


Abstract
In eukaryotic cells the CDC7/DBF4 kinase, also known as DBF4-dependent kinase (DDK), is required for the firing of DNA replication origins. CDC7 is also involved in replication stress responses and its depletion sensitises cells to drugs that affect fork progression, including Topoisomerase 2 poisons. Although CDC7 is an important regulator of cell division, relatively few substrates and bona-fide CDC7 phosphorylation sites have been identified to date in human cells. In this study, we have generated an active recombinant CDC7/DBF4 kinase that can utilize bulky ATP analogues. By performing in vitro kinase assays using benzyl-thio-ATP, we have identified TOP2A as a primary CDC7 substrate in nuclear extracts, and serine 1213 and serine 1525 as in vitro phosphorylation sites. We show that CDC7/DBF4 and TOP2A interact in cells, that this interaction mainly occurs early in S-phase, and that it is compromised after treatment with CDC7 inhibitors. We further provide evidence that human DBF4 localises at centromeres, to which TOP2A is progressively recruited during S-phase. Importantly, we found that CDC7/DBF4 down-regulation, as well S1213A/S1525A TOP2A mutations can advance the timing of centromeric TOP2A recruitment in S-phase. Our results indicate that TOP2A is a novel DDK target and have important implications for centromere biology.
INTRODUCTION
In order to divide, cells must completely and accurately replicate their DNA once every cell cycle. Incomplete or over-replication can lead to cell death or genomic instability, which is a major contributing factor in the development of cancer (1,2). As such, DNA replication is a tightly regulated and monitored process (reviewed in (3,4)). The DBF4-dependent kinase (DDK), which is a complex formed by the CDC7 catalytic subunit bound to either DBF4 or DBF4B (5,6) is involved in multiple aspects of the regulation of DNA replication. It is required for the firing of replication origins by phosphorylating multiple subunits of the MCM2-7 helicase complex (7–9). In addition, CDC7 kinase has important roles in the replication stress response and chromatin function. For instance, in human cells CDC7 phosphorylation of the mediator protein CLASPIN is important for full activation of CHK1 by ATR and for maintaining cell viability in the presence of drugs that affect replication fork progression (10–12,13). Also, CDC7 phosphorylation of RAD18 is required for the efficient recruitment of the translesion synthesis (TLS) polymerase Polη to stalled forks (14,15). In human cells, CDC7 kinase has also been shown to affect the function of the p150 Chromatin Assembly Factor 1 (CAF1) subunit (16), while in yeast it participates in the control of core histone levels (17), contributes to centromeric heterochromatin function (18), and directly phosphorylates Histone H3 at Ser45 during replication (19). Importantly, several laboratories have elucidated the role of CDC7 kinase in controlling the formation of DNA double strand breaks during meiotic DNA replication to promote meiotic recombination (20–25).
In recent years, a chemical genetics approach has been developed that provides a novel tool for inhibiting a specific kinase. The target kinase is mutated at a specific residue in the ATP binding pocket, termed a ‘gatekeeper’ residue, and this mutation enlarges the binding site sufficiently to allow entry and binding of novel small-molecule inhibitors, namely bulky pyrazolo pyrimidine compounds (PP1s) and novel staurosporine derivatives. Binding of these compounds can inhibit the engineered kinase but they are too bulky to enter the ATP pocket of other cellular kinases and are therefore unable to inhibit them (26,27). This approach has been widely used in a variety of organisms from budding yeast to cultured human cells and transgenic mice with the specific goal of studying the function of a given kinase (28,29). However, the identification of direct substrates and phosphorylation sites is still a challenging task. A recent important advance in this kinase chemical genetics approach was the finding that the engineered kinase can now accept and utilize an unnatural and bulky ATP analogue (N6-(benzyl)ATP). These analogues are very poor substrates for wild-type kinases, thus only the analogue-sensitive kinase (AS-kinase) can use the bulky ATP analogue. Further modification of this analogue into N6-(benzyl)ATP-γ-S (benzyl-thio-ATP) allows the transfer of a thio-phosphate reactive group as a distinctive molecular tag by the AS-kinase onto the substrate protein. Therefore, after an in vitro kinase reaction and further derivatization of these reactive groups with p-nitrobenzylmesylate (PNBM), labeled proteins that are direct substrates of the AS-kinase can be detected by western blotting (30). Furthermore, peptides containing this unique thio-phosphate modification can be specifically captured from digests of labeled protein mixtures; mass spectrometry is then used to reveal the identity of the corresponding protein species and the location of the phosphorylation site(s) (28,31–33).
In order to obtain further insights into possible substrates and roles of human DDK in the mitotic cell cycle, we have used the above-described approach and identified Topoisomerase 2 alpha (TOP2A) as a prime in vitro substrate of the kinase. Topoisomerase 2 (TOP2) enzymes resolve DNA catenates that form during DNA replication by catalysing the transient breakage and religation of duplex DNA, while allowing the passage of a second duplex through the gap (34,35). Of the two isoforms present in humans, only the alpha isoform, TOP2A, is essential for proliferation of cultured cells (36). Although reportedly dispensable for DNA replication, TOP2A is essential for proper chromosome condensation and sister chromatid separation, as TOP2A-deficient cells exhibit an increased number of amorphous and severely entangled chromosomes (36,37). TOP2A also plays an important role in resolving ultrafine anaphase DNA bridges arising from centromeric loci (38,39).
We have previously reported that treatment with etoposide, a TOP2 poison that prevents the religation step of the TOP2 catalytic cycle, thus stabilizing an intermediate TOP2-DNA covalent complex (40,41), induced more extensive cell death when cells were also depleted of CDC7 (10). The increased sensitivity to etoposide may be due to the roles of CDC7 in the general response to replication stress, or could perhaps reflect a functional interaction between CDC7 kinase and TOP2. In this study, we explored the latter hypothesis and we found that CDC7/DBF4 physically and functionally interacts with TOP2A in human cells.
MATERIALS AND METHODS
Cell culture and chemicals
Flp-In T-REx 293 cells (Invitrogen) and U2OS (obtained from the Centre for Chromosome Biology, NUI Galway, and validated by STR analysis), were grown in Dulbecco's modified Eagle's medium containing 10% fetal bovine serum and maintained at 37°C with 5% CO2 in a humidified atmosphere. Nocodazole (Sigma) was used at 1 μM, mimosine (Sigma) at 1 mM, PHA-767491 (Tocris) and XL413 (synthesized in house (11)) at 10 μM, DMSO was used as control. Plasmids and siRNA transfections were performed using jetPEI and jetPRIME (Polyplus Transfection) respectively according to manufacturer's instructions. siRNAs for CDC7 and DBF4 were obtained from Dharmacon. Control non-targeting siRNA AGUACUGCUUACGAUACGG from Ambion.
DNA plasmids
EGFP-DBF4 expression construct was obtained by cloning DBF4 coding sequence into vector pIC111 (42). N-terminal tagged (S-Tag-2xFLAG) TOP2A was obtained from Luo (43), and the coding sequence was PCR amplified and cloned into vector pcDNA3.1. The plasmid was subject to site-directed mutagenesis (Agilent, Quickchange II XL) to obtain constructs expressing S-Tag-2xFLAG-TOP2A WT, S1213A/S1525A and S1213D/S1525D mutants. All constructs were verified by sequencing.
Recombinant protein expression and purification
All recombinant proteins were expressed in Escherichia coli and purified using GSTrap FF or HisTrap FF columns (GE Healthcare) according to manufacturer's instructions. GST-tagged recombinant human CDC7–DBF4[197–333] complex and MCM2[Nterm] were eluted using PreScission protease to remove the GST-tag. The M118A, M134A and M118/M134A CDC7 was obtain by site directed mutagenesis of pGEX-CDC7/DBF4MC previously described (11).
Preparation of nuclear extract
Cells were resuspended in hypotonic buffer (10 mM HEPES pH 7.5, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM TCEP and protease inhibitors) and allowed to swell on ice for 20 min. An equal volume of lysis buffer (10 mM HEPES pH 7.5, 1.5 mM MgCl2, 10 mM KCl, 0.5 mM TCEP, 1.0% NP40 and protease inhibitors) was added to the cells and mixed by brief vortex. Nuclei were pelleted by centrifugation (450 × g, 5 min, 4°C) and lysed (50 mM HEPES pH 7.5, 150 mM NaCl, 1.5 mM MgCl2, 0.5 mM TCEP, 0.05% (v/v) Tween-20, protease inhibitors, and 0.25 U/μl Benzonase (Sigma) to digest chromatin).
In vitro thio-phosphorylation
Twenty microgram of nuclear extract was incubated with 200 ng of AS-CDC7/DBF4MC in the presence of ATP or 50 μM N6-(benzyl)ATP-γ-S, 1 mM GTP in kinase buffer (50 mM HEPES pH 7.5, 10 mM MgCl2, 0.5 mM TCEP and protease inhibitor) for 30 min at 30°C in a final volume of 20 μl. The reaction was terminated by adding EDTA to 20 mM. The mixture was alkylated with 2.5 mM p-nitrobenzylmesylate (PNBM) for 1.5 h at room temperature, heated in Laemmli buffer, and analyzed by western blot with anti-thiophosphate ester antibody (30).
