

Abstract
Transactivation by the ETS family of transcription factors, whose members share structurally conserved DNA-binding domains, is variably sensitive to methylation of their target genes. The mechanism by which DNA methylation controls ETS proteins remains poorly understood. Uncertainly also pervades the effects of hemi-methylated DNA, which occurs following DNA replication and in response to hypomethylating agents, on site recognition by ETS proteins. To address these questions, we measured the affinities of two sequence-divergent ETS homologs, PU.1 and Ets-1, to DNA sites harboring a hemi- and fully methylated CpG dinucleotide. While the two proteins bound unmethylated DNA with indistinguishable affinity, their affinities to methylated DNA are markedly heterogeneous and exhibit major energetic coupling between the two CpG methylcytosines. Analysis of simulated DNA and existing co-crystal structures revealed that hemi-methylation induced non-local backbone and groove geometries that were not conserved in the fully methylated state. Indirect readout of these perturbations was differentially achieved by the two ETS homologs, with the distinctive interfacial hydration in PU.1/DNA binding moderating the inhibitory effects of DNA methylation on binding. This data established a biophysical basis for the pioneering properties associated with PU.1, which robustly bound fully methylated DNA, but not Ets-1, which was substantially inhibited.
INTRODUCTION
The differentiation of distinct lineages of blood cells from a single progenitor species occurs in a multi-step process, termed hematopoiesis, that is tightly controlled at the transcriptional level (1,2). Members of the ETS family of transcription factors rank among the most essential hematopoietic regulators in ensuring the continued self-renewal of this progenitor, the hematopoietic stem cell (HSC), and its correct differentiation (3,4). During hematopoiesis, binding sites for ETS transcription factors in HSCs and derived cell types are closely correlated with genomic areas undergoing active changes in methylation status, suggesting a strong association between their activity and chromatin structure. For example, hypermethylated DNA in HSCs are enriched in binding sites for Ets-1 and sequence-similar ETS-family paralogs (5). While PU.1 binding is enriched in hypomethylated DNA in myeloid cells (6), it is associated with both hypermethylated and hypomethylated genes in osteoclastogenic monocytes (7). Current evidence therefore points to widespread heterogeneity in the interactions of ETS paralogs with methylated DNA. In addition, ETS members differ according to a hierarchy that distinguishes transcription factors in terms of their ‘pioneer’ ability to overcome chromatin restriction, induce chromatin opening, promote local nucleosomal modifications, and stimulate the expression of otherwise silenced target genes (8). By such criteria, functional and genomic studies (9–14) have established PU.1, but not Ets-1, as a pioneer transcription factor.
The contrasting functional differences among ETS members are presently confounded by the extensive structural homology of their eponymous DNA-binding domains. We have previously found that the ETS domains of PU.1 and Ets-1, which represent the extremes of sequence-divergent ETS paralogs (∼30% amino acid homology), nonetheless share superimposable backbone trajectories (15). Moreover, PU.1 and Ets-1 differ profoundly in their biophysical mechanisms of DNA recognition with respect to the role of hydration, electrostatics, and conformational dynamics in discriminating high- and low-affinity cognate DNA binding sites (15,16). As the molecular mechanisms that confer PU.1's distinctive pioneering properties remain unclear, we hypothesize that PU.1 and Ets-1 interact dissimilarly with methylated DNA, and moreover these differences are intrinsic to their respective ETS domains. Using a shared model DNA binding site that harbors a specific CpG dinucleotide, we tested this hypothesis by measuring its affinity for the ETS domains of both PU.1 and Ets-1 at all levels of CpG methylation. We found that the position and level of methylation strongly discriminated the two ETS domains, and in particular PU.1 retained robust affinity for the fully methylated DNA site while Ets-1 was substantially inhibited. Analysis of existing crystallographic and simulated structures revealed that DNA methylation disrupted site recognition by ETS proteins through their indirect readout of backbone and groove geometry. PU.1, whose DNA-binding interface is extensively hydrated relative to Ets-1, was significantly more tolerant with these perturbations. Our data provide a direct biophysical basis for PU.1 to engage methylated DNA autonomously in support of its status as a pioneer transcription factor, while Ets-1 is not.
MATERIALS AND METHODS
Proteins
Recombinant constructs representing the ETS domain of murine PU.1 (residues 167–272, designated PU.1ΔN167) and Ets-1 (residues 331–440, designated Ets-1ΔN331) were cloned with a thrombin-cleavable C-terminal 6xHis tag as described (15,16). A similarly tagged construct for auto-inhibited Ets-1 (residues 280–440, Ets-1ΔN280) was a gift from Dr Lawrence P. McIntosh (University of British Columbia). All constructs were over-expressed in Escherichia coli, purified by immobilized metal affinity chromatography, and polished by cation exchange chromatography as described (17). Purified protein was extensively dialyzed against binding buffer (10 mM Tris–HCl, pH 7.4 at 25°C, 150 mM NaCl). Buffers used with Ets-1 constructs, which harbored reduced cysteines, additionally contained 0.5 mM TCEP. Protein concentrations were determined by UV absorption at 280 nm using the following extinction coefficients (in M−1 cm−1): 22 460 (PU.1ΔN167), 32 430 (Ets-1ΔN331) and 39 880 (Ets-1ΔN280). The molecular weights of all constructs were verified by MALDI-ToF mass spectrometry (Figure S1, Supplemental Data).
Nucleic acids
Synthetic unmodified and site-specifically modified DNA oligos were purchased from Integrated DNA Technologies (Coralville, IA) and annealed to form duplex binding sites as described (18,19). Electrospray mass spectrometry confirmed the molecular weights of all oligos to be ±1 Da of expected values. Fluorescent DNA probes were constructed by annealing oligos harboring an internal Cy3 label with an unlabeled complementary strand as described (17). Oligo concentrations were determined spectrophotometrically using nearest-neighbor extinction coefficients (20).
Fluorescence polarization titrations
Protein binding to DNA was measured using a PerkinElmer LS 55 instrument as described (17). Briefly, we competitively titrated the Cy3-labeled DNA probe (1 nM) and purified protein (2 nM) with unmodified or methylated DNA sites and measured the reduction in steady-state fluorescence anisotropy (excitation/emission 522/563 nm). Poly(dI-dC)·poly(dI-dC) (Roche) was included at 10 μM bp to eliminate nonspecific binding. Each data point is represented as mean ± S.E. of five consecutive measurements as an indication of instrumental noise.
Anisotropy data was fitted with a mutually exclusive mechanistic model (17) to directly estimate absolute affinities. Equilibrium dissociation constants (KD) from independent experiments are given as mean ± S.E., and converted to standard free energy changes of association via 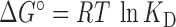 (R = 8.314 J mol−1 K−1, T = 298 K).
(R = 8.314 J mol−1 K−1, T = 298 K).
Molecular mechanics simulations
Full descriptions are given in Supplemental Methods. In brief, unmethylated and methylated DNA binding sites was examined in silico by all-atom energy minimization using the CHARMM36 force field in GROMACS 5.1.2. Duplex DNA containing the full experimental SC1 sequence (5′-CGGCCAAGCCGGAAGTGAGTGCC-3′), with or without site-specific 5-methylcytosine at the CpG dinucleotide in bold, were generated with fiber B-DNA geometry using 3DNA (21). Parameterization of 5-methylcytosine for CHARMM36 was taken as published (22). Each structure was hydrated with TIP3P water, neutralized with 0.15 M NaCl in a 1000-nm3 cubic box, and relaxed by steepest descent. Helical parameters of energy-minimized structures were fitted with Curves+ (23) and analyzed by principal component analysis using Origin 9.1 (Northampton, MA).
