34 and 9.15 µA/cm2) were higher than that observed in nasal epithelial from a CF subject tested concurrently (2.1 µA/cm2). The values in proband A are consistent with short circuit measures of nasal epithelia from other non-CF and CF subjects (14). Substantial reduction in the current of cells from proband A upon addition of GlyH-101 (−18.8 and −21.5 µA/cm2) is consistent with the chloride secretion being mediated by CFTR. Together, these findings suggest that loss of CA XII does not ablate CFTR-mediated chloride secretion across nasal respiratory epithelia.
P.His121Gln and p.Glu143Lys mutations cause near complete loss of enzyme activity of CAXII
As the missense variants permitted the generation of stable full-length protein, immunocytochemistry and confocal microscopy was utilized to test whether either variant affected the subcellular localization of CA XII. Expression in polarized epithelial Madin-Darby canine kidney (MDCK) cells revealed that WT CA XII localized to basolateral membranes (green, n = 7, Fig. 6, left panel). CA XII bearing p.His121Gln (green, n = 8, Fig. 6, middle panel) and p.Glu143Lys (green, n = 13, Fig. 6, right panel) showed basolateral localization indistinguishable from that of WT CA XII. Comparable staining patterns were observed when immunocytochemistry was performed with alternative CA XII antibodies (rabbit: Sigma Prestige; mouse: Novus) that detected different extracellular CA XII epitopes (data not shown). Since the p.His121Gln and p.Glu143Lys mutants were localized to plasma membranes, we tested the effect of each variant on CA function by measuring carbonic anhydrase enzyme activity. Carbonic anhydrase activity is the reversible rate of CO2 hydration. When assayed in a 2 mm NaCl solution, the activity of CA XII bearing p.His121Gln is reduced by 84.6 ± 3.6% compared to WT (n = 11) while the enzymatic activity of CA XII with p.Glu143Lys is reduced by 24.4 ± 4.9%, consistent with the approximate 30% reduction in activity previously reported under the same conditions (5) (Fig. 7). When assayed at physiological salt concentrations, the enzyme activity of p.His121Gln was reduced by 99.2 ± 0.5% compared to WT, and the activity of the p.Glu143Lys mutant was reduced by 97.1 ± 1.2% compared to WT (n = 9) (Fig. 7). The enzyme activities of p.His121Gln and p.Glu143Lys were not statistically different (P = 0.12; t-test) when assayed in the presence of 100 mm NaCl. These findings reveal a chloride-sensitive abolition of carbonic anhydrase enzyme activity for CA XII p.His121Gln and p.Glu143Lys mutants compared to WT.
Figure 6.
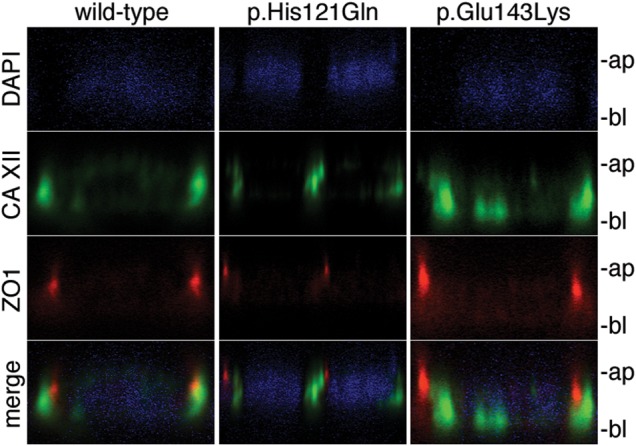
Subcellular localization of WT and mutant CA XII in polarized MDCK cells. Fluorescent co-staining of (left) WT CA XII (green), (center) p.His121Gln (green), and (right) p.Glu143Lys (green) with endogenous tight junction protein ZO1 (red) and nuclear stain DAPI (blue) in polarized MDCK cells imaged in the xz-plane. This micrograph reveals primarily lateral staining of CA XII; however, basal and lateral staining were observed for WT (n = 7 different micrographs), p.His121Gln (n = 8 different micrographs), and p.Glu143Lys (n = 13 different micrographs). The apical membrane is indicated by ‘ap’ and the basal membrane is indicated by ‘bl’.
Figure 7.