Covalent capture of thio-phosphorylated AS-CDC7 substrates
Eight milligrams of nuclear extract prepared from XL413-treated cells was incubated with purified recombinant AS-CDC7/DBF4MC to 1% by mass of total protein, 50 μM N6-(benzyl)ATP-γ-S, and 1.5 mM GTP in kinase buffer for 30 min at 30°C. The reactions were stopped by the addition of 20 mM EDTA. The proteins were precipitated with methanol/chloroform and digested with trypsin. The desalted peptides were added to iodoacetylagarose beads (SulfoLink gel, Pierce) in buffer (25 mM HEPES pH 7.0, BSA 25 μg/ml, TCEP 2 mM, 50% acetonitrile). The beads were incubated overnight in the dark with rotation, washed once with water, followed by 5 M NaCl, 50% acetonitrile, and 5% formic acid in water. The beads were then incubated in 1 ml 10 mM DTT prior to elution with 200 μl of freshly prepared 2 mg/ml Oxone. The phosphopeptides were desalted and enriched by Pierce™ Magnetic Titanium Dioxide Phosphopeptide Enrichment Kit (ThermoFisher Scientific). The recovered peptides were analyzed by LC–MS/MS.
Liquid chromatography–tandem MS (LC–MS/MS) analysis
Five microliters of desalted solution was injected in a capillary chromatographic system (Agilent 1100 Series, Agilent Technologies, Waldbronn, Germany). Peptide separations occurred on a RP homemade 15 cm reverse-phase spraying fused silica capillary column (75 μm i.d. × 15 cm), packed with 3 μm ReproSil C18 AQ (Dr. Maisch GmbH, Germany). A gradient of eluents A (5% acetonitrile, 0.1% formic acid) and B (acetonitrile, 0.1% formic acid) was used to achieve separation, from 8% B to 80% B in 48 min, at 0.3 μl/min flow rate. The LC system was connected to a Fourier transformed-LTQ Ultra mass spectrometer (FT-LTQ Ultra, Thermo Electron, San Jose, CA, USA) equipped with a nanoelectrospray ion source (Proxeon Biosystems, Odense, Denmark). Survey MS scans were acquired in the FT from m/z 350–1650 with 100 000 resolution. The six most intense doubly and triply charged ions were automatically selected for fragmentation. Target ions already selected for the MS/MS were dynamically excluded for 30s; the data-dependent neutral loss algorithm was enabled for each MS/MS spectra to trigger a MS3 scan (44). DATABASE SEARCHING–Raw MS files were converted using Raw2msm software (.msm files); MS2 and MS3 peak lists were analyzed using (Matrix Science, London, UK; version 2.3.02). Mascot was set up to search the UniProt_CP_Human_20150401 database (unknown version, 90 411 entries) with trypsin as digestion enzyme. Mascot was searched with a fragment ion mass tolerance of 0.50 Da and a parent ion tolerance of 10 PPM. Deamidation of asparagine and glutamine, oxidation and dioxidation of methionine, acetylation of the N-terminus, carbamidomethyl of cysteine, phosphorylation of serine, threonine and tyrosine were specified in Mascot as variable modifications. Criteria for protein identification—Scaffold (version Scaffold_4.4.3, Proteome Software Inc., Portland, OR, USA) was used to validate MS/MS based peptide and protein identifications. Peptide identifications were accepted if they could be established at >95.0% probability by the Peptide Prophet algorithm (45) with Scaffold delta-mass correction. Protein identifications were accepted if they could be established at greater than 99,0% probability and contained at least two identified peptides. Protein probabilities were assigned by the Protein Prophet algorithm (46). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped to satisfy the principles of parsimony.
Immunoprecipitations
Nuclei were isolated as described, and chromatin-bound proteins were released by Benzonase treatment in IP buffer (50 mM Tris pH 7.6, 150 mM NaCl, 2 mM MgCl2, 1 mM EDTA, 1% Triton X-100). IgG or anti-TOP2A antibody were prebound to protein A resin (GeneSpin) and equal amounts of precleared nuclear extract were immunoprecipitated for 2 h with rotation at 4°C. Following washes with IP buffer, proteins were recovered in 1× Laemmli buffer and analysed by SDS-PAGE and western blotting.
Antibodies and western blotting
Primary antibodies included: anti-CDC7 (MBL International), anti-TOP2A (Millipore), anti- NWSHPQFEK/Strep-tag (GenScript), anti-centromere antibodies (ACA) (Antibodies Incorporated), anti-MCM2 (AbD Serotec), anti-pSer40/41 MCM2 described in (7), β-actin (Sigma), anti-RPB1 (Santa-Cruz Biotechnology), anti-β-tubulin (Santa-Cruz Biotechnology), and anti-thiophosphate ester (Abcam). Equal amounts of cell extract were resolved on 7.5% or 10% gels for standard SDS-PAGE, or 5% gels with 5 μM Phos-tag acrylamide (Wako Pure Chemical Industries) and 10 μM MnCl2 for phosphorylation-dependent mobility shift detection, and subject to western blot analysis. IRDye® secondary antibodies for protein detection using the Odyssey infrared imaging system were used (Li-COR Biosciences). For dephosphorylation assays, 20 μg of extracts were incubated with 20 U of Lambda Phosphatase (Sigma) for 30 min before SDS-PAGE.
Immunofluorescence microscopy
Cells were grown on glass coverslips and treated as described in the text. Soluble proteins were pre-extracted on ice for 5 min with ice-cold HPEM buffer (30 mM HEPES, 65 mM PIPES, 100 mM NaCl, 10 mM EGTA, 2 mM MgCl2, 0.5% Triton X-100) and fixed with 4% PFA for 10 min at room temperature. Coverslips were incubated in blocking buffer (3% bovine serum albumin in PBS) for 30 min, followed by primary antibodies diluted in blocking buffer for 1 h. For EdU labeling, coverslips were then incubated in Click reaction solution (10 μM azide-fluor 545, 10 mM sodium-l-ascorbate, 2 mM CuSO4) for 30 min and washed. Finally, coverslips were incubated with the appropriate Alexa Fluor secondary antibodies (ThermoFisher Scientific), and DNA was counterstained with 4′,6-diamidino-2-phenylindole (DAPI). Images were captured using a DeltaVision Core system (Applied Precision) controlling an interline charge-coupled device camera (Coolsnap HQ2; Roper Technologies) mounted on an inverted microscope (IX-71; Olympus). Images were collected at 2× binning using a 60× or 100× oil objective. Z-series were collected using 0.2 μm intervals. Images were deconvolved using SoftWoRx (Applied Precision).
Flow cytometry
For the analysis of DNA content, cells were fixed with 70% ethanol/PBS, centrifuged, resuspended in PBS containing RNase A (2 μg/ml) and propidium iodide (5 μg/ml), and incubated for 30 min. Samples were analyzed using a BD FACS CantoA (BD Bioscences).
Chromatin immunoprecipitation (ChIP)
Cells were washed once with cold PBS, counted, and resuspended in PBS at room-temperature. Cross-linking was performed with 1.5 mM ethylene glycol bis (succinimidyl succinate) (EGS) from Thermo Fisher Scientific and 1% PFA for a total of 35 and 10 min, respectively. Fixation was quenched with 50 mM glycine. Nuclei were isolated by sequential incubation in Buffer A (100 mM Tris pH 8, 10 mM DTT), Buffer B (10 mM HEPES pH 7.5, 0.25% Triton X-100) and buffer C (10 mM HEPES pH 7.5, 200 mM NaCl). Chromatin was digested with MNase in digestion buffer (50 mM Tris pH 8, 5 mM CaCl2), to mostly mono and dinucleosomes with some trinucleosomes, and briefly sonicated to lyse nuclear membranes. Insoluble material was removed by centrifugation (16 000 × g, 10 min). Supernatant was diluted with ChIP IP buffer (20 mM Tris pH 8, 150 mM NaCl, 2.5 mM MgCl2, 0.5% Triton X-100, 0.1% Nonidet P-40) and incubated for 16 h with primary antibodies. Antibody complexes were recovered with Protein A/G beads (Thermo Fisher Scientific), and washed extensively with IP buffer, LiCl wash buffer (10 mM Tris pH 8, 1 mM EDTA, 250 mM LiCl2, 1% Nonidet P-40, 1% sodium deoxycholate) and TE buffer. Samples were eluted with elution buffer (0.1 M NaHCO3, 1% SDS) for 45 min at 65°C with constant agitation. Cross-links were reversed by incubation at 65°C for 18 h. Recovered DNA was analysed on a StepOnePlus Real-Time PCR System (Applied Biosystems). Primers were selected to amplify a 201 bp centromeric sequence specific to chromosome 1 (Fwd: GGCCTATGGCAGCAGAGGATATAACTGCC, Rev: GTGAGTTTTCTCCCGTATCCAACGAAATCC) (47), and a 165 bp non-centromeric sequence at the GAPDH promoter (sequences provided by Thermo Fisher Scientific; Fwd: TACTAGCGGTTTTACGGGCG, Rev: TCGAACAGGAGGAGCAGAGAGCGA).
Statistics
Graphs and statistical analyses were performed using GraphPad Prism software.
