


Abstract
Genome wide association studies (GWAS) provide a powerful approach for uncovering disease-associated variants in human, but fine-mapping the causal variants remains a challenge. This is partly remedied by prioritization of disease-associated variants that overlap GWAS-enriched epigenomic annotations. Here, we introduce a new Bayesian model RiVIERA (Risk Variant Inference using Epigenomic Reference Annotations) for inference of driver variants from summary statistics across multiple traits using hundreds of epigenomic annotations. In simulation, RiVIERA promising power in detecting causal variants and causal annotations, the multi-trait joint inference further improved the detection power. We applied RiVIERA to model the existing GWAS summary statistics of 9 autoimmune diseases and Schizophrenia by jointly harnessing the potential causal enrichments among 848 tissue-specific epigenomics annotations from ENCODE/Roadmap consortium covering 127 cell/tissue types and 8 major epigenomic marks. RiVIERA identified meaningful tissue-specific enrichments for enhancer regions defined by H3K4me1 and H3K27ac for Blood T-Cell specifically in the nine autoimmune diseases and Brain-specific enhancer activities exclusively in Schizophrenia. Moreover, the variants from the 95% credible sets exhibited high conservation and enrichments for GTEx whole-blood eQTLs located within transcription-factor-binding-sites and DNA-hypersensitive-sites. Furthermore, joint modeling the nine immune traits by simultaneously inferring and exploiting the underlying epigenomic correlation between traits further improved the functional enrichments compared to single-trait models.
INTRODUCTION
Genome wide association studies (GWAS) can help gain numerous insights on the genetic basis of complex diseases, and ultimately contribute to personalized risk prediction and precision medicine (1–4). However, fine-mapping the exact causal variants is challenging due to linkage disequilibrium (LD) and the lack of ability to interpret the function of noncoding variants, which contribute to ∼90% of the current GWAS catalog (40.7% intergenic and 48.6% intronic; (5)). On the other hand, several lines of evidence have been proposed to help interpret non-coding genetic signals, in order to gain insights into potential regulatory functions. In particular, epigenomic annotations can pinpoint locations of biochemical activity indicative of cis-regulatory functions (6,7). Indeed, comparison with genome-wide annotations of putative regulatory elements has shown enrichment of GWAS variants in enhancer-associated histone modifications, regions of open chromatin, and conserved non-coding elements (3,6,8–12), indicating they may play gene-regulatory roles. These enrichments have been used to predict relevant cell types and non-coding annotations for specific traits (6,9,13). Furthermore, many complex traits potentially share causal mechanisms such as autoimmune diseases (14,15) and psychiatric disorders (16,17). Thus, methods that jointly model the intrinsic comorbidity implicated in the GWAS summary statistics of the related traits may confer higher statistical power of causal variants detection.
Recently, several methods were developed to utilize the wealth of genome-wide annotations primarily provided by ENCODE consortium to predict causal variants and novel risk variants that are weakly associated in complex traits. Pickrell (23) developed a statistical approach called fgwas that models association statistics of a given trait and used regularized logistic function to simultaneously learn the relevant annotations. To account for LD, fgwas assumes at most one causal variants per locus via a softmax function. Kichaev et al. (18) recently developed a multivariate Gaussian framework called PAINTOR, which allows for more than one causal SNP but at most three to be located within a single locus by considering all of the combinatorial settings (18). Chung et al. (19) used a maximum likelihood framework called GPA to infer driver variants shared among multiple traits by modeling the corresponding GWAS P-values as Beta distributions with an option of using one or more sets of annotations to improve the power detecting causal variants (19). Although useful, these methods are often designed to simultaneously operate on a small number of independent annotations due to some computational constraints. Moreover, most methods only operate on one trait at a time whereas exploiting the correlation between traits at the epigenomic annotation level may prove useful for shared causal mechanisms that go beyond the level of individual variants.
In this article, we describe a novel Bayesian framework called RiVIERA (Risk Variant Inference using Epigenomic Reference Annotations) to model GWAS summary statistics in terms of P-values using large-scale reference datasets. The main novelty of RiVIERA is the ability to perform efficient Bayesian inference of the intrinsic causal signals across multiple traits while simultaneously inferring and exploiting enrichment signals and their correlation between traits over hundreds of tissue-specific epigenomic annotations. We achieve this efficiently via stochastic sampling of loci and powerful Hamiltonian Monte Carlo sampling of model parameters (20). We first use simulation to demonstrate the utility of RiVIERA in prioritizing driver variants and detecting functional epigenomic annotations. We then apply RiVIERA to some of the most well-powered GWAS datasets, consisting of 9 immunological disorders from ImmunoBase (15) and Schizophrenia 2014 data from Psychiatric Genomic Consortium (21). To infer tissue-specific epigenomic enrichments, we utilize the largest compendium of epigenomic annotations to date from ENCODE/Roadmap Consortia, consisting of 848 annotations including eight major epigenomic marks across 127 distinct cell types (7). This allows us to revisit the GWAS of these 10 common complex disorders by inferring their underlying regulatory variants implicated at the tissue-specific epigenomic contexts.
MATERIALS AND METHODS
GWAS summary statistics
The GWAS summary statistics for the nine immune diseases were obtained from ImmunoBase (17 March 2015) (15). The nine diseases are: Autoimmune Thyroid Disease (ATD), Celiac Disease (CEL), Juvenile Idiopathic Arthritis (JIA), Multiple Sclerosis (MS), Narcolepsy (NAR), Primary Biliary Cirrhosis (PBC), Psoriasis (PSO), Rheumatoid Arthritis (RA), Type 1 Diabetes (T1D). We imputed the P-values of un-genotyped SNPs using FAPI and 1000 Genome European data (Phase 1 version 3) (22). We then obtained the P-values of SNPs that fall within the pre-defined risk loci available from ImmunoBase for each of the nine immune traits. For all analyses, we filtered out risk loci or variants in the MHC regions or sex chromosomes X and Y. The Schizophrenia 2014 (SCZ2) summary data containing 642846 observed and imputed SNPs were obtained from Psychiatric Genomic Consortium (PGC) (21). Among these, 54 132 SNPs fall within the 105 SCZ-associated loci of the autosomes (chr 1-22) defined by PGC (we filtered out the 3 loci on chromosome X). Table 1 summarizes the total number of SNPs and risk loci for each individual GWAS that were subject to the proposed fine-mapping analyses.
Table 1. GWAS data summary.
| Abbrev | Trait | Total | Loci | gwSNPs | cSNP_st | cSNPs_mt |
|---|---|---|---|---|---|---|
| ATD | Autoimmune Thyroid Disease | 4206 | 8 | 630 | 38 | 49 |
| CEL | Celiac Disease | 29 784 | 39 | 2592 | 344 | 211 |
| JIA | Juvenile Idiopathic Arthritis | 13 427 | 22 | 3 | 440 | 223 |
| MS | Multiple Sclerosis | 61 360 | 104 | 2096 | 884 | 339 |
| NAR | Narcolepsy | 1316 | 3 | 62 | 22 | 16 |
| PBC | Primary Biliary Cirrhosis | 14 573 | 19 | 2498 | 172 | 111 |
| PSO | Psoriasis | 24 832 | 34 | 457 | 305 | 171 |
| RA | Rheumatoid Arthritis | 38 207 | 78 | 470 | 1978 | 719 |
| SCZ2 | Schizophrenia | 54 132 | 105 | 5217 | 2481 | NA |
| T1D | Type 1 Diabetes | 41 945 | 57 | 2832 | 826 | 327 |
We investigated 10 GWAS traits as listed above. Abbrev: abbreviation of the trait names; Total: total number of SNPs in the risk loci with imputed and observed summary statistics; Loci: total number of risk loci for each trait; gwSNPs: SNPs that pass GWAS cutoff P < 5e-8; cSNP_st: total number of SNPs that are included into the 95% credible set based on single-trait risk inference using RiVIERA; cSNP_mt: SNPs in 95% credible set constructed based on multi-trait joint risk inference using RiVIERAacross the nine immune traits (without SCZ2).