Enzymatic activity of CA XII proteins bearing p.His121Gln or p.Glu143Gly substitutions. Enzymatic activity of CA XII mutants p.His121Gln and p.Glu143Gly was determined by assaying the reversible rate of hydration of CO2 as previously described in the absence (left) or presence (right) of physiological concentration of NaCl and normalizing to wild-type. Boxes represent the interquartile range (IQR) and the horizontal bars within the boxplots represent the median. The top whisker represents the 75th percentile plus 1.5 times the IQR and the bottom whisker represents the 25th percentile minus 1.5 times the IQR. Circles represent statistical outliers. (Boxplot statistics calculated in R.).
Discussion
Each of the CA12 variants reported in the three individuals described here with elevated sweat chloride, recurrent hyponatremia and failure to thrive in infancy cause severe loss of CA XII activity. The two variants found in proband A generate mRNA transcripts that are missing nucleotides that form transmembrane domains and enable dimerization via a key glycine zipper motif (13). Translation of cDNA that replicate the mRNA transcripts identified in proband A generated unstable protein products in heterologous cells. On the other hand, CA XII proteins bearing the missense variant found in this study and the previously reported missense variant were stable and, in each case, localized to the basolateral membrane of MDCK cells, consistent with native CA XII location in kidney cells (15). Point mutation energy modeling by FoldX suggested that both missense variants should affect CA XII structure and catalytic function. Modeling indicated that the secondary amine of histidine 121 is essential for tetrahedral coordination of the zinc ion within the catalytic domain. In the WT conformation, the H121–zinc bond distance is ∼2.1Å (Supplementary Material, Fig. S1). When mutated to glutamine as in p.His121Gln, the distance from zinc to the hydroxyl of the carboxyl group is 3.042Å. This increased distance likely precludes formation of a coordinating bond, and the zinc ion is instead coordinated by a free hydroxide ion (red sphere). In the extracellular aqueous environment, this coordination would only be transient, potentially leading to poor catalytic activity. The distances of other residues, such as the highly conserved second shell glutamic acid 143, are also altered by p.His121Gln by a magnitude similar to that reported in previous studies of the Bedouin p.Glu143Lys mutation (4). CA XII bearing the p.Glu143Lys mutation was particularly sensitive to inhibition by chloride, as previously reported (5). Given that the active site of CA XII lies on the extracellular face of the basolateral compartment in the sweat duct, enzymatic activity of both mutants was assayed in the presence of increasing NaCl concentrations. The concentration of NaCl which most closely mimics the enzyme's native physiological environment is 100 mm NaCl (16). At this concentration, catalytic activity of CA XII bearing each missense mutation was reduced to less than 3% of WT activity. As the affected individuals are homozygous for the CA12 missense mutations, it is reasonable to conclude that the sweat gland dysfunction observed in each is due to near complete loss of CA XII activity. This conjecture is supported by the studies of proband A, where mutations in each CA12 gene lead to severe instability of CA XII protein.
Robust expression of CA XII in lung epithelia and the observation of bronchiectasis in proband A suggest a role for this protein in the maintenance of airways. Elevated sweat chloride concentration indicates aberrant chloride transport in the sweat duct and is a consistent feature of the three individuals carrying the loss of function CA12 variants reported here, as well as in 11 individuals homozygous for the p.Glu143Lys mutation reported previously (6). However, CFTR-mediated chloride transport appeared to be intact in the nasal respiratory epithelia as determined by in vitro and in vivo methods, suggesting that other pathways of ion transport in the airways might be disrupted by the loss of CA XII. Given the importance of CAs in the maintenance of pH via bicarbonate metabolism, the mechanism underlying airway damage could be related to aberrantly low pH of airway surface liquid (ASL) due to loss of bicarbonate production or transport. The pH of the ASL has been shown to be integral to the proper expansion and processing of mucins which play a key role in CF-related bronchiectasis (17). Alternatively, the bronchiectasis observed in proband A might be unrelated to the loss of CA XII function. Spirometry measurements in all tested individuals homozygous for the p.Glu143Lys mutation were reported as normal (6) and chest X-rays of proband B were reported to be normal at age 11; however, lung function measures and chest X-rays were also normal throughout the life of proband A up to her current age of 25. Detection of abnormal airway dilatation in proband A required high resolution CT scanning; therefore, it is possible that bronchiectasis remains undetected in the previously reported patients with loss of CA XII function and the affected individuals in pedigree B. Without comparison chest CTs from the affected individuals in pedigree B, it cannot be determined if proband B and his affected sister manifest bronchiectasis similar to that observed in proband A. Further, if loss of CA XII function does lead to bronchiectasis, the possibly ameliorating impact of CF-specific airway treatments is an important question. Proband A has undergone routine airway clearance, therapies, and antibiotic courses since being diagnosed with CF as an infant. It is possible that a lifetime of diligent pulmonary monitoring and treatments managed by an accredited CF care center minimized the effect of bronchiectasis upon pulmonary function. However, estimating the impact of respiratory therapy on degree of airway dilatation is difficult without study of additional CA XII-deficient individuals manifesting pulmonary disease. Indeed, the identification of additional individuals with loss of CA XII function and high resolution chest CT scanning will clarify the role of CA XII in the airways. Given the strong causative connection between aberrant chloride transport and bronchiectasis in CF, CA XII loss of function should be considered as a potential explanation for non-CF bronchiectasis.