RESULTS
Generation of an engineered CDC7 kinase and identification of TOP2A as an in vitro substrate
Based on sequence alignments and structural information, M118 and M134 appear to constrict the CDC7 ATP-binding pocket and could be considered the putative gatekeeper residues of the kinase (48,49). By site directed mutagenesis, we changed the codons for M118 and M134 either individually or in combination, in order to generate three different potential analogue sensitive (AS) alleles of CDC7, namely M118A, M134A and the double M118A/M134A. The wild type (WT) and the three potential AS-CDC7 proteins were then expressed in E. coli as GST-fusions together with a polypeptide corresponding to amino acids 197 to 334 of the DBF4 protein, which is sufficient to bind to and activate the kinase (11). Proteins were purified on GST affinity beads, GST-tags were cleaved, and their capability to transfer thio-phosphate groups from N6-(benzyl)ATP-γ-S to the MCM2 protein was tested in in vitro kinase reactions. We found that all the three putative AS-CDC7 proteins, but not WT CDC7 could thio-phosphorylate MCM2 and observed that M118A/M134A double mutant was the most efficient in catalyzing the reaction (Figure 1A). Thus, for all the subsequent experiments, we used the M118A/M134A double mutant as the AS-CDC7 protein kinase.
Figure 1.

Covalent capture of thio-phosphorylated peptides by AS-CDC7. (A) In vitro kinase assay with recombinant WT or M118A, M134A and M118A/M1134A CDC7/DBF4, recombinant MCM2 and N6-(benzyl)ATP-γ-S. Thio-phosphorylated MCM2 and CDC7 proteins are detected by western blotting. Ponceau-stained membrane shows equal amount of MCM2 substrate in reactions. (B) Western blot detection of thio-phosphorylated proteins after kinase reaction with nuclear extracts and N6-(benzyl)ATP in the presence or absence of AS-CDC7. (C) List of proteins and peptides identified after covalent capture of thio-phosphorylated peptides. The sites of modification are indicated in red.
In order to maximise the chances of identifying relevant cellular CDC7 substrates, we prepared large quantities of benzonase-treated nuclear extracts from HEK293 cells that had been challenged with the CDC7 inhibitor XL413 (50), thus leading to the in vivo dephosphorylation of relevant substrates by counteracting protein phosphatases and to the solubilization of chromatin-bound proteins. After in vitro kinase reactions with AS-CDC7 and N6-(benzyl)ATP-γ-S, we could detect several specific thio-phosphorylated bands by western blotting, with the most intense one migrating similarly to the recombinant AS-CDC7 kinase (Figure 1B and data not shown).
Proteins were then digested, thio-phosphorylated peptides captured on iodoacetylagarose beads, eluted as phosphopeptides by oxidation with Oxone, and identified by mass spectrometry. With this methodology, we identified several peptides derived from CDC7 and DBF4 proteins, indicating that the kinase undergoes extensive auto-phosphorylation. We also identified several phosphorylated peptides from cellular proteins, and most notably peptides derived from TOP2A phosphorylated at serine 1213 and serine 1525 (Figure 1C and Supplementary Figure S1).
These observations, together with the previous findings that CDC7 and TOP2 inhibitors synergise in causing cell death of cancer cells (10), lead us to postulate that the two proteins could functionally interact in cells.
CDC7/DBF4 and TOP2A interact in vivo
To begin assessing if active DDK physically interacts with TOP2 in cell extracts, we devised new tools to purify DBF4 and its associated proteins. Specifically, we used the Flp-In T-REx 293 system to generate a stable cell line with a single-site integration of DBF4 fused to tandem FLAG and Strep tags on the carboxyl-terminus (T-REx–DBF4). In this cell line, tagged DBF4 expression is normally repressed by upstream TetO2 sequences bound by the Tet repressor, but its expression is induced by the addition of doxycycline. A control cell line with integration of the empty vector (EV) was generated in parallel. We found that expression of tagged DBF4 did not obviously affect viability or cell cycle progression (Supplementary Figure S2A and S2B). Having assessed that TOP2A can only be solubilized by chromatin digestion when nuclear extracts are prepared at 150 mM NaCl, and knowing that CDC7 kinase under the same condition is also partially chromatin associated (Figure 2A and (10)) we then performed immunoprecipitations (IP) from benzonase-treated extracts prepared from both T-REx–DBF4 and T-REx–EV cells with anti-FLAG antibodies. After western blotting we observed that tagged DBF4 not only interacts with endogenous CDC7, but also co-immunoprecipitates with TOP2A (Figure 2B).
Figure 2.
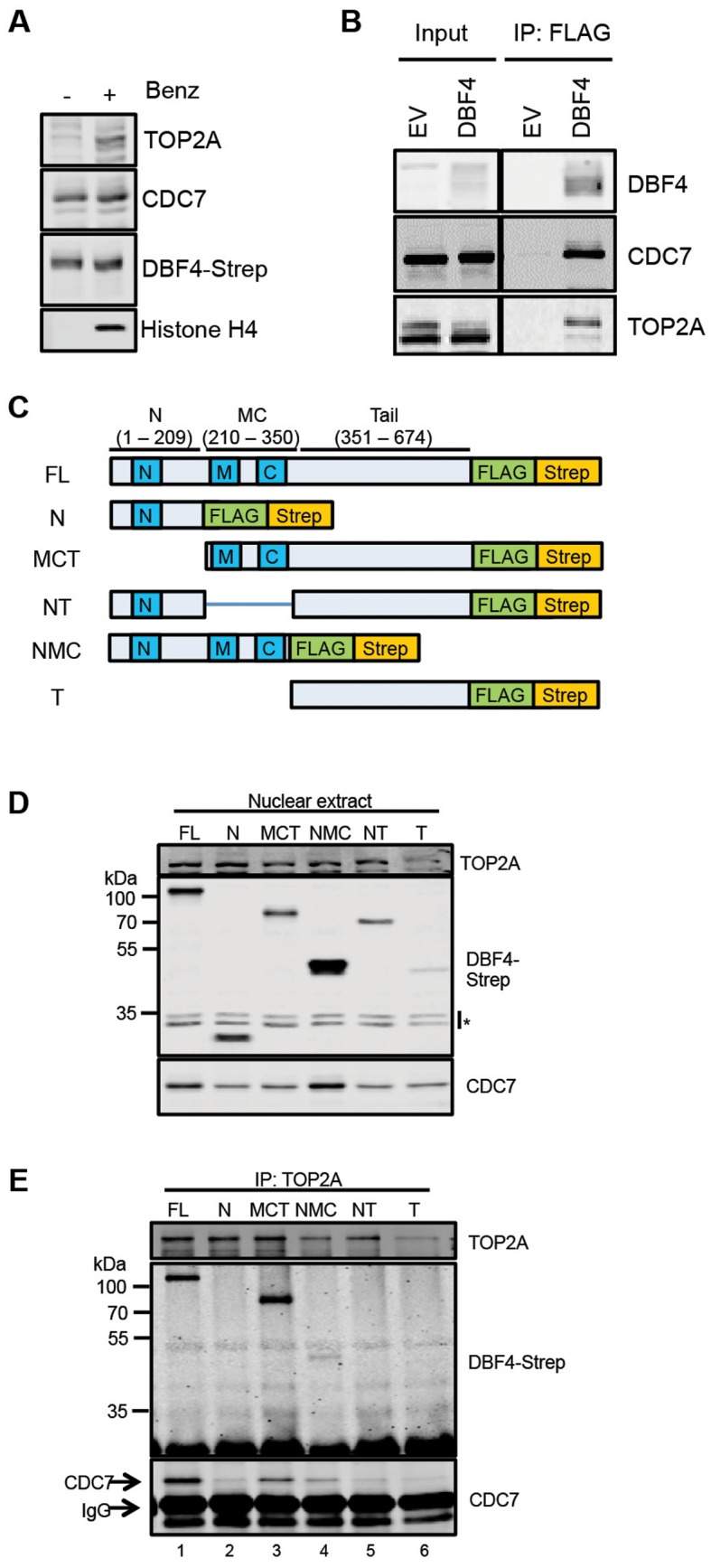
CDC7/DBF4 interacts with TOP2A. (A) Proteins extracted from nuclei with or without Benzonase were analysed by western blotting with the indicated antibodies. (B) Immunoprecipitations (IP) with anti-FLAG antibodies were performed from T-REx-EV and T-REx-DBF4 cells. (C) Schematic of DBF4 deletion mutant alleles used to generate Flp-In T-REx 293 conditionally expressing cell lines. (D) Detection of DBF4 fragments by western blotting in nuclear extracts prepared from Flp-In T-REx 293 cells upon addition of doxycycline. Asterisks indicate cross-reactive bands. (E) Endogenous TOP2A was immunoprecipitated from nuclear extracts and immunoprecpitated material was analysed by western blotting.
