


Abstract
The accumulation of somatic mitochondrial DNA (mtDNA) mutations contributes to the pathogenesis of human disease. Currently, mitochondrial mutations are largely considered results of inaccurate processing of its heavily damaged genome. However, mainly from a lack of methods to monitor mtDNA mutations with sufficient sensitivity and accuracy, a link between mtDNA damage and mutation has not been established. To test the hypothesis that mtDNA-damaging agents induce mtDNA mutations, we exposed MutaTMMouse mice to benzo[a]pyrene (B[a]P) or N-ethyl-N-nitrosourea (ENU), daily for 28 consecutive days, and quantified mtDNA point and deletion mutations in bone marrow and liver using our newly developed Digital Random Mutation Capture (dRMC) and Digital Deletion Detection (3D) assays. Surprisingly, our results demonstrate mutagen treatment did not increase mitochondrial point or deletion mutation frequencies, despite evidence both compounds increase nuclear DNA mutations and demonstrated B[a]P adduct formation in mtDNA. These findings contradict models of mtDNA mutagenesis that assert the elevated rate of mtDNA mutation stems from damage sensitivity and abridged repair capacity. Rather, our results demonstrate induced mtDNA damage does not readily convert into mutation. These findings suggest robust mitochondrial damage responses repress induced mutations after mutagen exposure.
INTRODUCTION
Maternally inherited mutations in the mitochondrial genome cause a diverse array of disorders, all of which are associated with defects in oxidative energy metabolism (1). Furthermore, emerging evidence implicates the accumulation of somatic mutations in mitochondrial DNA (mtDNA) as drivers of other complex traits, including neurodegenerative diseases, pathologies of aging and cancer (2–8). Yet, the mechanisms by which these mutations arise and contribute to the etiology of disease are poorly defined.
mtDNA readily reacts with exogenous chemicals (9–12), exhibiting lesion frequencies that are many hundreds of fold higher than those in nuclear DNA (nDNA) in the same cells following exposure. Thus, damage to mtDNA may underlie the vast majority of pathogenic mitochondrial mutations. Furthermore, the disparate induction of DNA damage that results between the nuclear and mitochondrial genomes may be due to the fact that many genotoxic substances preferentially concentrate within mitochondria (9,13). Other contributing factors that have been implicated in the magnitude of mtDNA damage include: a disputed protective function of mtDNA-packaging proteins (14–16), the proximity of mtDNA to reactive oxygen species (ROS) produced during oxidative phosphorylation, and the inherent susceptibility of mtDNA to adduct formation with genotoxic agents (11). As such, it is commonly theorized that mtDNA's inherent susceptibility to induced damage underlies its high rate of mutation, which is two to three orders of magnitude greater than nDNA (17–22). However, to our knowledge, this premise, i.e. that mtDNA is more susceptible to induced mutation, has never been tested.
Direct evidence linking lesion burden to mtDNA mutation remains scarce or contradictory (23–27), as previously existing assays have lacked sufficient accuracy and sensitivity to quantify de novo mtDNA mutations (28). Earlier mtDNA mutation detection technologies inherently suffer from assay-induced errors mediated by polymerase infidelity on damaged templates and by cloning artifacts (28,29). As such, we sought to improve upon these shortcomings by developing the Digital Random Mutation Capture (dRMC), a novel adaptation of the RMC assay (30) and Digital Deletion Detection (3D) (31) to track the accumulation of point and deletion mutations in mtDNA, respectively. In these approaches, enrichment for mutant mtDNA with restriction endonucleases precedes single molecule amplification, effectively eliminating issues with polymerase fidelity (30–32). dRMC and 3D couple the accuracy and increased throughput of droplet digital PCR (ddPCR) with the reporting specificity of molecular probe-based Taqman™ chemistry for accurate quantification of mtDNA mutation frequency at unprecedented sensitivity (30–32). With dRMC and 3D, it is possible to evaluate not only single point mutations, but also large deletions. To test the hypothesis that the high mutation rate of mtDNA stems from its well-documented sensitivity to DNA damage, we exploited the enhanced sensitivity of these assays to investigate the consequences of mutagen exposure on mtDNA mutagenesis in vivo, and the results are interpreted in the context of nuclear DNA mutation frequency in the same tissue samples.
MATERIALS AND METHODS
Animal treatment
Twenty to twenty-four week-old MutaTMMouse males were dosed via oral gavage in a single exposure or daily for 28 days with B[a]P dissolved in olive oil (75 mg/kg body weight in the single-exposure trial, 25, 50 or 75 mg/kg body weight/day in the subchronic trial). Fourteen to sixteen week-old males were dosed for 28 days with ENU dissolved in water (5 mg/kg body weight/day). B[a]P and ENU were obtained from Sigma-Aldrich Canada (Oakville, ON, USA). Each dose group, including vehicle control, contained four animals for the single-exposure study and five animals for the subchronic exposure study. Mice in the single-exposure study were anesthetized with isofluorane prior to cervical dislocation 24 h following B[a]P exposure. Tissues, including liver and bone marrow, were isolated, flash-frozen in liquid nitrogen and stored at −80°C until use. In the 28-day studies, mice were euthanized three days after the final treatment in the same manner as the single-exposure study, and tissues were obtained as described above. Mice were maintained under conditions approved by the Health Canada Ottawa Animal Care Committee. Food and water were available ad libitum for the duration of the experiment. The lacZ mutant frequencies in animals from the 28-day exposure to B[a]P have been described in Lemieux et al. 2011, however, here we present the lacZ data only from those mice where we conducted mtDNA analysis (those that had sufficient sample DNA quantity or quality).
Genomic DNA isolation
Bone marrow
Bone marrow cells were isolated and lysed according to previously published methods (33,34). Briefly, to collect bone marrow, femurs were flushed with PBS (Invitrogen Canada, Burlington, ON, USA), the solution was briefly centrifuged and the pellet was stored at –80°C. Cells were homogenized in 5 ml lysis buffer (1 mM Na2EDTA, 100 mM NaCl, 20 mM Tris-HCl, pH 7.4), supplemented with 1% SDS (w/v) and proteinase K (1 mg/ml, Invitrogen Canada, Burlington, ON, USA). The lysate was then incubated at 37°C overnight with gentle shaking. Genomic DNA was isolated the following day, using the phenol/chloroform extraction procedure described previously (35,36). Isolated DNA was dissolved in 50–100 μl TE buffer (10 mM Tris pH 7.6, 1 mM EDTA) and stored at 4°C until use. DNA was quantified using a NanoPhotometerTM (Implen, Westlake Village, CA, USA).
Liver
Liver tissue was thawed and homogenized on ice using a motor-driven conical tissue homogenizer in 5 ml TMST buffer (50 mM Tris pH 7.6, 3 mM magnesium acetate, 250 mM sucrose, 0.2% (v/v) Triton X-100). The liver homogenate was centrifuged for 6 min at 800 × g (4°C), the supernatant was discarded and the pellet was washed twice more with TMST buffer as before. The pellet was suspended in 5 ml lysis buffer (10 mM Tris pH 7.6, 10 mM EDTA, 150 mM NaCl, 1% (w/v) SDS and 1 mg/ml proteinase K (≥20 Units/mg). This suspension was incubated overnight at 37°C with gentle shaking. DNA was isolated and stored as described above.
TaqMan probe and primer design
The following primer/probe sets were used with murine total DNA for mtDNA mutation detection (designed using assembly GCA_000001635.6 for mus musculus). Control site: 5′- GAC ACA AAC TAA AAA GCT CA -3′ (forward primer), 5′- TAA GTG TCC TGC AGT AAT GT -3′ (reverse primer) and 5′-6FAM- CCA ATG GCA TTA GCA GTC CGG C -BHQ-1-3′ (probe). ND5 site: 5′- CCC ACT GTA CAC CAC CAC ATC AA -3′ (forward primer), 5′- TGT TGG CTG AGG TGA GGA TAA GCA -3′ (reverse primer) and 5′-6FAM- AAC CTG GCA CTG AGT CAC C -MGB-NFQ-3′ (probe). 12S rRNA site: 5′- GAC AGC TAA GAC CCA AAC TGG GAT -3′ (forward primer), 5′- CAT TGG CTA CAC CTT GAC CTA ACG -3′ (reverse primer) and 5′-6FAM- ACC GCC ATC TTC AGC A -MGB-NFQ-3′ (probe). Common deletion site: 5′- AGG CCA CCA CAC TCC TAT TG -3′ (forward primer), 5′- AAT GCT AGG CGT TTG ATT GG -3′ (reverse primer) and 5′-6FAM- AAG GAC TAC GAT ATG GTA TAA -MGB-NFQ-3′ (probe). RNaseP site for nuclear DNA quantification: 5′-GTG CTG CAG AAA GGG TAA GC-3′ (forward primer), 5′-CCA TCG GCA AAC AGT TAC AA-3′ (reverse primer) and 5′-VIC-TGG AAT ACT TTG TCC CAG CA-MGB-NFQ-3′ (probe).