Loss of CA XII function in a patient with respiratory disease in humans suggests a previously unsuspected role for this isozyme in the lung. Although each individual CA isozyme follows a tissue-specific expression pattern (18,19), RNA expression studies show that multiple isozymes can be expressed in certain tissues (20). Loss of function of one isozyme may be compensated for by other isozymes in certain tissues. This explanation is offered for the lack of a phenotype in patients with loss of function mutations in CA I (21). Our findings show that isozyme redundancy does not compensate for loss of CA XII function in the sweat gland. Further, although other transmembrane CA isozymes such as CA IV have been localized to the plasma face of lung microcapillaries, our results suggest that CA XII may also play an important role in the maintenance of the airways.
IHC of the sweat gland revealed CA XII to be highly expressed in the resorptive duct in basolateral distribution consistent with its location in other epithelia, namely endometrium, kidney, and large intestine (15,22,23). Faint staining was observed at the apical membrane which may be non-specific signal or evidence of dual CA XII localization. However, the pattern of CA XII staining was distinctly different for that of CA II, which was discretely localized near apical membranes of the ductal cells. CA XII was only found on basolateral membranes of polarized MDCK cells. In contrast, CA XII was discretely localized to the apical regions of normal airway. The distribution of CA XII does not appear to be due to non-specific signal as two antibodies that detect different antigenic regions of CA XII revealed the same immunolocalization pattern. Localization to the terminal bar is consistent with CA XII location in other tissues, including the bronchus and fallopian tubes (11). A possible factor in the different localization of CA XII could be its involvement in one of the transport metabolon complexes formed between CA isozymes and bicarbonate transporters that facilitate the exchange of bicarbonate across membranes (24). Bicarbonate transport metabolons comprised of CA isozymes and anion exchangers have so far been described for three of the transmembrane isozymes: CA IV associates with AE1 (24,25), CA IX associates with AE2 (26), and CA XIV associates with AE3 (27). AE1 and CA IV have been localized to both basolateral and apical membranes in different cell types in the kidney (25). Since the kidney can absorb and secrete ions including bicarbonate, it is possible that the different localization of the AE1/CA IV metabolon may be related to the direction of ion flow. However, to date, no metabolon interaction has been reported for the remaining transmembrane isozyme, CA XII.
Our study suggests a mechanism for the well-established salt wasting complication of topiramate, a CA inhibitor and anticonvulsant commonly prescribed to people suffering from epilepsy. Elevated sweat chloride concentration is an established phenomenon observed in epileptic children being treated with topiramate (28). In these children, CF was clinically and/or molecularly excluded as being the cause of this increase in sweat chloride value, and the effect disappeared when topiramate treatment ended. Investigations into the inhibition potency of topiramate across the α family of CAs have shown that topiramate is a strong inhibitor of CA XII, and not the other transmembrane isozymes CA IV, IX, and XIV (29,30). These pharmacologic observations are consistent with our hypothesis regarding the role of CA XII in the maintenance of proper ion composition in the sweat gland. Topiramate is also a strong inhibitor of the ubiquitous and highly active cytoplasmic isozyme CA II. Correspondingly, individuals being treated with topiramate have been observed to develop renal tubular acidosis (31), a hallmark feature of CA II deficiency syndrome.
In summary, the individuals studied here demonstrate that severe loss of function mutations in CA12 cause an autosomal recessive disorder affecting chloride and sodium resorption in the sweat duct. The observation of airway dilatation in proband A suggests a possible molecular etiology for some forms of non-CF bronchiectasis, a disease that affects over 110 000 individuals in the USA (32).
Materials and Methods
Recruitment
Family A was recruited and consented into the study Genome-wide Sequencing to Identify the Genes Responsible for Mendelian Disorders at Johns Hopkins University (IRB# NA_00045758). Family B was referred to Johns Hopkins University via private communication of Dr. Jozef Hertecant at Tawam Hospital, United Arab Emirates. All members of family A were consented into the Molecular Genetics of Cystic Fibrosis (IRB# NA_00050260).