Roadmap epigenome data
Roadmap epigenome data were obtained from Roadmap epigenomic web portal (March 2015). Peaks were defined if their P-values were below 0.01 (i.e. following the definition of ‘Narrow Peaks’ (7)). In total, there are 848 epigenome tracks, including eight epigenomic marks namely H3K4me1, H3K4me3, H3K36me3, H3K27me3, H3K9me3, H3K27ac, H3K9ac and DNase I in in 127 cell or tissue types, which were grouped into 19 categories (7). To associate each SNP with the annotations, we overlapped their genomic coordinates with each bigWig epigenome track making use of the R packages rtracklayer and GenomicAlignments. SNPs that fall within a peak of an annotations will have value 1 otherwise 0 for that annotation. The resulting matrix is a Vd × K input matrix containing the epigenomic values across K = 848 marks for each of the Vd SNPs in disease d.
Running existing fine-mapping software on simulated data
fgwas
The software fgwas (23) (version 0.3.4) were downloaded from GitHub. We prepared the input for fgwas (i) the z scores calculated as the t-statistics of the linear coefficients of the genotype of each variant fitted separately by least square regression on the simulated continuous phenotypes (Materials and methods) and (ii) 100 discretized epigenomic annotations at P < 0.01. To enable fine-mapping, we issued -fine flag and specify the region numbers for each SNP in the input file as required by the software. As part of the outputs from fgwas, we obtained ‘PPA’ and ‘estimate’ for the causal variants and influences of each epigenomic annotations, respectively.
GPA
GPA (0.9–3) (19) was downloaded from GitHub and run with default settings. Same as above, we set the annotations to one at P-value <0.01 and 0 otherwise. To test for trait-relevant annotations, we followed the package vignette. Briefly, we fit two GPA models with and without the annotation and compared the two models by aTest function from GPA, which performs likelihood-ratio (LR) test via χ2 approximation, and obtained the enrichment scores as the −log10 P-value.
PAINTOR
PAINTOR (version 2.1) was downloaded from GitHub (18). As suggested in the documentation, we prepared a list of input files for every locus including summary statistics as t-statistics, LD matrices, and binary epigenomic annotations. We ran the software with default setting with assumption of at most two causal variants per locus. We then extracted the ‘Posterior_Prob’ and ‘Enrichment.Values’ as the model predictions for causal variants and causal annotations, respectively.
Details of RiVIERA Bayesian model
Inference of empirical prior πvd
We first define the empirical prior function of a variant  being associated with disease d as a logistic function:
being associated with disease d as a logistic function:
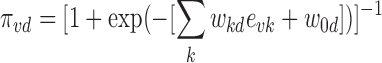 |
(1) |
where 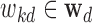 denotes the linear coefficient or the influence of the kth epigenomic mark affecting disease d and
denotes the linear coefficient or the influence of the kth epigenomic mark affecting disease d and 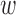 0d is the linear bias.
0d is the linear bias.
We assume that epigenomic causal effect 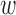 kd follows a multivariate Gaussian distribution with zero mean and unknown covariance:
kd follows a multivariate Gaussian distribution with zero mean and unknown covariance:
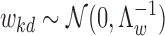 |
(2) |
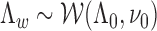 |
(3) |
where Λ is a D × D inverse covariance matrix
is a D × D inverse covariance matrix 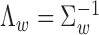 to model the pairwise epigenomic correlation among D diseases. It follows a Wishart distribution with identity matrix as prior (i.e. by default, we assume aprior no correlation between the target traits) and ν0 = 0 (i.e. by default, we did observe any samples aprior that are indicative of the correlation between any two diseases being modeled). The hyperparameters can be easily modified to incorporate prior belief on the correlation between any two diseases of interests.
to model the pairwise epigenomic correlation among D diseases. It follows a Wishart distribution with identity matrix as prior (i.e. by default, we assume aprior no correlation between the target traits) and ν0 = 0 (i.e. by default, we did observe any samples aprior that are indicative of the correlation between any two diseases being modeled). The hyperparameters can be easily modified to incorporate prior belief on the correlation between any two diseases of interests.
Additionally, the bias 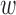 0d follows a Gaussian distribution with unknown variance and mean determined based on our prior belief of the causal fraction π0:
0d follows a Gaussian distribution with unknown variance and mean determined based on our prior belief of the causal fraction π0:
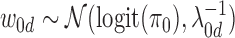 |
(4) |
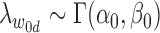 |
(5) |
where 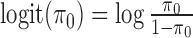 . By default, we set π0 to 0.01, implying that 1% of the SNPs in the risk loci are expected to be causal when no functional enrichment. We set α = 0.01 and β = 0.0001 to enable a broad hyperprior for
. By default, we set π0 to 0.01, implying that 1% of the SNPs in the risk loci are expected to be causal when no functional enrichment. We set α = 0.01 and β = 0.0001 to enable a broad hyperprior for 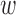 0d.
0d.
Notably, 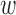 kd can be interpreted as enrichment coefficient for annotation k in disease d, where a positive
kd can be interpreted as enrichment coefficient for annotation k in disease d, where a positive 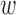 kd will increase the causal prior πvd when evk = 1. During the training, however,
kd will increase the causal prior πvd when evk = 1. During the training, however, 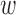 kd may become negative, which makes the interpretation difficult. Thus, we constrain
kd may become negative, which makes the interpretation difficult. Thus, we constrain 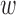 kd to be non-negative values, which involves imposing infinitely high potential energy for negative
kd to be non-negative values, which involves imposing infinitely high potential energy for negative 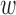 kd. More details are described in Supplementary Text 1.
kd. More details are described in Supplementary Text 1.
Inference of variant causality cvd given prior πvd and model parameters μd, ϕd
Because the target association variable avd for variant 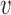 in disease d represents P-values, which are continuous and restricted to the interval (0, 1), we assume that it follows a Beta distribution with unknown mean μd and unknown precision ϕd:
in disease d represents P-values, which are continuous and restricted to the interval (0, 1), we assume that it follows a Beta distribution with unknown mean μd and unknown precision ϕd:
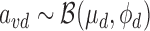 |
(6) |
Note that we re-parameterize Beta density function from the traditional ‘rate’ p and ‘shape’ q parameters, and instead use mean μ = p/(p + q) and precision ϕ = p + q, as per (24,25). Specifically, the density function of association variable avd is defined as follows:
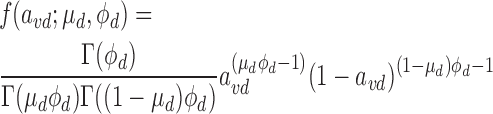 |
(7) |
Further, we let the mean μd and precision ϕd follow Beta and uniform prior, respectively:
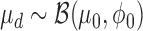 |
(8) |
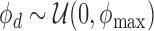 |
(9) |
where the hyperparameters (μ0, ϕ0) reflect apriori belief on the P-value signal of a causal variant. By default, we set μ0 = 0.1 and ϕ0 = 2. If ϕmax = ∞, ϕ follows an improper prior. Because it is unlikely to have a very large ϕ, by default, we set ϕmax to 1000. Notably, as long as ϕmax is large, the inference results remain the same with different ϕmax values.