Three short conserved motifs, named motifs N, M and C can be recognized in the DBF4 protein (6,51,52). The N terminal motif contains a BRCA1 C-terminal (BRCT) domain predicted to be involved in protein-protein interactions, while motifs M and C bind directly to the CDC7 subunit and activate the kinase (5,49). Human DBF4 additionally contains a large tail region, representing almost half of the protein, which contains no recognisable motifs. To map the DBF4 domains required for its interaction with TOP2A, we cloned several fragments of DBF4 open reading frames containing deletions of their N (residues 1–209), MC (210–350), or Tail (351–674) domains (Figure 2C). Flp-In T-REx cell lines were then generated as before, and the expression of the tagged DBF4 fragments were confirmed by western blotting although to different levels with the C-terminal tail expressing the least (Figure 2D). We then immunoprecipitated endogenous TOP2A from extracts prepared from these cell lines, and the co-purification of each of the DBF4 fragments was determined by western blotting. As expected, full-length DBF4 (FL) was co-immunoprecipitated with TOP2A (Figure 2E, lane 1). Of the fragments, only the DBF4 MCT (210–674) and NMC (1–350) were co-immunoprecipitated with TOP2A (Figure 2E, lanes 3 and 4), indicating that the N domain is dispensable and that the MC domains are required. While we could not detect a direct binding between TOP2A and the C-terminal tail of DBF4 in these experiments, we observed that the binding of the MCT fragment was more pronounced compared to the NMC, strongly suggesting that the tail contributes to this protein-protein interaction. This could be achieved either by stabilizing the overall structure of the CDC7 complex or by providing a second independent binding site.
CDC7/DBF4-TOP2A interaction occurs early in S-phase
In order to determine when, during the cell cycle, CDC7/DBF4 interacts with TOP2A, we immunoprecipitated TOP2A from T-REx-DBF4 cells that had been treated with mimosine or nocodazole. Mimosine blocks S-phase entry through activation of ATM checkpoint signaling and inhibition of the chromatin binding of key replication proteins (53,54), while nocodazole, a microtubule depolymerizing agent, prevents mitotic progression through activation of the spindle assembly checkpoint (Figure 3A) (55). We found that the amount of endogenous CDC7 and tagged DBF4 co-immunoprecipitated was higher in cells arrested with mimosine, and reduced in nocodazole-treated cells (Figure 3B). We noted that DBF4 migrates slower in SDS-PAGE when cells were treated with mimosine, possibly due to phosphorylation by either ATM or ATR (56).
Figure 3.

CDC7/DBF4 interacts with TOP2A predominantly in early S-phase. (A) T-REx-DBF4 cells were treated with mimosine or nocodazole for 19 h and DNA content analysed by Flow Cytometry. (B) Extracts were prepared from asynchronously growing, mimosine and nocodazole treated cells as in panel A and TOP2A was immunopreciptiated with specific antibodies. As control, unrelated mouse IgG was used. Immunoprecipitated proteins were then analysed by western blotting. (C) DNA content of cells treated with mimosine for 15 h and released into fresh media. Samples were collected at the indicated time-points. (D) TOP2A immunoprecipitates from extracts of cells released from mimosine block as in panel C.
To examine if the interaction between TOP2A and CDC7/DBF4 is modulated during progression through S-phase, T-REx-DBF4 cells were released from mimosine arrest and samples collected at 3-h intervals. DNA content analysis and EdU incorporation assay indicated that T-REx cells progressed slowly into S-phase (Figure 3C) in a manner that was independent from DBF4 overexpression (Supplementary Figure S3). After IP-Western, we found that the interaction of TOP2A with CDC7 and tagged DBF4 was significantly higher in mimosine-trapped cells compared to a control-treated asynchronous population (Figure 3D, lanes 2 and 3), that it peaked between 3 and 9 h after release, when most cells were in early to mid S-phase (Figure 3C and D lanes 4–6) and decreased significantly by 12 h when most cells were in mid and late S-phase (Figure 3C and D, lane 7).
CDC7 inhibition reduces TOP2A phosphorylation levels and TOP2A-DDK protein-protein interaction in vivo
Since TOP2A interacts with DDK during S-phase and it is a good in vitro substrate of the kinase, we asked if TOP2A is phosphorylated in a CDC7-dependent manner in a cellular context. For this purpose, T-REx-EV cells were treated for 3 h with XL413, a specific CDC7 inhibitor (49,50), or mock treated. Under these conditions, phosphorylation of MCM2 at Ser40/41, a reliable marker of cellular CDC7 activity (7), is strongly reduced (Figure 4A). When TOP2A was analyzed by western blot on standard SDS-PAGE we did not observe differences in the mobility of the protein in the two samples. Thus, to facilitate separation of differentially phosphorylated TOP2A species, Phos-tag acrylamide, which slows the migration of phosphorylated proteins relative to their unphosphorylated or less-phosphorylated forms (57), was added to the SDS-PAGE gels. In mock-treated cells, the TOP2A band becomes less distinct in the presence of the Phos-tag reagent, with slight smearing observed beneath the main band. Upon treatment with the CDC7 inhibitor, a second, faster migrating form was detected (Figure 4A), suggesting that at least some TOP2A phosphorylation could be attributed to CDC7 kinase.
Figure 4.

CDC7 inhibitors affect the phosphorylation levels of cellular TOP2A and its interaction with the kinase. (A) T-REx-EV cells were treated with 10 μM XL413 or DMSO for 3 h, and cell extracts were separated on a 5% SDS-PAGE supplemented with 5 μM Phos-tag acrylamide or on a standard 6% SDS-PAGE. (B) T-REx-DBF4 cells were treated with DMSO or XL413 for the indicated times in the presence of absence of proteasome inhibitor MG132. (C) Extracts prepared from T-REx-DBF4 cells were incubated in phosphatase reaction buffer in the presence or absence of λ-phosphatase. (D) T-REx-DBF4 cells were treated with mimosine for 12 h, then with 10 μM XL413 or DMSO for a further 3 h and immunoprecipitations were performed with anti-TOP2A antibody or control IgG. Immunoprecipiations were perfomed from 1.5 mg of extract and 20 μg of the input material (1.33%) was loaded on the gel. In all cases proteins were analyzed by western blotting; where indicated, low or high intensity scans of the same blot are shown.
During the course of these experiments, we observed that the electrophoretic mobility of DFB4-Strep protein was greatly altered by the CDC7 inhibitor (Figure 4B), indicating that DBF4 itself is phosphorylated by CDC7, consistent with the identification of thio-phosphorylated DBF4 peptides in our initial experiments (Figure 1C) as well as with increased mobility after phosphatase treatment of the extract (Figure 4C). Significantly, treatment with XL413 also caused an evident and sudden drop in DBF4 levels that were only marginally rescued by the co-treatment with the proteasome inhibitor MG132; yet, XL413 did not affect CDC7 levels (Figure 4B and D).
We then investigated if the TOP2A-DDK interaction was also affected by the compound. To this end, cells were treated with either DMSO or XL413 for 3 h, and TOP2A immunoprecipitation experiments were performed. Both CDC7 and DBF4 were detected in the immunoprecipitated material from the control-treated sample, but not in the XL413-treated sample (Figure 4D, lanes 5 and 6). Altogether these data show that inhibition of CDC7 kinase activity has multiple consequences on the phosphorylation, the levels, and the capability of CDC7/DBF4 to interact with and phosphorylate relevant substrates, including TOP2A.
DBF4 localises to centromeres throughout S-phase
Both CDC7 and DBF4 are known to be mostly nuclear proteins (5,6,58), but their sub-nuclear localization has not been well characterized, particularly in human cells due to the lack of sensitive and specific immunological reagents. We therefore examined the localization of tagged DBF4 by immunofluorescence microscopy with an anti-Strep antibody. Firstly, we confirmed that full-length tagged DBF4 is mostly confined to the nucleus, where it is largely diffused. However, extraction of soluble proteins before fixation revealed the presence of multiple DBF4 foci. By performing co-localization experiments, we observed that DBF4 foci did not obviously overlap with sites of active DNA replication, but co-staining with anti-centromere antibodies (ACA) showed that DBF4 foci could always be detected at centromeres (Figure 5A). DBF4 was present at centromeres at all stages of S-phase, and, interestingly, this was not affected by CDC7 kinase inhibition by XL413 (Supplementary Figure S4). Using the same DBF4 deletion mutants previously described, we investigated the domains required for its chromatin and centromeric localization. Expression plasmids for the DBF4 fragments were transfected into U2OS cells, and their localization detected by IF as before. We found that while the N-terminal domain alone was sufficient for chromatin binding, all three NMC domains are required for centromeric recruitment (Supplementary Figure S5).
Figure 5.

DBF4 is localized at centromeres throughout S-phase. (A) U2OS cells transiently expressing DBF4-FLAG-Strep were treated with 10 μM EdU for 15 min prior to pre-extraction and fixation. EdU-containing DNA is shown in magenta, DBF4 in green, and centromeres (ACA) in red. Early, mid or late S-phase cells were determined according to the pattern of EdU incorporation. Areas indicated in boxes are magnified next to the respective images. Scale bar = 10 μm. (B) U2OS cells were transfected with plasmid expressing EGFP-DBF4 fusion protein and analysed by fluorescence microscopy. EGFP signal is in green and ACA centromeric staining in red. (C) Chromatin immunoprecipitations (ChIP) were performed with anti-Strep and anti-RNA pol II (RPB1) antibodies from extracts of T-REx-EV and T-REx-DBF4 cells. The amounts of the non-centromeric GAPDH promoter and Chromosome 1 centromeric (Cent1) DNA recovered were determined by qPCR relative to the input samples. Bars represent the Mean ± S.D. of three technical replicates. Results are representative of three independent experiments.