Linkage exclusion assays
Three highly polymorphic deCODE STRs were selected for each locus to be excluded (CA12, and SCNN1B and SCNN1G, which lie in close proximity to one another) from the STS Marker track on the UCSC Genome Browser. Each marker was no more than 1 Mb from either end of the locus to be excluded. Primer sequences from the STS Marker track were run through BLAT to verify specificity. Oligonucleotides were synthesized by IDT. Forward primers were fluorescently labeled with 6-FAM. STRs were PCR amplified from genomic DNA and amplification products were separated on an ABI Prism 3100 Genetic Analyzer by automated capillary electrophoresis. Fragment sizing and visualization were performed using ABI GeneMapper software. Haplotype phasing was performed by manual inspection. If the proband and at least one unaffected sibling were found to be IBD2 for a locus, it was deemed to be excluded, given an assumption that the disease phenotype follows an autosomal recessive Mendelian inheritance pattern.
DNA sample acquisition, exome sequencing, and CA12 genotyping
Peripheral blood was obtained from all consented individuals in pedigree A and genomic DNA was extracted by a phenol/chloroform protocol. Exome capture was performed on all siblings within the pedigree using the Agilent SureSelect Human All Exon (51 Mb), and 100 bp paired-end reads were subsequently obtained from an Illumina HiSeq 2500 system as part of a study within the Baylor-Hopkins Center for Mendelian Genomics conducted by the Center for Inherited Disease Research. Reads were aligned to the hg19 reference genome using Burrows-Wheeler Aligner software (33) and subsequent alignment processing was completed using SAMtools (34), PicardTools, and GATK softwares (35,36) in a manner similar to (37). Filtering of variants was conducted via custom scripts, and variant prioritization was conducted using Enlis Genomic Research software. To verify mutations, a 654 bp region encompassing c.908-1 G>A and c.859_860insACCT was amplified from genomic DNA from the proband A and her mother by PCR using the following primers (IDT): 5′F GCCCTGTACTGCACACACAT and 3′R AGGATGATGCCCAGACTCAG. PCR products were purified using QIAquick PCR purification kit (Qiagen), and then sequenced using the Applied Biosystems 3730xl DNA Analyzer. The resulting sequences were analyzed via the Sequencher analysis suite (Gene Codes). Genomic DNA extracted from peripheral blood was obtained from all consented individuals in pedigree B. Exome capture was performed on all siblings using the Illumina Truseq Exome Enrichment kit (62 Mb), and 90 bp paired-end reads were subsequently obtained from an Illumina HiSeq 2000 system (Otogenetics). Exome sequencing data analysis was conducted in a manner similar to pedigree A, however, variant prioritization was conducted using VAAST software (38). CA12 (RefSeq# NM_001218.4) mutations and mode of inheritance were verified via Sanger sequencing. A 475 bp region encompassing c.363 C>A was amplified from genomic DNA of all siblings in pedigree B by PCRs using the following primers (IDT): 5′F GTCCCATGCTCTGGTGTTATC and 5′R CTTTCCAAGGTGAACCAAGAA. PCR products were purified, sequenced, and analyzed as described for pedigree A. The resulting sequences were analyzed via the Sequencher analysis suite (Gene Codes).
It should be noted that all variant nomenclature is specific to CA12 nucleotide and CA XII amino acid numbering and represent the minus strand sequence unless otherwise specified. All genomic coordinates are specific to hg19. All variants discovered in this study have been submitted to ClinVar.
IHC of CA XII in human sweat duct and lung
Frozen discarded unidentified skin and lung obtained from the Division of Surgical Pathology, Johns Hopkins Hospital, Baltimore, MD, were embedded in Optimal Cutting Temperature (OCT) compound and held at −70°C prior to sectioning. Six µm cryo-sections were mounted onto uncoated microscope slides. Staining with H&E (Sigma, St Louis, MO, USA) for 1 min was performed for morphological evaluation. The rest of the slides were stored at −70°C until use. Sections were fixed for 10 min in pre-cooled acetone followed by 5 min peroxidase block at room temperature to quench the endogenous peroxidase activity. Sections were further incubated in serum-free protein block (Dako # X0909) for 20 min at room temperature and incubated overnight at 4°C with the following primary antibodies: anti-rabbit CA12 (ProteinTech # 15180-1-AP) and anti-rabbit CA2 (LSBio # C138796). Relevant universal negative control antibodies: mouse (Dako # N1698) and rabbit (Dako # IR600) were used to ascertain non-specific staining. After washing, staining was performed using EnVision + System-HRP (AEC) kit from Dako (# K4008). Sections were covered with peroxidase-labeled polymer for 30 min. For visualization of the reaction, sections were developed in AEC+substrate-chromogen for 5–20 min. After washing, the sections were counterstained with hematoxylin (Dako # S3309) for 30 s, cleared, and mounted on Faramount aqueous mounting medium (Dako # S3025). Samples were analyzed under an Olympus BX51 microscope.