For lacZ mutation detection, primers were designed to the reference sequence GenBank:V00296.1. Control site: 5′- TAC GAT GCG CCC ATC TAC AC -3′ (forward primer), 5′-CAA ATT CAG ACG GCA AAC GA -3′ (reverse primer) and 5′-6FAM-CCT TCC TGT AGC CAG CTT TCA T-MGB-NFQ-3′(probe); TaqI site: 5′-TAC GCG TAG TGC AAC CGA AC -3′ (forward primer), 5′-AAG CCT GAC TGG CGG TTA AA-3′ (reverse primer) and 5′-6FAM-TGC AAA AAT CCA TTT CGC TGG T-MGB-NFQ -3′(probe).
For mitochondrial:nuclear copy number ratio analysis, primers were designed using the assembly GCA_000001635.6 for mus musculus, as above, with mitochondrial copies quantified using the control primer set and nuclear DNA copies quantified using primers directed toward murine RPP30 gene: 5′-GTG CTG CAG AAA GGG TAA GC-3′ (forward primer), 5′-CCA TCG GCA AAC AGT TAC AA-3′ (reverse primer) and 5′-VIC-TGG AAT ACT TTG TCC CAG CA-MGB-NFQ-3′ (probe).
Primers were designed using Primer3 specifications to limit off-target amplification within the murine genome (especially within nuclear mitochondrial segments, NUMTs, which are transpositions of mtDNA into the nucleus), and their specificity was confirmed using UCSC in silico PCR (http://rohsdb.cmb.usc.edu/GBshape/cgi-bin/hgPcr) and NCBI primer-BLAST (37). Additionally, melt peak analysis was performed in real-time PCR experiments along with agarose gel separation of PCR products to ensure single product amplification.
Mitochondrial DNA mutation detection
To measure point mutations in mouse mtDNA, we adapted the Random Mutation Capture (RMC) assay for the droplet digital PCR (ddPCR) platform, as detailed in the subsections below. Deletions were quantified in mtDNA extracted from mouse tissues using the Digital Deletion Detection (3D) method described previously by Taylor et al. (31).
TaqI digest
Rare mutation-bearing molecules were selectively enriched through endonucleolytic destruction of wild-type target sites. First, a 100 μl digestion reaction mixture was prepared containing 1 μg of genomic DNA, 1 μl (100 U) of TaqI (New England Biolabs, Ipswich, MA, USA) and TaqI reaction buffer (Fermentas, Vilnius, Lithuania). The reaction mixture was incubated at 65°C for 10 h, with an additional 100 U of TaqI added to each reaction every hour. After each TaqI addition, samples were thoroughly mixed and briefly centrifuged to ensure efficient digestion. Prior to ddPCR, complete cleavage of wild-type TaqI sites was verified by PCR amplification of the target regions followed by post-PCR restriction digest and agarose gel electrophoresis.
Droplet digital PCR (ddPCR)
The final concentration of digested DNA was adjusted to yield less than ∼3500 positive molecules per μl, which is within the range of linearity for the Poisson calculation (38). Reaction mixtures (25 μl) contained ddPCR Master Mix (Bio-Rad, Hercules, CA, USA), 250 nM TaqMan probe, 900 nM of each appropriate flanking primer and 0–100 ng of TaqI-digested DNA. Reaction droplets were made by applying 20 μl of each reaction mixture to a droplet generator DG8 cartridge (Bio-Rad) for use in the QX100 Droplet Generator (Bio-Rad). Following droplet generation, 38 μl of the droplet emulsion was carefully transferred to a Twin.tec semi-skirted 96-well PCR plate (Eppendorf, Hamburg, Germany), which was then heat-sealed with a pierceable foil sheet. To amplify the fragments, thermal cycling was carried out using the following protocol: initial denaturation step at 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 58°C for 1 min. The thermally cycled droplets were analyzed by flow cytometry in a QX100TM Droplet DigitalTM Reader (Bio-Rad) for fluorescence analysis and quantification of mutation frequencies.
Analysis of fluorescence amplitude and quantification
Following normal thermal cycling, droplets were individually scanned using the QX100™ Droplet Digital™ PCR system (Bio-Rad). Positive (mutation-bearing) and negative droplets were distinguished on the basis of fluorescence amplitude using a global threshold. The number of mutant genomes per droplet was calculated automatically by the accompanying software (QuantaSoft, Bio-Rad) using Poisson statistics as described elsewhere (39). Quantification of point mutation frequency requires ddPCR amplification using two primer sets. The first primer set flanks the test region and measures the concentration of mutation-bearing molecules. The second primer set flanks a region in the mitochondrial genome that bears no restriction recognition sites. This control set measures the concentration of all mtDNA genomes. Because de novo point mutations are so rare, reactions using the different primer sets must be run using different dilutions of the digested DNA, and the results are normalized against undiluted concentrations during downstream calculations. Mutation frequency per base pair is calculated by taking the ratio of the normalized concentrations of mutation-bearing mtDNA molecules to the total mtDNA molecules screened, divided by the number of bases per target site. Reactions that yielded <5 positive droplets per well were scored conservatively as having no positives above background, though in rare cases technical replicates were pooled to achieve higher droplet counts (38). At least two technical repeats were performed per biological sample, and the average result presented.
Nuclear mutation assessment with dRMC
The methodology for quantifying nDNA mutations follows that for mtDNA mutations (as above) with a few changes: input DNA was scaled to 10 μg per mouse sample; 20 1-hr rounds of TaqI digestion were required for this amount of DNA; and ddPCR cycling parameters consisted of an initial denaturation step at 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 60°C for 1 min.
Mitochondrial:nuclear DNA copy number ratio
ddPCR reactions were performed with TaqI-digested genomic DNA as above, using the mitochondrial control primer/probe set (within the ATP6 gene) for mtDNA copy quantification, and the RNAseP (RPP30) primer/probe set for nDNA copy quantification (both detailed above), with an initial denaturation step at 95°C for 10 min, followed by 40 cycles of 94°C for 30 s and 60°C for 1 min. After droplet processing, mtDNA:nDNA copy number ratio was calculated by dividing the concentration of mtDNA molecules detected by the nDNA molecules determined by ddPCR.
Quantitative long-range real-time PCR
The induced lesion frequency of B[a]P in the single-exposure mice was quantified using long-range quantitative real-time PCR amplification, as described previously (40). One microgram of each DNA sample for mitochondrial amplification was digested with NotI prior to amplification, as this was seen to increase amplification efficiency (41). No NotI sites are found in the target amplicons. Primers for mouse mtDNA short (mtDNA control primers above) and long (forward 5′-CCA CCG CGG TCA TAC GAT TA-3′, reverse 5′-CGA TGT CTC CGA TGC GGT TA-3′ 11.9 kb) amplicons and mouse nDNA short (nDNA control primers) and long (forward 5′- CGC CGC CTT GCC CTC GTC T-3′, reverse 5′-AGC TCC GCA AAT TCG CCT ACA C-3′ 12.5 kb, localized to the lambda transgene) amplicons were used to amplify DNA isolated from mouse liver and bone marrow in reaction mixtures of 0.05 U/μl JumpStartTM AccuTaqTM polymerase (Sigma), JumpStartTM AccuTaqTM polymerase buffer (working concentrations of 50 mM Tris-Hcl, 15 mM ammonium sulphate, pH 9.3, adjusted with NH4OH, 2.5 mM MgCl2 and 1% Tween-20), 500 μM dNTP mix (Promega) and 1 µl of a 1:8 dilution of EvaGreen® in water. Full-strength EvaGreen® inhibited long-range product formation. Samples containing 50–200 ng of DNA were amplified in triplicate using a C1000 series CF96® Real-Time PCR Detection System (Bio-Rad) with detection in the SYBR® Green fluorescence channel. Cycling parameters for the 12 kb mtDNA and nDNA fragments were as follows: initial denaturation at 96°C for 30 s, followed by 40 cycles of denaturation at 94°C for 15 s and extension at 65°C for 16 min. A final extension step of 30 min at 65°C was performed after cycling, prior to a melting curve analysis to verify correct product amplification—a ramp from 58°C to 96°C where fluorescence amplitude was measured at 0.5°C intervals—with subsequent sample cooling to 4°C. Short-range amplicons were amplified as above, though with extension periods of 1 min during cycling and a 3 min final extension, at 58°C and 62°C for mtDNA and nDNA primer sets, respectively. Primers were designed with the aid of NCBI's primer-BLAST (37) and UCSC In-Silico PCR (http://rohsdb.cmb.usc.edu/GBshape/cgi-bin/hgPcr), as with the primer/probes used in ddPCR. Standard curves and melt temperature analysis are included in Supplementary Figures S7–S10.