To confirm DBF4 localization at centromeres, we first expressed a EGFP-DBF4 fusion protein in U2OS cells; again, we observed that this protein was largely diffused in the nucleus but also accumulated in foci that, to great extent, overlapped with ACA foci (Figure 5B). Secondly, we used chromatin immunoprecipitation (ChIP) technique. qPCR primers were selected to amplify a centromere-specific target sequence on Chromosome 1 (Cent1) (47) and a non-centromeric sequence corresponding to the GAPDH promoter close to the transcription start site. We performed ChIP using a long spacer cross-linker and anti-Strep and anti-RNA polymerase II (RPB1) antibodies. As the baseline control for background antibody binding, ChIP from T-REx-EV cells was performed alongside T-REx-DBF4 cells. By qPCR analysis, the abundance of each target locus recovered was calculated relative to the 2% input sample (Figure 5C). As the positive control for the ChIP protocol, occupancy of RNA polymerase II at the GAPDH promoter was used. ChIP with the anti-Strep antibody showed increased occupancy of tagged DBF4 at centromeric compared to a non-centromeric locus, again supporting the idea that DBF4 accumulates at centromeres.
TOP2A recruitment at centromeres occurs throughout S-phase
We then investigated if CDC7/DBF4 affects either the activity or subcellular localization of TOP2A. To detect catalytically active TOP2A, we used the Differential Retention of Topoisomerase (DRT) assay (37). Briefly, asynchronously growing U2OS cells were treated with the TOP2A catalytic inhibitor ICRF-187/dexrazoxane, which prevents the release of catalytically-committed TOP2A from DNA (34,35). Soluble proteins, including TOP2A molecules not involved in strand passage activity during the time of treatment, are then salt-and detergent-extracted prior to formaldehyde fixing and immunofluorescence microscopy; for reference, centromeres were detected using anti-centromere antibodies. As previously reported, TOP2A was detected along the chromosome axes and concentrated at centromeres of mitotic chromosomes (Supplementary Figure S6 and (37,59)). Interestingly, we also observed centromeric localization in a number of interphase cells. In contrast to mitotic cells, however, not all interphase centromeres co-localized with TOP2A, and the proportion of TOP2A positive centromeres differed between cells. To further stratify the interphase cells within the cell cycle, a brief treatment with EdU was introduced, and cells were determined to be in early, mid, or late S-phase according to the pattern of EdU incorporation observed (60). The proportion of TOP2A positive centromeres per cell appeared to correlate with S-phase progression, low in early S-phase and increasing in late S-phase (Figure 6A). To quantify this result, the total number of discernible centromeres in a single optical section of a randomly selected cell was counted, and the percentage of centromeres that were also co-stained with TOP2A was determined (Figure 6B). A significantly higher proportion of TOP2A positive centromeres was found in late S-phase cells compared to early S-phase. Thus, we conclude that TOP2A recruitment to centromeres is a gradual process that occurs throughout S-phase.
Figure 6.

TOP2A is recruited to, and is active at centromeres in mid to late S-phase. (A) U2OS cells were treated with 10 μM EdU and 50 μg/ml ICRF-187 for 15 min prior to pre-extraction, fixation and immunofluorecence microscopy analysis. On top the pattern of EdU incorporation is shown in grey while on the bottom TOP2A is shown in green and centromeres (ACA) in red. Areas indicated in boxes are magnified below the respective images. Scale bar = 10 μm. (B) Cells in early and late S-phase were randomly selected and the proportion of centromeres co-incident with TOP2A foci were manually quantified. Each point on the graph represents data from 1 cell. Bars represent the mean ± S.D. ****P < 0.0001, Student's t-test.
CDC7/DBF4 limits TOP2A recruitment in early S-phase
Since both CDC7/DBF4 and TOP2A are localized at centromeres in S-phase, we postulated that CDC7/DBF4 might regulate the centromeric recruitment or activity of TOP2A at these locations. Using the DRT assay we therefore examined the localization of TOP2A after 48 h of transfection with siRNA targeting CDC7 and/or DBF4. While endogenous CDC7 can be easily detected by western blotting, the detection of endogenous DBF4 in these cells was problematic, therefore in these experiments, the biological readout of the efficiency of siRNA-mediated depletion of DDK subunits was also determined indirectly through a reduction in MCM2 phosphorylation, either as a mobility shift or at a specific CDC7–dependent phosphorylation site Ser40/41 (Figure 7A). After 48 h of transfection of siRNAs a large proportion of the cells were still actively proliferating despite low DDK activity, and importantly, in those cells that were actively undergoing DNA synthesis, we observed an increased proportion of centromeres marked with active TOP2A. This was particularly striking in cells with an early S-phase replication pattern (Figure 7B), where the number of TOP2A positive centromeres is typically low in unperturbed conditions (Figure 6). Quantification of randomly selected early S-phase cells confirmed a large increase in TOP2A positive centromeres caused by CDC7 kinase depletion (Figure 7C). Notably, combined depletion of CDC7 and DBF4 did not result in an additive effect on TOP2A centromeric recruitment. Similarly to siRNA depletion of the protein, a 3-h treatment with CDC7 inhibitors, XL413 or PHA767491, also caused increased localization of active TOP2A at centromeres in early S-phase cells (Figure 7D). These data are consistent with either TOP2A activity at centromeres being directly regulated through TOP2A phosphorylation by CDC7/DBF4, or indirectly through a different mechanism.
Figure 7.

CDC7/DBF4 inhibition increases the recruitment of TOP2A to centromeres. U2OS cells were transfected with siRNA against CDC7 and/or DBF4 for 48 h. Cell extrects were then prepared and analysed by western blotting (A) or cells were treated with 10 μM EdU and 50 μg/ml ICRF-187 for 15 min prior to pre-extraction, fixation and immunofluorecence microscopy analysis (B). Representative images of early S-phase cells are shown. (C) Quantification was performed as in Figure 6. ***P < 0.001, Student's t-test. Scale bar = 10 μm. (D) U2OS cells were challenged with 10 μM XL413, 10 μM PHA767491 or with DMSO as control. 15 min before harvesting cells were also treated with 10 μM EdU and 50 μg/ml ICRF-187 and analysed as above. *P < 0.05, Student's t-test.
In order to discriminate between these alternative models, we expressed either FLAG- tagged TOP2A WT, a phosphorylation-deficient TOP2A protein carrying alanine substitutions in the two serines that were identified as CDC7 phospho-sites in vitro, namely S1213A/S1525A, or the corresponding potential phospho-mimetic protein S1213D/S1525D. All these proteins were catalytically active in the cells as they could be trapped on chromatin with ICRF187 although we noticed their expression level was reproducibly different, with S1213A/S1525A protein accumulating to higher levels (Figure 8A). This may be due to protein or mRNA stabilization caused by the single S1525A substitution, as an independent report showed that a S1525A TOP2A was consistently expressed at a higher level than the WT protein (43). When we analysed centromeric occupancy in early S-phase cells, we found that the ectopically expressed FLAG-tagged TOP2A WT, just like endogenous TOP2A or the S1213D/S1525D TOP2A, was detected at low frequency at centromeres, while, in contrast, the phospho-deficient S1213A/S1525A protein displayed increased occupancy, to a level similar to what was observed with endogenous TOP2A when CDC7 kinase was depleted (Figure 8B and C). Since the amount of TOP2A active at centromeres is a very small fraction of the total TOP2A pool in each cell analysed, it is unlikely that centromeric recruitment is driven solely by mass action equilibrium suggesting instead that phosphorylation at these sites regulates TOP2A centromeric function.
Figure 8.

Phosphorylation at Ser1213 and S1525 controls the timing of TOP2A recruitment at centromeres. U2OS cells were tranfected with constructs expressing either WT, S1213A/S1525A or S1213D/S1525D FLAG-tagged TOP2A. After 48 h, cell extracts were prepared and analysed by western blotting (A) or cells were treated with 10 μM EdU and 50 μg/ml ICRF-187 for 15 min prior to pre-extraction, fixation and immunofluorecence microscopy analysis (B). Representative images of early S-phase cells are shown. (C) Quantification was performed as in Figures 6 and 7. ***P < 0.001, Student's t-test. Scale bar = 10 μm.
DISCUSSION
In this work, we have used a chemical genetics approach to identify potential substrates of CDC7 kinase from nuclear extracts. During this process we have validated the idea that two methionines, M118 and M134, can act as gatekeeper residues, and their substitution with a smaller amino acid such as alanine, either in combination or on their own, allows the bulky ATP analogue N6-(benzyl)ATP to bind and to be used as a cofactor instead of ATP for both auto- and trans-phosphorylation of proteins. Our attempts to label and capture CDC7 substrates from nuclear extracts have led to the identification of TOP2A as a main substrate and to the identification of S1213 and S1525 as CDC7 in vitro phospho-sites. However, the list of potential substrates is not exhaustive, as we did not retrieve known CDC7 substrates, such as the MCM complex or CLASPIN. This could be due to a combination of technical reasons, including the use of an engineered kinase that lacks portions of DBF4 sequence that could be relevant to promote substrate recognition. We also mapped seven sites of autophosphorylation on the CDC7 catalytic subunit, and three on DBF4. All the mapped CDC7 sites reside in the large insert domain, for which structural information is lacking. This insert domain interrupts the contiguity of the canonical kinase domain structure, a feature which is peculiar to CDC7 and has been shown to mediate protein-protein interactions (61). Interestingly, several of these sites were also previously identified in large phosphoproteomics studies and have been annotated in the PhosphositePlus database (62), suggesting that CDC7 auto-phosphorylation can occur in vivo.