With the prior 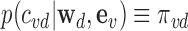 and likelihood p(avd|μd, ϕd) ≡ f(avd; μd, ϕd) established, the posterior probability of association (PPA) (26) of variant
and likelihood p(avd|μd, ϕd) ≡ f(avd; μd, ϕd) established, the posterior probability of association (PPA) (26) of variant 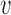 being causal for disease d then follows:
being causal for disease d then follows:
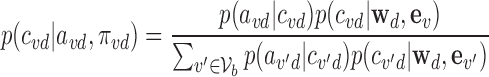 |
(10) |
where 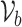 represent all variants within locus b. The 95% credible set
represent all variants within locus b. The 95% credible set 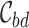 for each locus b is the minimal number of SNPs
for each locus b is the minimal number of SNPs 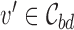 in the locus such that
in the locus such that 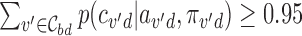 .
.
Joint posterior distribution
The complete likelihood density function treating cvd as missing values is defined as:
 |
(11) |
The logarithmic joint posterior density function is then:
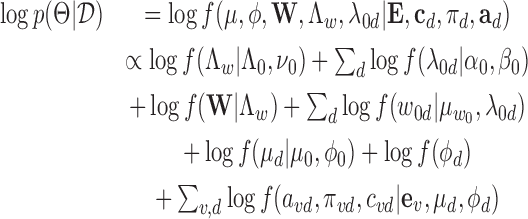 |
(12) |
In principle, causality is inferred by integrating out all nuisance parameters:
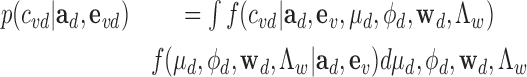 |
(13) |
which is not tractable. We employ Markov Chain Monte Carlo (MCMC) to sample from the joint posterior.
Markov Chain Monte Carlo
We use Gibbs sampling (27) to sample the precision matrix Λ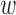 of epigenomic effects from the posterior distribution. Specifically, Gibbs sampling requires a closed form posterior distribution. Due to the conjugacy of the Wishart prior of epigenomic precision Λ
of epigenomic effects from the posterior distribution. Specifically, Gibbs sampling requires a closed form posterior distribution. Due to the conjugacy of the Wishart prior of epigenomic precision Λ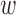 to the multivariate normal distribution of epigenomic effect
to the multivariate normal distribution of epigenomic effect 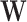 , the posterior of the epigenomic precision matrix Λ
, the posterior of the epigenomic precision matrix Λ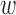 also follows Wishart distribution (28):
also follows Wishart distribution (28):
 |
(14) |
where  is the sample variance of
is the sample variance of 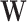 , i.e.
, i.e. 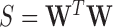 .
.
Similarly, we sample λ0d from Gamma posterior distribution:
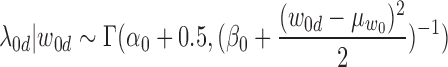 |
(15) |
To sample epigenomic effects 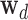 , prior bias
, prior bias 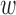 0d, causal mean μd, causal precision ϕd for disease d = 1, …, D, we employ a more powerful gradient-based sampling scheme namely Hamiltonian Monte Carlo (also known as hybrid Monte Carlo) (HMC) (20,29), exploiting the fact that the joint posterior of our model is differentiable with respect to the model parameters μd, ϕd,
0d, causal mean μd, causal precision ϕd for disease d = 1, …, D, we employ a more powerful gradient-based sampling scheme namely Hamiltonian Monte Carlo (also known as hybrid Monte Carlo) (HMC) (20,29), exploiting the fact that the joint posterior of our model is differentiable with respect to the model parameters μd, ϕd, 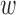 kd,
kd, 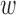 0d (Supplementary Text S1). Finally, after discarding t% models accepted before the burn-in period (default: t = 20%), we obtain the Bayesian estimates of PPA by averaging the corresponding values computed over the T′ individual models accepted throughout the T MCMC runs.
0d (Supplementary Text S1). Finally, after discarding t% models accepted before the burn-in period (default: t = 20%), we obtain the Bayesian estimates of PPA by averaging the corresponding values computed over the T′ individual models accepted throughout the T MCMC runs.
Bayesian fold-enrichment tests for epigenomic annotations
Due to co-linearity among the epigenomic annotations, directly using 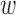 kd to assess the epigenomic enrichment for annotation k may be misleading. We propose an heuristic approach to assess the log fold-enrichment of the full prior model over the alternative prior with the effect of annotation k for disease d removed (i.e.
kd to assess the epigenomic enrichment for annotation k may be misleading. We propose an heuristic approach to assess the log fold-enrichment of the full prior model over the alternative prior with the effect of annotation k for disease d removed (i.e. 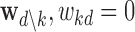 ):
):
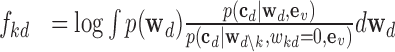 |
(16) |
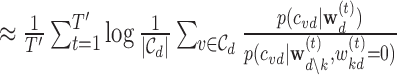 |
(17) |
where 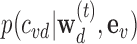 is the logistic prior based on Eq 1,
is the logistic prior based on Eq 1, 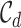 is the union of all the 95% credible sets across loci for disease d:
is the union of all the 95% credible sets across loci for disease d: 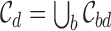 . Notably, under the optional constraint that
. Notably, under the optional constraint that 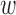 kd ≥ 0, fkd is always positive, which directly translates to fold-enrichment of annotation k conditioned on all the other annotations k′ ≠ k. The 95% Bayesian credible interval for fkd are obtained from the T′ MCMC runs. The significance of each annotation k is determined based on the ranking of its lower bound fkd (i.e. the 2.5% quantile of fkd).
kd ≥ 0, fkd is always positive, which directly translates to fold-enrichment of annotation k conditioned on all the other annotations k′ ≠ k. The 95% Bayesian credible interval for fkd are obtained from the T′ MCMC runs. The significance of each annotation k is determined based on the ranking of its lower bound fkd (i.e. the 2.5% quantile of fkd).
Alternatively, we can estimate the fold-enrichment for each annotation simply based on the ratio of estimated fraction of causal variants in an annotation evk over the fraction of all of the variants in that annotation  , where c
, where c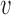 is the PPA for SNP
is the PPA for SNP 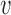 . This is more efficient and accurate when the underlying causal variants were randomly sampled from the annotations as done in the simulation.
. This is more efficient and accurate when the underlying causal variants were randomly sampled from the annotations as done in the simulation.
Stochastic gradient updates per locus
Directly updating model parameters based on the gradients of all GWAS loci at each MCMC iteration is inefficient and results in poor HMC acceptance rate. Instead, at each MCMC update, we randomly sample one locus and update the model parameters (which are shared across loci) based on that locus. We find this approach quite efficient in capturing meaningful causal properties such as causal signals and relevant epigenomes that are shared across all risk loci. Together, we outline the overall algorithm of the proposed Bayesian model in Algorithm S2 (Supplementary Text S1).
GWAS simulation
To assess the power of the proposed fine-mapping model in identifying causal variants and compare it with existing methods, we implemented a simulation pipeline adapted from (18). Briefly, the simulation can be divided into three stages (i) simulate genotypes based on the haplotypes from 1000 Genome European data (phase 1 version 3) using HapGen2 (30) (Supplementary Figure S1); (ii) simulate epigenomic enrichments and subsequently sample causal variants accordingly using 100 Roadmap annotations selected from each of the 19 categories of primary tissue/cell types (Supplementary Figure S1); (iii) simulate liability phenotype plus the random noise to obtain the desired heritability (fixed at 0.25) and subsequently the GWAS summary statistics in terms p-values and z-scores via ordinary least square regression. Details are described in Supplementary Text.