Determination of induced lesion frequency
Lesion frequency was calculated using methods similar to those previously described (40,41). Amplification products were quantified from the EvaGreen® fluorescence amplitude, with regression analysis (CFX Manager, Bio-Rad) and a standard curve for each sample set, which was performed simultaneously with sample amplification using the same prepared reagent mix along with no template controls. Assuming a random distribution of lesions within the amplicon, the Poisson equation [f(x) = e−λ λx/x!] for undamaged templates is f(0) = e− λ. The average lesion frequency for each amplicon (λ) is calculated by dividing the normalized (long-range over short-range ratio of input copies determined by standard curve) amplification in DNA extracted from treated mice by the same in DNA from untreated mice (thus lesion frequency is equivalent to 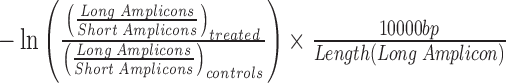 . Values reported reflect the average of individual samples assayed in triplicate, and then pooled for lesion frequency (n = 4–5 for each group).
. Values reported reflect the average of individual samples assayed in triplicate, and then pooled for lesion frequency (n = 4–5 for each group).
lacZ mutation evaluation
The frequency of lacZ transgene mutants in genomic DNA isolated from liver and bone marrow was assessed using the phenyl-β-D-galactopyranoside (P-Gal) positive selection assay as previously described. λgt10lacZ DNA was rescued from genomic DNA using the TranspackTM lambda packaging system (Agilent, Mississauga, ON, USA). Packaged phage particles were then mixed with the host bacterium (Escherichia coli lacZ−, galE−, recA−, pAA119 with galT and galK), plated on minimal medium containing 0.3% (w/v) P-Gal and incubated overnight at 37°C. Total plaque-forming units (pfu) were measured on concurrent titer plates that did not contain P-Gal. Mutant frequency is expressed as the ratio of mutant pfu to total pfu.
Statistical analysis
All reported results represent at least three biological replicates with 2 technical replicates. Error reported represents s.e.m. Concentration- and dose-response data were analyzed using both non-parametric (e.g. chi-square and Fisher's exact tests) and parametric methods (e.g. analysis of variance, ANOVA; Welch's t-test). Non-parametric methods are frequently employed to analyze experimental data sets where the response variable is a discrete dichotomous outcome (i.e. mutant versus normal). The non-parametric analyses of lacZ mutant frequency employed 2 × 2 or 2 × 4 contingency tables with chi-square and Fisher's exact tests to assess the degree of association between treatment and response at each dose/concentration. In addition, a one-way Cochran-Armitage test was employed to determine whether increasing doses or concentrations yielded an increasing likelihood of recovering mutant copies of lacZ. The parametric methods analyzed frequency values (mutant lacZ per 105 plaque forming units, mtDNA mutation frequencies and mtDNA damage), and employed one-way ANOVA (B[a]P treatment samples, 4 treatment groups) or unpaired t-test with Welch's correction (for the two ENU treatment groups) to investigate the relationship between dose/concentration and mutant frequency (MF). Mutation frequencies in mtDNA and lesion frequencies in mtDNA and nDNA were Poisson transformed during analysis from droplet and RT-PCR data, respectively, and thus are not compatible with chi-square tests. For each one-way ANOVA, a one-tailed, post-hoc Dunnett's test was employed to subsequently compare responses at individual doses/concentrations to matched controls. Values of P < 0.05 were considered statistically significant.
RESULTS
We used benzo[a]pyrene (B[a]P) to investigate the relationship between induced mtDNA damage and mutation. B[a]P is an established mutagen that has been shown to induce 40- to 90-fold more lesions in mtDNA than in nDNA (9,19). B[a]P is present in a wide range of combustion products, including tobacco smoke, coal tar and vehicular exhaust (13), and requires metabolic activation by cytochrome P450 isozymes, followed by epoxide hydrolase, to form mutagenic metabolites (e.g. benzo(a)pyrene-7,8-diol-9,10-epoxide, BPDE), which form bulky helix-distorting lesions by covalently modifying DNA.
To maximize the likelihood of mtDNA damage, we implemented a 28-day sub-chronic dosing regimen, which included three concentrations of B[a]P (25, 50 or 75 mg/kg body weight/day). As B[a]P requires metabolic conversion to a DNA-reactive substance (i.e. BPDE), we elected to study mtDNA mutagenesis in the liver, a well-known site of this activation (42–44). Additionally, to assess the impact of mutagen treatment in a highly proliferative tissue, bone marrow was also included in our study. Both tissues have been shown to be exceptionally sensitive to DNA-damaging agents in nuclear and mitochondrial DNA, including B[a]P-induced damage. Moreover, previous work has shown that B[a]P significantly increases mutations in nuclear DNA, at similar doses, and in the same tissues tested as in the present study (34). However, the potential effect of B[a]P exposure on the induction of mtDNA mutations in vivo has not been examined.
To this end, we extracted DNA from liver and bone marrow cells to explore the possible effect of B[a]P treatment on mtDNA mutagenesis using the dRMC assay that builds upon the RMC methodology, and 3D assay (30,31). The dRMC and 3D assays (Figure 1) have been used to quantify point mutations and deletion mutations in both humans and mice previously (30,45,46)(Supplementary Figure S1).
Figure 1.

Illustrated overview of the 3D and dRMC assays for the quantification of mitochondrial mutations. (1) Whole cell DNA is extracted. (2) mtDNA is incubated with TaqI restriction endonuclease, which recognizes 5′-TCGA-3′ sites. mtDNA that are wild-type at TaqI sites (WT, blue), will be cleaved, whereas mtDNA with a mutation in the mutation target site (red) will be resistant to cleavage. A control region devoid of TaqI site(s) (purple) is used to quantify total mtDNA copies interrogated. (3) Digested DNA is added to a PCR mastermix with site-specific primers which flank the mutational target and Taqman probes, and then partitioned into thousands of 1 nl droplets in an oil immersion. The control region and mtDNA with mutations in the target site act as substrates for amplification, whereas mtDNA which are WT at the mutational target are not. (4) Droplets are thermal cycled to amplify target DNA as well as release the Taqman probe fluorophore from its quencher through Taq polymerase's inherent exonuclease activity. The ongoing rounds of amplification displace and cleave more probe, accumulating fluorescence. (5) Post-amplification, droplets are detected and their fluorescence is quantified. Mutation frequency is calculated by dividing the mutant concentration by the concentration of the control region.
Effect of B[a]P exposure on the frequency of mtDNA point mutation
Whole-cell DNA was extracted from frozen bone marrow and liver for mtDNA mutation analysis from B[a]P-exposed and control mice after 28 days of daily treatment and 3 post-exposure rest days. In bone marrow mtDNA isolated from B[a]P-treated mice, the mutation burdens ordered by increasing daily dose of B[a]P, were 3.8 ± 1.1, 4.2 ± 1.2, 3.8 ± 1.4 × 10−6 bp and 2.6 ± 0.5, 2.4 ± 0.6, 2.0 ± 1.0 × 10−6 bp (Figure 2A), for the 12S rRNA region and ND5 site, respectively. In untreated mice, bone marrow mtDNA mutation frequencies were 3.5 ± 0.7 × 10−6 bp and 1.7 ± 0.2 × 10−6 bp, at the 12S and ND5 sites, respectively. No significant increases, or dose-dependent changes (ANOVA, multiple-comparisons corrected t-test), were observed between control (Figure 2A) and treatment groups.
Figure 2.

B[a]P treatment does not increase the frequency of mitochondrial point mutations. Mice were treated daily with B[a]P or vehicle for 28 consecutive days and tissues collected three days later. After DNA extraction, mutation frequency per bp (± s.e.m.) was determined via dRMC within the 12S rRNA and ND5 genes in mouse mtDNA. B[a]P did not induce mutations in (A) bone marrow (P = 0.66, 12S rRNA locus; P = 0.21, ND5 locus; one-way ANOVA) or (B) liver isolates of mice treated (P = 0.98, 12S rRNA locus; P = 0.98, ND5 locus; one-way ANOVA).
mtDNA isolates from the liver of B[a]P-treated mice displayed mutation frequencies, ordered by increasing dose, of 2.3 ± 0.5, 2.4 ± 0.1, 2.5 ± 0.2 × 10−6 bp and 2.0 ± 0.8, 2.1 ± 0.9, and 2.0 ± 0.7 × 10−6 bp for 12S rRNA and ND5 sites (Figure 2B), respectively. The mutation frequency of untreated liver mtDNA at the 12S rRNA and ND5 sites were 2.6 ± 1.0 × 10−6 bp and 2.2 ± 1.00 × 10−6 bp. As with bone marrow, liver mtDNA mutation frequency was unaffected by B[a]P exposure. Thus, in both tissues, B[a]P exposure did not affect the frequency of point mutations in mtDNA.