We have demonstrated that DBF4 can be detected at centromeres. To our knowledge, this is the first evidence that DBF4 is recruited to centromeres in human cells. We found that dual crosslinking with formaldehyde and ethylene glycol bis (succinimidyl succinate) (EGS) was necessary to immunoprecipitate DBF4-associated chromatin. EGS is a protein-protein cross-linking agent with a long spacer arm that, in combination with formaldehyde, stabilises proteins that are not directly bound to DNA (63). This suggests that CDC7/DBF4 is instead recruited through other protein factor(s). We have also found that the N-terminal 1–209 residues of DBF4 are sufficient for chromatin binding. This region remains poorly characterized, but it contains a BRCT domain proposed to be involved in protein–protein interactions. Interestingly, the equivalent domain of DBF4 in budding yeast, Saccharomyces cerevisiae, has been shown to be required for interaction with several replication and checkpoint proteins, including ORC, RAD53 and RIF1 (64–66). The mechanism regulating CDC7/DBF4 centromeric recruitment, however, is likely to be distinct, as the DBF4 M and C domains, in an addition to the N domain, are also required for this.
It was recently reported that in S. cerevisiae, CDC7/DBF4 is recruited to kinetochores where it promotes the replication of centromeric regions early in S-phase through the recruitment of the essential replication proteins SLD3 and SLD7 (Treslin/TICRR and MTBP in humans) to pericentromeric origins (67). Differently from budding yeast, however, human centromeric DNA is replicated asynchronously between mid to late S-phase (68,69) while here DBF4 was detected at all centromeres from early S-phase, inconsistent with normal centromeric replication timing. As our experiments rely on the overexpression of DBF4, future studies would be necessary to verify if the overexpression of DBF4 is sufficient to alter centromeric DNA replication timing. In the same study, the authors also reported that centromeric ScCDC7/DBF4 independently recruits the SCC2–SCC4 cohesin loading complex for the establishment of robust pericentromeric sister chromatid cohesion (67). X. laevis CDC7/DBF4B, which is the predominant CDC7 kinase complex in egg extracts, also physically interacts with SCC2-SCC4 and is required for its chromatin binding (70). Thus, it is plausible that human CDC7/DBF4 may also be involved in the establishment of pericentromeric sister chromatid cohesion, and it would be interesting to verify if the homologous complex in humans, NIPBL-MAU2, is also regulated by CDC7 kinase in a similar manner.
CDC7/DBF4 interacts with and phosphorylates TOP2A in vitro. The interaction likely occurs on chromatin, requires a region of DBF4 that is sufficient for CDC7 activation, is further stabilized by the C-terminal tail of DBF4 and, interestingly, it is abolished by a very specific CDC7 ATP-competitive inhibitor XL413. At present, it is not fully clear if this interaction is solely mediated by CDC7 or if it is phosphorylation-dependent, as CDC7 kinase inhibition has dramatic effects on both CDC7 and DBF4 phosphorylation levels as well as causing a rapid drop in DBF4 levels. TOP2A is a highly modified protein with many residues phosphorylated in its C-terminus (62) and several kinases have been implicated in its regulation during the cell cycle including CK2 and proline directed kinases (71) which have mainly been studied using in vitro assays. As the treatment with XL413 causes a reduction in the overall levels of TOP2A phosphorylation, we propose that CDC7 is also directly involved in the regulation in vivo. Future studies, involving the development and thorough validation of multiple anti-phosphopeptide antibodies will be important to determine the contribution of CDC7 and other kinases to TOP2A phosphorylation at different sites.
Our work has also revealed that active TOP2A can be detected at an increasing number of centromeres as cell progress through S-phase. We observed that TOP2A activity at centromeric regions does not always overlap with ongoing DNA synthesis at these sites, and that the timing of TOP2A centromeric recruitment is advanced by CDC7 depletion. Similarly, the timing of centromeric recruitment of a phosphorylation-deficient TOP2A at two key serines, putative CDC7 target sites S1213 and S1525, is advanced.
Based on these results, we propose that phosphorylation of TOP2A by CDC7/DBF4 in early S-phase prevents its localization and/or activity at centromeres, and inhibition of TOP2A function could be relevant to prevent premature separation of centromeric DNA.
CDC7 regulation of TOP2A at centromeres may have important significance with respect to centromeric replication and cohesion, and could also explain the increased sensitivity to TOP2 poisons in cells lacking CDC7 activity.
Supplementary Material
Acknowledgments
Ed Luk, Stony Brook University, for FLAG IP protocol, Adrian Bracken, Trinity College Dublin, for RPB1 antibody and advice on ChIP, Kuntian Luo for TOP2A plasmid, Enda O'Connell, NUIG, for advice on qPCR protocol and Peter Cherepanov for insight on CDC7 structural features before publication, Elaine Dunleavy for help with microscopy, Sandra Healy and Bob Lahue for critical reading of the manuscript, Gemma O'Brien and all the members of the Santocanale laboratory for support and discussion. We also thank the NCBES Flow Cytometry core facility at NUIG, a facility that is funded by NUIG and the Irish Government's Programme for Research in Third Level Institutions, Cycles 4 and 5, National Development Plan 2007–2013.
Author's contribution: K.W. designed and performed most of the experiments, G.N.W. and J.F. developed covalent capture methodology with AS-CDC7, H.Q. generated cell lines and performed experiments in Figure 4B and Supplementary Figure S2, M.D.R. generated TOP2A mutants, A.C. and A.B. performed mass spectrometry analysis and data analysis, C.S. directed research, K.W. and C.S. wrote the manuscript.
Footnotes
Present addresses:
Kevin Z. L. Wu, MRC ProteinPhosphorylation and Ubiquitylation Unit, University of Dundee, Dundee, UK.
Jennifer Fitzgerald, Eli Lilly S.A. Irish Branch, Kinsale, Ireland.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Science Foundation Ireland (SFI) [08/IN.1/B2064 to C.S.]; Irish Research Council postdoctoral fellowship (to G.N.W.); Molecular Medicine Ireland fellowship (to H.Q.).
Conflict of interest statement. None declared.
REFERENCES
- 1.Zeman M.K., Cimprich K.A. Causes and consequences of replication stress. Nat. Cell Biol. 2014;16:2–9. doi: 10.1038/ncb2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Hills S.A., Diffley J.F.X. DNA replication and oncogene-induced replicative stress. Curr. Biol. 2014;24:R435–R444. doi: 10.1016/j.cub.2014.04.012. [DOI] [PubMed] [Google Scholar]
- 3.Symeonidou I.-E., Taraviras S., Lygerou Z. Control over DNA replication in time and space. FEBS Lett. 2012;586:2803–2812. doi: 10.1016/j.febslet.2012.07.042. [DOI] [PubMed] [Google Scholar]
- 4.Sclafani R.A., Holzen T.M. Cell cycle regulation of DNA replication. Annu. Rev. Genet. 2007;41:237–280. doi: 10.1146/annurev.genet.41.110306.130308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jiang W., McDonald D., Hope T.J., Hunter T. Mammalian Cdc7-Dbf4 protein kinase complex is essential for initiation of DNA replication. EMBO J. 1999;18:5703–5713. doi: 10.1093/emboj/18.20.5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kumagai H., Sato N., Yamada M., Mahony D., Seghezzi W., Lees E., Arai K., Masai H. A novel growth- and cell cycle-regulated protein, ASK, activates human Cdc7-related kinase and is essential for G1/S transition in mammalian cells. Mol. Cell. Biol. 1999;19:5083–5095. doi: 10.1128/mcb.19.7.5083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Montagnoli A., Valsasina B., Brotherton D., Troiani S., Rainoldi S., Tenca P., Molinari A., Santocanale C. Identification of Mcm2 phosphorylation sites by S-phase-regulating kinases. J. Biol. Chem. 2006;281:10281–10290. doi: 10.1074/jbc.M512921200. [DOI] [PubMed] [Google Scholar]
- 8.Masai H., Taniyama C., Ogino K., Matsui E., Kakusho N., Matsumoto S., Kim J.M., Ishii A., Tanaka T., Kobayashi T., et al. Phosphorylation of MCM4 by Cdc7 kinase facilitates its interaction with Cdc45 on the chromatin. J. Biol. Chem. 2006;281:39249–39261. doi: 10.1074/jbc.M608935200. [DOI] [PubMed] [Google Scholar]