RESULTS
ETS transcription factors characteristically recognize 10-bp DNA sequences harboring a central 5′-GGAA/T-3′ consensus. While mutation of the consensus results in a nonspecific site due to the loss of core interactions in the major groove, variation in the flanking positions generates a palette of cognate sites that span over 100-fold in binding affinities (24). As a result, a single cognate sequence is recognizable to multiple ETS proteins. One such co-cognate sequence is SC1 (5′-GCCGGAAGTG-3′; CpG in bold and central consensus underlined), which was originally identified as a high-affinity site for Ets-1 (25) but subsequently shown to be site-specifically recognized by PU.1 as well (26). The CpG dinucleotide in the SC1 site represents the most common position for such a motif in ETS binding sites (27). We took advantage of this shared DNA target and measured the affinities of Ets-1 and PU.1 for the SC1 sequence at different levels of methylation.
To generate hemi- and fully methylated DNA binding sites, we annealed SC1-based oligos harboring 5′-GCMeCGGAAGTG-3′ and 5′-CACTTCCMeGGC-3′ with each other or unmodified counterparts. Thus, SC1 sites with mono-methylated 5′-GGAA-3′ (forward) or 5′-TTCC-3′ (reverse) strands represent two distinct hemi-methylated sites while the dimethylated SC1 site corresponds to full methylation of the CpG dinucleotide. Binding affinity for these sites were measured by competitive fluorescence polarization titrations using a Cy3-labeled DNA probe (Figure S2, Supplemental Data). Specifically, we titrated fixed concentrations of DNA probe and ETS domains of Ets-1 or PU.1 with the SC1 sites and measured the drop in fluorescence anisotropy of the probe from the ETS-bound to unbound state. Duplex poly(dI-dC) was included to abolish nonspecific binding. When fitted with a mechanistic model that accounts for probe depletion (17), the titration data (Table 1) afforded an unambiguous determination of absolute affinities (not IC50) of each protein for the variously methylated and unlabeled SC1 sites, free from interference from the fluorescent label if the titrations were carried out as direct binding to the probe.
Table 1. Effect of hemi- and full methylation on the DNA site recognition by the ETS domains of PU.1 and Ets-1.
| KD, nM; ΔG°, kJ/mol | ||||||
|---|---|---|---|---|---|---|
| PU.1ΔN167 | Ets-1Δ331 | Ets-1Δ280 | ||||
| Unmethylated | 0.32 ± 0.17 (6) | −54.15 ± 1.29 | 0.14 ± 0.03 (3) | −56.23 ± 0.61 | 1.3 ± 0.5 (3) | −51.04 ± 1.12 |
| Hemi-methylated | ||||||
| 5′-CMeGGAA-3′ | 1.5 ± 0.9 (4) | −50.34 ± 1.54 | >104 (3) | >−28 | >104 (3) | >−28 |
| 5′-TTCCMeG-3′ | 74 ± 17 (5) | −40.69 ± 0.56 | 0.28 ± 0.14 (3) | −54.50 ± 1.22 | 3.5 ± 1.9 (3) | −48.21 ± 1.30 |
| Fully methylated | 3.9 ± 1.4 (5) | −47.96 ± 0.88 | 23 ± 6 (3) | −43.59 ± 0.63 | (3.6 ± 1.2) × 103 (3) | −31.06 ± 0.79 |
The affinities of the ETS domains of PU.1 (ΔN167) and Ets-1 (minimal domain, ΔN331 and auto-inhibited domain ΔN280) for the cognate DNA binding site SC1 (5′-GCCGGAAGTG-3′) were determined by fluorescence polarization titrations under physiologic conditions, as described in Materials and Methods and illustrated in Figure 1. Parametric values of the equilibrium dissociation constants (KD) are given as ± S.E. of N replicate experiments in parentheses. Standard free energies of binding were calculated from KD values at 25°C.
The ETS domains of PU.1 and Ets-1 differ markedly in their recognition of methylated DNA specific sites
Under identical physiologic solution conditions, the ETS domain of both Ets-1 and PU.1 bound unmethylated SC1 tightly with similar sub-nM affinities (Figures 1A and E). However, the two homologs differed markedly in their affinities for the hemi-methylated SC1 sites. For PU.1 (PU.1ΔN167), hemi-methylation at the 5′-GGAA-3′ strand reduced binding affinity about five-fold (Figure 1B), but hemi-methylation at the 5′-TTCC-3′ strand caused over 200-fold inhibition (Figure 1C). In contrast, Ets-1 (Ets-1ΔN331) exhibited the opposite behavior: hemi-methylated SC1 at the 5′-GGAA-3′ strand failed to compete at up to 1 μM (Figure 1F), but hemi-methylation at the 5′-TTCC-3′ strand was only marginally inhibitory (Figure 1G). We found that the weak but detectable displacement of PU.1ΔN167 by forward hemi-methylated SC1 was highly reproducible (Figure S3, Supplemental Data). In addition, the dispersion of affinities was not attributable to impurities in the recombinant protein preparations, which were homogenous as judged by mass spectrometry (Supplementary Figure S1), and therefore reflected the intrinsic behavior encoded in the two sequence-divergent but structurally conserved ETS domains.
Figure 1.

The ETS domain of PU.1 and Ets-1 respond disparately to hemi- and fully methylated site-specific DNA. The affinities of the ETS domains of PU.1 (ΔN167, Panels A to D) and Ets-1 (minimal domain, ΔN331, Panels E to H; auto-inhibited domain, ΔN280, Panels I to L) for the cognate DNA binding site SC1 (5′-GCCGGAAGTG-3′) were determined by fluorescence polarization titrations under physiologic solution conditions, as described in Materials and Methods. In brief, fixed concentrations of a Cy3-labeled DNA probe (1 nM) and protein (2 nM) were titrated by (unlabeled) un-, hemi-, or fully methylated SC1 sites. Poly(dI-dC)·poly(dI-dC) was present at 10 μM bp to eliminate nonspecific binding. The data was fitted by a mechanistic model that accounts for titrant depletion and the affinity of each protein for the probe (Figure S2, Supplemental Data) to directly estimate absolute affinities of each SC1 competitor (not IC50). Note that the starting anisotropy reflected the extent of probe saturation, effects of binding on the photophysical properties of the Cy3 label, as well as the rotational correlation of the complex. Experimentally, the titrations were constrained by the high affinity of the probe for the proteins (dissociation constants <1 nM) and the concentrations of both components required for robust anisotropy measurements. As a result, the desired (competitor) SC1 dissociation constants deviated significantly from the concentrations at half-maximal inhibition (i.e., IC50).
PU.1 robustly binds fully CpG-methylated site-specific DNA
The fully methylated SC1 site bound both PU.1 and Ets-1 with affinities intermediate of their hemi-methylated counterparts (Figure 1D and H). Notably, the inhibition on PU.1 was modest (12-fold relative to unmethylated) relative to Ets-1 (>160-fold). Thus, PU.1 robustly bound fully methylated site-specific DNA, while Ets-1 was significantly inhibited. Taken together with the hemi-methylation data, while site-specific recognition by the ETS domains of PU.1 and Ets-1 was sensitive to CpG methylation with respect to both the extent and position of methylation, PU.1 was generally less inhibited by DNA methylation than Ets-1.