Nasal epithelial culture and Ussing chamber studies
Nasal epithelia from the proband of pedigree A were expanded and cultured using previous described method (39). Cells were mounted in Ussing chambers and studied as previously described (40). Apical and basolateral chambers contained the same bathing solution with symmetrical Cl− concentrations. CFTR-mediated Cl− current were measured using a previously described protocol (40).
Identification of CA12 mutant transcripts
RNA was isolated from expanded nasal brushings of proband A by standard Trizol-chloroform method. cDNA was made using RT-PCR (Qiagen iScript) and served as template for CA12 amplification. PCR products of CA12 transcript isoforms were separated by 3% agarose gel electrophoresis and subject to Sanger sequencing.
Development of mutant CA12 expression vectors
Full length wild-type (WT) CA12 cDNA in bacterial pBS II expression vector was generously provided by Dr. William Sly. The proband B variant c.363 C>A (p.His121Gln) was introduced into full length WT CA12 cDNA in bacterial pBSII expression vector using the QuikChange II XL Site-Directed Mutagenesis kit reagents and protocol (Agilent). Mutagenesis products were confirmed by Sanger sequencing. Each cDNA was removed from the bacterial expression vector using restriction enzymes KpnI and BamHI, purified by gel electrophoresis, and ligated into the eukaryotic pcDNA5 FRT expression vector. Subcloning was confirmed by Sanger sequencing. Proband A variant c.859_860insACCT was introduced into pcDNA5 FRT expression vector bearing CA12 cDNA using the QuikChange II XL Site-Directed Mutagenesis kit reagents and protocol (Agilent). A previously described CA12 missense variant, c.427 G>A (p.Glu143Lys), deemed a moderate hypomorph and reported in a large consanguineous Bedouin kindred manifesting a highly similar phenotype (4,5), was also introduced into the full length CA12 cDNA. To replicate the consequences of the splice acceptor variant c.908-1 G>A found in proband A, CA12 cDNAs lacking exon 10 and lacking exons 9 and 10 were custom synthesized (GeneWiz, South Plainfield, NJ, USA). Since exon 10 skipping was predicted to result in a frameshift and subsequent read-through of the natural termination codon, 287 bp of the CA12 3′ UTR were included in each construct. Both constructs were removed from the bacterial pUC57 expression vector using restriction enzymes KpnI and EcoRV, purified by gel electrophoresis, and ligated into eukaryotic pcDNA5 FRT expression vector. Subcloning was confirmed by Sanger sequencing.
Expression of WT CA XII and mutant proteins in HEK293 cells
HEK293 cells were transiently transfected with 500 ng of WT and mutant CA12 vectors using Lipofectamine 2000 Reagent and standard protocol (Invitrogen). Cells were lysed 24 h post-transfection. Western blotting of cell lysates was performed using anti-CA XII antibody (Novus #NBP1-81668), and loading control GAPDH antibody (Sigma #G9545).
Immunocytochemistry of CA XII in a polarized epithelial cell line
Madin-Darby canine kidney (MDCK) cells were transiently transfected with 1.6 µg of CA12 cDNA using 3.2 µl Lipofectamine 2000 Transfection Reagent and protocol (Invitrogen). Cells were fixed one day post-transfection with 4% paraformaldehyde for 20 min and rinsed with 1X PBS. Cells were permeabilized with 0.5% Triton X-100 for 5 min, then rinsed with 1X PBS, and blocked overnight at 4C with 2.5% goat serum. Cells were immunostained using rabbit anti-CA XII primary antibody (ProteinTech #15180-1-AP) diluted 1:200 and anti-ZO1 primary antibody (Invitrogen) with a conjugated anti-mouse red fluorophore diluted 1:200, followed by incubation in anti-rabbit secondary antibody diluted 1:50. Findings were validated by staining with different primary anti-CA XII antibodies from Sigma Prestige (mouse) and Novus (rabbit). All antibodies were diluted in 2.5% goat serum blocking solution. Cells were washed in 1X PBS three times for 10 min following the 90 min primary antibody incubation. Cells were washed in 1X PBS four times for 15 min following the 30 min secondary antibody incubation. Cells were mounted on microscope slides with Molecular Probes ProLong Gold Antifade Reagent with DAPI and viewed with the Zeiss LSM510-Meta single-point confocal laser-scanning microscope and Zen imaging software.