- 9.Sheu Y.-J., Stillman B. Cdc7-Dbf4 phosphorylates MCM proteins via a docking site-mediated mechanism to promote S phase progression. Mol. Cell. 2006;24:101–113. doi: 10.1016/j.molcel.2006.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tenca P., Brotherton D., Montagnoli A., Rainoldi S., Albanese C., Santocanale C. Cdc7 is an active kinase in human cancer cells undergoing replication stress. J. Biol. Chem. 2007;282:208–215. doi: 10.1074/jbc.M604457200. [DOI] [PubMed] [Google Scholar]
- 11.Rainey M.D., Harhen B., Wang G.-N., Murphy P.V., Santocanale C. Cdc7-dependent and -independent phosphorylation of Claspin in the induction of the DNA replication checkpoint. Cell Cycle. 2013;12 doi: 10.4161/cc.24675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J.M., Kakusho N., Yamada M., Kanoh Y., Takemoto N., Masai H. Cdc7 kinase mediates Claspin phosphorylation in DNA replication checkpoint. Oncogene. 2008;27:3475–3482. doi: 10.1038/sj.onc.1210994. [DOI] [PubMed] [Google Scholar]
- 13.Suzuki T., Tsuzuku J., Hayashi A., Shiomi Y., Iwanari H., Mochizuki Y., Hamakubo T., Kodama T., Nishitani H., Masai H., et al. Inhibition of DNA damage-induced apoptosis through Cdc7-mediated stabilization of Tob. J. Biol. Chem. 2012;287:40256–40265. doi: 10.1074/jbc.M112.353805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Day T.A., Palle K., Barkley L.R., Kakusho N., Zou Y., Tateishi S., Verreault A., Masai H., Vaziri C. Phosphorylated Rad18 directs DNA polymerase η to sites of stalled replication. J. Cell Biol. 2010;191:953–966. doi: 10.1083/jcb.201006043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Yamada M., Watanabe K., Mistrik M., Vesela E., Protivankova I., Mailand N., Lee M., Masai H., Lukas J., Bartek J. ATR-Chk1-APC/CCdh1-dependent stabilization of Cdc7-ASK (Dbf4) kinase is required for DNA lesion bypass under replication stress. Genes Dev. 2013;27:2459–2472. doi: 10.1101/gad.224568.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gérard A., Koundrioukoff S., Ramillon V., Sergère J.-C., Mailand N., Quivy J.-P., Almouzni G. The replication kinase Cdc7-Dbf4 promotes the interaction of the p150 subunit of chromatin assembly factor 1 with proliferating cell nuclear antigen. EMBO Rep. 2006;7:817–823. doi: 10.1038/sj.embor.7400750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Takayama Y., Mamnun Y.M., Trickey M., Dhut S., Masuda F., Yamano H., Toda T., Saitoh S. Hsk1- and SCF(Pof3)-dependent proteolysis of S. pombe Ams2 ensures histone homeostasis and centromere function. Dev. Cell. 2010;18:385–396. doi: 10.1016/j.devcel.2009.12.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bailis J.M., Bernard P., Antonelli R., Allshire R.C., Forsburg S.L. Hsk1-Dfp1 is required for heterochromatin-mediated cohesion at centromeres. Nat. Cell Biol. 2003;5:1111–1116. doi: 10.1038/ncb1069. [DOI] [PubMed] [Google Scholar]
- 19.Baker S.P., Phillips J., Anderson S., Qiu Q., Shabanowitz J., Smith M.M., Yates J.R., Hunt D.F., Grant P.A. Histone H3 Thr 45 phosphorylation is a replication-associated post-translational modification in S. cerevisiae. Nat. Cell Biol. 2010;12:294–298. doi: 10.1038/ncb2030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo H.-C., Kunz R.C., Chen X., Marullo A., Gygi S.P., Hollingsworth N.M. Cdc7-Dbf4 is a gene-specific regulator of meiotic transcription in yeast. Mol. Cell. Biol. 2012;32:541–557. doi: 10.1128/MCB.06032-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matos J., Lipp J.J., Bogdanova A., Guillot S., Okaz E., Junqueira M., Shevchenko A., Zachariae W. Dbf4-dependent CDC7 kinase links DNA replication to the segregation of homologous chromosomes in meiosis I. Cell. 2008;135:662–678. doi: 10.1016/j.cell.2008.10.026. [DOI] [PubMed] [Google Scholar]
- 22.Valentin G., Schwob E., Della Seta F. Dual role of the Cdc7-regulatory protein Dbf4 during yeast meiosis. J. Biol. Chem. 2006;281:2828–2834. doi: 10.1074/jbc.M510626200. [DOI] [PubMed] [Google Scholar]
- 23.Wan L., Niu H., Futcher B., Zhang C., Shokat K.M., Boulton S.J., Hollingsworth N.M. Cdc28-Clb5 (CDK-S) and Cdc7-Dbf4 (DDK) collaborate to initiate meiotic recombination in yeast. Genes Dev. 2008;22:386–397. doi: 10.1101/gad.1626408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sasanuma H., Hirota K., Fukuda T., Kakusho N., Kugou K., Kawasaki Y., Shibata T., Masai H., Ohta K. Cdc7-dependent phosphorylation of Mer2 facilitates initiation of yeast meiotic recombination. Genes Dev. 2008;22:398–410. doi: 10.1101/gad.1626608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Murakami H., Keeney S. Temporospatial coordination of meiotic DNA replication and recombination via DDK recruitment to replisomes. Cell. 2014;158:861–873. doi: 10.1016/j.cell.2014.06.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bishop A.C., Ubersax J.A., Petsch D.T., Matheos D.P., Gray N.S., Blethrow J., Shimizu E., Tsien J.Z., Schultz P.G., Rose M.D., et al. A chemical switch for inhibitor-sensitive alleles of any protein kinase. Nature. 2000;407:395–401. doi: 10.1038/35030148. [DOI] [PubMed] [Google Scholar]
- 27.Alaimo P.J., Shogren-Knaak M.A., Shokat K.M. Chemical genetic approaches for the elucidation of signaling pathways. Curr. Opin. Chem. Biol. 2001;5:360–367. doi: 10.1016/s1367-5931(00)00215-5. [DOI] [PubMed] [Google Scholar]
- 28.Koch A., Hauf S. Strategies for the identification of kinase substrates using analog-sensitive kinases. Eur. J. Cell Biol. 2010;89:184–193. doi: 10.1016/j.ejcb.2009.11.024. [DOI] [PubMed] [Google Scholar]
- 29.Elphick L.M., Lee S.E., Gouverneur V., Mann D.J. Using chemical genetics and ATP analogues to dissect protein kinase function. ACS Chem. Biol. 2007;2:299–314. doi: 10.1021/cb700027u. [DOI] [PubMed] [Google Scholar]
- 30.Allen J.J., Li M., Brinkworth C.S., Paulson J.L., Wang D., Hübner A., Chou W.-H., Davis R.J., Burlingame A.L., Messing R.O., et al. A semisynthetic epitope for kinase substrates. Nat. Methods. 2007;4:511–516. doi: 10.1038/nmeth1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Allen J.J., Lazerwith S.E., Shokat K.M. Bio-orthogonal affinity purification of direct kinase substrates. J. Am. Chem. Soc. 2005;127:5288–5289. doi: 10.1021/ja050727t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Blethrow J.D., Glavy J.S., Morgan D.O., Shokat K.M. Covalent capture of kinase-specific phosphopeptides reveals Cdk1-cyclin B substrates. Proc. Natl. Acad. Sci. U.S.A. 2008;105:1442–1447. doi: 10.1073/pnas.0708966105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Carlson S.M., White F.M. Labeling and identification of direct kinase substrates. Sci. Signal. 2012;5:pl3. doi: 10.1126/scisignal.2002568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Nitiss J.L. DNA topoisomerase II and its growing repertoire of biological functions. Nat. Rev. Cancer. 2009;9:327–337. doi: 10.1038/nrc2608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pommier Y., Leo E., Zhang H., Marchand C. DNA topoisomerases and their poisoning by anticancer and antibacterial drugs. Chem. Biol. 2010;17:421–433. doi: 10.1016/j.chembiol.2010.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gonzalez R.E., Lim C.-U., Cole K., Bianchini C.H., Schools G.P., Davis B.E., Wada I., Roninson I.B., Broude E.V. Effects of conditional depletion of topoisomerase II on cell cycle progression in mammalian cells. Cell Cycle. 2011;10:3505–3514. doi: 10.4161/cc.10.20.17778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Agostinho M., Rino J., Braga J., Ferreira F., Steffensen S., Ferreira J. Human topoisomerase II alpha: Targeting to subchromosomal sites of activity during interphase and mitosis. Mol. Biol. Cell. 2004;15:2388–2400. doi: 10.1091/mbc.E03-08-0558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rouzeau S., Cordelières F.P., Buhagiar-Labarchède G., Hurbain I., Onclercq-Delic R., Gemble S., Magnaghi-Jaulin L., Jaulin C., Amor-Guéret M. Bloom's syndrome and PICH helicases cooperate with topoisomerase IIα in centromere disjunction before anaphase. PLoS ONE. 2012;7:e33905. doi: 10.1371/journal.pone.0033905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Broderick R., Nieminuszczy J., Blackford A.N., Winczura A., Niedzwiedz W. TOPBP1 recruits TOP2A to ultra-fine anaphase bridges to aid in their resolution. Nat. Commun. 2015;6:6572. doi: 10.1038/ncomms7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Meresse P., Dechaux E., Monneret C., Bertounesque E. Etoposide: discovery and medicinal chemistry. Curr. Med. Chem. 2004;11:2443–2466. doi: 10.2174/0929867043364531. [DOI] [PubMed] [Google Scholar]