ETS auto-inhibition in methylated DNA binding
In many ETS-family transcription factors, such as Ets-1, their ETS domains are flanked by ‘auto-inhibitory’ elements that allosterically reduce DNA binding affinity by unfolding in the DNA-bound state (28). Since the magnitude of auto-inhibition varies with DNA sequence (29), and has not been characterized with methylated DNA, we measured the binding of methylated SC1 to an auto-inhibited Ets-1 construct harboring additional residues 280 to 330 (termed Ets-1ΔN280; 30), under the same conditions as the minimal ETS domain (Ets-1ΔN331) and the PU.1 ETS domain (PU.1 ΔN167). Full-length PU.1 is not auto-inhibited.
Our solution titrations showed that auto-inhibition reduced the affinity of Ets-1 for unmethylated SC1 by ∼80-fold (Figure 1I), in agreement with previous electrophoretic mobility shift and filter binding measurements (16,28,30). As with the minimal ETS domain of Ets-1 (Ets-1ΔN331), hemi-methylated SC1 in the 5′-GGAA-3′ failed to compete for auto-inhibited Ets-1 (Ets-1ΔN280) at up to 1 μM, but hemi-methylation at the 5′-TTCC-3′ strand was only marginally (∼3-fold) inhibitory (Figure 1J and K). Similarly, the affinity of Ets-1ΔN280 for fully methylated SC1 was intermediate between the two hemi-methylated states (Figure 1L). In addition, the magnitude of auto-inhibition jumped from 13-fold for the reverse hemi-methylated site to almost 160-fold for the fully methylated site (Table 1). Thus, auto-inhibition was modulated by the methylation state of ETS binding sites. Crucially, since Ets-1ΔN280 is representative of full-length Ets-1, the data indicate that PU.1, which lacks auto-inhibition, has over 900-fold higher affinity for fully methylated DNA than Ets-1.
The 5-methylcytosines in a CpG dinucleotide are strongly coupled in ETS/DNA recognition
The intermediate affinities of both PU.1 and Ets-1 for fully methylated SC1 relative to the two hemi-methylated sites indicated non-additive effects by the two 5-methyl substituents on site-specific ETS/DNA binding. To gain more insight into the perturbations of CpG methylation, we analyzed the energetics of ETS binding to methylated SC1 sites in terms of thermodynamic cycles. Specifically, describing the free energy of association to fully methylated SC1 as a stepwise progression from binding to unmethylated SC1, followed by either of the two hemi-methylated sites, to fully methylated DNA gives rise to a (second-order) coupling term, 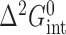 , that captures the non-additive interactions of the two 5-methyl substitutions (Figure 2):
, that captures the non-additive interactions of the two 5-methyl substitutions (Figure 2):
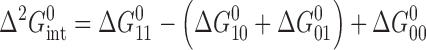 |
(1) |
Figure 2.

Free energy analysis of PU.1 and Ets-1 binding to methylated site-specific DNA. Experimental affinities of the ETS domains of PU.1 (ΔN167, blue) and Ets-1 (minimal domain, ΔN331, red; and auto-inhibited domain, ΔN280, magenta) to the SC1 binding site were analyzed in terms of a thermodynamic cycle of standard free energy changes in kJ/mol. The incremental change for each experimentally measured step (ΔΔG°) is also shown. The coupling free energy (Δ2Gint°) was computed according to Equation (1). Error bars for ΔΔG° and Δ2Gint° are propagated from the S.E. of experimental ΔG° values. The assigned value of ΔG10° for the Ets1-based constructs, which was not measurable in our experiment, was -28.52 ± 0.50 kJ/mol (see text).
The subscripts on the right side refer to the fully [11], two hemi- [10 and 01], and unmethylated [00] SC1 sites. We have previously used this approach to study the long-range effects of base mutations in the binding affinity of PU.1 to sequence-specific binding sites (24). In the case of methylated sites, the coupling free energies for the minimal domains of both PU.1 and Ets-1 (Δ331) were below −10 kJ/mol and significantly larger in magnitude than values typically observed for base mutations (∼−5 kJ/mol), indicating that methylation was more perturbative to DNA site recognition than changes in base identity. Interestingly, coupling was significantly weaker for Ets-1ΔN280 than the minimal Ets-1Δ331. Since competition by the hemi-methylated (5′-GGAA-3′) SC1 site for Ets-1 was not detectable, we assigned a lower limit, based on our titration range, for the dissociation constant at 10 μM (ΔG°10 ∼ −28 kJ/mol; Table 1). If the actual affinity was even weaker, the values of the coupling free energy for both Ets-1 constructs would be higher than shown in Figure 2.
Structural basis of the strand-dependent sensitivity to hemi-methylated DNA by the ETS domains of PU.1 and Ets-1
The opposite behavior of the ETS domains of PU.1 and Ets-1 towards hemi-methylated SC1 sites was unexpected in light of the strong backbone homology of the two ETS domains (15,16) observed in their co-crystal structures with high-affinity DNA (31,32). Despite a plethora of high-affinity ETS/DNA structures, no experimental structure of an ETS complex with methylated DNA sites is currently available. To better understand the structural origin of ETS inhibitory by methylated DNA, we used the ETS/DNA co-crystal structures of PU.1 and Ets-1 as templates, and mutated the bound DNA with position-matched methylated SC1 sequences. The bases were replaced while maintaining the original backbone deoxyribose and phosphate coordinates, assuming negligible differences in the induced DNA backbone conformation from the original sequences. Our assumption was grounded in the conserved DNA backbone conformation observed among high-resolution binary ETS/DNA complex structures, involving divergent DNA sequences, in which neutralization of phosphates on one side of the DNA double helix causes the DNA to asymmetrically collapse over the protein (33,34). Indeed, indirect readout of exclusively backbone atoms of the target DNA gives rise to the overlapping sequence recognition among ETS homologs (as exemplified here by PU.1 and Ets-1), and the hypothesis that selectivity is derived in part by the sequence-dependent propensity of the DNA target to adopt to the ETS-induced curvature (35).
Examination of the hemi-methylated protein/DNA contact interfaces (Figure 3) suggests that differences in interfacial hydration between the two complexes in the major groove account for their distinct behavior towards hemi-methylated DNA. In both proteins, an essential, conserved arginine anchors four interactions involving the DNA targets and other sidechains. Whereas three out of the four contacts are water-mediated for PU.1, only one is water-mediated for Ets-1. Although the 5-methylcytosine base in the forward strand (5′-CMeGGAA-3′) does not participate directly in these interactions, its C5-methyl is poised to clash (∼3 Å between heavy atoms) with the conserved arginine of both ETS domains (Figure 3A and B). The accommodation required to relieve this clash is expected to be significantly more perturbative for Ets-1, whose arginine contacts its partners directly and is more strongly inhibited by forward hemi-methylated SC1, than for PU.1 whose arginine forms primarily water-mediated contacts.
Figure 3.