Incidence of mtDNA deletions following mutagen exposure
The bulky adducts induced by B[a]P may underlie the lack of conversion into mtDNA point mutations, as the strand-distorting lesion produced by B[a]P strongly inhibits mitochondrial replication and thus lesion bypass (47). Polymerase stalling, however, has been hypothesized to cause deletion mutations (47,48). The observed lack of point mutation induction (Figure 2) in mtDNA may be predicated upon blocked replication instead of error-prone polymerase bypass or DNA repair processes. Thus, we speculated that this would lead to polymerase stalling at the sites of damaged bases and, potentially, induce the formation of large mtDNA deletions.
To examine this possibility, we employed the 3D assay (31) to quantify mtDNA deletions following exposure to B[a]P. 3D can evaluate the presence of deletions in the mitochondrial genome, such as the ‘common deletion’: a 3.8 kb region in mouse mtDNA that shows preferential excision and end-joining due to sequence microhomology (49,50). The frequency of mtDNA deletions in our control mice was 1.1 ± 0.4 and 111.4 ± 31.0 deletions per 107 genomes for bone marrow and liver, respectively. These values are complementary to those found in previous studies for similarly aged mice, where liver showed the highest frequency of deletions (49). Deletion frequencies in treated mice, ordered by increasing doses of B[a]P were: 1.1 ± 0.50, 0.9 ± 0.40 and 1.1 ± 0.2 copies per 107 genomes in bone marrow (Figure 3A); and, 71.4 ± 15.5, 47.6 ± 14.3 and 84.1 ± 33.2 deletions per 107 genomes, in liver (Figure 3B). As with point mutations, B[a]P exposure did not significantly change the frequency of deletions at any dose or in either tissue (ANOVA, multiple-comparisons adjusted t-test).
Figure 3.

B[a]P treatment does not induce mitochondrial deletions. Following 28 days of treatment with B[a]P no significant induction of deletion mutation frequency, (± s.e.m.) per mitochondrial genome was determined via 3D in (A) bone marrow (P = 0.94; one-way ANOVA) and (B) liver mtDNA(P = 0.37; one-way ANOVA).
B[a]P adducts in mtDNA and nDNA
Although the induction of mtDNA damage induced by B[a]P is extensively described, the unexpected lack of mutation induction in mtDNA following B[a]P exposure prompted us to address the possibility that damage was not induced in our test animals. To quantify the potential induction of B[a]P induced damage, we extracted DNA from bone marrow and liver tissues 24 h post-treatment with an acute dose of 75 mg B[a]P/kg body weight. As adducts formed by B[a]P inhibit polymerase extension, we quantified their presence via long-range quantitative PCR (13,51). This sensitive assay quantifies lesions that inhibit polymerase extension, and is not specific to one species of DNA adduct or lesion. B[a]P induced 0.29 ± 0.10 lesions per 10 kb (Figure 4A, P < 0.05, one-tailed Welch's t-test) and 0.26 ± 0.09 lesions per 10 kb (Figure 4B, P < 0.05, one-tailed Welch's t-test) in bone marrow and liver mtDNA, respectively. DNA samples were also processed for nDNA lesions using quantitative PCR directed to portions of the lacZ transgene. B[a]P induced 1.27 ± 0.40 lesions per 10 kb (Supplementary Figure S2A, P < 0.01, one-tailed Welch's t-test) and 0.66 ± 0.11 lesions per 10 kb (Supplementary Figure S2B, P < 0.05, one-tailed Welch's t-test) in bone marrow and liver nDNA, respectively. Thus, the lack of induced point and deletion mutations in the mitochondrial genome following 28 days of daily B[a]P exposures cannot be explained by the absence of damage induction. A single exposure of 75 mg B[a]P/kg body weight introduced 29 lesions per 106 bp in mtDNA with the potential to stall or inhibit polymerase extension. Yet despite the abundance of B[a]P-induced DNA lesions, no significant induction of mutation is observed in the mitochondrial genome following 28 days of daily B[a]P exposures.
Figure 4.

B[a]P treatment induces mtDNA adducts. The presence of lesions (± s.e.m.) in mouse bone marrow and liver mtDNA was enumerated by quantitative PCR. Mice were treated with a single, acute dose of B[a]P at either 0 or 75 mg/kg body weight. DNA was extracted from bone marrow and liver tissues 24 h after treatment. B[a]P induces significant adduct burden in each tissue mtDNA (* indicates P < 0.05; one-tailed Welch's t-test). (A) Lesions formed in bone marrow. (B) Lesions formed in liver.
Nuclear B[a]P-induced mutagenesis
Mutation and damage burdens in mtDNA are typically described with comparisons to nDNA. Therefore, we sought to place the observed mitochondrial resistance to mutation in the context of the nuclear genome. We had selected B[a]P as our test mutagen, as previous reports had demonstrated preferential B[a]P adduct formation in mtDNA compared to nDNA (9). As such, we had hypothesized the mitochondrial genome would be more sensitive to B[a]P-induced mutation than the nuclear genome. In our evaluation of induced nDNA mutation and damage, we utilized the MutaTMMouse transgenic rodent, which harbors a stably integrated lacZ transgene incorporated into a recoverable lambda phage shuttle vector. The shuttle vector can readily be recovered by packaging in phage particles that are subsequently used to infect galactose-sensitive bacteria (52,53). In the presence of P-gal, only those phages that receive a mutant lacZ can form plaques, allowing quantification of the mutant frequency in the nDNA (33,54). The mutant frequency in untreated animals was 4.3 ± 0.9 × 10−5 in bone marrow, and 6.3 ± 0.6 × 10−5 in liver. Contrary to our observations in the mitochondrial genome, B[a]P exposure resulted in a dose-dependent increase in nuclear mutant frequencies in both tissues, with 203 ± 35.8, 344 ± 75.0 and 679 ± 59.8 mutants × 10−5 in bone marrow and 26 ± 4.8, 96 ± 14.5 and 219.0 ± 59.8 mutants × 10−5 in liver samples (Figure 5A, P ≤ 0.0001 in bone marrow; Figure 5B, P ≤ 0.001 in liver, chi-square test). Additionally, we were able to confirm increased mutant frequencies using a nuclear version of the dRMC that quantifies mutations within the lacZ transgene. Mutation frequencies in untreated animals were 2.9 ± 1.7 × 10−6 bp in bone marrow and 1.6 ± 0.4 × 10−6 bp in liver tissues, whereas mutation frequency in mice exposed to 75 mg/kg body weight/day B[a]P was 35.2 ± 14.4 × 10−6 bp in bone marrow and 29.5 ± 8.3 × 10−6 bp in liver (Supplementary Figure S3, P ≤ 0.05 for bone marrow and liver, one-tailed Welch's-adjusted t-test). These results show a clear differential response between mtDNA and nDNA to B[a]P-induced mutagenesis.
Figure 5.

B[a]P treatment results in a dose-dependent increases in the frequency of nuclear DNA transgene (lacZ) mutants. After 28 days of daily treatment with B[a]P, DNA was extracted from mouse tissues 3 days post-exposure. Mutant frequency (± s.e.m.) in mouse nuclear DNA displayed significant, dose-dependent increases (**P < 0.01; ***P < 0.001; ****P < 0.0001; Welch's-adjusted t-test). (A) Nuclear mutant frequency induced in bone marrow isolates (χ2 = 4898, P ≤ 0.0001; Fisher's Exact, P ≤ 0.0001). (B) Nuclear mutant frequency induced in liver nuclear DNA (χ2 = 897.2, P ≤ 0.0001; Fisher's Exact, P ≤ 0.0001).
