

Abstract
Background
Autophagy is a cellular degradation process that is highly evolutionarily-conserved in yeast, plants, and animals. In plants, autophagy plays important roles in regulating intracellular degradation and recycling of amino acids in response to nutrient starvation, senescence, and other environmental stresses. Foxtail millet (Setaria italica) has strong resistance to stresses and has been proposed as an ideal material for use in the study of the physiological mechanisms of abiotic stress tolerance in plants. Although the genome sequence of foxtail millet (Setaria italica) is available, the characteristics and functions of abiotic stress-related genes remain largely unknown for this species.
Results
A total of 37 putative ATG (autophagy-associated genes) genes in the foxtail millet genome were identified. Gene duplication analysis revealed that both segmental and tandem duplication events have played significant roles in the expansion of the ATG gene family in foxtail millet. Comparative synteny mapping between the genomes of foxtail millet and rice suggested that the ATG genes in both species have common ancestors, as their ATG genes were primarily located in similar syntenic regions. Gene expression analysis revealed the induced expression of 31 SiATG genes by one or more phytohormone treatments, 26 SiATG genes by drought, salt and cold, 24 SiATG genes by darkness and 25 SiATG genes by nitrogen starvation. Results of qRT-PCR showing that among 37 SiATG genes, the expression level of SiATG8a was the highest after nitrogen starvation treatment 24 h, suggesting its potential role in tolerance to nutrient starvation. Moreover, the heterologous expression of SiATG8a in rice improved nitrogen starvation tolerance. Compared to wild type rice, the transgenic rice performed better and had higher aboveground total nitrogen content when the plants were grown under nitrogen starvation conditions.
Conclusions
Our results deepen understanding about the characteristics and functions of ATG genes in foxtail millet and also identify promising new genetic resources that should be of use in future efforts to develop varieties of foxtail millet and other crop species that have resistance to nitrogen deficiency stress.
Electronic supplementary material
The online version of this article (doi:10.1186/s12864-016-3113-4) contains supplementary material, which is available to authorized users.
Keywords: Foxtail millet, Autophagy-associated genes, Expression pattern, Functional identification, Nitrogen starvation
Background
The yield and quality of crops are adversely influenced under adverse environmental conditions, such as high salt, drought, and nutrient starvation. In order to survive, plants have evolved various capacities that enable them to resist or adapt to environmental changes. Autophagy is a highly evolutionarily-conserved cellular degradation process that is induced by nutrient starvation and it is essential for recycling of the cellular cytoplasmic contents and the breakdown of damaged proteins [1, 2]. In autophagy, the macromolecules or organelles are sequestered into a double-membraned vesicle termed an autophagosome. The content of autophagosome ranges from damaged proteins to obsolete organelles. This content is finally delivered into a vacuole (in yeast and plants) or a lysosome (animals) for degradation and nutrient recycling during periods of stress and starvation [1, 2]. Autophagy maintains cellular homeostasis during stressful conditions and plays important roles in various cellular processes. Therefore, the molecular and physiological mechanisms of plant autophagy are attracting more and more research attention.
Identification of genes that participate in autophagy should establish a foundation for furthering our understanding of the molecular mechanism and the potential roles of autophagy in different physiological processes. So far, most of our knowledge about autophagy comes from studies of yeast. In yeast, the number of ATG genes currently stands at 41 [3–7]. A group of autophagy-defective mutants have been isolated and used to study the autophagy process in yeast [8–12]. The studies of these yeast mutants have revealed the molecular mechanisms underlying autophagy [13]. The plant and mammalian homologues of the yeast ATG genes have been found, and these show conservation of the core ATG mechanism during evolution. Recently, some ATG homologues have been found and functionally analyzed in the genomes of Arabidopsis, rice, and maize [14–17]. Through genome sequencing, a total of 33 ATG homologues are known to exist in the rice (Oryza sativa) genome, and more than 30 ATG genes have been identified in Arabidopsis thaliana, tobacco, maize [16–19]. Autophagy is involved in a variety of processes in plant metabolism and development, including apoptotic processes, senescence, nutrient starvation, vacuole biogenesis, and seed development. Autophagy is related to responses to biotic and abiotic stresses such as viral infection and oxidative, salt, and drought stress [1, 20–22]. Under normal conditions, autophagy is kept at a low level, but it can be activated by nutrient starvation, which is known to induce the expression of some ATG genes [23]. The rice mutant Osatg7-1 has reduced biomass production and nitrogen use efficiency compared with wild type plants [24]. Under nitrogen starvation, the maize atg12 mutant shows enhanced leaf senescence and stunted ear development. Nitrogen partitioning studies revealed that seed yield and the nitrogen-harvest index were significantly decreased in the atg12 mutant [17]. Some Arabidopsis autophagy-defective mutants are also known to be particularly sensitive to nutrient starvation. Transgenic Arabidopsis plants of an ATG-knockdown line, RNAi-ATG18a, have been shown to be more sensitive than wild type plants to nutrient-limiting conditions [25]. The Arabidopsis atg mutants, such as atg4a4b-1, atg5-1, atg7-1, atg9-1 and atg10-1 exhibited abnormal phenotypes (chlorosis, decreased bolting time, and early senescence) under nutrient starvation [21, 22, 25–29]. ATG8, one of the conserved autophagy proteins, is a lipid-conjugated ubiquitin-like protein that functions as a scaffold for membrane expansion during autophagosome formation [30, 31]. In contrast to the single ATG8 gene present in the genome of budding yeast, there are nine and six ATG8 orthologues present in the Arabidopsis and rice genomes, respectively [16, 32]. In plant cells, AtAtg8 synthetic substrates can be efficiently incorporated into autophagosomes, and this processing is known to require AtAtg4s. Synthetic AtAtg8 substrate could be processed efficiently in an AtAtg4-dependent manner. These results indicate that, in vivo, the synthetic AtAtg8 substrate is used efficiently in the biogenesis of autophagosomes [33]. Under nitrogen starvation, the expression and transcription patterns of OsATG8 homologues were different in various tissues, indicating that the functions among homologues maybe different [16]. In plants, ATG8 genes are known to function efficiently in cellular processes in young, non-senescing tissues, under both favorable growth conditions and under starvation stress [34]. TdATG8, from wild emmer wheat, may play a role in a drought tolerance mechanism and TdATG8 is known to be a positive regulator of osmotic and drought stress responses [35]. Expression of AtATG8 in Arabidopsis increased plant sensitivity to mild salt and osmotic stress [36]. Heterologous expression of GmATG8c in Arabidopsis conferred tolerance to nitrogen deficiency and increased yields [37]. To date, plant autophagy research has been concentrated on model plant species, few studies have explored the functions of ATG-like genes in non-model crop species.
Foxtail millet (Setaria italica L.) is a member of the Poaceae grass family and is an ancient crop in China. Foxtail millet is an important food and fodder crop in arid regions because of its characteristics such as resistance to drought and nutrition deficiency stress. Published plant genome sequences have facilitated research on foxtail millet [38–40]. However, there have been few articles reporting studies related to stress-related gene function in foxtail millet so far. In this study, 37 ATG genes in the foxtail millet genome (internally annotated as Setaria italica autophagy- associated genes; SiATGs) were identified. We determined their chromosomal locations, predicted the protein structures and analyzed the expression patterns of these 37 SiATG genes with qRT-PCR. The results showed that among SiATG genes the expression level of SiATG8a was the highest after low nitrogen treatment 24 h and overexpression of SiATG8a enhanced the tolerance of transgenic rice to nitrogen starvation stress. Our results deepen understanding about the characteristics and functions of ATG genes in foxtail millet and also identify promising new genetic resources for foxtail millet and other crop species that have resistance to nitrogen deficiency stress.
Results
Bioinformatics analysis of 37 SiATG genes: identification and chromosomal distribution
A total of 37 putative ATG genes were identified in foxtail millet via genome-wide analysis (Additional file 1: Table S1). Their domains were further confirmed with the Pfam database (http://pfam.xfam.org/). The lengths of the amino acid sequences, the predicted molecular weights, and the predicted isoelectric points (pI) (Additional file 1: Table S1 and S2) differed greatly among the 37 SiATG proteins. The coding sequences, genomic sequences, protein sequences and nucleotide sequences upstream of the initiation codon for transcription for the 37 SiATGs were all downloaded from (www.phytozome.net/). The deduced SiATG protein sequences ranged from 91 amino acids (SiATG12) to 2474 amino acids (SiTOR), and the corresponding molecular weights varied from 10 to 278 kDa. The predicted isoelectric points varied from 4.53 (SiATG3a) to 9.51 (SiATI12a). According to information on the phytozome website, ten proteins were found to be splice variants of primary transcripts encoding autophagy-associated proteins: SiATG1, SiATG5, SiATG8b, SiATG8d, SiATG10, SiATG16, SiTOR, SiVPS15, SiVPS34, and SiNBR1 (Additional file 1: Table S1).
Foxtail millet has nine chromosomes, ranging in size from 35.9 Mb (chromosome 6) to 58.9 Mb (chromosome 9). The physical map positions of the 37 SiATG genes in the nine chromosomes of foxtail millet are presented in Fig. 1. The specific location of each SiATG gene on the chromosomes is given in Additional file 1: Table S1. Subcellular localization predictions revealed that most of the SiATG proteins were predicted to be localized to the cytoplasm or the nucleus (Additional file 1: Table S2).
Fig. 1.