Structural basis of differential inhibition of PU.1 and Ets-1 binding at hemi-methylated site-specific DNA. The high-affinity DNA sequences in co-crystal structures of the ETS domain of PU.1 (PDB: 1PUE) and Ets-1 (1K79) were mutated to methylated SC1 without modification of backbone coordinates. The protein/DNA interface near the methylated CpG dinucleotide is shown. The peptide backbones of PU.1 (Panels A and B) and Ets-1 (Panels C and D) are shown in gold and green, respectively, and only protein sidechains involved in the indicated interactions are rendered for clarity. Hydrogen bonds (2.8 ± 0.6 Å) emanating from the essential arginine (Arg235 in PU.1, Arg394 in Ets-1) at the recognition helix are shown in yellow. The closest heavy atom from the 5-methyl (colored green) of 5-methylcytosine (5MC) is joined by a red dash with distance in Å. For the PU.1/DNA complex, only the ordered waters of first contact are shown in Panel A, and only one hydration ‘train’ is shown in Panel C; note the same ordered waters marked ‘1’ and ‘3’ in the two panels. For the Ets-1/DNA complex, only ordered water participating in H-bonding with the protein is shown in Panels B and D.
In the reverse strand, ordered water also appears to differentiate the sensitivity of PU.1 and Ets-1 to CpG methylation. Although the leading C in the CpG dinucleotide formally overlaps with the central consensus (5′-TTCCMeG-3′), it does not contact the protein at all in the Ets-1/DNA complex, and its 5-methyl is over 7 Å away from the protein (Figure 3C). In contrast, the same position in the PU.1/DNA complex is part of a contiguous hydration network that anchors other protein and DNA residues (Figure 3D). The 5-methyl in the reverse hemi-methylated CpG, which is positioned ∼3 Å from the nearest ordered water molecule, is poised to disrupt this hydration network, resulting in the significant inhibition of reverse hemi-methylation on PU.1, while Ets-1 bound the same site almost as well as unmethylated SC1 (c.f. Table 1).
Molecular mechanics simulations revealed that stepwise methylation of a CpG dinucleotide is a structurally non-additive modification
While the co-crystal ETS/DNA structures for PU.1 and Ets-1 bound to unmethylated DNA account for the differential sensitivity of the two ETS domains to hemi-methylated DNA, it does not explain their intermediate behavior toward fully methylated DNA. In addition, thermodynamic cycle analysis (cf. Figure 2) indicates a substantial coupling free energy between the two methyl substituents. Both structural and thermodynamic considerations therefore strongly argue against the conformation for fully methylated DNA as the summed modifications of its hemi-methylated states. Given the role of structural propensity in the indirect readout of the target DNA by ETS proteins (35), we hypothesize that conformations that deviate most severely from the unmethylated state (i.e. the most ETS-favored conformation) are expected to be the most inhibitory to ETS binding. More specifically, the negative coupling free energies observed for all three ETS constructs suggest a compensatory effect in which fully methylated SC1 may be closer in conformation to the unmethylated state than either of hemi-methylated sites. To test this hypothesis, we simulated the structures of all four methylation states of the full 23-bp SC1 sequence used in our titration experiments, using parameters that mostly closely approximated our experimental conditions (Figure 4). While all energy-minimized structures were substantially homologous, as expected, the fully methylated SC1 site aligned more closely to the unmethylated site than either of the hemi-methylated counterparts.
Figure 4.

Simulated structures of unmethylated and methylated cognate ETS-binding sites. A duplex DNA harboring the same SC1 sequence as used in the titration experiments (5′-CGGCCAAGCGGAAGTGAGTGCC-3′) at various methylation states was simulated by all-atom energy minimization at physiological ionic strength using the CHARMM36 force field as described in Materials and Methods. The RMSDs of pairwise structural alignments are tabulated. The structures are convergent as judged by the change in total potential energies (Figure S4, Supplemental Data). The CpG dinucleotide is bracketed and 5-methyl substituents are colored in gold.
To analyze the structures in greater detail, standard helical parameters for each energy-minimized structures were determined to compare the conformational perturbations due to hemi- and full-methylation of the CpG dinucleotide in the SC1 site. The helical parameters were categorized as bp-axis (translation of the bases towards and perpendicular to the grooves: Xdisp, Ydisp; rotations around the short and long axes of the base pairs: inclination and tip), intra-bp (three translations: shear, stretch, and stagger; three rotations: buckle, propeller and opening), inter-bp (three translations: shift, slide and rise; three rotations: tilt, roll and twist), backbone (torsional angles along each phosphodiester chain and pseudorotation of the sugar ring) and groove geometry (widths and depths of the major and minor grooves) (23). In terms of molecular recognition, intra-bp, and inter-bp parameters primarily relate to direct readout of nucleobases, backbone and groove geometry reflects indirect readout of DNA shape and curvature, and bp-axis parameters represent a hybrid category.
Comparison of the four structures’ helical parameters showed complex changes along the DNA sequence that were not localized to the CpG dinucleotide. To intrepret the underlying perturbations, we performed principal component analysis (PCA) on each of the 39 helical parameters. PCA unfurls high-dimensional data by projecting the data on a reduced set of principal components (PC) that capture most of the variance. We found that the first two principal components (PC1 and PC2) accounted for a combined variance from 79% to 97% (≥90% in 31 parameters) in the data (Figure 5A), and tested for statistical differences among the PCs (Bartlett's test). As the first two principal components covered most the variance in the data, we constructed PC1–PC2 loading plots to evaluate the correlation of the four minimized structures with respect to each helical parameter (Table 2). Thus, highly correlated structures which were defined similarly in principal component space should cluster in the same quadrant in the loading plot, while structures whose principal component vectors occupied different quadrants would be more divergent from each other.
Figure 5.

Principal component analysis (PCA) of simulated un-, hemi-, and fully methylated structures of a CpG-containing ETS-binding site. The energy-minimized structures of the experimental SC1 sequence (cf. Figure 4) were analyzed by PCA as described in Materials and Methods. (A) Summed fractional variance of the first and second principal components for the 39 helical parameters. To assess the relative significance of the principal components (PCs), Bartlett's test was performed to identify parameters for which PC3 and PC4 (+), or PC2 to PC4 (*), were not significantly different; parameters whose four PCs were all significantly different were unmarked (all P < 0.05). (B) Major groove width (MGW) and roll, as representative parameters for which the fully methylated site was uncorrelated and correlated with either of the hemi-methylated sequence, respectively (cf. Table 2). Top: Variation in the parameters along the DNA sequence (00: unmethylated; 10: forward hemi-methylated; 01: reverse hemi-methylated; 11: fully methylated). Bottom: biplots scoring each base step (half step in the case of MGW) as points on principal component axes (% of the total variance in parenthesis), and each methylation state as vectors. To disperse the data, the points and vectors are plotted on different scales, and the CpG nucleotide (C10, G11) plus its nearest neighbors (C9, G12) are highlighted in orange (numbers denote position along the DNA sequence). Note the general lack of clustering of positions proximal to the CpG nucleotide with the methylation states, reflecting delocalized heterogeneity in the conformation of the four structures.
Table 2. Summary of principal component analysis (PCA) of the helical parameters of un-, hemi- and fully-methylated, energy-minimized SC1 DNA.