Gene ontology enrichment analysis
We obtained the latest gene annotations from Ensembl database (version 80) programmatically via biomaRt package (31), which resulted in 10,801 gene ontology (GO) terms in biological processes (BP). To assign SNPs to genes, we performed lift-over to map the SNPs from hg19 to hg38 using rtracklayer (32) and assigned each SNP to a gene if it is located within 35 kb up and 10 kb downstream of that gene. The resulting Ensemble gene identifiers were matched with those genes in each GO-BP category. We then performed hypergeometric tests on each GO-BP term for all of potential in-cis target genes of the SNPs in each trait and adjusted for multiple testings using Benjamini-Hocherg family-wise Type I error correction method (33). For the 9 immune traits, the enrichment signals are strong so we set the cutoff at FDR < 0.005; for Schizophrenia, we set FDR < 0.2.
RiVIERA software
RiVIERA is available as an open-source R package with documented functions and walk-through examples described in the vignette. Most functions were implemented in C++ by integrating Rcpp and RcppArmadillo libraries (34). These libraries enabled us to apply RiVIERA to large matrices very efficiently with complied code and having much lesser memory overhead than a naïve R implementation. RiVIERA is available at Github (https://yueli-compbio.github.io/RiVIERA-beta).
RESULTS
RiVIERA model overview
The fundamental hypothesis of our model is that non-coding disease associations are driven by disruption of regulatory elements of common activity patterns (e.g. motifs of sequence-specific regulators), thus leading to gene expression changes and ultimately phenotypic changes at the cellular or organism level between case and control individuals. Our RiVIERA Bayesian model aims to infer the probability that a given variant 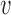 is a driver for disease d by modeling the corresponding GWAS association statistic for that variant using a vector of genome-wide epigenomic annotations (
is a driver for disease d by modeling the corresponding GWAS association statistic for that variant using a vector of genome-wide epigenomic annotations (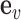 ). Given a set of B risk loci, the inputs to RiVIERA are GWAS summary statistics in terms of p-values and a set of discrete or continuous epigenomic annotations (Figure 1A). In this study, we used binary signals to ease interpretation of the functional enrichments. We train RiVIERA by repeatedly sampling one locus at each iteration to efficiently learn the intrinsic (i.e. locus-independent) causal signals. Figure 1B depicts RiVIERA as probabilistic graphical model (35). The observed variable of our model is the GWAS association values (in terms of p-values) avd for each variant
). Given a set of B risk loci, the inputs to RiVIERA are GWAS summary statistics in terms of p-values and a set of discrete or continuous epigenomic annotations (Figure 1A). In this study, we used binary signals to ease interpretation of the functional enrichments. We train RiVIERA by repeatedly sampling one locus at each iteration to efficiently learn the intrinsic (i.e. locus-independent) causal signals. Figure 1B depicts RiVIERA as probabilistic graphical model (35). The observed variable of our model is the GWAS association values (in terms of p-values) avd for each variant 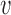 in each disease d. We assume that avd follows a Beta distribution with unknown mean and dispersion parameters. The effect of each annotation on each trait is learned as global annotation-by-disease weight matrix
in each disease d. We assume that avd follows a Beta distribution with unknown mean and dispersion parameters. The effect of each annotation on each trait is learned as global annotation-by-disease weight matrix 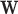 , which follows a D-dimensional multivariate normal distribution with zero mean and D × D disease-disease covariance Λ
, which follows a D-dimensional multivariate normal distribution with zero mean and D × D disease-disease covariance Λ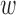 . The prior probability πvd that a variant
. The prior probability πvd that a variant 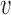 is causal in disease d is essentially a linear combination of the weighted genomic annotations
is causal in disease d is essentially a linear combination of the weighted genomic annotations 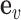 , which reflects the disease-associated active histone marks and DNA accessibility in the 127 cell types (Materials and methods). The outputs of the model (Figure 1C) are (a) posterior probability of association (PPA) cvd that variant
, which reflects the disease-associated active histone marks and DNA accessibility in the 127 cell types (Materials and methods). The outputs of the model (Figure 1C) are (a) posterior probability of association (PPA) cvd that variant 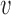 is causal in disease d; (b) the Bayesian fold-enrichment estimates fkd based on the ratio between the full prior model with all annotations over the null prior model with all annotations except for annotation k.
is causal in disease d; (b) the Bayesian fold-enrichment estimates fkd based on the ratio between the full prior model with all annotations over the null prior model with all annotations except for annotation k.
Figure 1.

RiVIERA model overview. (A) Inputs to RiVIERA are GWAS summary statistics and epigenomic annotations for B risk loci. At a given iteration, the model samples one locus and tries to learn the intrinsic causal signals implicated in the corresponding GWAS summary data and epigenomic profiles. Highlighted variant is the causal variant based on the simulated data. (B) The probabilistic graphical model representation of RiVIERA (35). Variables for which distribution is defined are in circle. Epigenomic profiles are treated as observed values with no circle. The variable in shaded circle are observed (i.e. GWAS association avd and variables in unshaded circle are unobserved. The variables in red are observed and variables in blue are the variables of interest (i.e. causal indicator). The two colored plates represent K annotations (red) and V variants (blue). We model the GWAS association avd of variant 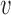 in terms of P-value sampled from Beta distribution with unknown precision ϕd and mean μd, which respectively follow an uninformative prior and a Beta distribution with hyperparameters μ0, ϕ0. The latent variable cvd indicates whether variant
in terms of P-value sampled from Beta distribution with unknown precision ϕd and mean μd, which respectively follow an uninformative prior and a Beta distribution with hyperparameters μ0, ϕ0. The latent variable cvd indicates whether variant 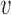 is causal in disease d. On top of it, we dedicate an empirical prior as a linear combination of the epigenomic profile evk weighted by the epigenomic influence
is causal in disease d. On top of it, we dedicate an empirical prior as a linear combination of the epigenomic profile evk weighted by the epigenomic influence 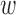 kd, which follows multivariate normal with zero mean and a D × D inverse covariance or precision matrix
kd, which follows multivariate normal with zero mean and a D × D inverse covariance or precision matrix 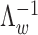 , where D is the number of traits that are being modeled. The linear bias
, where D is the number of traits that are being modeled. The linear bias 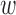 0d expresses the prior belief of the causal fraction π0 (default: 0.01). (C) Outputs from the model are posterior probabilities of association (PPA) for each variant in each locus, the 95% credible set containing the minimal number of SNPs whose PPA sum to 0.95 or greater, and the Bayesian estimates of the fold-enrichment of each annotation.
0d expresses the prior belief of the causal fraction π0 (default: 0.01). (C) Outputs from the model are posterior probabilities of association (PPA) for each variant in each locus, the 95% credible set containing the minimal number of SNPs whose PPA sum to 0.95 or greater, and the Bayesian estimates of the fold-enrichment of each annotation.
Method comparison using GWAS simulation
The goal of the simulation is to evaluate the model's power to predict (i) causal variants in each locus; () the relevant annotations that determine which variants are causal. To this end, we simulated GWAS summary statistics based on 1000 Genome European data (Phase 1 release 3) (Supplementary Figure S1) and 100 representative epigenomic annotations (Supplementary Figure S1) (Materials and methods). We performed a series of power analyses over 500 simulation runs.