- 41.Pommier Y. Drugging topoisomerases: lessons and challenges. ACS Chem. Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Cheeseman I.M., Desai A. A combined approach for the localization and tandem affinity purification of protein complexes from metazoans. Sci. STKE. 2005:pl1. doi: 10.1126/stke.2662005pl1. [DOI] [PubMed] [Google Scholar]
- 43.Luo K., Yuan J., Chen J., Lou Z. Topoisomerase II alpha controls the decatenation checkpoint. Nat. Cell Biol. 2009;11:U204–U196. doi: 10.1038/ncb1828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gruhler A., Olsen J.V., Mohammed S., Mortensen P., Faergeman N.J., Mann M., Jensen O.N. Quantitative phosphoproteomics applied to the yeast pheromone signaling pathway. Mol. Cell Proteomics. 2005;4:310–327. doi: 10.1074/mcp.M400219-MCP200. [DOI] [PubMed] [Google Scholar]
- 45.Keller A., Nesvizhskii A.I., Kolker E., Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 46.Nesvizhskii A.I., Keller A., Kolker E., Aebersold R. A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem. 2003;75:4646–4658. doi: 10.1021/ac0341261. [DOI] [PubMed] [Google Scholar]
- 47.Dunham I., Lengauer C., Cremer T., Featherstone T. Rapid generation of chromosome-specific alphoid DNA probes using the polymerase chain reaction. Hum. Genet. 1992;88:457–462. doi: 10.1007/BF00215682. [DOI] [PubMed] [Google Scholar]
- 48.Swords R., Mahalingam D., O'Dwyer M., Santocanale C., Kelly K., Carew J., Giles F. Cdc7 kinase - a new target for drug development. Eur. J. Cancer. 2010;46:33–40. doi: 10.1016/j.ejca.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 49.Hughes S., Elustondo F., Di Fonzo A., Leroux F.G., Wong A.C., Snijders A.P., Matthews S.J., Cherepanov P. Crystal structure of human CDC7 kinase in complex with its activator DBF4. Nat. Struct. Mol. Biol. 2012 doi: 10.1038/nsmb.2404. doi:10.1038/nsmb.2404. [DOI] [PubMed] [Google Scholar]
- 50.Koltun E.S., Tsuhako A.L., Brown D.S., Aay N., Arcalas A., Chan V., Du H., Engst S., Ferguson K., Franzini M., et al. Discovery of XL413, a potent and selective CDC7 inhibitor. Bioorg. Med. Chem. Lett. 2012 doi: 10.1016/j.bmcl.2012.04.024. doi:10.1016/j.bmcl.2012.04.024. [DOI] [PubMed] [Google Scholar]
- 51.Ogino K., Takeda T., Matsui E., Iiyama H., Taniyama C., Arai K., Masai H. Bipartite binding of a kinase activator activates Cdc7-related kinase essential for S phase. J. Biol. Chem. 2001;276:31376–31387. doi: 10.1074/jbc.M102197200. [DOI] [PubMed] [Google Scholar]
- 52.Montagnoli A., Bosotti R., Villa F., Rialland M., Brotherton D., Mercurio C., Berthelsen J., Santocanale C. Drf1, a novel regulatory subunit for human Cdc7 kinase. EMBO J. 2002;21:3171–3181. doi: 10.1093/emboj/cdf290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Park S.-Y., Im J.-S., Park S.-R., Kim S.-E., Wang H.-J., Lee J.-K. Mimosine arrests the cell cycle prior to the onset of DNA replication by preventing the binding of human Ctf4/And-1 to chromatin via Hif-1α activation in HeLa cells. Cell Cycle. 2012;11:761–766. doi: 10.4161/cc.11.4.19209. [DOI] [PubMed] [Google Scholar]
- 54.Kubota S., Fukumoto Y., Ishibashi K., Soeda S., Kubota S., Yuki R., Nakayama Y., Aoyama K., Yamaguchi N., Yamaguchi N. Activation of the prereplication complex is blocked by mimosine through reactive oxygen species-activated ataxia telangiectasia mutated (ATM) protein without DNA damage. J. Biol. Chem. 2014;289:5730–5746. doi: 10.1074/jbc.M113.546655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zieve G.W., Turnbull D., Mullins J.M., McIntosh J.R. Production of large numbers of mitotic mammalian cells by use of the reversible microtubule inhibitor nocodazole. Nocodazole accumulated mitotic cells. Exp. Cell Res. 1980;126:397–405. doi: 10.1016/0014-4827(80)90279-7. [DOI] [PubMed] [Google Scholar]
- 56.Lee A.Y.-L., Chiba T., Truong L.N., Cheng A.N., Do J., Cho M.J., Chen L., Wu X. Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) protein to regulate intra-S-phase checkpoint. J. Biol. Chem. 2012;287:2531–2543. doi: 10.1074/jbc.M111.291104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kosako H. Phos-tag Western blotting for detecting stoichiometric protein phosphorylation in cells. Protocol Exchange. 2009 doi:10.1038/nprot.2009.170. [Google Scholar]
- 58.Jiang W., Hunter T. Identification and characterization of a human protein kinase related to budding yeast Cdc7p. Proc. Natl. Acad. Sci. U.S.A. 1997;94:14320–14325. doi: 10.1073/pnas.94.26.14320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tavormina P.A., Côme M.-G., Hudson J.R., Mo Y.-Y., Beck W.T., Gorbsky G.J. Rapid exchange of mammalian topoisomerase II alpha at kinetochores and chromosome arms in mitosis. J. Cell Biol. 2002;158:23–29. doi: 10.1083/jcb.200202053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dimitrova D.S., Berezney R. The spatio-temporal organization of DNA replication sites is identical in primary, immortalized and transformed mammalian cells. J. Cell. Sci. 2002;115:4037–4051. doi: 10.1242/jcs.00087. [DOI] [PubMed] [Google Scholar]
- 61.Kim B.J., Kim S.-Y., Lee H. Identification and characterization of human Cdc7 nuclear retention and export sequences in the context of chromatin binding. J. Biol. Chem. 2007;282:30029–30038. doi: 10.1074/jbc.M703705200. [DOI] [PubMed] [Google Scholar]
- 62.Hornbeck P.V., Zhang B., Murray B., Kornhauser J.M., Latham V., Skrzypek E. PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015;43:D512–D520. doi: 10.1093/nar/gku1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zeng P.-Y., Vakoc C.R., Chen Z.-C., Blobel G.A., Berger S.L. In vivo dual cross-linking for identification of indirect DNA-associated proteins by chromatin immunoprecipitation. BioTechniques. 2006;41:694–698. doi: 10.2144/000112297. [DOI] [PubMed] [Google Scholar]
- 64.Duncker B.P., Shimada K., Tsai-Pflugfelder M., Pasero P., Gasser S.M. An N-terminal domain of Dbf4p mediates interaction with both origin recognition complex (ORC) and Rad53p and can deregulate late origin firing. Proc. Natl. Acad. Sci. U.S.A. 2002;99:16087–16092. doi: 10.1073/pnas.252093999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Matthews L.A., Jones D.R., Prasad A.A., Duncker B.P., Guarné A. Saccharomyces cerevisiae Dbf4 has unique fold necessary for interaction with Rad53 kinase. J. Biol. Chem. 2012;287:2378–2387. doi: 10.1074/jbc.M111.233973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hiraga S.-I., Alvino G.M., Chang F., Lian H.-Y., Sridhar A., Kubota T., Brewer B.J., Weinreich M., Raghuraman M.K., Donaldson A.D. Rif1 controls DNA replication by directing Protein Phosphatase 1 to reverse Cdc7-mediated phosphorylation of the MCM complex. Genes Dev. 2014;28:372–383. doi: 10.1101/gad.231258.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Natsume T., Müller C.A., Katou Y., Retkute R., Gierliński M., Araki H., Blow J.J., Shirahige K., Nieduszynski C.A., Tanaka T.U. Kinetochores coordinate pericentromeric cohesion and early DNA replication by cdc7-dbf4 kinase recruitment. Mol. Cell. 2013;50:661–674. doi: 10.1016/j.molcel.2013.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ten Hagen K.G., Gilbert D.M., Willard H.F., Cohen S.N. Replication timing of DNA sequences associated with human centromeres and telomeres. Mol. Cell. Biol. 1990;10:6348–6355. doi: 10.1128/mcb.10.12.6348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Erliandri I., Fu H., Nakano M., Kim J.H., Miga K.H., Liskovykh M., Earnshaw W.C., Masumoto H., Kouprina N., Aladjem M.I., et al. Replication of alpha-satellite DNA arrays in endogenous human centromeric regions and in human artificial chromosome. Nucleic Acids Res. 2014;42:11502–11516. doi: 10.1093/nar/gku835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Takahashi T.S., Basu A., Bermudez V., Hurwitz J., Walter J.C. Cdc7-Drf1 kinase links chromosome cohesion to the initiation of DNA replication in Xenopus egg extracts. Genes Dev. 2008;22:1894–1905. doi: 10.1101/gad.1683308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Isaacs R.J., Davies S.L., Sandri M.I., Redwood C., Wells N.J., Hickson I.D. Physiological regulation of eukaryotic topoisomerase II. Biochim. Biophys. Acta. 1998;1400:121–137. doi: 10.1016/s0167-4781(98)00131-6. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.