|
The loading plots in Table 2 showed that the vectors for all but two of the helical parameters (the torsional angles β on the 5′-GGAA-3′ strand and ϵ on the 5′-TTCC-3′ strand) occupied one side of PC1, reflecting the absence of overt irregularities in the minimized structures (cf. Figure 4). Here, we were specifically interested in how the conformation of the fully methylated SC1 site correlated with its hemi-methylated counterparts, by identifying helical parameters for which the PC vector for fully methylated SC1 occupied the same quadrant as either or both vectors representing the hemi-methylated SC1 sites. Strikingly, whereas 15 out of 17 helical parameters related to direct base readout (bp-axis, intra-bp and inter-bp) showed correlations between fully and hemi-methylated SC1, only 2 out of 22 parameters related to indirect readout (backbone and groove geometry) exhibited such correlations (Table 2). Correlation among the fully and hemi-methylated DNA in the base-centric parameters persisted even after a third PC was introduced (where statistically justified, i.e. those parameters marked ‘+’ in Figure 5A). For the two backbone/groove geometric parameters showing correlation, addition of PC3 resolved the correlation in the reverse (5′-TTCC-3′) glycosidic torsional angle χ (Figure S5, Supplemental Data); the same procedure on minor groove depth was not supported by Bartlett's test. Thus, fully methylated SC1 diverged substantially, specifically in backbone and groove geometry, from its hemi-methylated counterparts.
Interestingly, scoring of the helical parameters at each base position in principal component space showed no clustering with methylation state regardless of overall correlation among the fully and hemi-methylated states (Figure 5B), confirming that the structural perturbations were delocalized over the DNA duplex. In addition, of the 22 parameters in which fully methylated SC1 was uncorrelated to hemi-methylated SC1, 14 showed correlations between the fully methylated and unmethylated DNA (i.e. occupying the same quadrant, while the two hemi-methylated versions cluster in another), indicating substantial similarity in those parameters between fully and unmethylated DNA.
Finally, we examined the effect of methylation on the electrostatic properties of the ETS binding site (Figure S6, Supplemental Data). The van der Waals surface potential showed a highly localized patch of positive charge at 5-methylcytosines, due to increased exposure of the C6 and C2’ positions, that was associated with a slight deepening of the major groove (up to 0.5 Å). Although these patches were observed in all the methylated structures, their effect on the overall electrostatics was negligible as PCA of the per-atom electrostatic energies showed that their variance was dominated (>98%) by the first principal component.
In summary, principal component analysis of the helical parameters indicated that fully methylated SC1 was substantially different from hemi-methylated DNA in backbone and groove geometry. Moreover, the reversion of many of these parameters that are essential for indirect readout in fully methylated SC1 toward values most favored for ETS recognition, as embodied by unmethylated SC1, provided a biophysical basis for the nonadditivity in affinities among the fully and hemi-methylated DNA.
DISCUSSION
The nature of the interactions of ETS proteins with epigenetically modified DNA is currently controversial. Early studies, focusing on Ets-1 and sequence-similar (Class I) paralogs such as Ets-2 and GABPα, reported strong inhibition at fully methylated DNA binding sites (36,37), which were subsequently confirmed (38–40). Similar inhibition at DNA sites harboring fully methylated CpG dinucleotides was recently shown for another Class I ETS member, ETV1 (41). Thus, Class I members appeared to be strongly inhibited by fully methylated CpG-bearing binding sites. Nonetheless, some ETS-dependent promoters are highly methylated yet aberrantly active (42,43). Since only 16 of the 28 known human ETS members are Class I paralogs (44), and ETS members are heterogeneously competent in initiating transcription at nucleosomes (14,45), uncertainty in the methylation sensitivity of sequence-divergent ETS paralogs, such as PU.1 (a Class III member) represents a fundamental gap in our understanding of epigenetic control of ETS transcription factors in vivo.
Robust recognition of methylated DNA by PU.1: relevance to PU.1 as a pioneer transcription factor
Gaston and Fried first reported a difference in sensitivity of ETS proteins, including Ets-2 and PEA3 (ETV4), to hemi-methylated DNA sites (46). The inhibitory effects of hemi- and full methylation of the SC1 site on binding to Ets-1 agreed with their observations taken at a single concentration. Unlike Ets-1, PU.1 was more strongly inhibited by hemi-methylation in the 5′-TTCC-3′ strand than the 5′-GGAA-3′ strand. The difference in the two ETS domains’ sensitivity to hemi-methylation was explained by the presence of ordered interfacial water that was abundant in PU.1, but sparse in Ets-1 in their respective co-crystal structures (cf. Figure 3). PU.1 also bound fully methylated SC1 with 10-fold higher affinity than Ets-1, and over 900-fold when auto-inhibition was included, the latter corresponding to an affinity on the same order as binding to noncognate DNA (47). These contrasting differences therefore establish a biophysical basis for PU.1, but not Ets-1, as a pioneer transcription factor that capable of occupying methylated cognate DNA sites in vivo (48). PU.1 target genes that harbor a CpG dinucleotide at the same position as SC1 include CXCR1 (49), secretory IL-1 (50), tec (51), LIMD1 (52) and L-PGDS (53). While epigenetic control of ETS factors involves additional regulation from histone modifications (54,55), intrinsic robustness in binding methylated DNA should endow PU.1 and other master transcription factors a major advantage in transcriptional initiation over other regulators that do not. In addition, the relative competence of PU.1 to engage fully methylated DNA relative to Ets-1 (and by extension, other Class I paralogs) suggests a potential for functional inhibition of non-pioneer ETS paralogs by occupying their methylated binding sites.
Molecular mechanics, methylation status, and ETS/DNA affinity
Principal component analysis (PCA) corroborated the closer alignment of fully methylated SC1 with unmethylated DNA than either of the hemi-methylated sites (cf. Figure 4), and established the parametric basis of these deviations. Thus, while CpG methylation induced delocalized pertubrations in most DNA helical parameters along the entire DNA sequence, progression from hemi- to full methylation of a CpG dinucleotide altered ETS recognition principally through changes in backbone and groove conformation, rather than perturbations in base-pairing geometries. This demarcation was particularly striking given the correlations frequently reported between bp- and backbone-centric parameters in unmodified DNA (56,57). In terms of DNA recognition by ETS proteins, the greater deviation in backbone and groove conformation in hemi- over fully methylated DNA gave rise to the compensatory coupling free energy (cf. Figure 2) and the intermediate affinities with which the ETS domains bound fully methylated SC1 site. More generally, the PCA results showed that recognition of methylated DNA sites by ETS proteins operated via the indirect readout mechanism, formulated on the basis of unmethylated DNA (35), in which DNA shape represented a primary determinant of sequence selectivity.
In our analysis, consideration of energy-minimized DNA structures was sufficient to capture the relevant conformational determinants for ETS binding to fully methylated DNA. The PCA approach, which integrates over the entire DNA target in terms of helical parameters, was relatively coarse-grained in structure but could be addressed by including more experimentally tested DNA targets. Our simulations also did not include molecular dynamics and therefore any additional contributions from DNA flexibility or potential capture of specific conformations to the binding free energy (58,59). Previously, molecular mechanics succeeded in uncovering the relationship between the helical properties of unbound DNA and recognition of CpG-methylated DNA by DNase I (60). As in the present study, methylation substantially perturbed DNase's sequence preference, suggesting a similar interplay between DNA shape and methylation status as we observed for ETS/DNA binding.
Why does PU.1 bind fully methylated DNA more robustly than Ets-1?