Evaluation of ENU as a mitochondrial DNA mutagen
To explore whether the resistance of mtDNA to mutagenesis is unique to chemicals that induce bulky adducts, we investigated the potential of N-ethyl-N-nitrosourea (ENU) to induce mtDNA mutation. ENU is an alkylating agent that acts by transferring its ethyl group to oxygen or nitrogen radicals in nucleic acids (55). This primarily induces base mis-pairing and misincorporation by replicative polymerases without substantial stalling, which we hypothesized would encourage polymerase bypass errors rather than inhibit replication (56). Similar to our protocol for B[a]P exposure, we employed a 28 day, sub-chronic dosing regimen of 5 mg/kg body weight/day of ENU. As with the B[a]P-treated cohort, we evaluated mitochondrial point mutations and large deletions with dRMC and 3D, and quantified nDNA mutagenesis. In bone marrow, control and ENU-treated mouse mtDNA, point mutation frequencies were: 4.6 ± 1.5 × 10−6 bp versus 4.5 ± 0.5 for the 12S rRNA locus; and, 1.6 ± 0.3 × 10−6 bp versus 1.9 ± 0.4 × 10−6 bp for ND5 site (Figure 6A). In liver, these frequencies were: 7.9 ± 4.8 × 10−6 bp versus 14.8 ± 4.8 for the 12S rRNA site; and 3.2 ± 0.4 × 10−6 bp, and 4.4 ± 1.19 × 10−6 for the ND5 site (Figure 6B). Thus, mirroring our B[a]P results, the mtDNA point mutation frequency was not significantly altered at either mtDNA target and in either tissue after ENU exposure (bone marrow: P = 0.97, 12S rRNA locus; P = 0.90, ND5 locus; and in liver: P = 0.29, 12S rRNA locus; P = 0.09, ND5 locus, Welch's unpaired t-test). 3D quantification of mtDNA ‘common’ deletions revealed 1.6 ± 0.3 and 1.5 ± 0.3 deletions per 107 genomes in untreated and treated bone marrow, respectively (Figure 7A). In liver, these frequencies were 12.5 ± 3.8 deletions per 107 genomes and 20.4 ± 5.5 (Figure 7B). In summary, regardless of the tissue of origin, ENU did not induce mtDNA point or deletion mutations.
Figure 6.

Subchronic ENU treatment does not increase the frequency of mitochondrial point mutations. Mice were treated daily with vehicle or 5 mg/kg ENU for 28 consecutive days. Three days following treatment, DNA was extracted from bone marrow and liver. Mutation frequency per bp mtDNA (± s.e.m.) was determined via dRMC at TaqI restriction sites within the 12S rRNA and ND5 genes in mouse mitochondrial DNA. (A) Bone marrow mutation frequency (P = 0.97, 12S rRNA locus; P = 0.90, ND5 locus; Welch's unpaired t-test). (B) Liver mutation frequency (P = 0.29, 12S rRNA locus; P = 0.09, ND5 locus; Welch's unpaired t-test).
Figure 7.

Subchronic ENU treatment does not induce deletions in mouse bone marrow and liver mtDNA. Deletion frequency per mitochondrial genome (± s.e.m.) was determined via 3D. (A) Bone marrow deletion frequency in mice treated with ENU (P = 0.82, two-tailed Welch's unpaired t-test). (B) Liver deletion frequency in mice treated with ENU (P = 0.09, two-tailed Welch's unpaired t-test).
Nuclear ENU-induced mutagenesis
Similar to our observations with B[a]P, quantification of nuclear mutants following ENU exposure showed, as expected, that ENU significantly induced mutant frequencies in both bone marrow and liver tissues. Specifically, we observed lacZ mutant frequencies of 5.0 ± 1.2 × 10−5 in the untreated mice bone marrow, whereas 155.0 ± 11.1 mutants × 10−5 were recovered from the ENU-treated mice. In liver, we quantified 3.0 ± 0.7 mutants × 10−5 in the untreated cohort, and 27.0 ± 3.3 mutants × 10−5 from their ENU-exposed counterparts (Supplementary Figure S4). The lacZ dRMC assay performed on these samples confirmed mutation induction by ENU in the nuclear genome. In these mice, mutant frequency of untreated animals was 1.1 ± 0.8 × 10−6 bp in bone marrow and 1.6 ± 0.7 × 10−6 bp in liver tissues, whereas mutation frequency in mice exposed to 5 mg/kg body weight/day ENU was 15.5 ± 4.6 × 10−6 bp in bone marrow and 10.2 ± 4.67 × 10−6 bp in liver (Supplementary Figure S5, P ≤ 0.05 for bone marrow and liver, one-tailed Welch's-adjusted t-test). As with the results of B[a]P exposure, these findings show a clear difference in the sensitivity of mtDNA and nDNA to ENU-induced mutagenesis. Intriguingly, although mtDNA copies per nuclear genome were unaffected by either B[a]P or ENU exposure in bone marrow (B[a]P, P = 0.37; ENU, P = 0.40, both Welch's-adjusted t-test), recovered liver tissue posted increased mtDNA copy number in B[a]P-exposed mice (ANOVA, P = 0.06; 0 versus 75 mg/kg body weight/day P = 0.03, Welch's-adjusted t-test) and decreased mtDNA copy number in ENU-exposed mice (P = 0.03, Welch's-adjusted t-test), suggesting tissue- and compound-specific responses to mutagens that do not produce changes in the relative abundance of mtDNA copies (Supplementary Figure S6).
DISCUSSION
Diverse theories have emerged to explain the remarkably greater frequency of somatic mutations in mtDNA, as compared to the nuclear genome (3,6,57,58). One popular model, which developed from observations of increased damage burdens in mtDNA following genotoxin exposures (9,10,19,21,59), posits that this damage readily drives mutagenesis and thus mutation accumulation (60,61). This paradigm expands foundational concepts in nuclear mutagenesis to the mitochondrial genome: i.e. unrepaired DNA lesions, when encountered during replication, can promote error-prone trans-lesion synthesis or block polymerase extension, which result in point and deletion mutations (62). In essence, this model proposes the susceptibility of mtDNA to damage largely underlies its high rate of mutation.
However, testing the validity of this hypothesis has remained impractical, as the accurate quantification of de novo mtDNA mutations has been hampered by technical limitations (63); notably, the inability to distinguish true mutations from DNA damage (28,64). To overcome this and other impediments to accurate mutation quantification, we employed our droplet digital PCR assays, dRMC and 3D, to ascertain the degree to which exposure to damaging agents influences the frequency of mitochondrial and nuclear mutagenesis. In the dRMC assay, the frequency of DNA mutations is evaluated per base pair by leveraging the fact that single base mutations in the 4-bp recognition motif of TaqI restriction enzyme (5′-TCGA-3′) are sufficient to shield sites from endonuclease activity. In the 3D assay, TaqI sites present in wild-type mtDNA are removed by deletion events, maintaining the deletion-bearing genome as a substrate for amplification and subsequent detection. The dRMC and 3D assays permit accurate resolution of a single point (32,65) or deletion (31) mutations, respectively, in a background of hundreds of millions of wild-type genomes, and enabled our investigation into mtDNA mutagenesis.
To test the hypothesis that DNA-damaging compounds induce mtDNA point and deletion mutations, we first exposed mice to one of two model mutagens, B[a]P or ENU. The bulky adduct created by BPDE, the active metabolite of B[a]P, can stall both mitochondrial (47) and nuclear replication (66) and was expected to initiate mtDNA mutagenesis, similarly to the nuclear genome; in a study of human mitochondrial DNA polymerase γ (pol γ) tolerance of B[a]P adducts, polymerase extension ceased after error-prone incorporation of a single base-pair adjacent to the adduct (47). In contrast to B[a]P, ENU-induced damage is often bypassed by DNA polymerases, but the alkylated base modification alters DNA-polymerase interactions and can result in polymerase misincorporation during trans-lesion synthesis in the nucleus (67). In concert with these compounds’ proven mutagenic effects in nDNA, previous studies have demonstrated that the mitochondrial genome is a focal point for B[a]P (9,13,19) and ENU (21,59) adduct formation. Therefore, following mutagen exposure we anticipated robust conversion of DNA damage to mutation and a rise in the mtDNA mutation frequency, which would drastically exceed the level of induced mutation in the nucleus. However, against expectations, our results demonstrate that this was not the case. Rather, although sub-chronic exposures to B[a]P or ENU increase mutations in nDNA, they did not increase mtDNA mutagenesis, suggesting that DNA adducts are not readily converted into mutations in the mitochondrial genome.
These results could imply that adducts are managed through efficient DNA repair. Though the list of DNA repair pathways identified in the mitochondrion is greatly expanded from earlier assertions (68), deficiencies in lesion processing capability persist and may shape uniquely mitochondrial responses to DNA damage. For example, although a considerable amount of data asserts the susceptibility of mtDNA to adduct formation and damage (18–22), there is no known nucleotide-excision repair (NER) pathway in mitochondria for resolving bulky adducts such as those produced by B[a]P (68,69). No clear mitochondrial DNA repair mechanism has been identified to alleviate these lesions. As for management of ENU-induced DNA alkylation damage, mitochondrial versions of mismatch repair (MMR) (70,71), base-excision repair (BER) (72,73) and alkylation-specific DNA repair enzymes have been described (74). Thus, it remains possible that mitochondria safeguard against converting DNA lesions into mutation via robust mechanisms for mtDNA repair.