Carbonic anhydrase enzyme activity assay
Cell pellets were lysed by sonication in 300 µl lysis buffer (PBS containing protease inhibitors, 1 mm each of PMSF, o-phenanthroline, EDTA, benzamidine-hydrochloride and iodoacetamide plus 1% NP-40) and left on ice. The media were centrifuged to remove dead cells. The protein concentration of cell lysates was determined by microLowry's procedure using bovine serum albumin as a standard (41). The carbonic anhydrase activity was determined using Maren's procedure (42) as described (43). To reflect extracellular physiological chloride concentration (16), 100 mm NaCl was utilized.
Supplementary Material
Funding
This work was supported by the National Institutes of Health [R01 DK044003]; and the Cystic Fibrosis Foundation [CUTTIN13A2].
Supplementary Material
Acknowledgements
This study would not be possible without the participation of the patients and families described in this manuscript. The authors would like to acknowledge Diane Acquazzino for acquisition of patient records.
Conflict of Interest statement. None declared.
References
- 1.Groman J.D., Meyer M.E., Wilmott R.W., Zeitlin P.L., Cutting G.R. (2002) Variant cystic fibrosis phenotypes in the absence of CFTR mutations. N. Engl. J. Med., 347, 401–407. [DOI] [PubMed] [Google Scholar]
- 2.Sheridan M.B., Fong P., Groman J.D., Conrad C., Flume P., Diaz R., Harris C., Knowles M., Cutting G.R. (2005) Mutations in the beta subunit of the epithelial Na+ channel in patients with a cystic fibrosis-like syndrome. Hum. Mol. Genet., 14, 3493–3498. [DOI] [PubMed] [Google Scholar]
- 3.Groman J.D., Karczeski B., Sheridan M., Robinson T.E., Fallin M.D., Cutting G.R. (2005) Phenotypic and genetic characterization of patients with features of ‘nonclassic’ forms of cystic fibrosis. J. Pediatr., 146, 675–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Muhammad E., Leventhal N., Parvari G., Hanukoglu A., Hanukoglu I., Chalifa-Caspi V., Feinstein Y., Weinbrand J., Jacoby H., Manor E. et al. (2011) Autosomal recessive hyponatremia due to isolated salt wasting in sweat associated with a mutation in the active site of Carbonic Anhydrase 12. Hum. Genet., 129, 397–405. [DOI] [PubMed] [Google Scholar]
- 5.Feldshtein M., Elkrinawi S., Yerushalmi B., Marcus B., Vullo D., Romi H., Ofir R., Landau D., Sivan S., Supuran C.T. et al. (2010) Hyperchlorhidrosis caused by homozygous mutation in CA12, encoding carbonic anhydrase XII. Am J. Hum. Genet., 87, 713–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feinstein Y., Yerushalmi B., Loewenthal N., Alkrinawi S., Birk O.S., Parvari R., Hershkovitz E. (2014) Natural history and Clinical manifestations of hyponatremia and hyperchlorhidrosis due to carbonic anhydrase XII deficiency. Horm. Res. Paediatr., 81, 336–342. [DOI] [PubMed] [Google Scholar]
- 7.Riepe F.G., Van Bemmelen M.X., Cachat F., Plendl H., Gautschi I., Krone N., Holterhus P.M., Theintz G., Schild L. (2009) Revealing a subclinical salt-losing phenotype in heterozygous carriers of the novel S562P mutation in the α subunit of the epithelial sodium channel. Clin. Endocrinol., 70, 252–258. [DOI] [PubMed] [Google Scholar]
- 8.Lek M., Karczewski K., Minikel E., Samocha K., Banks E., Fennell T., O'Donnell-Luria A., Ware J., Hill A., Cummings B. et al. (2015) Analysis of protein-coding genetic variation in 60,706 humans. bioRxiv, 030338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mosler K., Coraux C., Fragaki K., Zahm J.-M., Bajolet O., Bessaci-Kabouya K., Puchelle E., Abély M., Mauran P. (2008) Feasibility of nasal epithelial brushing for the study of airway epithelial functions in CF infants. J. Cyst. Fibros., 7, 44–53. [DOI] [PubMed] [Google Scholar]