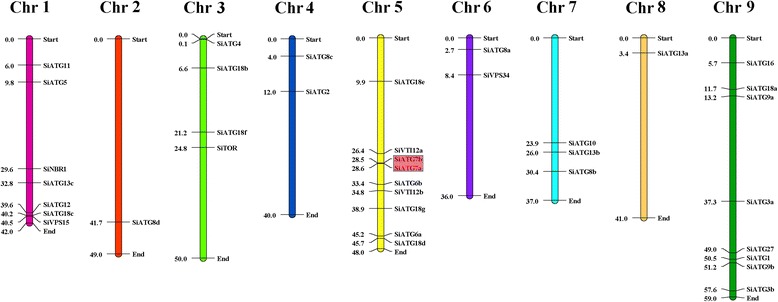
Distribution of 37 SiATG genes onto the nine foxtail millet chromosomes. Localization of the foxtail millet ATG genes on the foxtail millet chromosomes. Tandem duplicated genes on a particular chromosome are indicated with colored boxes. Chromosomal distances are given in Mb
Phylogenetic analysis of foxtail millet ATGs with ATGs of other plant species, and gene structure analysis
In recent years, the autophagy-associated genes in multiple plant species have been identified. We chose several representational plants for analysis, including Arabidopsis thaliana (At), Oryza sativa (Os), Nicotiana tabacum (Nt) and Zea mays (Zm) and constructed the phylogenetic trees of the autophagy-associated genes in these species, in addition to Setaria italica (Si). The multiple sequence alignments of the ATG protein sequences were conducted with the ClustalX 1.81 program, using the default multiple alignment parameters. The unrooted phylogenetic trees were constructed using MEGA5.0 software by maximum likelihood method with 1000 bootstrap replicates to compare the evolutionary relationships. As shown in Fig. 2, the phylogenetic analysis suggested that each SiATG protein sequence was highly similar to their homologues in other plant species. Since a good number of the internal branches were observed to have high bootstrap values, it clearly shows the derivation of statistically reliable pairs of possible orthologues proteins sharing similar functions from a common ancestor. The phylogenetic tree also revealed that majority of foxtail millet SiATG families distribution predominates with species bias, they are more closely related to those in grass species (O. sativa and Z. mays,). These results are consistent with the present understanding of plant evolutionary history [41]. Close association of SiATG families with their counter-parts in other plants, expressions or functions for some of which have reported, may be an implication of sequence conservation and evidence to their similar biological roles. Being a rational systematic approach, such phylogeny-based function prediction has been applied for prediction of stress-responsive proteins in other species like rice, Arabidopsis and maize.
Fig. 2.
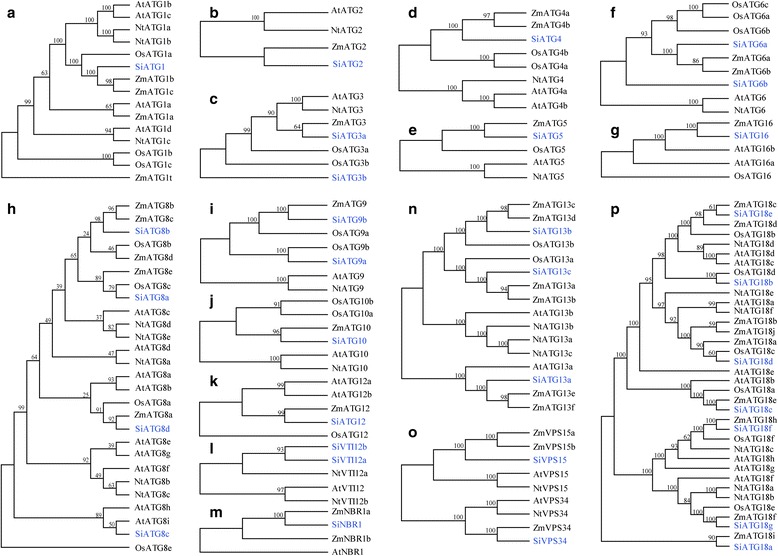
Phylogenetic analysis of foxtail millet and other plant ATGs. Phylogenetic relationships of ATGs from S. italica (Si) with those of N. tabacum (Nt), A. thaliana (At), O. sativa (Os), and Z. mays (Zm). The tree was constructed from an analysis conducted with MEGA 5.1 software using a maximum likelihood method. The names in blue color are the foxtail millet ATGs
As with other plants, some foxtail millet ATG members are encoded by small gene families, implying redundancy and subfunctionalization (ATG8, ATG13, ATG18). Other foxtail millet ATG members are encoded by a single gene (ATG1, ATG2, ATG4, ATG5, ATG10, ATG11, ATG12, ATG16,ATG27, TOR, VPS15, VPS34,and NBR1), making them attractive research materials for reverse genetics studies [17].
Exon-intron structural divergence within gene families can play pivotal roles in the evolution of multi-gene families [42]. Exon-intron structural analysis revealed that members of some ATG subfamilies have the same number of exons and similar exon-intron structures. Examples of this scenario include SiATG9a/ SiATG9b and SiATG8a/ SiATG8b/ SiATG8c/ SiATG8d (Fig. 3). To further explore the origins and evolutionary processes of the foxtail millet ATG genes, a comparative synteny map between the foxtail millet and rice genomes was constructed (Fig. 4). Rice is an important model plant species, and the genome-wide identification and expression analysis of the rice ATG genes has been reported [16]. Thus, through comparative genomics analysis, the probable functions of the foxtail millet ATG genes can be inferred. Synteny analysis indicated that 59 % (22/37) of the SiATG genes showed synteny with their orthologues in the rice genome, and there were 28 pairs that unambiguously exist in both the foxtail millet and rice genomes (e.g.,SiATG3a/OsATG3b, SiATG5/OsATG5, and SiATG7a/OsATG7). Although there is a close evolutionary relationship between foxtail millet and rice, some SiATG genes were not mapped onto any colinear blocks with rice (e.g., SiATG8a and SiATG8c). This result suggests that the chromosomes of foxtail millet have undergone extensive rearrangements during genomic evolution.
Fig. 3.
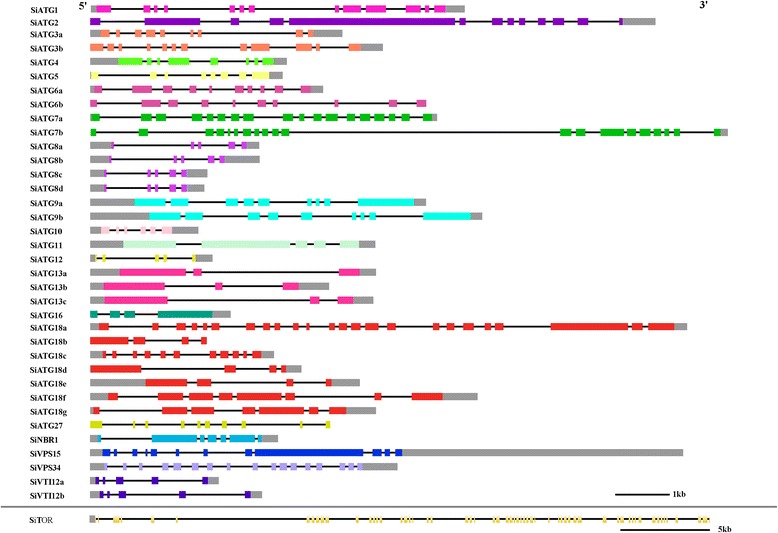
Exon–intron structure of the foxtail millet ATG genes. The exon-intron structures of the SiATG genes were determined by comparing the coding sequences and the corresponding genomic sequences using the Gene Structure Display Server (GSDS) program. Gray and colored boxes represent untranslated upstream / downstream regions and coding regions, respectively. Lines indicate introns
Fig. 4.
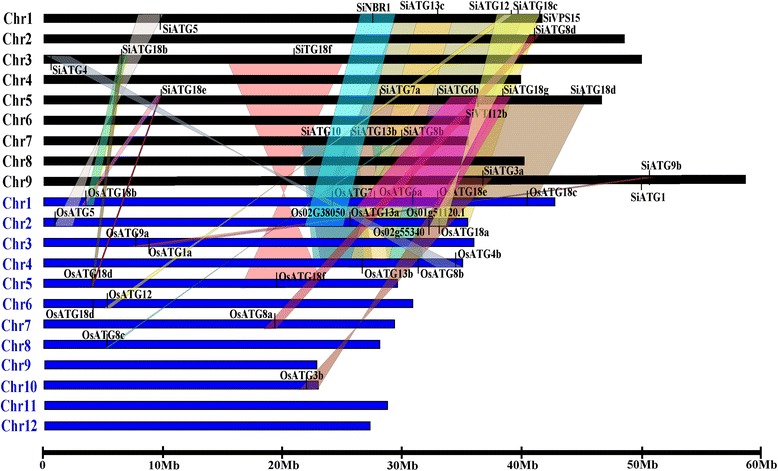
Synteny analysis of the ATG genes between foxtail millet and rice. Foxtail millet and rice chromosomes are depicted as black and blue bars, respectively. Foxtail millet and rice ATG genes are indicated by vertical black lines. Colored bars denote syntenic regions between foxtail millet and rice chromosomes. A twisted colored bar indicates that syntenic regions have opposite orientations
Duplication and divergence rates of the SiATG genes
The distribution of the 37 SiATG genes on the nine foxtail millet chromosomes was asymmetric. Only one pair of SiATG genes (SiATG7a /SiATG7b) occurred as tandem repeats. Another 10 genes (SiATG13b/ SiATG13c, SiATG9a/ SiATG9b, SiATG6a/ SiATG6b, SiATG8a/ SiATG8b, SiATG18b/ SiATG18e) occurred as five pairs of segmentally-duplicated genome regions (Table 1). The Ka/Ks ratios of these five segmental duplicated gene pairs ranged from 0.028 to 0.420, with an average of 0.210, whereas the Ka/Ks ratio of the aforementioned tandem duplication gene pair was 0.340 (Table 1). One tandem repeat and five segmental duplicated genome regions belonged to paralogous pairs, and among six paralogous pairs, four pairs showed Ks values from 0.932 to 1.674, suggesting that these duplications might have occurred 71.7 million year ago (MYA) in the foxtail millet genome (Table 1). Based on the Ka/Ks ratios, the dates of the four paralogous pair duplication events were calculated to have occurred from between 71.72 to 128.76 MYA (Table 1). The Ks values of the SiATG7a/ SiATG7b and SiATG6a/ SiATG6b paralogous pairs were 0.0679 and 0.3522, respectively, suggesting that the dates of these two paralogous pair duplication events might have occurred about 5.22 and 27 MYA, respectively. We also calculated the Ka/Ks ratios of the duplicated genes between orthologous gene pairs of foxtail millet and maize (31 pairs) (Additional file 1: Table S4), rice (28 pairs) (Additional file 1: Table S5), and sorghum (29 pairs) (Additional file 1: Table S6).