In addition to DNA repair, studies have proposed that selective destruction of mitochondrial genomes eliminates the potential for mutation conversion from damaged mtDNA (75,76). Such degradation would be reflected via a decrease in mitochondrial genomes copy number. In support of targeted degradation of damaged mtDNA, mitochondrial genome copy-number was reduced in the liver tissues of ENU-exposed mice; although bone marrow samples from the same mice exhibited no differences between treated and untreated cohorts. Exposure to B[a]P produced a dose-dependent increase in liver mtDNA copy-number, while copy-number was unchanged in bone marrow. Thus, our results do not support a role for targeted degradation of damaged of mitochondrial genomes in mtDNA mutation repression, though the activities of such a pathway might be masked by other phenomena.
For example, it is possible that upon encountering B[a]P-induced lesions, terminal pausing of pol γ produces incomplete extension products of linear mtDNA, which, if not repaired, are likely targeted for destruction (76). If these linear products include the control region amplified by our primers, and they are not degraded, they would serve as template for our mtDNA:nDNA copy number assay and thus inflate the mtDNA:nDNA ratio. Contrasting the dynamic liver mtDNA:nDNA ratios, our analysis observed bone marrow tissues had no mtDNA copy-number changes in either mutagen exposure cohort. While these results may hint at mechanisms for mutation avoidance in mtDNA, as our results are inconsistent across mutagen and tissue type, we can draw no unifying conclusions. Thus, untangling a pathway by which mtDNA avoids mutation following damage remains an intriguing area of future investigation.
As mtDNA adducts do not appear to contribute appreciably to the induction of point and deletion mutations, the generation of mtDNA mutations must be ascribed to other sources. We demonstrate that B[a]P and ENU exposures produced no significant increases in mtDNA point mutation or deletion frequencies, while inducing mutation in nDNA. Regardless of the mechanism by which damage-induced mutagenesis in the mitochondrial genome is suppressed, these findings highlight that the elevated frequency of somatic mtDNA mutation is not likely a byproduct of broad DNA damage sensitivity (24,77). Thus, as exogenous damage to the mitochondrial genome appears to be a negligible source of induced point and deletion mutations, the majority of mutations induced in mtDNA are likely consequences of endogenous sources of error (78). Indeed, the most reliable models of increased mtDNA mutation frequency employ functional mutants of pol γ (4,79). The burden of mtDNA mutations in mice deficient in the proofreading domain of pol γ, so-called ‘mutator mice’, can be hundreds- to thousands-fold higher than wild-type littermates (80). Curiously, the mutation spectrum of mtDNA in mutator mice is inconsistent with the expected spectra of pol γ misincorporation on undamaged template DNA (46,81), and expression of a mitochondrial-targeted human catalase in these mice, which reduces the ROS hydrogen peroxide, also reduced their mutation frequency (46). Thus, although synthesis by pol γ is fairly accurate on undamaged template in vitro (81,82), the presence of naturally-occurring mitochondrial ROS may contribute to the elevated spontaneous mutation frequency of the mitochondrial genome (60). Byproducts of mitochondrial metabolism, reactive oxygen species are recurrently associated with organismal aging and mtDNA mutagenesis (4,6,83,84). Recent appraisals of the mitochondrial mutation spectrum in aging and in models of attenuated oxidative damage repair, although, have concluded that oxidative damage imparts minimal contributions to mtDNA mutation frequency (85,86). Importantly, these assertions rely upon a narrowly-defined, unverified consensus signature of oxidative damage and induced mutagenesis in mitochondria. The lesions generated by reactive oxygen species range in severity from the subtle, 8-oxo-dG, to the obvious, strand breaks (87); consequently, the imputed mutation ‘signature’ of oxidative DNA damage has developed as the amalgam of results derived from mutagenesis studies using defined lesions, often pursued in vitro, and not necessarily in the context of the mitochondrial replisome (88–92). Given the varied lesions formed by oxidative DNA damage (87), a direct assessment of mutation frequency and spectrum in mtDNA following oxidative damage is warranted, as these results may identify lesions which contrast in mutagenic potential with the adducts induced in our system. Additionally, the ostensible capacity of pol γ for lesion recognition and aborted synthesis (47,79,93–96) appears a probable mechanism for mutation suppression in mtDNA. Future studies examining the factors that repress the conversion of mtDNA damage to mutations may elucidate these mechanisms, and could identify interventions to augment their activity, hopefully with advances for our understanding of pathologies in which somatic mtDNA mutations are implicated, such as aging and cancer (3,97).
Supplementary Material
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of Environmental Health Sciences [R01ES019319 to J.H.B.]; National Institutes of Aging [T32AG000057 to W.J.V.]; National Cancer Institute [F30CA200247]; Health Canada Intramural funding [to F.M., A.S.L. and P.A.W.]. Funding for open access charge: National Institute of Environmental Health Sciences [R01ES019319].
Conflict of interest statement. None declared.
REFERENCES
- 1.Schon E.A., DiMauro S., Hirano M. Human mitochondrial DNA: roles of inherited and somatic mutations. Nat. Rev. Genet. 2012;13:878–890. doi: 10.1038/nrg3275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bender A., Krishnan K.J., Morris C.M., Taylor G.A., Reeve A.K., Perry R.H., Jaros E., Hersheson J.S., Betts J., Klopstock T., et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006;38:515–517. doi: 10.1038/ng1769. [DOI] [PubMed] [Google Scholar]
- 3.Taylor R.W., Turnbull D.M. Mitochondrial DNA mutations in human disease. Nat. Rev. Genet. 2005;6:389–402. doi: 10.1038/nrg1606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J.N., Rovio A.T., Bruder C.E., Bohlooly Y.M., Gidlof S., Oldfors A., Wibom R., et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature. 2004;429:417–423. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 5.Kujoth G.C., Hiona A., Pugh T.D., Someya S., Panzer K., Wohlgemuth S.E., Hofer T., Seo A.Y., Sullivan R., Jobling W.A., et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science. 2005;309:481–484. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 6.Schriner S.E., Linford N.J., Martin G.M., Treuting P., Ogburn C.E., Emond M., Coskun P.E., Ladiges W., Wolf N., Van Remmen H., et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science. 2005;308:1909–1911. doi: 10.1126/science.1106653. [DOI] [PubMed] [Google Scholar]
- 7.Imanishi H., Hattori K., Wada R., Ishikawa K., Fukuda S., Takenaga K., Nakada K., Hayashi J. Mitochondrial DNA mutations regulate metastasis of human breast cancer cells. PloS One. 2011;6:e23401. doi: 10.1371/journal.pone.0023401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee H.-C., Chang C.-M., Chi C.-W. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res. Rev. 2010;9(Suppl.):S47–S58. doi: 10.1016/j.arr.2010.08.009. [DOI] [PubMed] [Google Scholar]
-
9.Backer J.M., Weinstein I.B. Interaction of Benzo(a)pyrene and its Dihydrodiol-Epoxide derivative with nuclear and mitochondrial DNA in C3H10T
 cell cultures. Cancer Res. 1982;42:2764–2769. [PubMed] [Google Scholar]