- 10.Haapasalo J., Hilvo M., Nordfors K., Haapasalo H., Parkkila S., Hyrskyluoto A., Rantala I., Waheed A., Sly W.S., Pastorekova S. (2008) Identification of an alternatively spliced isoform of carbonic anhydrase XII in diffusely infiltrating astrocytic gliomas. Neuro Oncol., 10, 131–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Uhlen M., Fagerberg L., Hallstrom B.M., Lindskog C., Oksvold P., Mardinoglu A., Sivertsson A., Kampf C., Sjostedt E., Asplund A. et al. (2015) Proteomics. Tissue-based map of the human proteome. Science, 347, 1260419. [DOI] [PubMed] [Google Scholar]
- 12.Maquat L.E. (2004) Nonsense-mediated mRNA decay: splicing, translation and mRNP dynamics. Nat. Rev. Mol. Cell Biol., 5, 89–99. [DOI] [PubMed] [Google Scholar]
- 13.Whittington D.A., Waheed A., Ulmasov B., Shah G.N., Grubb J.H., Sly W.S., Christianson D.W. (2001) Crystal structure of the dimeric extracellular domain of human carbonic anhydrase XII, a bitopic membrane protein overexpressed in certain cancer tumor cells. Proc. Natl. Acad. Sci. USA, 98, 9545–9550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Mall M., Wissner A., Seydewitz H.H., Hübner M., Kuehr J., Brandis M., Greger R., Kunzelmann K. (2000) Effect of genistein on native epithelial tissue from normal individuals and CF patients and on ion channels expressed in Xenopus oocytes. Br. J. Pharmacol., 130, 1884–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Parkkila S., Parkkila A.-K., Saarnio J., Kivelä J., Karttunen T.J., Kaunisto K., Waheed A., Sly W.S., Türeci Ö., Virtanen I. (2000) Expression of the membrane-associated carbonic anhydrase isozyme XII in the human kidney and renal tumors. J. Histochem. Cytochem., 48, 1601–1608. [DOI] [PubMed] [Google Scholar]
- 16.Lee M. (2009) Basic Skills in Interpreting Laboratory Data. ASHP. [Google Scholar]
- 17.Quinton P.M. (2008) Cystic fibrosis: impaired bicarbonate secretion and mucoviscidosis. Lancet, 372, 415–417. [DOI] [PubMed] [Google Scholar]
- 18.Scriver C.R., Kaufman S. (2001) The Carbonic Anhydrase II Deficiency Syndrome: Osteopetrosis with Renal Tubular Acidosis and Cerebral Calcification. In Scriver C.R., Beaudet A.L., Valle D., Sly W.S. (eds), Metabolic and Molecular Bases Inherited Disease. McGraw-Hill, Inc., NY, Vol. IV pp. 1667–1724. [Google Scholar]
- 19.Sly W.S., Shah G. (2001) Renal Tubular Acidosis. In Scriver C.R., Beaudet A.L., Sly W.S., Valle D. (eds), The Metabolic and Molecular Bases Inherited Disease. McGraw-Hill, Inc., NY, Vol. IV, pp. 5331–5343. [Google Scholar]
- 20.Gilmour K. (2010) Perspectives on carbonic anhydrase. Comp. Biochem. Physiol. A Mol. Integr. Physiol., 157, 193–197. [DOI] [PubMed] [Google Scholar]
- 21.Tashian R.E., Hewett-Emmet D., Dodgson S.J., Forster R.E., Sly W.S. (1984) The value of inherited deficiencies of human carbonic anhydrase isozymes in understanding their cellular roles. Ann. N.Y. Acad. Sci., 429, 262–275. [DOI] [PubMed] [Google Scholar]
- 22.Kivelä A., Parkkila S., Saarnio J., Karttunen T.J., Kivelä J., Parkkila A.-K., Waheed A., Sly W.S., Grubb J.H., Shah G. (2000) Expression of a novel transmembrane carbonic anhydrase isozyme XII in normal human gut and colorectal tumors. Am. J. Pathol., 156, 577–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Karhumaa P., Parkkila S., Türeci Ö., Waheed A., Grubb J., Shah G., Parkkila A.-K., Kaunisto K., Tapanainen J., Sly W. (2000) Identification of carbonic anhydrase XII as the membrane isozyme expressed in the normal human endometrial epithelium. Mol. Hum. Reprod., 6, 68–74. [DOI] [PubMed] [Google Scholar]
- 24.McMurtrie H., Cleary H., Alvarez B., Loiselle F., Sterling D., Morgan P., Johnson D., Casey J. (2004) The bicarbonate transport metabolon. J. Enzyme Inhib. Med. Chem., 19, 231–236. [DOI] [PubMed] [Google Scholar]
- 25.Sterling D., Alvarez B.V., Casey J.R. (2002) The extracellular component of a transport metabolon. Extracellular loop 4 of the human AE1 Cl-/HCO3- exchanger binds carbonic anhydrase IV. J. Biol. Chem., 277, 25239–25246. [DOI] [PubMed] [Google Scholar]