Both PU.1 and Ets-1 bind the SC1 site, a representative high-affinity ETS binding site, with experimentally indistinguishable affinity. However, PU.1 binds fully methylated SC1 more strongly than the minimal ETS domain of Ets-1. The observation that hemi-methylation induces more perturbative changes in DNA backbone and groove geometry than full methylation provides an insight to this difference. Instead of absolute free energy changes [cf. Equation (1)], the thermodynamic cycle connecting the methylation states in Figure 2 may be reformulated in terms of differential free energy changes as follows:
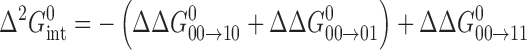 |
(2) |
Thus, the negative coupling free energy (Δ2G°int) quantifies the partial reversion of the fully methylated DNA towards the ETS-favored unmethylated state 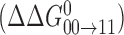 from the more disruptive backbone and groove conformations found in the hemi-methylated states
from the more disruptive backbone and groove conformations found in the hemi-methylated states 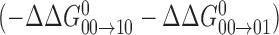 . For PU.1Δ167, both sets of terms, and the resultant coupling energy, are significantly smaller in magnitude than Ets-1Δ311 (cf. Figure 2). Physically, we propose that the hydration network in the PU.1/DNA interface acts as a moderating mechanism against the conformational perturbations of methylation, as may be achieved through alternative water-mediated contact arrangements. We have previously found that, unlike Ets-1, high-affinity PU.1/DNA binding in solution is tightly coupled to net hydration of the complex and highly sensitive to osmotic stress (16), consistent with the difference in ordered interfacial water observed in co-crystal structures for the two proteins (cf. Figure 3).
. For PU.1Δ167, both sets of terms, and the resultant coupling energy, are significantly smaller in magnitude than Ets-1Δ311 (cf. Figure 2). Physically, we propose that the hydration network in the PU.1/DNA interface acts as a moderating mechanism against the conformational perturbations of methylation, as may be achieved through alternative water-mediated contact arrangements. We have previously found that, unlike Ets-1, high-affinity PU.1/DNA binding in solution is tightly coupled to net hydration of the complex and highly sensitive to osmotic stress (16), consistent with the difference in ordered interfacial water observed in co-crystal structures for the two proteins (cf. Figure 3).
Biological relevance of hemi-methylated DNA in gene transactivation
Hemi-methylation arises in vivo through the semi-conservative nature of DNA replication: as incorporated nucleotides are unmethylated, the DNA daughter strands are hemi-methylated until fully re-methylated by DNA methyl transferase I (DNMT1) (61). Inhibition of DNMT1 leads to passive demethylation as hemi-methylated DNA accumulates (62), and is gaining attention due to the growing clinical use of DNMT1 inhibitors (‘hypomethylating agents’, e.g. azacitidine and decitabine) for the treatment of hematologic malignancies. The markedly different sensitivities of ETS transcription factors to hemi-methylation of CpG-bearing binding sites, as demonstrated presently by PU.1 and Ets-1, suggest how hemi-methylation may alter patterns of gene expression, by asymmetrically inhibiting transactivation at ETS-dependent promoters depending on the orientation of the ETS binding site. Thus, hemi-methylation may be an important mechanism by which heterogeneity in a tissue's transcriptome can be transiently (e.g., post-DNA replication) or persistently (e.g. in hypomethylation therapy) generated. To our knowledge, such a question has not been previously addressed.
CONCLUSION
Despite sharing superimposable backbone trajectories in their DNA-bound states, the ETS domains of PU.1 and Ets-1 encode markedly different sensitivity to DNA methylation. In particular, PU.1's robustness towards fully methylated DNA offers a biophysical basis for its functional status as a pioneer transcription factor. Moreover, hemi-methylated CpG states present distinct excursions in backbone and groove geometry that, contrary to expectations, are not intermediate of full methylation. Hemi-methylated sites are therefore distinct targets to ETS proteins (and presumably other DNA-binding proteins) for which indirect readout of backbone and groove elements plays a major role in DNA site recognition.
Supplementary Material
Acknowledgments
We thank Dr Donald Hamelberg for helpful discussion on molecular simulations and Mr Shingo Esaki for ETS protein purification.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Science Foundation [MCB 15451600]; National Institutes of Health [R21 HL129063]. Funding for open access charge: NSF[MCB 15451600]; NIH [R21 HL129063].
Conflict of interest statement. None declared.
REFERENCES
- 1.Zhu J., Emerson S.G. Hematopoietic cytokines, transcription factors and lineage commitment. Oncogene. 2002;21:3295–3313. doi: 10.1038/sj.onc.1205318. [DOI] [PubMed] [Google Scholar]
- 2.Orkin S.H. Transcription factors and hematopoietic development. J. Biol. Chem. 1995;270:4955–4958. doi: 10.1074/jbc.270.10.4955. [DOI] [PubMed] [Google Scholar]
- 3.Ciau-Uitz A., Wang L., Patient R., Liu F. ETS transcription factors in hematopoietic stem cell development. Blood Cells Mol. Dis. 2013;51:248–255. doi: 10.1016/j.bcmd.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 4.Scott E.W., Simon M.C., Anastasi J., Singh H. Requirement of transcription factor PU.1 in the development of multiple hematopoietic lineages. Science. 1994;265:1573–1577. doi: 10.1126/science.8079170. [DOI] [PubMed] [Google Scholar]
- 5.Hogart A., Lichtenberg J., Ajay S.S., Anderson S., Margulies E.H., Bodine D.M. Genome-wide DNA methylation profiles in hematopoietic stem and progenitor cells reveal overrepresentation of ETS transcription factor binding sites. Genome Res. 2012;22:1407–1418. doi: 10.1101/gr.132878.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pham T.H., Benner C., Lichtinger M., Schwarzfischer L., Hu Y., Andreesen R., Chen W., Rehli M. Dynamic epigenetic enhancer signatures reveal key transcription factors associated with monocytic differentiation states. Blood. 2012;119:e161–e171. doi: 10.1182/blood-2012-01-402453. [DOI] [PubMed] [Google Scholar]
- 7.de la Rica L., Rodriguez-Ubreva J., Garcia M., Islam A.B., Urquiza J.M., Hernando H., Christensen J., Helin K., Gomez-Vaquero C., Ballestar E. PU.1 target genes undergo Tet2-coupled demethylation and DNMT3b-mediated methylation in monocyte-to-osteoclast differentiation. Genome Biol. 2013;14:R99. doi: 10.1186/gb-2013-14-9-r99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Iwafuchi-Doi M., Zaret K.S. Pioneer transcription factors in cell reprogramming. Genes Dev. 2014;28:2679–2692. doi: 10.1101/gad.253443.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Tagore M., McAndrew M.J., Gjidoda A., Floer M. The Lineage-Specific Transcription Factor PU.1 Prevents Polycomb-Mediated Heterochromatin Formation at Macrophage-Specific Genes. Mol. Cell Biol. 2015;35:2610–2625. doi: 10.1128/MCB.00027-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.DiSpirito J.R., Fang B., Wang F., Lazar M.A. Pruning of the Adipocyte Peroxisome Proliferator-Activated Receptor γ Cistrome by Hematopoietic Master Regulator PU.1. Mol. Cell. Biol. 2013;33:3354–3364. doi: 10.1128/MCB.00599-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L., et al. Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 2010;32:317–328. doi: 10.1016/j.immuni.2010.02.008. [DOI] [PubMed] [Google Scholar]