First, we examined how well the posterior probabilities were calibrated by taking the credible SNPs that contribute to 95% posterior mass inferred by each method (Supplementary Figure S2). As expected, when our model assumption of single-causal variant per locus holds, we observe that our model is well calibrated (Supplementary Figure S2), where the 95% credible SNPs indeed correspond to approximately 95% of the causal variants. When there are more than one causal variants per locus, the 95% credible SNPs include on average 50% the true causal SNPs (Supplementary Figure S2)
Because the number of variants within the credible set differs depending on the concentration of the posterior probabilities inferred by each method, we sought to control that bias by evaluating the proportion of identified causal variants as a function of the absolute number of selected variants. When the assumption of one-causal-variant-per-locus holds, we observed comparable or better performance of RiVIERA compared to existing methods (Figure 2). As expected, when the assumption is violated, our current model is second to PAINTOR, which is able to infer multiple causal variants per locus (Supplementary Figure S3). We also examined the correlation between the functional enrichments estimated by each method and the underlying epigenomic enrichments that were used to simulate the causal variants. The performance of the four methods are comparable with the proposed model achieving a slightly better correlation (Supplementary Figure S4).
Figure 2.

Model performance on simulated datasets. Proportion of causal variants were identified by each method as a function of increasing number of top variants selected.
Applications to immune and psychiatric disorders
To demonstrate RiVIERA in a real-world application, we used it to investigate 10 complex diseases including nine immune diseases with summary statistics obtained from ImmunoBase (15) and Schizophrenia from Psychiatric Genomic Consortium (PGC) (21) (Table 1). We used 848 epigenomic annotations from ENCODE/Roadmap consortium (Materials and methods) to build a functional prior for each trait to aid fine-mapping and conduct cell-type specific epigenomic enrichment analyses (7). We first applied RiVIERA to the 10 traits separately to examine individual causal signals and then demonstrated RiVIERA's capability to operate on the nine immune traits and the improved detection power compared to the single-trait model.
RiVIERA detected meaningful tissue-specific enhancers in test GWAS traits
We first sought to confirm the validity of the model through its ability to identify meaningful cell-types or tissues for each trait. To this end, we selected the top 5% (i.e. the top 43) of the 848 annotations for each disease based on the corresponding Bayesian estimates of the lower bounds of the 95% credible interval (Supplementary Table S1; Materials and methods). We then performed hypergeometric tests on enrichments of each of the 19 categories grouped by Roadmap consortium based on the cell types and tissues (7). Indeed, we observed a significant enrichment for Blood & T-cell for all nine immune disorders but not for Schizophrenia, which exhibits exclusive epigenomic enrichments in the Brain category (Hypergeometric adjusted P-values < 0.05) (Figure 3A). Additionally, we also observed modest enrichments for B-cell and Thymus tissue in the nine immune traits. We then examined the enrichment status for the eight epigenomic marks. Indeed, enhancer marks namely H3me4me1 and/or H3K27ac are most significantly enriched among all eight marks (q-values < 0.05). In addition, H3K4me3 associated with promoter is also enriched in most immune traits. Interestingly, we also observed a modest enrichment of H3K9me3 in Schizophrenia but not in the immune traits. We further ascertained the enrichment results by re-running RiVIERA on the permuted data matrix and observed diminishment of the meaningful enrichment observed above (Supplementary Figure S5).
Figure 3.

Predicted tissue-specific epigenomic enrichments in the 10 GWAS traits. (A). Hypergeometric enrichment for each of the 19 primary tissue categories using the top 5% or 43 annotations out of the 848 annotations in total for each trait based on the lower bound of the 95% credible interval of the Bayesian fold-enrichment estimates by our RiVIERA; model; (B) enrichments for the eight epigenomic marks among the top 43 annotations for each trait. Y-axis is the logarithmic q-values, which are the corrected P-values from the hypergeometric tests for multiple testing across traits and tissue groups or marks by Benjamini–Hochberg method (33). On both plots, horizontal dashed bars indicate standard statistical threshold of FDR < 0.05.
SNPs in the credible set exhibit promising regulatory potentials
The variants in the credible set are more enriched for functional elements. Inspired by the promising tissue-specific enhancer enrichment results obtained above, we refined our RiVIERA model by re-training it on the top 5% (or 43) annotations on each trait using the same GWAS data. For each locus in each trait, we then constructed 95% credible set (Supplementary Table S2; Materials and methods). On average, we were able to construct a rather small credible set ranging from 4 to 25 SNPs per locus for the 10 traits (Table 1). As a comparison, we extracted the same number of SNPs with the most significant GWAS P-values from each locus. For ease of reference, we named our SNPs in the credible set as ‘credible SNP’ and the GWAS counterpart as ‘GWAS SNP’. Compared to GWAS SNPs, the credible SNPs exhibit substantially higher averaged placental conservation scores (phastCons46way obtained from UCSC database) across most traits (Figure 4 CONS).
Figure 4.

Functional enrichments of credible SNPs. The top left panel displays the averaged phastCons46way conservation scores for variants in the 95% credible set (cred_snp) and the same number of SNPs chosen based on GWAS P-values (gwas_snp). The three other panels illustrate hypergeometric enrichments in terms of the -log10q-values corrected for multiple testing over the 10 traits of the selected variants for GTEx whole blood eQTL located within transcription factor binding sites based on sequence motif (TFBS) (eQTL + TFBS) and genomic digital footprint (DGF) (eQLT + DGF) and eQTL in both TFBS and DGF (eQTL+TFBS+DGF).
Moreover, the credible SNPs were significantly enriched for expression quantitative trait loci (eQTL) that are in the regulatory regions. Specifically, we obtained in total 806 847 GTEx whole-blood eQTL-SNPs (version 6) (36) and retained 122 549 and 23 973 eQTL-SNPs that overlap with transcription factor binding sites derived from 1772 TF recognition motifs (37) and digital genomic footprints (DGF) at 6-bp resolution derived from DNaseI data in CD cells using method described in (38), respectively as well as 6743 eQTL-SNPs that overlapped with both the TFBS and DGF regions. We then performed hypergeometric tests to assess the significance of overlap between the credible/GWAS SNPs and the regulatory-eQTL SNPs. Indeed, our credible SNPs exhibit much higher enrichments for those eQTL-SNPs, suggesting their regulatory potentials elucidated based on the enhancer activities by our proposed RiVIERA model (Figure 4; Supplementary Table S3).
Gene-centric analysis revealed enrichment for meaningful biological processes
Genes adjacent to the SNPs in credible sets are significantly enriched for disease-specific biological processes. In particular, we observed significant enrichments of many immune-related processes for the in-cis genes for which the SNPs in the credible set are within 35 kb upstream or 10 kb downstream (Figure 5; Supplementary Table S4; Materials and methods). For instance, regulation of T cell homeostatic proliferation, regulation of interferon-gamma-mediated signaling pathway, and regulation of type I interferon-mediated signal pathways are among the most significantly enriched GO terms in five or six out of the nine immune traits. In contrast, the enrichments for Schizophrenia are dominated by GO terms involving synaptic processes and neuronal differentiation/development. The enrichment results are mostly consistent between the credible genes and the genes derived from the same number of SNPs chosen based on the GWAS P-values (GWAS-genes).
Figure 5.