- 26.Morgan P.E., Pastorekova S., Stuart-Tilley A.K., Alper S.L., Casey J.R. (2007) Interactions of transmembrane carbonic anhydrase, CAIX, with bicarbonate transporters. Am. J. Physiol. Cell Physiol., 293, C738–C748. [DOI] [PubMed] [Google Scholar]
- 27.Casey J.R., Sly W.S., Shah G.N., Alvarez B.V. (2009) Bicarbonate homeostasis in excitable tissues: role of AE3 Cl−/HCO3− exchanger and carbonic anhydrase XIV interaction. Am. J. Physiol. Cell Physiol., 297, 1091–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guglani L., Sitwat B., Lower D., Kurland G., Weiner D.J. (2012) Elevated sweat chloride concentration in children without cystic fibrosis who are receiving topiramate therapy. Pediatr. Pulmonol., 47, 429–433. [DOI] [PubMed] [Google Scholar]
- 29.Winum J.Y., Poulsen S.A., Supuran C.T. (2009) Therapeutic applications of glycosidic carbonic anhydrase inhibitors. Med. Res. Rev., 29, 419–435. [DOI] [PubMed] [Google Scholar]
- 30.Supuran C.T. (2008) Carbonic anhydrases: novel therapeutic applications for inhibitors and activators. Nat. Rev. Drug Discov., 7, 168–181. [DOI] [PubMed] [Google Scholar]
- 31.Mirza N., Marson A.G., Pirmohamed M. (2009) Effect of topiramate on acid–base balance: extent, mechanism and effects. Br. J. Clin. Pharmacol., 68, 655–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weycker D., Edelsberg J., Oster G., Tino G. (2005) Prevalence and economic burden of bronchiectasis. Clin. Pulm. Med., 12, 205–209. [Google Scholar]
- 33.Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R. (2009) The Sequence Alignment/Map format and SAMtools. Bioinformatics, 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M. (2010) The Genome Analysis Toolkit: a MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res., 20, 1297–1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Depristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., Del Angel G., Rivas M.A., Hanna M. et al. (2011) A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet., 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ng S.B., Turner E.H., Robertson P.D., Flygare S.D., Bigham A.W., Lee C., Shaffer T., Wong M., Bhattacharjee A., Eichler E.E. et al. (2009) Targeted capture and massively parallel sequencing of 12 human exomes. Nature, 461, 272–276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Yandell M., Huff C.D., Hu H., Singleton M., Moore B., Xing J., Jorde L.B., Reese M.G. (2011) A probabilistic disease-gene finder for personal genomes. Genome Res., 21, 1529–1542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li X., Rossen N., Sinn P.L., Hornick A.L., Steines B.R., Karp P.H., Ernst S.E., Adam R.J., Moninger T.O., Levasseur D.N. et al. (2013) Integrin alpha6beta4 identifies human distal lung epithelial progenitor cells with potential as a cell-based therapy for cystic fibrosis lung disease. PLoS One, 8, e83624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li X., Comellas A.P., Karp P.H., Ernst S.E., Moninger T.O., Gansemer N.D., Taft P.J., Pezzulo A.A., Rector M.V., Rossen N. et al. (2012) CFTR is required for maximal transepithelial liquid transport in pig alveolar epithelia. Am. J. Physiol. Lung Cell. Mol. Physiol., 303, L152–L160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Peterson G.L. (1979) Review of the Folin phenol protein quantitation method of Lowry, Rosebrough, Farr and Randall. Anal. Biochem., 100, 201–220. [DOI] [PubMed] [Google Scholar]
- 42.Maren T.H. (1960) A simplified micromethod for the determination of carbonic anhydrase and its inhibitors. J. Pharmacol. Exp. Ther., 130, 26–29. [PubMed] [Google Scholar]
- 43.Sundaram V., Rumbolo P., Grubb J., Strisciuglio P., Sly W.S. (1986) Carbonic anhydrase II deficiency: diagnosis and carrier detection using differential enzyme inhibition and inactivation. Am. J. Hum. Genet., 38, 125. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.