cell cultures. Cancer Res. 1982;42:2764–2769. [PubMed] [Google Scholar] - 10.Yakes F.M., Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. U.S.A. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hunter S.E., Jung D., Di Giulio R.T., Meyer J.N. The QPCR assay for analysis of mitochondrial DNA damage, repair, and relative copy number. Methods. 2010;51:444–451. doi: 10.1016/j.ymeth.2010.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hunter S.E., Gustafson M.A., Margillo K.M., Lee S.A., Ryde I.T., Meyer J.N. In vivo repair of alkylating and oxidative DNA damage in the mitochondrial and nuclear genomes of wild-type and glycosylase-deficient Caenorhabditis elegans. DNA Repair (Amst) 2012;11:857–863. doi: 10.1016/j.dnarep.2012.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Le Goff J., Gallois J., Pelhuet L., Devier M.H., Budzinski H., Pottier D., Andre V., Cachot J. DNA adduct measurements in zebra mussels, Dreissena polymorpha, Pallas. Potential use for genotoxicant biomonitoring of fresh water ecosystems. Aquat. toxicol. 2006;79:55–64. doi: 10.1016/j.aquatox.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 14.Guliaeva N.A., Kuznetsova E.A., Gaziev A.I. [Proteins associated with mitochondrial DNA protect it against the action of X-rays and hydrogen peroxide] Biofizika. 2006;51:692–697. [PubMed] [Google Scholar]
- 15.Canugovi C., Maynard S., Bayne A.C., Sykora P., Tian J., de Souza-Pinto N.C., Croteau D.L., Bohr V.A. The mitochondrial transcription factor A functions in mitochondrial base excision repair. DNA Repair (Amst) 2010;9:1080–1089. doi: 10.1016/j.dnarep.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Huang J.C., Zamble D.B., Reardon J.T., Lippard S.J., Sancar A. HMG-domain proteins specifically inhibit the repair of the major DNA adduct of the anticancer drug cisplatin by human excision nuclease. Proc. Natl. Acad. Sci. U.S.A. 1994;91:10394–10398. doi: 10.1073/pnas.91.22.10394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcelino L.A., Thilly W.G. Mitochondrial mutagenesis in human cells and tissues. Mutat. Res. 1999;434:177–203. doi: 10.1016/s0921-8777(99)00028-2. [DOI] [PubMed] [Google Scholar]
- 18.Yang Z., Schumaker L.M., Egorin M.J., Zuhowski E.G., Guo Z., Cullen K.J. Cisplatin preferentially binds mitochondrial DNA and voltage-dependent anion channel protein in the mitochondrial membrane of head and neck squamous cell carcinoma: possible role in apoptosis. Clin. Cancer Res. 2006;12:5817–5825. doi: 10.1158/1078-0432.CCR-06-1037. [DOI] [PubMed] [Google Scholar]
- 19.Backer J.M., Weinstein I.B. Mitochondrial DNA is a major cellular target for a Dihydrodiol-Epoxide derivative of Benzo[a]pyrene. Science. 1980;209:297–299. doi: 10.1126/science.6770466. [DOI] [PubMed] [Google Scholar]
- 20.Niranjan B.G., Bhat N.K., Avadhani N.G. Preferential attack of mitochondrial DNA by aflatoxin B1 during hepatocarcinogenesis. Science. 1982;215:73–75. doi: 10.1126/science.6797067. [DOI] [PubMed] [Google Scholar]
- 21.Wunderlich V., Schutt M., Bottger M., Graffi A. Preferential alkylation of mitochondrial deoxyribonucleic acid by N-methyl-N-nitrosourea. Biochem. J. 1970;118:99–109. doi: 10.1042/bj1180099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Murata T., Hibasami H., Maekawa S., Tagawa T., Nakashima K. Preferential binding of cisplatin to mitochondrial DNA and suppression of ATP generation in human malignant melanoma cells. Biochem. Int. 1990;20:949–955. [PubMed] [Google Scholar]
- 23.Mita S., Monnat R.J., Jr, Loeb L.A. Resistance of HeLa cell mitochondrial DNA to mutagenesis by chemical carcinogens. Cancer Res. 1988;48:4578–4583. [PubMed] [Google Scholar]
- 24.Mambo E., Gao X., Cohen Y., Guo Z., Talalay P., Sidransky D. Electrophile and oxidant damage of mitochondrial DNA leading to rapid evolution of homoplasmic mutations. Proc. Natl. Acad. Sci. U.S.A. 2003;100:1838–1843. doi: 10.1073/pnas.0437910100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guzzella L., Monarca S., Zani C., Feretti D., Zerbini I., Buschini A., Poli P., Rossi C., Richardson S.D. In vitro potential genotoxic effects of surface drinking water treated with chlorine and alternative disinfectants. Mutat. Res. 2004;564:179–193. doi: 10.1016/j.mrgentox.2004.08.006. [DOI] [PubMed] [Google Scholar]
- 26.Berneburg M., Plettenberg H., Medve-Konig K., Pfahlberg A., Gers-Barlag H., Gefeller O., Krutmann J. Induction of the photoaging-associated mitochondrial common deletion in vivo in normal human skin. J. Investig. Dermatol. 2004;122:1277–1283. doi: 10.1111/j.0022-202X.2004.22502.x. [DOI] [PubMed] [Google Scholar]
- 27.Gebhard D., Mahler B., Matt K., Burger K., Bergemann J. Mitochondrial DNA copy number – but not a mitochondrial tandem CC to TT transition – is increased in sun-exposed skin. Exp. Dermatol. 2014;23:209–211. doi: 10.1111/exd.12327. [DOI] [PubMed] [Google Scholar]
- 28.Kraytsberg Y., Nicholas A., Caro P., Khrapko K. Single molecule PCR in mtDNA mutational analysis: Genuine mutations vs. damage bypass-derived artifacts. Methods. 2008;46:269–273. doi: 10.1016/j.ymeth.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rothfuss O., Gasser T., Patenge N. Analysis of differential DNA damage in the mitochondrial genome employing a semi-long run real-time PCR approach. Nucleic Acids Res. 2010;38:e24. doi: 10.1093/nar/gkp1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Vermulst M., Bielas J.H., Loeb L.A. Quantification of random mutations in the mitochondrial genome. Methods. 2008;46:263–268. doi: 10.1016/j.ymeth.2008.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Taylor S.D., Ericson N.G., Burton J.N., Prolla T.A., Silber J.R., Shendure J., Bielas J.H. Targeted enrichment and high-resolution digital profiling of mitochondrial DNA deletions in human brain. Aging Cell. 2014;13:29–38. doi: 10.1111/acel.12146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ericson N.G., Kulawiec M., Vermulst M., Sheahan K., O'Sullivan J., Salk J.J., Bielas J.H. Decreased mitochondrial DNA mutagenesis in human colorectal cancer. PLoS Genet. 2012;8:e1002689. doi: 10.1371/journal.pgen.1002689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brault D., Renault D., Tombolan F., Thybaud V. Kinetics of induction of DNA damage and lacZ gene mutations in stomach mucosa of mice treated with beta-propiolactone and N-methyl-N'-nitro-N-nitrosoguanidine, using single-cell gel electrophoresis and MutaMouse models. Environ. Mol. Mutagen. 1999;34:182–189. [PubMed] [Google Scholar]
- 34.Lemieux C.L., Douglas G.R., Gingerich J., Phonethepswath S., Torous D.K., Dertinger S.D., Phillips D.H., Arlt V.M., White P.A. Simultaneous measurement of benzo[a]pyrene-induced Pig-a and lacZ mutations, micronuclei and DNA adducts in Muta Mouse. Environ. Mol. Mutagen. 2011;52:756–765. doi: 10.1002/em.20688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Douglas G.R., Gingerich J.D., Gossen J.A., Bartlett S.A. Sequence spectra of spontaneous lacZ gene mutations in transgenic mouse somatic and germline tissues. Mutagenesis. 1994;9:451–458. doi: 10.1093/mutage/9.5.451. [DOI] [PubMed] [Google Scholar]
- 36.Vijg J., Douglas G.R. Bacteriophage lambda and plasmid LacZ transgenic mice for studying mutations in vivo. NY: Plenum Press; 1996. [Google Scholar]
- 37.Ye J., Coulouris G., Zaretskaya I., Cutcutache I., Rozen S., Madden T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinformatics. 2012;13:134. doi: 10.1186/1471-2105-13-134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pinheiro L.B., Coleman V.A., Hindson C.M., Herrmann J., Hindson B.J., Bhat S., Emslie K.R. Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 2011;84:1003–1011. doi: 10.1021/ac202578x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hindson B.J., Ness K.D., Masquelier D.A., Belgrader P., Heredia N.J., Makarewicz A.J., Bright I.J., Lucero M.Y., Hiddessen A.L., Legler T.C., et al. High-throughput droplet digital PCR system for absolute quantitation of DNA copy number. Anal. Chem. 2011;83:8604–8610. doi: 10.1021/ac202028g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bielas J.H. Non-transcribed strand repair revealed in quiescent cells. Mutagenesis. 2006;21:49–53. doi: 10.1093/mutage/gei073. [DOI] [PubMed] [Google Scholar]