Gene ontology enrichments across the 10 traits. Rows are the GO biological processes and columns are the 10 traits. Color intensities in each cell reflect the significance level in terms of -log10P-value. Asterisks indicate q-values above significant cutoff after correcting for multiple testings (FDR < 0.2). GO names that match the pattern ‘synap|neuro|nerve‘ are colored blue to highlight their exclusive association with ‘Schizophrenia’ (also in blue). Notably, GO terms ‘keratinization‘ and ‘epidermis development’ (highlighted in the red box) are exclusively enriched for Psoriasis. Diseases were ordered based on hierarchical clustering based on the Pearson correlation of their GO enrichment scores.
Intriguingly, we observed a highly significant enrichment for keratinization (GO:0031424) and epidermis (e.g. skin) development (GO:0008544) exclusively for Psoriasis. In particular, 17 genes among the 241 credible genes belong to keratinization and epidermis development, which contain in total 49 and 121 genes, respectively (q < 9 × 10−18, q < 2 × 10−10). Indeed, Psoriasis is mainly characterized as a chronic skin disease with epidermal hyper-proliferation (39,40). In contrast, there are only six out of 157 GWAS-genes are defined in each of two GO categories (q < 0.001).
To further ascertain the RiVIERA fine-mapping results, we created a visualization scheme for each of the 469 risk loci across 10 traits examined (Supplementary Figure S6). Figure 6 displays two example loci for Type 1 diabetes (chr17: 37383069-38239012) and Schizophrenia (chr7: 104598324-105062839). The upper panel displays the RiVIERA model prior, the genetic signals from GWAS −log P-values, and RiVIERA PPA. Red colored and diamond shape points are GTEx whole-blood eQTL SNP and top SNPs included into 60% credible set (we used 60% to not clutter the plot with the remaining SNPs in the 95% credible set that exhibit low PPA). Intuitively, SNPs with high PPA exhibit both prominent genetic and epigenetic signals. Thus, to infer causal variants, RiVIERA efficiently took into account not only the GWAS signals derived from the genetic data but also the prior signals mainly driven by the weighted epigenomic profiles. The middle panel illustrates the cumulative density for each epigenomic profiles weighted by the tissue-specific enrichment estimates.
Figure 6.

Visualization of fine-mapping results. Top track: the upper panel display the RiVIERA prior, genetic signals of GWAS −log10P-values, and RiVIERA PPA; the middle track illustrates the cumulative density of weighted epigenomic profiles colored based on the epigenomic group; the bottom tracks shows the conservation, gene annotations (Gencode 19), transcription factor binding sites (TFBS), and SNP positions. The red colored and bigger diamond plots indicate whole-blood GTEx eQTL SNPs and SNPs included into the 60% credible set, respectively. For illustration purpose, only one risk locus for Type 1 diabetes and one for Schizophrenia are shown above. The full display of 469 risk loci were in Supplementary Figure S6.
Consistent with the overall enrichment results (Figure 3), we observed prominent enrichments for the enhancer regions predominantly in blood T cells for all of the nine immune traits and brain tissue for Schizophrenia. The bottom tracks display the external functional information (i.e. not in the training data) including conservation score, genes, transcription factor binding sites based on motif matches that may further aid variant selection for downstream experimental validation (please refer to Supplementary Table S2 for detailed information). We also visualized the signals within the of Psoriasis-associated risk region ch1:152536784–152785170, which harbors genes involved in keratinization and epidermis development as mentioned above. Interestingly, as an exception of most other immune-susceptible loci, the underlying epigenomic profiles exhibit prominent signals not only in blood T-cell but also in epithelia enhancer regions (Supplementary Figure S6). However, the associated SNPs exhibit rather weak genetic signal perhaps due to lower allele frequencies.
Multi-trait causal inference improved functional enrichments in most immune traits
Exploiting epigenomic correlation between highly related immune diseases improved functional enrichments in several traits. We performed multi-trait causal inference over all of the nine autoimmune traits by jointly applying our RiVIERA to 364 risk loci concatenated together from the nine immune traits using 174 epigenomic annotations which was a union of unique annotations from the top 43 annotations for each individual trait. We focused only on the nine immune GWAS (i.e. leaving out Schizophrenia) because they commonly utilized the same genotyping platform namely ImmunoChip. The multi-trait GWAS summary statistics triggered RiVIERA to infer the disease covariance matrix and sample disease-specific epigenomic weights from the joint posterior with modified zero-mean multivariate normal prior that takes into account the sampled disease covariance (Materials and methods). As a results, RiVIERA sampled correlated epigenomic weights between traits more frequently compared to the single-trait model.
We constructed the 95% credible sets for each trait using the disease-specific PPA derived from the joint model and assessed the functional enrichments as above (Supplementary Table S6). Notably, the cross-trait model exploited 174 annotations as apposed to 43 annotations used by the single-trait model. To examine whether the improved enrichments were due to the increased number of annotations, we re-ran a single-trait model for each of the nine traits separately each using the 174 annotations. Compared to the 95% credible set constructed based on the single-trait causal inference using the top 43 annotations, we observed smaller 95% credible sets for eight out of the nine immune traits (Table 1), suggesting that the mulit-trait joint inference further improved the model confidence in some of the highly related traits.
More importantly, we observed a much more improved enrichments for the GTEx whole-blood eQTL SNPs located within open chromatin regions or digital genomic footprints in most of the immune traits (Figure 7; Supplementary Table S5). Thus, the joint inference further improved the regulatory potential through following the Hamiltonian trajectory that is more consistent with the epigenomic correlation pattern between the related immune traits. We also repeated the GO enrichment analysis on the 95% credible set and found that the enriched GO terms were mostly immune-specific biological processes and consistent with the above single-trait analyses (Supplementary Figure S7; Supplementary Table S7).
Figure 7.

Enrichments for eQLT using credible SNPs constructed from multi-trait joint inference. Credible SNPs for each trait were constructed based on PPA inferred by the joint RiVIERA model over the 9 immune traits using 174 annotations, which are the union of the top 43 annotations detected from each trait individually. We then assessed the hypergeometric enrichments of the 95% credible sets for the GTEx whole-blood eQTL that are within DNA hypersensitive sites as defined by the genomic digital footprint data (38). We compared these enrichment scores derived from the multi-trait model (cred_snp_mt) to the enrichments derived from the single-trait models either running on 43 annotations (cred_snp_st43) or on the 174 annotations (cred_snp_st174). The latter was included to control for the improvements due to the increased number of annotations (from 43 to 174).
DISCUSSION
Understanding the genetic basis of complex traits hinges upon powerful integrative methods to map genotypes to phenotypes (41). Fine-mapping causal variants has been a highly active and fruitful research in the past several years (9,18,42–44). However, most existing methods typically operate solely on genetic data by estimating each SNP of being causal conditioned on the lead index SNPs in the same LD block, which are typically approximated by the 1000 Genome data (9,15,45,46). With the recent availability of large-scale functional genomic data provide by ENCODE/Roadmap consortia, there is an urgent need to incorporate these valuable information in a principled way as a form of Bayesian prior. In this article, we describe a novel Bayesian fine-mapping method RiVIERA to re-prioritize GWAS summary statistics based on their epigenomic contexts. The main contribution of RiVIERA is the ability to systematically construct from a targeted set of susceptible loci a Bayesian credible set of SNPs, which exhibit plausible tissue-specific regulatory properties implicated in the large epigenomic data compendium either in a single trait or across multiple traits.