Table 1.
Ka/Ks analysis and estimation of the absolute dates for the duplication events between the duplicated SiATG homologues
| Duplicated pair | Ks | Ka | Ka/Ks | Duplicate type | Purifying selection | Date (million years) |
|---|---|---|---|---|---|---|
| SiATG6a/SiATG6b | 0.3522 | 0.1482 | 0.4208 | Segmental | YES | 27.09 |
| SiATG8a/SiATG8b | 0.9323 | 0.0265 | 0.0284 | Segmental | YES | 71.72 |
| SiATG9a/SiATG9b | 1.0214 | 0.2045 | 0.2002 | Segmental | YES | 78.57 |
| SiATG13b/SiATG13c | 1.0042 | 0.213 | 0.2121 | Segmental | YES | 77.25 |
| SiATG18b/SiATG18e | 1.6740 | 0.3135 | 0.1873 | Segmental | YES | 128.76 |
| SiATG7a/SiATG7b | 0.0679 | 0.0231 | 0.3402 | Tandem | YES | 5.22 |
Among the orthologous gene pairs of SiATG with other grass species, the highest average Ka/Ks value was between sorghum and foxtail millet (0.2061) and the lowest average Ka/Ks value were between maize and foxtail millet (0.1826). The rate of Ks of ATG genes between rice and foxtail millet was relatively higher than between sorghum and maize, with the divergence between foxtail millet and rice probably occurring between 36-95 MYA (Additional file 1: Tables S4–S6). Conversely, the Ka/Ks ratio of the ATG gene pairs between foxtail millet and maize (Ka/Ks = 0.1826) was lower than that of sorghum and foxtail millet (average Ka/Ks = 0.2061) and that of foxtail millet and rice (Ka/Ks = 0.1834). These results suggest that intense purifying selection between foxtail millet and maize was more significant than that of sorghum and foxtail millet, and foxtail millet and that of rice. The divergence time between foxtail millet and maize was at the latest17–82 MYA.
Differential expression profiles of the foxtail millet ATG genes under phytohormone, abiotic stress, and nitrogen and carbon starvation treatments
To investigate the tissue-specific expression of the 37 SiATG genes in foxtail millet, total RNA from roots, stems, and leaves were prepared and analyzed by qRT-PCR. As shown in Additional file 1: Figure S1, the expression of these SiATG genes was very low under normal conditions. The expression levels of the SiATGs in response to various treatments were quantified with the goal of identifying some key ATGs involved in responses to stresses (Figs. 5, 6 and 7 and Additional file 1: Table S8). All of the SiATG genes showed differential expression profiles in response to the various phytohormone treatments (Fig. 5). In particular, the expression levels of SiATG7a and SiATG7b increased significantly under MeJA (methyl jasmonate) treatment. Often, multiple members of the same subfamilies had similar expression patterns, for example: SiATG9a and SiATG9b.
Fig. 5.
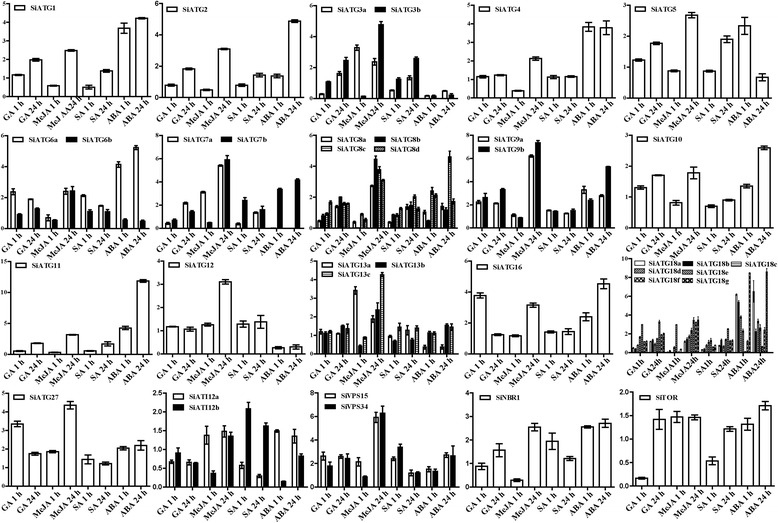
Expression profiles of 37 SiATG genes in response to treatment with various hormones. Relative expression levels of each ATG in foxtail millet seedlings under various hormones were normalized to Actin mRNA (AF288226.1). 3-week-old seedlings treated with 5μM gibberellic acid (GA), 5μM methyl jasmonate (MeJA), 5μM salicylic acid (SA) and 5μM abscisic acid (ABA) for 1 h and 24 h, respectively. The expression level of each ATG was calculated and compared with those of seedlings under normal growth conditions
Fig. 6.
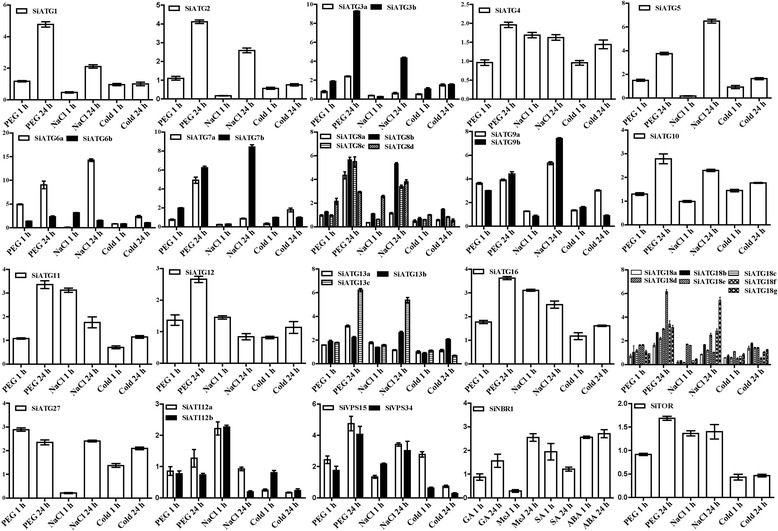
Expression profiles of 37 SiATG genes in response to various abiotic stresses. The relative expression levels of each ATG gene in foxtail millet seedlings under drought, salt, and cold stresses were normalized to Actin mRNA (AF288226.1). 3-week-old seedlings treated with high salt (100mM NaCl), drought (6 % PEG 6000) stresses, and cold stress (4 °C) for two time durations (1 h and 24 h). The expression level of each ATG gene was calculated and compared with that of seedlings grown under normal growth conditions
Fig. 7.
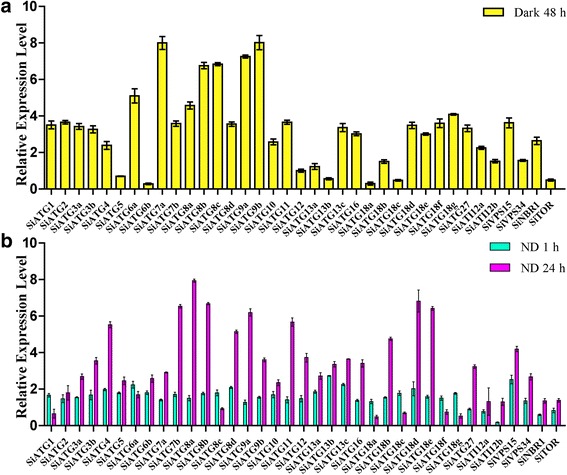
Expression analysis of 37 SiATG genes in response to nitrogen deficiency and darkness treatment using qRT-PCR. a 3-week old foxtail millet seedlings were kept in darkness for 48 h. b foxtail millet seeds were surfaced sterilized, and then seeds were germinated on MS and cultured for 14 days. On day 15, some seedlings were cultured on nitrogen-deficient liquid medium for 1 h or 24 h. The relative expression levels of each ATG gene in seedlings grown under darkness and nitrogen starvation were normalized to the Actin mRNA level (AF288226.1). The expression level of each ATG gene was calculated and compared with that in seedlings under normal growth conditions. ND: N deficient
To investigate the effect of different abiotic stresses treatments on the expression the foxtail millet ATG genes, we used qRT-PCR to monitor the expression patterns of the 37ATG genes in plants grown under drought, salt, and cold treatment (both 1 h and 24 h duration treatments). As shown in Fig. 6, the expression of most of the SiATG genes was either induced or suppressed as a result of these abiotic stress treatments. Although it was the case that the expression of most SiATG homologues was altered during the early stages of drought treatment, the expression of all 37 SiATG genes had clearly increased during the later stages of treatment. The expression of most of the SiATG genes was down-regulated during the early cold stress treatment, although the expression of seven genes was increased slightly (e.g.,SiATG10, SiATG16, and SiATG27).