- 12.Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Barozzi I., Simonatto M., Bonifacio S., Yang L., Rohs R., Ghisletti S., Natoli G. Coregulation of Transcription Factor Binding and Nucleosome Occupancy through DNA Features of Mammalian Enhancers. Mol. Cell. 2014;54:844–857. doi: 10.1016/j.molcel.2014.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sherwood R.I., Hashimoto T., O'Donnell C.W., Lewis S., Barkal A.A., van Hoff J.P., Karun V., Jaakkola T., Gifford D.K. Discovery of directional and nondirectional pioneer transcription factors by modeling DNase profile magnitude and shape. Nat. Biotechnol. 2014;32:171–178. doi: 10.1038/nbt.2798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.He G., Tolic A., Bashkin J.K., Poon G.M. Heterogeneous dynamics in DNA site discrimination by the structurally homologous DNA-binding domains of ETS-family transcription factors. Nucleic Acids Res. 2015;43:4322–4331. doi: 10.1093/nar/gkv267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wang S., Linde M.H., Munde M., Carvalho V.D., Wilson W.D., Poon G.M. Mechanistic heterogeneity in site recognition by the structurally homologous DNA-binding domains of the ETS family transcription factors Ets-1 and PU.1. J. Biol. Chem. 2014;289:21605–21616. doi: 10.1074/jbc.M114.575340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Stephens D.C., Kim H.M., Kumar A., Farahat A.A., Boykin D.W., Poon G.M. Pharmacologic efficacy of PU.1 inhibition by heterocyclic dications: a mechanistic analysis. Nucleic Acids Res. 2016;44:4005–4013. doi: 10.1093/nar/gkw229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Poon G.M.K. Sequence discrimination by DNA-binding domain of ETS family transcription factor PU.1 is linked to specific hydration of protein-DNA interface. J. Biol. Chem. 2012;287:18297–18307. doi: 10.1074/jbc.M112.342345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Poon G.M.K. DNA binding regulates the self-association of the ETS domain of PU.1 in a sequence-dependent manner. Biochemistry. 2012;51:4096–4107. doi: 10.1021/bi300331v. [DOI] [PubMed] [Google Scholar]
- 20.Tataurov A.V., You Y., Owczarzy R. Predicting ultraviolet spectrum of single stranded and double stranded deoxyribonucleic acids. Biophys. Chem. 2008;133:66–70. doi: 10.1016/j.bpc.2007.12.004. [DOI] [PubMed] [Google Scholar]
- 21.Lu X.J., Olson W.K. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang P., Brank A.S., Banavali N.K., Nicklaus M.C., Marquez V.E., Christman J.K., MacKerell A.D. Use of oligodeoxyribonucleotides with conformationally constrained abasic sugar targets to probe the mechanism of base flipping by HhaI DNA (cytosine C5)-methyltransferase. J. Am. Chem. Soc. 2000;122:12422–12434. [Google Scholar]
- 23.Lavery R., Moakher M., Maddocks J.H., Petkeviciute D., Zakrzewska K. Conformational analysis of nucleic acids revisited: Curves+ Nucleic Acids Res. 2009;37:5917–5929. doi: 10.1093/nar/gkp608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Poon G.M., Macgregor R.B., Jr Base coupling in sequence-specific site recognition by the ETS domain of murine PU.1. J. Mol. Biol. 2003;328:805–819. doi: 10.1016/s0022-2836(03)00362-0. [DOI] [PubMed] [Google Scholar]
- 25.Nye J.A., Petersen J.M., Gunther C.V., Jonsen M.D., Graves B.J. Interaction of murine ets-1 with GGA-binding sites establishes the ETS domain as a new DNA-binding motif. Genes Dev. 1992;6:975–990. doi: 10.1101/gad.6.6.975. [DOI] [PubMed] [Google Scholar]
- 26.Graves B.J., Gillespie M.E., McIntosh L.P. DNA binding by the ETS domain. Nature. 1996;384:322. doi: 10.1038/384322a0. [DOI] [PubMed] [Google Scholar]
- 27.Wei G.H., Badis G., Berger M.F., Kivioja T., Palin K., Enge M., Bonke M., Jolma A., Varjosalo M., Gehrke A.R., et al. Genome-wide analysis of ETS-family DNA-binding in vitro and in vivo. EMBO J. 2010;29:2147–2160. doi: 10.1038/emboj.2010.106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen J.M., Skalicky J.J., Donaldson L.W., McIntosh L.P., Alber T., Graves B.J. Modulation of transcription factor Ets-1 DNA binding: DNA-induced unfolding of an alpha helix. Science. 1995;269:1866–1869. doi: 10.1126/science.7569926. [DOI] [PubMed] [Google Scholar]
- 29.Wang H., McIntosh L.P., Graves B.J. Inhibitory module of Ets-1 allosterically regulates DNA binding through a dipole-facilitated phosphate contact. J. Biol. Chem. 2002;277:2225–2233. doi: 10.1074/jbc.M109430200. [DOI] [PubMed] [Google Scholar]
- 30.Jonsen M.D., Petersen J.M., Xu Q.P., Graves B.J. Characterization of the cooperative function of inhibitory sequences in Ets-1. Mol. Cell. Biol. 1996;16:2065–2073. doi: 10.1128/mcb.16.5.2065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kodandapani R., Pio F., Ni C.Z., Piccialli G., Klemsz M., McKercher S., Maki R.A., Ely K.R. A new pattern for helix-turn-helix recognition revealed by the PU.1 ETS-domain-DNA complex. Nature. 1996;380:456–460. doi: 10.1038/380456a0. [DOI] [PubMed] [Google Scholar]
- 32.Garvie C.W., Hagman J., Wolberger C. Structural studies of Ets-1/Pax5 complex formation on DNA. Mol. Cell. 2001;8:1267–1276. doi: 10.1016/s1097-2765(01)00410-5. [DOI] [PubMed] [Google Scholar]
- 33.Strauss-Soukup J.K., Maher L.J., 3rd Role of asymmetric phosphate neutralization in DNA bending by PU.1. J. Biol. Chem. 1997;272:31570–31575. doi: 10.1074/jbc.272.50.31570. [DOI] [PubMed] [Google Scholar]
- 34.Okonogi T.M., Alley S.C., Harwood E.A., Hopkins P.B., Robinson B.H. Phosphate backbone neutralization increases duplex DNA flexibility: a model for protein binding. Proc. Natl. Acad. Sci. U.S.A. 2002;99:4156–4160. doi: 10.1073/pnas.072067799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Szymczyna B.R., Arrowsmith C.H. DNA binding specificity studies of four ETS proteins support an indirect read-out mechanism of protein-DNA recognition. J. Biol. Chem. 2000;275:28363–28370. doi: 10.1074/jbc.M004294200. [DOI] [PubMed] [Google Scholar]