One benefit of using the raw epigenomic annotations rather than using the inferred signals such as ChromHMM (7) or GenoSkyline (47) states derived from the annotations is that it eases the interpretation of the actual relevant epigenomic marks in the relevant tissue types and facilitates downstream experimental efforts to assay the specific marks in the disease-specific cell types. However, the correlation of the epigenomic marks will make difficult estimating the underlying functional enrichments. Moreover, we choose to model the summary statistics rather than genotypes because it is not always possible to obtain individual-level phenotype-genotype data particularly for large-scale meta-analysis. Thus, effective methods based on summary statistics may benefit wider research communities than methods that solely operate on individual-level genotype data (18,19,23). Moreover, our model requires only p-values because it uses Beta distribution to model the likelihood. In contrast, fgwas requires both the z-scores and the standard error from the linear regression used in the GWAS to estimate the Wakefield approximate Bayes factors. While some recent GWAS summary statistics include those information, there are many do not have z-scores and/or standard error of the linear model but only P-values (e.g. the ImmunoChip data we used in our studies for the nine immune traits). When the standard error is not available in a given GWAS summary statistics, fgwas needs to be estimate it from the minor allele frequency of a reference panel such as 1000 Genome, which may not be accurate depending the study cohorts. Additionally, modeling P-values via Beta density function only has more relaxing model assumption than modeling z-scores via normal density although both methods are highly effective in practice.
Overall, SNPs included into the credible set exhibit both significant GWAS signal and high prior. In some cases, however, SNPs that were added to the credible set in each locus do not exhibit significant GWAS p-values (Supplementary Tables S2 and S6). This generally occurs when the genetic signals in those loci are weak relative to the SNPs in other loci for the same trait, and the model functional prior eventually dominates the SNP prioritization. Thus, we recommend considering these variants cautiously when designing downstream experiments.
One important assumption of our model is that there is one causal variant per locus, which is reflected by the normalization of variants within each locus so that they sum to 1 in order to obtain PPA and construct 95% credible sets (23). When this assumption holds, the posterior probabilities are well calibrated (Supplementary Figure S2). However, as demonstrated in our simulation, when this assumption is violated, the PPA is not well calibrated (Supplementary Figures S2 and S3). Other existing method such as PAINTOR (18) and CAVIAR (48) employ multivariate normal distribution to model all of the variants within a locus using LD reference panel estimated from 1000 Genome data as the covariance matrix, which allows inferring more than one causal variants per locus. While CAVIER used only summary statistics, PAINTOR is able to employ functional annotations to aid fine-mapping. Both methods require computing the likelihood density across a combinatorial set of causal configurations and therefore still needs to assume at most an arbitrarily small number of causal variants, typically below 10 causal SNPs per locus.
As future works, we will explore potential ways to enable efficient inference of more than one causal variants per locus. Furthermore, we will also explore the potential gain of incorporating trans-ethnic data, which was effectively demonstrated by the trans-ethnic version of the PAINTOR model (49). Moreover, in addition to modeling the epigenomic correlation between traits, variant prioritization may further benefit by jointly inferring the comorbidity at the individual SNP level (19), gene level (50), and/or pathway level (17). Together, we believe that RiVIERA will serve as a valuable tool complementary to the existing methods in identifying novel risk variants through tissue-specific epigenome-aware fine-mapping as well as aiding the selection of the appropriate cell types and epigenomic marks for more focused investigations of the disruptions of chromatin states by the disease-specific causal variants.
Supplementary Material
Acknowledgments
We thank Yongjin Park, Gerald Quon, Abhishek Sarkar, and Zhizhuo Zhang for the helpful discussions.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institutes of Health (NIH) [R01-HG004037, RC1-HG005334, R01-HG008155]. Funding for open access charge: NIH [R01 HG004037].
Conflict of interest statement. None declared.
REFERENCES
- 1.Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., et al. Genome-wide association study of 14,000 cases of seven common diseases and 3,000 shared controls. Nature. 2007;447:661–678. doi: 10.1038/nature05911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wray N.R., Goddard M.E., Visscher P.M. Prediction of individual genetic risk of complex disease. Curr. Opin. Genet. Dev. 2008;18:257–263. doi: 10.1016/j.gde.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 3.Hindorff L.A., Sethupathy P., Junkins H.A., Ramos E.M., Mehta J.P., Collins F.S., Manolio T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. U.S.A. 2009;106:9362–9367. doi: 10.1073/pnas.0903103106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Visscher P.M., Brown M.A., McCarthy M.I., Yang J. Five years of GWAS discovery. Am. J. Hum. Genet. 2012;90:7–24. doi: 10.1016/j.ajhg.2011.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Welter D., MacArthur J., Morales J., Burdett T., Hall P., Junkins H., Klemm A., Flicek P., Manolio T., Hindorff L., et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014;42:D1001–D1006. doi: 10.1093/nar/gkt1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., et al. Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 2011;473:43–49. doi: 10.1038/nature09906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Consortium R.E., Kundaje A., Meuleman W., Ernst J., Bilenky M., Yen A., Heravi-Moussavi A., Kheradpour P., Zhang Z., Wang J., et al. Integrative analysis of 111 reference human epigenomes. Nature. 2015;518:317–330. doi: 10.1038/nature14248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ward L.D., Kellis M. Interpreting noncoding genetic variation in complex traits and human disease. Nat. Biotechnol. 2012;30:1095–1106. doi: 10.1038/nbt.2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trynka G., Sandor C., Han B., Xu H., Stranger B.E., Liu X.S., Raychaudhuri S. Chromatin marks identify critical cell types for fine mapping complex trait variants. Nat. Genet. 2013;45:124–130. doi: 10.1038/ng.2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurano M.T., Humbert R., Rynes E., Thurman R.E., Haugen E., Wang H., Reynolds A.P., Sandstrom R., Qu H., Brody J., et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337:1190–1195. doi: 10.1126/science.1222794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hnisz D., Abraham B.J., Lee T.I., Lau A., Saint-André V., Sigova A.A., Hoke H.A., Young R.A. Super-enhancers in the control of cell identity and disease. Cell. 2013;155:934–947. doi: 10.1016/j.cell.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lindblad-Toh K., Garber M., Zuk O., Lin M.F., Parker B.J., Washietl S., Kheradpour P., Ernst J., Jordan G., Mauceli E., et al. A high-resolution map of human evolutionary constraint using 29 mammals. Nature. 2011;478:476–482. doi: 10.1038/nature10530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schaub M.A., Boyle A.P., Kundaje A., Batzoglou S., Snyder M. Linking disease associations with regulatory information in the human genome. Genome Res. 2012;22:1748–1759. doi: 10.1101/gr.136127.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., et al. Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 2007;39:1329–1337. doi: 10.1038/ng.2007.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Onengut-Gumuscu S., Chen W.