To study the expression patterns of the 37 SiATG genes in response to carbon starvation stress, foxtail millet plants were kept in darkness for 48 h. As shown in Fig. 7a, seven SiATG genes (SiATG6a, SiATG7a, SiATG8a, SiATG8b, SiATG8c, SiATG9a and SiATG9b) showed highly expressed in darkness. The expression of the other SiATG genes was not changed significantly under this treatment. To monitor the response of SiATG genes in foxtail millet during nitrogen starvation, the expression of the 37 SiATG genes was analyzed in plants that had been subjected to nitrogen-free treatment for 1 h and for 24 h (Fig. 7b). After 1 h of treatment, the expression of most of the 37 SiATG genes was up-regulated, although the expression of SiATG27 was down-regulated. After 24 h of starvation, the expression of many genes was elevated. Among 37 SiATG genes the expression level of SiATG8a was the highest after low nitrogen treatment 24 h, suggesting its potential role in tolerance to nutrient starvation.
Overexpression of SiATG8a in rice conferred improved tolerance to nitrogen starvation
We investigated the tolerance to low nitrogen stress in transgenic rice expressing SiATG8a (Fig. 8a, 8b). The heterologous expression of SiATG8a in three transgenic rice lines was confirmed. The transgenic rice plants showed markedly higher transcript levels (Additional file 1: Figure S2). Three T2 generation transgenic lines, referred to as L1, L2, and L3, were obtained and grown hydroponically under nitrogen-free conditions. After 21 days of nitrogen starvation, it was clear that the three transgenic lines had higher survival rates than the control rice plants (Fig. 8i). Although the total protein concentration did not differ between the transgenic plants and control rice under normal conditions, it was significantly decreased in the transgenic plants that were grown under nitrogen starvation conditions (Fig. 8c). Additionally, the nitrogen content in the aboveground parts of the plants and the fresh weight of the three transgenic lines were higher than in the control rice (Fig. 8d, f). We also found that the root total nitrogen content in the three transgenic rice lines was slightly reduced compared to the wild type, but the results of variance analysis showed that these differences were not statistically significant between transgenic plants and wild type under N-free conditions (Fig. 8e). Under nitrogen starvation treatment, the transgenic plants were, on average, taller than the control rice plants (Fig. 8g). qRT-PCR analysis of the 18 endogenous OsATGs showed that the expression of these genes did not differ between the wild type and transgenic plants (Additional file 1: Figure S3A, B). For the low nitrogen starvation treatment, 40 millet varieties were randomly chosen. As shown in Additional file 1: Figure S4, the expression levels of the ATG8a gene in 40 foxtail millet varieties were associated with plant height under nitrogen starvation. We also observed that nitrogen deficiency stress inhibited the growth of the foxtail millet above ground, but the range of the declines of the individual varieties was different. The foxtail millet variety #17 and 18 (Longgu 10 and Chihegu) had the highest values for these traits, but the expression of the SiATG8a gene was low (Additional file 1: Figure S4). The effect of gene expression and phenotype in the different millet varieties is also of relevance under nitrogen starvation.
Fig. 8.
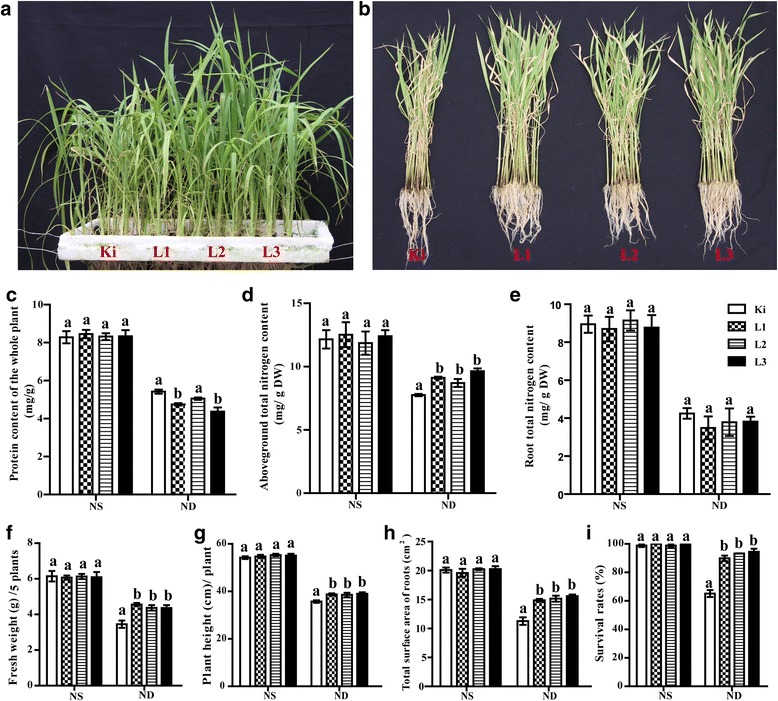
Heterologous expression of SiATG8a in rice enhanced tolerance to nitrogen starvation conditions. a and b 3-week old seedlings of control rice plants and transgenic plants grown in normal Hoagland solution were sub cultured in N-deficient liquid medium for 21 days. Statistical analysis of data for transgenic plants and wild type are shown for (c) protein content of the whole plant. d Aboveground total nitrogen content. e Root total nitrogen content. f Fresh weight. g Plant height. h Total surface area of roots. i Survival rate. For statistical analyses, the SAS 9.3 software package was used. Data presented in (c-i) are means ± SD from three independent experiments, and different letters above the columns indicate significant differences at the P ≤ 0.05 level. NS: N sufficient, ND: N deficient
Discussion
Heterologous expression of SiATG8a in rice enhances tolerance to nitrogen starvation
The Arabidopsis autophagy mutants, such as atg4a4b-1, atg5-1, atg7-1, atg9-1, atg10-1 and ATG18aRNAi plants exhibited abnormal phenotypes compared to the wild-type plant (chlorosis, decreased bolting time, and early senescence) under nutrient starvation, which confirmed the important roles of autophagy in nutrient recycling [21, 22, 25–29]. Constitutive expression of GmATG8c in soybean callus cells enhanced nitrogen starvation tolerance and accelerated the growth of the calli. Transgenic Arabidopsis over-expressing GmATG8c increased the yield under the nitrogen starvation compared to wild type plants [37]. In our study, expression level of SiATG8a was the highest among 37 SiATG genes under nitrogen starvation condition, indicating its potential role in nutrient starvation responses. The SiATG8a transgenic plants appeared to be more robust than the wild type, indicating SiATG8a conferred transgenic plants tolerance to low nitrogen supply. N utilization is a complex process that consists of several steps, including uptake, translocation, assimilation, and remobilization [43]. Many results indicate that nutrient recycling could be precisely controlled by altering the timing and strength of autophagic processes, which would offer novel strategies for the improvement of N utilization in other crops [17, 24, 44]. Leaf soluble proteins can be rapidly degraded and are a major source of N in plants. Autophagy has recently been reported to be implicated in efficient nitrogen remobilization in senescent leaves, it facilitates protein degradation for nitrogen recycling in these organs [24, 45]. Tolerance to low nitrogen of SiATG8a might be related to the presumed enhanced protein degradation and utilization ability in senescent leaves. We found that the total nitrogen content of the aboveground parts in transgenic plants is significantly higher than that in control plants under low nitrogen condition (Fig. 8d). We also measured the total nitrogen content of roots and found that the total root nitrogen content in the three transgenic rice lines was slightly reduced compared to the wild type rice, but the difference was not statistically significant (Fig. 8e). There were no significant differences in the total nitrogen content for whole plants between transgenic and control plants (Additional file 1: Figure S5). These results indicated that the translocation of nitrogen from roots to aboveground organs might be enhanced in transgenic plants, which lead to strong tolerance to low nitrogen stress of transgenic plants. Further study will be necessary to characterize the molecular basis of how the SiATG8a promote influences nitrogen translocation from roots to aboveground organs in plants.
The analysis of genes structure, phylogenetic classification, duplication and divergence rates of the SiATG genes
Many genes are known to be involved in autophagy. In yeast, 41 ATGs genes have been identified and confirmed to function in autophagy. In recent years, the autophagy-associated genes in multiple plant species have been identified. More than 30 ATG genes have been identified in Arabidopsis thaliana, Zea mays, Oryza sativa and Nicotiana tabacum genomes, demonstrating the evolutionarily conservation of the core ATG mechanism in different species [16–19]. A comprehensive search for autophagy- associated gene superfamily members from the PHYTOZOME database has revealed the presence of 37 SiATG genes, which is similar to the number of such genes in rice, tobacco and maize [16, 17, 19]. Unlike yeast where there is only a single copy, multiple copies of autophagy genes such as ATG8, exist in other species. For example, there are five, nine and eleven copies of ATG8 in tobacco, Arabidopsis, and soybean, respectively [16, 19, 32]. The unrooted phylogenetic trees suggested that each SiATG protein sequence was highly similar to their homologues in other plant species, it clearly shows the derivation of statistically reliable pairs of possible orthologues proteins sharing similar functions from a common ancestor. These results are consistent with the present understanding of plant evolutionary history [41]. Close association of SiATG families with their counter-parts in other plants, expressions or functions for most of which have reported, may be an implication of sequence conservation and evidence to their similar biological roles. Such phylogeny-based function prediction has been applied for prediction of stress-responsive proteins in other species like rice, Arabidopsis and maize. Although SiATG proteins tended to cluster together in the tree with respect to their type and they were not equally distributed in the clades which may be due to the occurrence of duplication and divergence of SiATG genes.