- 36.Yokomori N., Kobayashi R., Moore R., Sueyoshi T., Negishi M. A DNA methylation site in the male-specific P450 (Cyp 2d-9) promoter and binding of the heteromeric transcription factor GABP. Mol. Cell. Biol. 1995;15:5355–5362. doi: 10.1128/mcb.15.10.5355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Umezawa A., Yamamoto H., Rhodes K., Klemsz M.J., Maki R.A., Oshima R.G. Methylation of an ETS site in the intron enhancer of the keratin 18 gene participates in tissue-specific repression. Mol. Cell. Biol. 1997;17:4885–4894. doi: 10.1128/mcb.17.9.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lucas M.E., Crider K.S., Powell D.R., Kapoor-Vazirani P., Vertino P.M. Methylation-sensitive Regulation of TMS1/ASC by the Ets Factor, GA-binding Protein-α. J. Biol. Chem. 2009;284:14698–14709. doi: 10.1074/jbc.M901104200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nickel J., Short M.L., Schmitz A., Eggert M., Renkawitz R. Methylation of the mouse M-lysozyme downstream enhancer inhibits heterotetrameric GABP binding. Nucleic Acids Res. 1995;23:4785–4792. doi: 10.1093/nar/23.23.4785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Polansky J.K., Schreiber L., Thelemann C., Ludwig L., Kruger M., Baumgrass R., Cording S., Floess S., Hamann A., Huehn J. Methylation matters: binding of Ets-1 to the demethylated Foxp3 gene contributes to the stabilization of Foxp3 expression in regulatory T cells. J. Mol. Med. (Berl.) 2010;88:1029–1040. doi: 10.1007/s00109-010-0642-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cooper C.D., Newman J.A., Aitkenhead H., Allerston C.K., Gileadi O. Structures of the Ets protein DNA-binding domains of transcription factors Etv1, Etv4, Etv5, and Fev: determinants of DNA binding and redox regulation by disulfide bond formation. J. Biol. Chem. 2015;290:13692–13709. doi: 10.1074/jbc.M115.646737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Toyota M., Shen L., Ohe-Toyota M., Hamilton S.R., Sinicrope F.A., Issa J.-P.J. Aberrant methylation of the cyclooxygenase 2 CpG island in colorectal tumors. Cancer Res. 2000;60:4044–4048. [PubMed] [Google Scholar]
- 43.Asting A.G., Caren H., Andersson M., Lonnroth C., Lagerstedt K., Lundholm K. COX-2 gene expression in colon cancer tissue related to regulating factors and promoter methylation status. BMC Cancer. 2011;11:238. doi: 10.1186/1471-2407-11-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hollenhorst P.C., McIntosh L.P., Graves B.J. Genomic and biochemical insights into the specificity of ETS transcription factors. Annu. Rev. Biochem. 2011;80:437–471. doi: 10.1146/annurev.biochem.79.081507.103945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wang J., Zhuang J., Iyer S., Lin X., Whitfield T.W., Greven M.C., Pierce B.G., Dong X., Kundaje A., Cheng Y., et al. Sequence features and chromatin structure around the genomic regions bound by 119 human transcription factors. Genome Res. 2012;22:1798–1812. doi: 10.1101/gr.139105.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gaston K., Fried M. CpG methylation has differential effects on the binding of YY1 and ETS proteins to the bi-directional promoter of the Surf-1 and Surf-2 genes. Nucleic Acids Res. 1995;23:901–909. doi: 10.1093/nar/23.6.901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goetz T.L., Gu T.L., Speck N.A., Graves B.J. Auto-inhibition of Ets-1 is counteracted by DNA binding cooperativity with core-binding factor alpha2. Mol. Cell. Biol. 2000;20:81–90. doi: 10.1128/mcb.20.1.81-90.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pham T.H., Minderjahn J., Schmidl C., Hoffmeister H., Schmidhofer S., Chen W., Langst G., Benner C., Rehli M. Mechanisms of in vivo binding site selection of the hematopoietic master transcription factor PU.1. Nucleic Acids Res. 2013;41:6391–6402. doi: 10.1093/nar/gkt355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wilkinson N.C., Navarro J. PU.1 regulates the CXCR1 promoter. J. Biol. Chem. 1999;274:438–443. doi: 10.1074/jbc.274.1.438. [DOI] [PubMed] [Google Scholar]
- 50.Smith M.F., Jr, Carl V.S., Lodie T., Fenton M.J. Secretory interleukin-1 receptor antagonist gene expression requires both a PU.1 and a novel composite NF-kappaB/PU.1/ GA-binding protein binding site. J. Biol. Chem. 1998;273:24272–24279. doi: 10.1074/jbc.273.37.24272. [DOI] [PubMed] [Google Scholar]
- 51.Honda H., Ozawa K., Yazaki Y., Hirai H. Identification of PU.1 and Sp1 as essential transcriptional factors for the promoter activity of mouse tec gene. Biochem. Biophys. Res. Commun. 1997;234:376–381. doi: 10.1006/bbrc.1997.6650. [DOI] [PubMed] [Google Scholar]
- 52.Foxler D.E., James V., Shelton S.J., Vallim T.Q., Shaw P.E., Sharp T.V. PU.1 is a major transcriptional activator of the tumour suppressor gene LIMD1. FEBS Lett. 2011;585:1089–1096. doi: 10.1016/j.febslet.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Joo M., Kwon M., Cho Y.J., Hu N., Pedchenko T.V., Sadikot R.T., Blackwell T.S., Christman J.W. Lipopolysaccharide-dependent interaction between PU.1 and c-Jun determines production of lipocalin-type prostaglandin D synthase and prostaglandin D2 in macrophages. Am. J. Physiol. Lung Cell Mol. Physiol. 2009;296:L771–L779. doi: 10.1152/ajplung.90320.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Zhang J.A., Mortazavi A., Williams B.A., Wold B.J., Rothenberg E.V. Dynamic transformations of genome-wide epigenetic marking and transcriptional control establish T cell identity. Cell. 2012;149:467–482. doi: 10.1016/j.cell.2012.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.van Riel B., Rosenbauer F. Epigenetic control of hematopoiesis: the PU.1 chromatin connection. Biol. Chem. 2014;395:1265–1274. doi: 10.1515/hsz-2014-0195. [DOI] [PubMed] [Google Scholar]
- 56.Packer M.J., Hunter C.A. Sequence-dependent DNA structure: the role of the sugar-phosphate backbone. J. Mol. Biol. 1998;280:407–420. doi: 10.1006/jmbi.1998.1865. [DOI] [PubMed] [Google Scholar]
- 57.Bharanidharan D., Gautham N. Principal component analysis of DNA oligonucleotide structural data. Biochem. Biophys. Res. Commun. 2006;340:1229–1237. doi: 10.1016/j.bbrc.2005.12.127. [DOI] [PubMed] [Google Scholar]
- 58.Carvalho A.T.P., Gouveia L., Kanna C.R., Wärmländer S.K.T.S., Platts J.A., Kamerlin S.C.L. Understanding the structural and dynamic consequences of DNA epigenetic modifications: Computational insights into cytosine methylation and hydroxymethylation. Epigenetics. 2014;9:1604–1612. doi: 10.4161/15592294.2014.988043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Pérez A., Castellazzi C.L., Battistini F., Collinet K., Flores O., Deniz O., Ruiz M.L., Torrents D., Eritja R., Soler-López M., et al. Impact of methylation on the physical properties of DNA. Biophys. J. 2012;102:2140–2148. doi: 10.1016/j.bpj.2012.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lazarovici A., Zhou T., Shafer A., Dantas Machado A.C., Riley T.R., Sandstrom R., Sabo P.J., Lu Y., Rohs R., Stamatoyannopoulos J.A., et al. Probing DNA shape and methylation state on a genomic scale with DNase I. Proc. Natl. Acad. Sci. U.S.A. 2013;110:6376–6381. doi: 10.1073/pnas.1216822110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Jones P.A., Liang G. Rethinking how DNA methylation patterns are maintained. Nat. Rev. Genet. 2009;10:805–811. doi: 10.1038/nrg2651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jones P.A., Taylor S.M. Cellular differentiation, cytidine analogs and DNA methylation. Cell. 1980;20:85–93. doi: 10.1016/0092-8674(80)90237-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.