-M., Burren O., Cooper N.J., Quinlan A.R., Mychaleckyj J.C., Farber E., Bonnie J.K., Szpak M., Schofield E., et al. Fine mapping of type 1 diabetes susceptibility loci and evidence for colocalization of causal variants with lymphoid gene enhancers. Nat. Genet. 2015;47:381–386. doi: 10.1038/ng.3245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cross-Disorder Group of the Psychiatric Genomics Consortium. Identification of risk loci with shared effects on five major psychiatric disorders: a genome-wide analysis. Lancet. 2013;381:1371–1379. doi: 10.1016/S0140-6736(12)62129-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.O'Dushlaine C., Rossin L., Lee P.H., Duncan L., Parikshak N.N., Newhouse S., Ripke S., Neale B.M., Purcell S.M., Posthuma D., et al. Psychiatric genome-wide association study analyses implicate neuronal, immune and histone pathways. Nat. Neurosci. 2015;18:199–209. doi: 10.1038/nn.3922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kichaev G., Yang W.-Y., Lindstrom S., Hormozdiari F., Eskin E., Price A.L., Kraft P., Pasaniuc B. Integrating functional data to prioritize causal variants in statistical fine-mapping studies. PLoS Genet. 2014;10:e1004722. doi: 10.1371/journal.pgen.1004722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chung D., Yang C., Li C., Gelernter J., Zhao H. GPA: a statistical approach to prioritizing GWAS results by integrating pleiotropy and annotation. PLoS Genet. 2014;10:e1004787. doi: 10.1371/journal.pgen.1004787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Duane S., Kennedy A.D., Pendleton B.J., Roweth D. Hybrid monte carlo. Phys. Lett. B. 1987;195:216–222. [Google Scholar]
- 21.Ripke S., Neale B.M., Corvin A., Walters J. T.R., Farh K.-H., Holmans P.A., Lee P., Bulik-Sullivan B., Collier D.A., Huang H., et al. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511:421–427. doi: 10.1038/nature13595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kwan J.S., Li M.-X., Deng J.-E., Sham P.C. FAPI: Fast and Accurate P-value Imputation for genome-wide association study. Eur. J. Hum. Genet. 2015;24:761–766. doi: 10.1038/ejhg.2015.190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pickrell J.K. Joint analysis of functional genomic dataand genome-wide association studies of 18 human traits. Am. J. Hum. Genet. 2014;94:559–573. doi: 10.1016/j.ajhg.2014.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ferrari S., Cribari-Neto F. Beta regression for modelling rates and proportions. J. Appl. Stat. 2004;31:799–815. [Google Scholar]
- 25.Bayes C.L., Bazán J.L., García C. A new robust regression model for proportions. Bayesian Anal. 2012;7:841–866. [Google Scholar]
- 26.Stephens M., Balding D.J. Bayesian statistical methods for genetic association studies. Nat. Rev. Genet. 2009;10:681–690. doi: 10.1038/nrg2615. [DOI] [PubMed] [Google Scholar]
- 27.Geman S., Geman D. Stochastic relaxation, Gibbs distributions, and the Bayesian restoration of images. Pattern Anal. Mach. Intell. IEEE Trans. 1984;6:721–741. doi: 10.1109/tpami.1984.4767596. [DOI] [PubMed] [Google Scholar]
- 28.Bernardo J.M., Smith A.F. Bayesian Theory. Vol. 405. Chichester: John Wiley & Sons; 2009. [Google Scholar]
- 29.Neal R.M. MCMC using Hamiltonian dynamics. Handbook of Markov Chain Monte Carlo. 2011;2 http://www.mcmchandbook.net/HandbookChapter5.pdf [Google Scholar]
- 30.Su Z., Marchini J., Donnelly P. HAPGEN2: simulation of multiple disease SNPs. Bioinformatics. 2011;27:2304–2305. doi: 10.1093/bioinformatics/btr341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Durinck S., Spellman P.T., Birney E., Huber W. Mapping identifiers for the integration of genomic datasets with the R/Bioconductor package biomaRt. Nat. Protoc. 2009;4:1184–1191. doi: 10.1038/nprot.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lawrence M., Gentleman R., Carey V. rtracklayer: an R package for interfacing with genome browsers. Bioinformatics. 2009;25:1841–1842. doi: 10.1093/bioinformatics/btp328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Stat. Soc. Ser. B (Methodological) 1995;57:289–300. [Google Scholar]
- 34.Eddelbuettel D., François R., Allaire J., Chambers J., Bates D., Ushey K. Rcpp: seamless R and C++ integration. J. Stat. Softw. 2011;40:1–18. [Google Scholar]
- 35.Koller D., Friedman N. Probabilistic Graphical Models: Principles and Techniques. Cambridge: MIT press; 2009. [Google Scholar]
- 36.Ardlie K.G., Deluca D.S., Segre A.V., Sullivan T.J., Young T.R., Gelfand E.T., Trowbridge C.A., Maller J.B., Tukiainen T., Lek M., et al. The genotype-tissue expression (GTEx) pilot analysis: Multitissue gene regulation in humans. Science. 2015;348:648–660. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kheradpour P., Kellis M. Systematic discovery and characterization of regulatory motifs in ENCODE TF binding experiments. Nucleic Acids Res. 2014;42:2976–2987. doi: 10.1093/nar/gkt1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neph S., Vierstra J., Stergachis A.B., Reynolds A.P., Haugen E., Vernot B., Thurman R.E., John S., Sandstrom R., Johnson A.K., et al. An expansive human regulatory lexicon encoded in transcription factor footprints. Nature. 2012;489:83–90. doi: 10.1038/nature11212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Genetic Analysis of Psoriasis Consortium & the Wellcome Trust Case Control Consortium 2. A genome-wide association study identifies new psoriasis susceptibility loci and an interaction between HLA-C and ERAP1. Nat. Genet. 2010;42:985–990. doi: 10.1038/ng.694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tsoi L.C., Spain S.L., Knight J., Ellinghaus E., Stuart P.E., Capon F., Ding J., Li Y., Tejasvi T., Gudjonsson J.E., et al. Identification of 15 new psoriasis susceptibility loci highlights the role of innate immunity. Nat. Genet. 2012;44:1341–1348. doi: 10.1038/ng.2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ritchie M.D., Holzinger E.R., Li R., Pendergrass S.A., Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015;16:85–97. doi: 10.1038/nrg3868. [DOI] [PubMed] [Google Scholar]
- 42.Maller J.B., McVean G., Byrnes J., Vukcevic D., Palin K., Su Z., Howson J.M., Auton A., Myers S., Morris A., et al. Bayesian refinement of association signals for 14 loci in 3 common diseases. Nat. Genet. 2012;44:1294–1301. doi: 10.1038/ng.2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wen X., Luca F., Pique-Regi R. Cross-population joint analysis of eQTLs: fine mapping and functional annotation. PLoS Genet. 2015;11:e1005176. doi: 10.1371/journal.pgen.1005176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wallace C., Cutler A.J., Pontikos N., Pekalski M.L., Burren O.S., Cooper J.D., Garcia A.R., Ferreira R.C., Guo H., Walker N.M., et al. Dissection of a complex disease susceptibility region using a Bayesian stochastic search approach to fine mapping. PLoS Genet. 2015;11:e1005272. doi: 10.1371/journal.pgen.1005272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.The 1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farh K.K.-H., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R. J.H., Shishkin A.A., et al. Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 2015;518:337–343. doi: 10.1038/nature13835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lu Q., Powles R.L., Wang Q., He B.J., Zhao H. Integrative tissue-specific functional annotations in the human genome provide novel insights on many complex traits and improve signal prioritization in genome wide association studies. PLoS Genet. 2016;12:e1005947. doi: 10.1371/journal.pgen.1005947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chen W., Larrabee B.R., Ovsyannikova I.G., Kennedy R.B., Haralambieva I.H., Poland G.A., Schaid D.J. Fine Mapping Causal Variants with an Approximate Bayesian Method Using Marginal Test Statistics. Genetics. 2015;200:719–736. doi: 10.1534/genetics.115.176107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kichaev G., Pasaniuc B. Leveraging functional-annotation data in trans-ethnic fine-mapping studies. Am. J. Hum. Genet. 2015;97:260–271. doi: 10.1016/j.ajhg.2015.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hormozdiari F., Kichaev G., Yang W.-Y., Pasaniuc B., Eskin E. Identification of causal genes for complex traits. Bioinformatics. 2015;31:i206–i213. doi: 10.1093/bioinformatics/btv240. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.