In this study, the exon-intron structures of ATG genes identified in the foxtail millet indicated that the exon-intron structures were conserved in foxtail millet. The results of inter-genomic analyses between rice, sorghum, maize, and foxtail millet revealed that there is highly conserved colinearity, which supports a close evolutionary relationship among these grasses [39]. To further explore the origin and evolutionary process of SiATG genes, a comparative synteny map between foxtail millet and rice genomes was produced which showed that there are 22 genes unambiguously existing in both the foxtail millet and rice genomes. These results suggest that the majority of SiATG genes share a common ancestor with OsATG genes counterparts. Despite the close evolutionary relationship between the foxtail millet and rice, the chromosomes of grasses have undergone extensive rearrangement. There are some SiATG genes that were not mapped onto any colinear blocks with rice, such as SiATG8a. A possible explanation for this is that foxtail millet and rice chromosomes have undergone extensive rearrangements and fusions that led to selective gene loss, which makes the identification of chromosomal syntenies difficult [38]. It does not mean that these ATG genes from foxtail millet and rice do not share a common ancestor but may be linked to environmental conditions in which some millet genes might be more appropriate and result in a better performance than rice. Gene duplication events play a key role in the expansion of gene families [46]. Many genes are known to be involved in autophagy most of the foxtail millet duplications were generated in the whole genome duplication (WGD) event similar to that in Brachypodium, rice, sorghum and maize, this was estimated to have occurred approximately 70 MYA [39]. In this study, gene duplication analysis revealed that segmental and tandem duplicated events resulted in the generation of multiple copies of SiATG genes, suggesting that these events play a significant role in the expansion of the SiATG gene family. In addition, some duplicate genes are important in the evolution of new traits and in speciation [47]. In the long-term evolution process, some genes may have a relatively more important role. In genetics, the calculation of the Ka/Ks ratio is an important parameter when deciding whether Darwinian positive selection was involved in gene divergence [48]. In all organisms, the majority of non-synonymous substitutions are harmful mutation; only a minority neutral or favorable mutations. Darwinian positive selection will retain the benefit of the non-synonymous mutations, whereas, for the harmful non-synonymous mutations, purifying selection will remove them gradually. In the present study, the average ratio of Ka/Ks for segmentally duplicated gene pairs is 0.210, whereas the Ka/Ks value for tandemly duplicated gene pairs was 0.34, strongly indicating that the SiATG gene family underwent a strong purifying selection pressure as the Ka/Ks ratios of six duplicated pairs were <1. Comparative genomic analysis is a quick way to extrapolate genomic knowledge acquired in one taxon to a less-well-studied species [49]. Duplicate genes tend to diverge in coding and regulatory regions. Divergences in coding regions can change the function of the gene or may lead to the acquisition of new functions as a result of amino acid-altering substitutions and/or alterations in exon-intron structure, divergence in regulatory regions can result in change in expression pattern [50]. Previous research has revealed that the foxtail millet genome underwent whole-genome duplication similar to other grasses about 70 MYA [39]. The relatively higher rate of synonymous substitution between rice and foxtail millet ATG genes may point towards their earlier divergence around 36–95 MYA as compared to sorghum and maize ATG genes.
Expression of SiATG genes in response to stress treatments
Under adverse environmental conditions, plants adopt various strategies to respond to particular stresses to enable growth and survival [51]. Many studies have shown that ATG proteins play key roles in plant responses to various environmental stresses [1, 20–22]. Cell autophagy generally remains at a low level in unstressed plants but can be induced when plants are subjected to adverse external stress. During such stress, plants require several phytohormone, including ABA, GA, MeJA and SA, which play key regulatory roles in different plant processes [52]. In our study, expression profile analysis demonstrated that all 37 SiATGs genes displayed variations in their expression behavior in response to one or more stresses, as shown in Additional file 1: Table S8. The complex expression patterns in response to one or more hormones implied that these 37 SiATGs may play different roles in response to multiple environmental stimuli. Oxidative, high salt and osmotic stress conditions can induce autophagy in plants. High salinity causes ion stress and drought usually leads to osmotic stress in the plant [53]. The accumulation of ROS (reactive oxygen species) and oxidized proteins in plant cells increased in plants grown under drought and salt stress conditions [54]. Autophagy is a macromolecular degradation pathway through which oxidized proteins can be voided. Transgenic RNAi-AtATG18a plants are more sensitive to ROS, salt and drought conditions than are wild type plants [25]. AtATG8 and OsATG10b have been reported to play important roles in responses to salt and osmotic stresses [36, 55]. Recent studies have shown that autophagy plays an important role in nitrogen remobilization and seed production in Arabidopsis [44, 45]. During leaf senescence, nitrogen and fixed-carbon limitations, the levels of maize ATG transcripts and/ or the ATG8-lipid adduct were increased indicating that autophagy plays important role in nutrient remobilization [15]. In this study, most of the 37 SiATGs responded differentially to at least one stress (Fig. 6). SiATG6a, SiATG8a, SiATG8c and SiATG9a showed increased expression during carbohydrate starvation and SiATG8a, SiATG8c and SiATG9a were up-regulated under nitrogen starvation (Fig. 7a, b). Those results indicated that ATG genes are widely involved in various abiotic stress responses of plants in addition to low nitrogen stress in plants.
Conclusion
A total of 37 putative SiATG genes were identified in foxtail millet through genome-wide analysis. We analyzed the chromosomal distribution, intron-exon structures, duplication and divergence rates, differential expression profiles under phytohormone, abiotic stresses, and nitrogen and carbon starvation treatments. Heterologous expression of SiATG8a in rice enhanced tolerance to nitrogen starvation in this study.
Methods
Plant materials and growth conditions
Seeds of foxtail millet were soaked in water and germinated at 28 °C for 2 days and then grown in Hoagland solution for 3 weeks. For the stress treatments, 3-week-old seedlings were exposed to high salt (100mM NaCl), drought (6 % PEG 6000), or hormones (5μM abscisic acid (ABA), 5μM salicylic acid (SA), 5μM methyl jasmonate (MeJA), and 5μM gibberellic acid (GA)) for 1 h and 24 h, respectively [56–58]. The carbon starvation and cold treatments were achieved, respectively, by keeping the seedlings in the dark for 48 h and keeping the seedlings at 4 °C for 1 h and 24 h. For the nitrogen-free treatment, foxtail millet seeds were surface sterilized and then germinated on MS medium (0.8 %w/v agar, 3 %sucrose) and cultured under a 16/8 h light/ dark photoperiod for 14 days. On day 15, seedlings were cultured on nitrogen-deficient liquid medium (modified MS medium with 5 mM KCl, 3 mM CaCl2, 1.5 mM MgSO4, 1.25mM KH2PO4, pH 5.8) for 1 h and 24 h. Control seedlings were grown in normal MS medium. For the nitrogen starvation treatment, 40 millet varieties were randomly chosen, the relative reduction values for the three replicates are presented. The list of the foxtail millet varieties examined in this study is shown in Additional file 1: Table S7. At the end of the treatments, all of the samples were frozen immediately in liquid nitrogen and stored at -80 °C prior to analysis.
Bioinformatics analysis of the autophagy gene family in foxtail millet
Some ATG-like genes (SiATGs) were identified in the foxtail millet genomic database (PHYTOZOME v9.0 database, www.phytozome.net/) using ‘autophagy’ as a key word. Other foxtail millet ATGs, together with ATG proteins from yeast, rice, Arabidopsis, tobacco, and maize (sources detailed subsequently), were identified in the PHYTOZOME v9.0 database via BLASP using the ATG domain as a query. The coding sequences, genomic sequences, protein sequences and nucleotide sequences upstream of the initiation codon for transcription for the 37 SiATGs were all downloaded from (www.phytozome.net/). Yeast ATG protein sequences were downloaded from Pfam26.0 (http://pfam.sanger.ac.uk/) [3–7]. The Arabidopsis ATG protein sequences were downloaded from the Arabidopsis information resource (https://www.arabidopsis.org/). The NtATG, ZmATG, and OsATG protein sequences were downloaded from (www.phytozome.net/) [16–19]. All redundant sequences were removed using the ‘decrease redundancy’ tool at http://web.expasy.org/decrease_redundancy/. Each putative SiATG gene sequence was checked by SMART (http://smart.embl-heidelberg.de/) to confirm the presence of the ATG domain. For nomenclature, the prefix `Si` for Setaria italica was used, followed by ‘ATG’. The serial number for each foxtail millet autophagy-associated genes member was assigned according to their relationship with the members of rice, Arabidopsis, tobacco, and maize autophagy-associated genes family. The subcellular localizations of the SiATG proteins were predicted using YLoc (http://abi.inf.uni-tuebingen.de/Services/YLoc/webloc.cgi), an interpretable web server for predicting the subcellular localization of proteins [59, 60].
Phylogenetic analysis of ATG genes
To further investigate the evolutionary relationships of among the ATG proteins in various plants species, the phylogenetic trees of the ATG genes were constructed. Multiple sequence alignment of ATG protein sequences were conducted with the ClustalX 1.81 program using the default multiple alignment parameters. The unrooted phylogenetic tree were constructed using MEGA5.0 software with a maximum likelihood method using sequences from S. italica (Si), N. tabacum (Nt), A. thaliana (At), O. sativa (Os), and Z. mays (Zm) [16–19, 61]. Bootstrap analysis was performed with 1000 replicates to obtain a support value for each branch.
Chromosomal location, gene structure analysis, and estimation of genomic distribution
All SiATG genes were mapped onto the nine foxtail millet chromosomes according to their ascending order of physical position (bp), from the short arm telomere to the long arm telomere, these were visualized using MapChart [62]. The exon-intron structures of the SiATG genes were determined by comparing the coding sequences (CDS) with their corresponding genomic sequences using the Gene Structure Display Server (GSDS) (http://gsds.cbi.pku.edu.cn/). Sequence duplications in foxtail millet, and the synteny between foxtail millet and rice, were established using the SynMap tool (https://genomevolution.org/CoGe/SynMap.pl) with the following settings: Blast Algorithm-Last, DAG Chainer options, and Merge syntenic Blocks, using the recommended parameters. Existing tandem duplications of the ATG genes in the foxtail millet genome were identified by manually evaluating the physical locations of genes within a region of 200 kb.
Estimation of the synonymous and non-synonymous substitution rates of the ATG genes
Synonymous (Ks) and non-synonymous (Ka) substitution rates were estimated according to previously-described criteria [63]. Ks and Ka substitution rates were calculated using the CODEML program and confirmed with the GEvo tool (https://genomevolution.org/CoGe/SynMap.pl). The time (million years ago, MYA) of duplication and divergence time (T) was calculated using a synonymous mutation rate of λ substitutions per synonymous site per year as T = Ks/2λ (λ = 6.5 × 10−9) [64, 65].
Total RNA isolation and quantitative real-time PCR
Total RNA from foxtail millet was isolated using Trizol reagent (Invitrogen, USA) according to the manufacturer’s instructions. The integrity of the RNA was confirmed by electrophoresis with 1.2 % agarose gels. The relative expression levels of the SiATG genes under various treatments were examined using quantitative real-time PCR (qRT-PCR). For each plant sample, 5μg of total RNA was reverse transcribed to cDNA in a 50μl reaction system using a PrimeScript 1st Strand cDNA Synthesis Kit (Transgen Biotech. Beijing, China). The primers for the qRT-PCR analysis were designed from a non-conserved region using Primer-BLAST (http://www.ncbi.nlm.nih.gov/tools/primer-blast/). Reverse primers were designed preferentially from the 3’-untranslated region wherever possible, as these regions are, in general, both relatively more unique than the coding sequence and closer to the reverse transcriptase (RT) start site. qRT-PCR was performed with three biological replicates, with three technical replicates per biological replicate. The primers used in these experiments are detailed in Additional file 1: Table S9.
Construction of the SiATG8a gene vector and rice transformation
The SiATG8a coding region was amplified by PCR using the primers F1 (BamH I) and R1 (Sac I). The PCR product was then digested and ligated into the binary vector pMWB014 (driven by a ubiquitin promoter) to obtain the construct pMWB014-SiATG8a. This construct was transformed into Oryza sativa cv. Kitaake using Agrobacterium-mediated transformation [66]. Fifty transgenic rice lines were generated and confirmed by PCR using the F2 and R2 primers. Nine transgenic lines were analyzed for phenotypes. Similar phenotypes were observed for these nine lines, and data from three of these selected lines were chosen for presentation here. The transcription level of SiATG8a in the three selected transgenic rice lines was analyzed via qRT-PCR. The primers used in this analysis are detailed in Additional file 1: Table S10. The transcription levels of the endogenous ATG (i.e., OsATG) in the three selected transgenic rice lines and in wild-type rice were verified by qRT-PCR. The primers used in this analysis are detailed in Additional file 1: Table S11.
Phenotypic analysis and measurement of nitrogen and protein content in transgenic rice plants
Seedlings of transgenic rice were grown in the greenhouse under a 16/8 photoperiod at 30 °C. For the nitrogen-free treatment experiment, 3-week-old seedlings of control rice and transgenic rice plants grown in normal Hoagland solution conditions were sub-cultured in nitrogen-deficient liquid medium (Hoagland hydroponic solution (pH 5.5), 5 mM KNO3, 5 mM Ca(NO3)2, 2 mM MgSO4, 1 mM NH4NO3). The nitrogen-free solution was prepared by replacing KNO3 and Ca(NO3)2 with KCl and CaCl2, respectively. After nitrogen starvation, plants that had green and healthy young leaves were considered to have survived, and the survival rate was calculated, using two green leaves as the standard for survival estimate. All experiments were repeated independently three times, and data were analyzed by Dunnett’s Test at the P ≤ 0.05 level. Total nitrogen content was determined by the Kjeldahl method [67]. Protein content was measured using the Bradford method [68].
Acknowledgements
The foxtail millet varieties were kindly provided by Prof. Diao Xian-Min, Institute of Crop sciences, Chinese Academy of Agricultural Sciences, Beijing, China. We are also grateful to Dr. John Hugh Snyder for help revise the manuscript.
Funding
This work was supported by the National Key Project for Research on Transgenic Biology [2016ZX08002-005], Beijing science and technology project [Z141100002314018] and the Graduate Innovation Fund of Harbin Normal University [HSDBSCX2012-04]. MJH and Rothamsted Research are supported via the 20:20 Wheat® Programme by the UK Biotechnology and Biological Sciences Research Council.
Availability of data and materials
All relevant data of this article are available within the manuscript and its additional files. Nucleic acid sequence of SiATG8a has been deposited in NCBI GenBank under accessions number KX539300 and the accession numbers of other putative protein sequences using analysis are available in the NCBI database (http://www.ncbi.nlm.nih.gov/protein/) and provided in Additional file 1: Table S1.
Authors’ contributions
WW L conducted the bioinformatic work, generated and analyzed data, and wrote the manuscript. MC conceived and designed experiments, and edited the manuscript. EHW, LQH, LZ, and ZC helped to carried out experiments and analyzed data. MH participated in the study design and helped to draft the manuscript. ZSX, LCL, and YBZ provided reagents. CHG and YZM contributed with valuable discussions. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Consent for publication
Not applicable.
Ethics approval and consent to participate
Not applicable.
Additional file
Expression analysis of autophagy-associated gene (ATG) in various foxtail millet organs. Figure S2. The transcription levels of SiATG8a in three transgenic rice lines relative to wild type rice. Figure S3. The transcription levels of 18 endogenous ATG in the three transgenic rice lines. Figure S4. SiATG8a expression and phenotype analysis in multiple foxtail millet varieties grown under nitrogen starvation conditions. Figure S5. The total nitrogen content for whole plants under normal and starvation conditions. Table S1. The autophagy-associated gene (ATG) homologue superfamily in foxtail millet. Information includes common names and locus names of the putative SiATGs in different versions of PHYTOZOME. TableS2. The autophagy-associated gene (ATG) homologue superfamily in foxtail millet. pI: isoelectric point, M: molecular weight. Table S3. Conserved motifs identified in the foxtail millet ATG family proteins using MEME software. Table S4. The Ka/Ks ratios and estimated divergence times for orthologous ATG proteins between foxtail millet and maize. Table S5. The Ka/Ks ratios and estimated divergence times for orthologous ATG proteins between foxtail millet and rice. Table S6. The Ka/Ks ratios and estimated divergence times for orthologous ATG proteins between foxtail millet and sorghum. Table S7. Foxtail millet varieties examined in this study. Table S8. Overview of the expression of ATGs in foxtail millet in response to treatment with various stresses. “+” and “-”indicate that the relative expression level of a given ATGs was up-regulated or down-regulated, respectively, in response to a given stress. The number represents the difference in expression expressed as a fold change. 0 means the absolute value are between 0 and 1;1 means ≥ 1 fold change; 2 means ≥ 2 fold change etc. Table S9. Primers used for the qRT-PCR analysis of the 37 SiATG genes. Table S10. A list of the primer sequences used for the cloning of SiATG8a and the PCR analysis of SiATG8a in transgenic rice. Table S11. Primers used for the for qRT-PCR analysis of endogenous ATG genes of rice. (DOCX 1530 kb)
Contributor Information
Weiwei Li, Email: liweiwei.0304@163.com.
Ming Chen, Email: chenming02@caas.cn.
Erhui Wang, Email: 603712884@qq.com.
Liqin Hu, Email: 1069423177@qq.com.
Malcolm J. Hawkesford, Email: malcolm.hawkesford@rothamsted.ac.uk
Li Zhong, Email: 494628745@qq.com.
Zhu Chen, Email: 709269930@qq.com.
Zhaoshi Xu, Email: xuzhaoshi@caas.cn.
Liancheng Li, Email: liliancheng@caas.cn.
Yongbin Zhou, Email: zhouyongbin@caas.cn.
Changhong Guo, Email: kaku3008@126.com.
Youzhi Ma, Email: mayouzhi@caas.cn.
References
- 1.Thompson AR, Vierstra RD. Autophagic recycling: lessons from yeast help define the process in plants. Curr Opin Plant Biol. 2005;8(2):165–173. doi: 10.1016/j.pbi.2005.01.013. [DOI] [PubMed] [Google Scholar]
- 2.Yoshimoto K, Takano Y, Sakai Y. Autophagy in plants and phytopathogens. FEBS Lett. 2010;584(7):1350–1358. doi: 10.1016/j.febslet.2010.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Kanki T, Wang K, Baba M, Bartholomew CR, Lynch-Day MA, Du Z, Geng JF, Mao K, Yang ZF, Yen WL, et al. A Genomic Screen for Yeast Mutants Defective in Selective Mitochondria Autophagy. Mol Biol Cell. 2009;20(22):4730–4738. doi: 10.1091/mbc.E09-03-0225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yao ZY, Delorme-Axford E, Backues SK, Klionsky DJ. Atg41/Icy2 regulates autophagosome formation. Autophagy. 2015;11(12):2288–2299. doi: 10.1080/15548627.2015.1107692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mizushima N. Autophagy: process and function. Gene Dev. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 6.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell. 2009;17(1):87–97. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 7.Suzuki K, Kondo C, Morimoto M, Ohsumi Y. Selective transport of alpha-mannosidase by autophagic pathways identification of a novel receptor, Atg34p. J Biol Chem. 2010;285(39):30019–30025. doi: 10.1074/jbc.M110.143511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–174. doi: 10.1016/0014-5793(93)80398-E. [DOI] [PubMed] [Google Scholar]
- 9.Thumm M, Egner R, Koch B, Schlumpberger M, Straub M, Veenhuis M, Wolf DH. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349(2):275–280. doi: 10.1016/0014-5793(94)00672-5. [DOI] [PubMed] [Google Scholar]
- 10.Harding TM, Morano KA, Scott SV, Klionsky DJ. Isolation and Characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131(3):591–602. doi: 10.1083/jcb.131.3.591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Noda T, Suzuki K, Ohsumi Y. Yeast autophagosomes: de novo formation of a membrane structure. Trends Cell Bio. 2002;12(5):231–235. doi: 10.1016/S0962-8924(02)02278-X. [DOI] [PubMed] [Google Scholar]
- 12.Wang CW, Klionsky DJ. The molecular mechanism of autophagy. Mol Med. 2003;9(3–4):65–76. [PMC free article] [PubMed] [Google Scholar]
- 13.Klionsky DJ, Cregg JM, Dunn WA, Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, et al. A unified nomenclature for yeast autophagy-related genes. Dev Cell. 2003;5(4):539–545. doi: 10.1016/S1534-5807(03)00296-X. [DOI] [PubMed] [Google Scholar]
- 14.Su W, Ma HJ, Liu C, Wu JX, Yang JS. Identification and characterization of two rice autophagy associated genes, OsAtg8 and OsAtg4. Mol Biol Rep. 2006;33(4):273–278. doi: 10.1007/s11033-006-9011-0. [DOI] [PubMed] [Google Scholar]
- 15.Chung T, Suttangkakul A, Vierstra RD. The ATG autophagic conjugation system in maize: ATG transcripts and abundance of the ATG8-lipid adduct are regulated by development and nutrient availability. Plant Physiol. 2009;149(1):220–234. doi: 10.1104/pp.108.126714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xia KF, Liu T, Ouyang J, Wang R, Fan T, Zhang MY. Genome-wide identification, classification, and expression analysis of autophagy-associated gene homologues in rice (Oryza sativa L.) DNA Res. 2011;18(5):363–377. doi: 10.1093/dnares/dsr024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li FQ, Chung T, Pennington JG, Federico ML, Kaeppler HF, Kaeppler SM, Otegui MS, Vierstra RD. Autophagic Recycling Plays a Central Role in Maize Nitrogen Remobilization. Plant Cell. 2015;27(5):1389–1408. doi: 10.1105/tpc.15.00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Il Kwon S, Park OK. Autophagy in plants. J Plant Biol. 2008;51(5):313–320. doi: 10.1007/BF03036132. [DOI] [Google Scholar]
- 19.Zhou XM, Zhao P, Wang W, Zou J, Cheng TH, Peng XB, Sun MX. A comprehensive, genome-wide analysis of autophagy-related genes identified in tobacco suggests a central role of autophagy in plant response to various environmental cues. DNA Res. 2015;22(4):245–257. doi: 10.1093/dnares/dsv012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu Y, Schiff M, Czymmek K, Talloczy Z, Levine B, Dinesh-Kumar SP. Autophagy regulates programmed cell death during the plant innate immune response. Cell. 2005;121(4):567–577. doi: 10.1016/j.cell.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 21.Xiang Y, Contento AL, Bassham D. Disruption of autophagy results in constitutive oxidative stress in Arabidopsis. Autophagy. 2007;3(3):257–258. doi: 10.4161/auto.3847. [DOI] [PubMed] [Google Scholar]
- 22.Xiong Y, Contento AL, Nguyen PQ, Bassham DC. Degradation of oxidized proteins by autophagy during oxidative stress in Arabidopsis. Plant Physiol. 2007;143(1):291–299. doi: 10.1104/pp.106.092106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chung T, Phillips AR, Vierstra RD. ATG8 lipidation and ATG8-mediated autophagy in Arabidopsis require ATG12 expressed from the differentially controlled ATG12A and ATG12B loci. Plant J. 2010;62(3):483–493. doi: 10.1111/j.1365-313X.2010.04166.x. [DOI] [PubMed] [Google Scholar]
- 24.Wada S, Hayashida Y, Izumi M, Kurusu T, Hanamata S, Kanno K, Kojima S, Yamaya T, Kuchitsu K, Makino A, et al. Autophagy Supports Biomass Production and Nitrogen Use Efficiency at the Vegetative Stage in Rice. Plant Physiol. 2015;168(1):60–U721. doi: 10.1104/pp.15.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xiong Y, Contento AL, Bassham DC. AtATG18a is required for the formation of autophagosomes during nutrient stress and senescence in Arabidopsis thaliana. Plant J. 2005;42(4):535–546. doi: 10.1111/j.1365-313X.2005.02397.x. [DOI] [PubMed] [Google Scholar]
- 26.Doelling JH, Walker JM, Friedman EM, Thompson AR, Vierstra RD. The APG8/12-activating enzyme APG7 is required for proper nutrient recycling and senescence in Arabidopsis thaliana. J Biol Chem. 2002;277(36):33105–33114. doi: 10.1074/jbc.M204630200. [DOI] [PubMed] [Google Scholar]
- 27.Thompson AR, Doelling JH, Suttangkakul A, Vierstra RD. Autophagic nutrient recycling in Arabidopsis directed by the ATG8 and ATG12 conjugation pathways. Plant Physiol. 2005;138(4):2097–2110. doi: 10.1104/pp.105.060673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bassham DC. Plant autophagy-more than a starvation response. Curr Opin Plant Biol. 2007;10(6):587–593. doi: 10.1016/j.pbi.2007.06.006. [DOI] [PubMed] [Google Scholar]
- 29.Hanaoka H, Noda T, Shirano Y, Kato T, Hayashi H, Shibata D, Tabata S, Ohsumi Y. Leaf senescence and starvation-induced chlorosis are accelerated by the disruption of an Arabidopsis autophagy gene. Plant Physiol. 2002;129(3):1181–1193. doi: 10.1104/pp.011024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xie ZP, Nair U, Klionsky DJ. Atg8 controls phagophore expansion during autophagosome formation. Mol Biol Cell. 2008;19(8):3290–3298. doi: 10.1091/mbc.E07-12-1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nakatogawa H, Ichimura Y, Ohsumi Y. Atg8, a ubiquitin-like protein required for autophagosome formation, mediates membrane tethering and hemifusion. Cell. 2007;130(1):165–178. doi: 10.1016/j.cell.2007.05.021. [DOI] [PubMed] [Google Scholar]
- 32.Yoshimoto K, Hanaoka H, Sato S, Kato T, Tabata S, Noda T, Ohsumi Y. Processing of ATG8s, ubiquitin-like proteins, and their deconjugation by ATG4s are essential for plant autophagy. Plant Cell. 2004;16(11):2967–2983. doi: 10.1105/tpc.104.025395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Woo J, Park E, Dinesh-Kumar SP. Differential processing of Arabidopsis ubiquitin-like Atg8 autophagy proteins by Atg4 cysteine proteases (vol 111, pg 863, 2013) Proc Natl Acad Sci U S A. 2014;111(25):9325. doi: 10.1073/pnas.1318207111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Slavikova S, Shy G, Yao YL, Giozman R, Levanony H, Pietrokovski S, Elazar Z, Galili G. The autophagy-associated Atg8 gene family operates both under favourable growth conditions and under starvation stresses in Arabidopsis plants. J Exp Bot. 2005;56(421):2839–2849. doi: 10.1093/jxb/eri276. [DOI] [PubMed] [Google Scholar]
- 35.Kuzuoglu-Ozturk D, Yalcinkaya OC, Akpinar BA, Mitou G, Korkmaz G, Gozuacik D, Budak H. Autophagy-related gene, TdAtg8, in wild emmer wheat plays a role in drought and osmotic stress response. Planta. 2012;236(4):1081–1092. doi: 10.1007/s00425-012-1657-3. [DOI] [PubMed] [Google Scholar]
- 36.Slavikova S, Ufaz S, Avin-Wittenberg T, Levanony H, Galili G. An autophagy-associated Atg8 protein is involved in the responses of Arabidopsis seedlings to hormonal controls and abiotic stresses. J Exp Bot. 2008;59(14):4029–4043. doi: 10.1093/jxb/ern244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Xia TM, Xiao D, Liu D, Chai WT, Gong QQ, Wang NN. Heterologous expression of ATG8c from soybean confers tolerance to nitrogen deficiency and increases yield in Arabidopsis. Plos One. 2012;7(5):e37217. doi: 10.1371/journal.pone.0037217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bennetzen JL, Schmutz J, Wang H, Percifield R, Hawkins J, Pontaroli AC, Estep M, Feng L, Vaughn JN, Grimwood J, et al. Reference genome sequence of the model plant Setaria. Nat Biotechnol. 2012;30(6):555–561. doi: 10.1038/nbt.2196. [DOI] [PubMed] [Google Scholar]
- 39.Zhang GY, Liu X, Quan ZW, Cheng SF, Xu X, Pan SK, Xie M, Zeng P, Yue Z, Wang WL, et al. Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat Biotechnol. 2012;30(6):549–554. doi: 10.1038/nbt.2195. [DOI] [PubMed] [Google Scholar]
- 40.Jia GQ, Huang XH, Zhi H, Zhao Y, Zhao Q, Li WJ, Chai Y, Yang LF, Liu KY, Lu HY, et al. A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica) Nat Genet. 2013;45(8):957–961. doi: 10.1038/ng.2673. [DOI] [PubMed] [Google Scholar]
- 41.Kellogg EA. Evolutionary history of the grasses. Plant Physiol. 2001;125(3):1198–1205. doi: 10.1104/pp.125.3.1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang YC, Mao LY, Wang H, Brocker C, Yin XJ, Vasiliou V, Fei ZJ, Wang XP. Genome-wide identification and analysis of grape aldehyde dehydrogenase (ALDH) gene superfamily. Plos One. 2012;7(2):e32153. doi: 10.1371/journal.pone.0032153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chardon F, Noel V, Masclaux-Daubresse C. Exploring NUE in crops and in Arabidopsis ideotypes to improve yield and seed quality. J Exp Bot. 2012;63(9):3401–3412. doi: 10.1093/jxb/err353. [DOI] [PubMed] [Google Scholar]
- 44.Guiboileau A, Yoshimoto K, Soulay F, Bataille MP, Avice JC, Masclaux-Daubresse C. Autophagy machinery controls nitrogen remobilization at the whole-plant level under both limiting and ample nitrate conditions in Arabidopsis. New Phytol. 2012;194(3):732–740. doi: 10.1111/j.1469-8137.2012.04084.x. [DOI] [PubMed] [Google Scholar]
- 45.Guiboileau A, Avila-Ospina L, Yoshimoto K, Soulay F, Azzopardi M, Marmagne A, Lothier J, Masclaux-Daubresse C. Physiological and metabolic consequences of autophagy deficiency for the management of nitrogen and protein resources in Arabidopsis leaves depending on nitrate availability. New Phytol. 2013;199(3):683–694. doi: 10.1111/nph.12307. [DOI] [PubMed] [Google Scholar]
- 46.Vision TJ, Brown DG, Tanksley SD. The origins of genomic duplications in Arabidopsis. Science. 2000;290(5499):2114–2117. doi: 10.1126/science.290.5499.2114. [DOI] [PubMed] [Google Scholar]
- 47.Pennisi E. Evolutionary biology - Twinned genes live life in the fast lane. Science. 2000;290(5494):1065–1066. doi: 10.1126/science.290.5494.1065a. [DOI] [PubMed] [Google Scholar]
- 48.Juretic N, Hoen DR, Huynh ML, Harrison PM, Bureau TE. The evolutionary fate of MULE-mediated duplications of host gene fragments in rice. Genome Res. 2005;15(9):1292–1297. doi: 10.1101/gr.4064205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lyons E, Pedersen B, Kane J, Alam M, Ming R, Tang HB, Wang XY, Bowers J, Paterson A, Lisch D, et al. Finding and comparing syntenic regions among Arabidopsis and the outgroups papaya, poplar, and grape: CoGe with rosids. Plant Physiol. 2008;148(4):1772–1781. doi: 10.1104/pp.108.124867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu G, Guo C, Shan H, Kong H. Divergence of duplicate genes in exon-intron structure. Proc Natl Acad Sci U S A. 2012;109(4):1187–1192. doi: 10.1073/pnas.1109047109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Ahuja I, de Vos RC, Bones AM, Hall RD. Plant molecular stress responses face climate change. Trends Plant Sci. 2010;15(12):664–674. doi: 10.1016/j.tplants.2010.08.002. [DOI] [PubMed] [Google Scholar]
- 52.Puranik S, Sahu PP, Mandal SN, Suresh BV, Parida SK, Prasad M. Comprehensive Genome-wide survey, genomic constitution and expression profiling of the NAC transcription factor family in foxtail millet (Setaria italic L.) Plos One. 2013;8(5):e64594. doi: 10.1371/journal.pone.0064594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhu JK. Cell signaling under salt, water and cold stresses. Curr Opin Plant Biol. 2001;4(5):401–406. doi: 10.1016/S1369-5266(00)00192-8. [DOI] [PubMed] [Google Scholar]
- 54.Tsugane K, Kobayashi K, Niwa Y, Ohba Y, Wada K, Kobayashi H. A recessive arabidopsis mutant that grows photoautotrophically under salt stress shows enhanced active oxygen detoxification. Plant Cell. 1999;11(7):1195–1206. doi: 10.1105/tpc.11.7.1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin JH, Yoshimoto K, Ohsumi Y, Jeon JS, An G. OsATG10b, an autophagosome component, is needed for cell survival against oxidative stresses in rice. Mol Cells. 2009;27(1):67–74. doi: 10.1007/s10059-009-0006-2. [DOI] [PubMed] [Google Scholar]
- 56.Rose TL, Bonneau L, Der C, Marty-Mazars D, Marty F. Starvation-induced expression of autophagy-related genes in Arabidopsis. Biol Cell. 2006;98(1):53–67. doi: 10.1042/BC20040516. [DOI] [PubMed] [Google Scholar]
- 57.Puranik S, Bahadur RP, Srivastava PS, Prasad M. Molecular cloning and characterization of a membrane associated NAC family gene, SiNAC from foxtail millet [Setaria italica (L.) P. Beauv.] Mol Biotechnol. 2011;49(2):138–150. doi: 10.1007/s12033-011-9385-7. [DOI] [PubMed] [Google Scholar]
- 58.Puranik S, Jha S, Srivastava PS, Sreenivasulu N, Prasad M. Comparative transcriptome analysis of contrasting foxtail millet cultivars in response to short-term salinity stress. J Plant Physiol. 2011;168(3):280–287. doi: 10.1016/j.jplph.2010.07.005. [DOI] [PubMed] [Google Scholar]
- 59.Briesemeister S, Rahnenfuhrer J, Kohlbacher O. Going from where to why-interpretable prediction of protein subcellular localization. Bioinformatics. 2010;26(9):1232–1238. doi: 10.1093/bioinformatics/btq115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Briesemeister S, Rahnenfuhrer J, Kohlbacher O. YLoc-an interpretable web server for predicting subcellular localization. Nucleic Acids Res. 2010;38:W497–W502. doi: 10.1093/nar/gkq477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tamura K, Peterson D, Peterson N, Stecher G, Nei M, Kumar S. MEGA5: Molecular evolutionary genetics analysis using maximum likelihood, evolutionary distance, and maximum parsimony methods. Mol Biol Evol. 2011;28(10):2731–2739. doi: 10.1093/molbev/msr121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Voorrips RE. MapChart: Software for the graphical presentation of linkage maps and QTLs. J Hered. 2002;93(1):77–78. doi: 10.1093/jhered/93.1.77. [DOI] [PubMed] [Google Scholar]
- 63.Chen Z, Chen M, Xu ZS, Li LC, Chen XP, Ma YZ. Characteristics and expression patterns of the aldehyde dehydrogenase (ALDH) gene superfamily of foxtail millet (Setaria italica L.) Plos One. 2014;9(7):e101136. doi: 10.1371/journal.pone.0101136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290(5494):1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- 65.Yang ZF, Gu SL, Wang XF, Li WJ, Tang ZX, Xu CW. Molecular evolution of the CPP-like gene family in plants: Insights from comparative genomics of Arabidopsis and rice. J Mol Evol. 2008;67(3):266–277. doi: 10.1007/s00239-008-9143-z. [DOI] [PubMed] [Google Scholar]
- 66.Hiei Y, Ohta S, Komari T, Kumashiro T. Efficient transformation of rice (Oryza sativa L.) mediated by Agrobacterium and sequence analysis of the boundaries of the T-DNA. Plant J. 1994;6(2):271–282. doi: 10.1046/j.1365-313X.1994.6020271.x. [DOI] [PubMed] [Google Scholar]
- 67.Li YL, Fan XR, Shen QR. The relationship between rhizosphere nitrification and nitrogen-use efficiency in rice plants. Plant Cell Environ. 2008;31(1):73–85. doi: 10.1111/j.1365-3040.2007.01737.x. [DOI] [PubMed] [Google Scholar]
- 68.Carlsson N, Borde A, Wolfel S, Akerman B, Larsson A. Quantification of protein concentration by the Bradford method in the presence of pharmaceutical polymers. Anal Biochem. 2011;411(1):116–121. doi: 10.1016/j.ab.2010.12.026. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All relevant data of this article are available within the manuscript and its additional files. Nucleic acid sequence of SiATG8a has been deposited in NCBI GenBank under accessions number KX539300 and the accession numbers of other putative protein sequences using analysis are available in the NCBI database (http://www.ncbi.nlm.nih.gov/protein/) and provided in Additional file 1: Table S1.