- 41.Furda A., Santos J.H., Meyer J.N., Van Houten B. Quantitative PCR-based measurement of nuclear and mitochondrial DNA damage and repair in mammalian cells. Methods Mol. Biol. 2014;1105:419–437. doi: 10.1007/978-1-62703-739-6_31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Long A.S., Lemieux C.L., Arlt V.M., White P.A. Tissue-specific in vivo genetic toxicity of nine polycyclic aromatic hydrocarbons assessed using the Muta™Mouse transgenic rodent assay. Toxicol. Appl. Pharmacol. 2016;290:31–42. doi: 10.1016/j.taap.2015.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Miller K.P., Ramos K.S. Impact of cellular metabolism on the biological effects of benzo[a]pyrene and related hydrocarbons. Drug Metab. Rev. 2001;33:1–35. doi: 10.1081/dmr-100000138. [DOI] [PubMed] [Google Scholar]
- 44.Choudhary D., Jansson I., Schenkman J.B., Sarfarazi M., Stoilov I. Comparative expression profiling of 40 mouse cytochrome P450 genes in embryonic and adult tissues. Arch. Biochem. Biophys. 2003;414:91–100. doi: 10.1016/s0003-9861(03)00174-7. [DOI] [PubMed] [Google Scholar]
- 45.Karunadharma P.P., Basisty N., Dai D.F., Chiao Y.A., Quarles E.K., Hsieh E.J., Crispin D., Bielas J.H., Ericson N.G., Beyer R.P., et al. Subacute calorie restriction and rapamycin discordantly alter mouse liver proteome homeostasis and reverse aging effects. Aging Cell. 2015;14:547–557. doi: 10.1111/acel.12317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Vermulst M., Bielas J.H., Kujoth G.C., Ladiges W.C., Rabinovitch P.S., Prolla T.A., Loeb L.A. Mitochondrial point mutations do not limit the natural lifespan of mice. Nat. Genet. 2007;39:540–543. doi: 10.1038/ng1988. [DOI] [PubMed] [Google Scholar]
- 47.Graziewicz M.A., Sayer J.M., Jerina D.M., Copeland W.C. Nucleotide incorporation by human DNA polymerase γ opposite benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyguanosine and deoxyadenosine. Nucleic Acids Res. 2004;32:397–405. doi: 10.1093/nar/gkh213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Krishnan K.J., Reeve A.K., Samuels D.C., Chinnery P.F., Blackwood J.K., Taylor R.W., Wanrooij S., Spelbrink J.N., Lightowlers R.N., Turnbull D.M. What causes mitochondrial DNA deletions in human cells. Nat. Genet. 2008;40:275–279. doi: 10.1038/ng.f.94. [DOI] [PubMed] [Google Scholar]
- 49.Tanhauser S.M., Laipis P.J. Multiple deletions are detectable in mitochondrial DNA of aging mice. J. Biol. Chem. 1995;270:24769–24775. doi: 10.1074/jbc.270.42.24769. [DOI] [PubMed] [Google Scholar]
- 50.Nie H., Shu H., Vartak R., Milstein A.C., Mo Y., Hu X., Fang H., Shen L., Ding Z., Lu J., et al. Mitochondrial common deletion, a potential biomarker for cancer occurrence, is selected against in cancer background: a meta-analysis of 38 studies. PloS One. 2013;8:e67953. doi: 10.1371/journal.pone.0067953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jung D., Cho Y., Meyer J.N., Di Giulio R.T. The long amplicon quantitative PCR for DNA damage assay as a sensitive method of assessing DNA damage in the environmental model, Atlantic killifish (Fundulus heteroclitus) Comp. Biochem. Physiol. Toxicol. Pharmacol. 2009;149:182–186. doi: 10.1016/j.cbpc.2008.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lambert I.B., Singer T.M., Boucher S.E., Douglas G.R. Detailed review of transgenic rodent mutation assays. Mutat. Res. 2005;590:1–280. doi: 10.1016/j.mrrev.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 53.Gossen J.A., de Leeuw W.J., Tan C.H., Zwarthoff E.C., Berends F., Lohman P.H., Knook D.L., Vijg J. Efficient rescue of integrated shuttle vectors from transgenic mice: a model for studying mutations in vivo. Proc. Natl. Acad. Sci. U.S.A. 1989;86:7971–7975. doi: 10.1073/pnas.86.20.7971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hakura A., Tsutsui Y., Sonoda J., Tsukidate K., Mikami T., Sagami F. Comparison of the mutational spectra of the lacZ transgene in four organs of the MutaMouse treated with benzo[a]pyrene: target organ specificity. Mutat. Res. 2000;447:239–247. doi: 10.1016/s0027-5107(99)00213-4. [DOI] [PubMed] [Google Scholar]
- 55.Justice M.J., Noveroske J.K., Weber J.S., Zheng B., Bradley A. Mouse ENU mutagenesis. Hum. Mol. Genet. 1999;8:1955–1963. doi: 10.1093/hmg/8.10.1955. [DOI] [PubMed] [Google Scholar]
- 56.Noveroske J.K., Weber J.S., Justice M.J. The mutagenic action of N-ethyl-N-nitrosourea in the mouse. Mamm. Genome. 2000;11:478–483. doi: 10.1007/s003350010093. [DOI] [PubMed] [Google Scholar]
- 57.Taylor R.W., Barron M.J., Borthwick G.M., Gospel A., Chinnery P.F., Samuels D.C., Taylor G.A., Plusa S.M., Needham S.J., Greaves L.C., et al. Mitochondrial DNA mutations in human colonic crypt stem cells. J. Clin. Invest. 2003;112:1351–1360. doi: 10.1172/JCI19435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Khaidakov M., Heflich R.H., Manjanatha M.G., Myers M.B., Aidoo A. Accumulation of point mutations in mitochondrial DNA of aging mice. Mutat. Res. 2003;526:1–7. doi: 10.1016/s0027-5107(03)00010-1. [DOI] [PubMed] [Google Scholar]
- 59.LeDoux S.P., Wilson G.L., Beecham E.J., Stevnsner T., Wassermann K., Bohr V.A. Repair of mitochondrial DNA after various types of DNA damage in Chinese hamster ovary cells. Carcinogenesis. 1992;13:1967–1973. doi: 10.1093/carcin/13.11.1967. [DOI] [PubMed] [Google Scholar]
- 60.Pinz K.G., Shibutani S., Bogenhagen D.F. Action of mitochondrial DNA polymerase gamma at sites of base loss or oxidative damage. J. Biol. Chem. 1995;270:9202–9206. doi: 10.1074/jbc.270.16.9202. [DOI] [PubMed] [Google Scholar]
- 61.Chen J.Z., Kadlubar F.F. Mitochondrial mutagenesis and oxidative stress in human prostate cancer. J. Environ. Sci. Health C Environ. Carcinog. Ecotoxicol. Rev. 2004;22:1–12. doi: 10.1081/GNC-120037931. [DOI] [PubMed] [Google Scholar]
- 62.Friedberg E.C., Walker G.C., Siede W., Wood R.D. DNA Repair Mutagenesis. 2nd edn. American Society for Microbiology Press; 2006. [Google Scholar]
- 63.Jacobs H.T. The mitochondrial theory of aging: Dead or alive. Aging Cell. 2003;2:11–17. doi: 10.1046/j.1474-9728.2003.00032.x. [DOI] [PubMed] [Google Scholar]
- 64.Greaves L.C., Beadle N.E., Taylor G.A., Commane D., Mathers J.C., Khrapko K., Turnbull D.M. Quantification of mitochondrial DNA mutation load. Aging Cell. 2009;8:566–572. doi: 10.1111/j.1474-9726.2009.00505.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wright J.H., Modjeski K.L., Bielas J.H., Preston B.D., Fausto N., Loeb L.A., Campbell J.S. A random mutation capture assay to detect genomic point mutations in mouse tissue. Nucleic Acids Res. 2011;39:e73. doi: 10.1093/nar/gkr142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Busbee D.L., Joe C.O., Norman J.O., Rankin P.W. Inhibition of DNA synthesis by an electrophilic metabolite of benzo[a]pyrene. Proc. Natl. Acad. Sci. U.S.A. 1984;81:5300–5304. doi: 10.1073/pnas.81.17.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Shrivastav N., Li D., Essigmann J.M. Chemical biology of mutagenesis and DNA repair: cellular responses to DNA alkylation. Carcinogenesis. 2010;31:59–70. doi: 10.1093/carcin/bgp262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kazak L., Reyes A., Holt I.J. Minimizing the damage: Repair pathways keep mitochondrial DNA intact. Nat. Rev. Mol. Cell Biol. 2012;13:659–671. doi: 10.1038/nrm3439. [DOI] [PubMed] [Google Scholar]
- 69.Croteau D.L., Stierum R.H., Bohr V.A. Mitochondrial DNA repair pathways. Mutat. Res. 1999;434:137–148. doi: 10.1016/s0921-8777(99)00025-7. [DOI] [PubMed] [Google Scholar]
- 70.Mason P.A., Matheson E.C., Hall A.G., Lightowlers R.N. Mismatch repair activity in mammalian mitochondria. Nucleic Acids Res. 2003;31:1052–1058. doi: 10.1093/nar/gkg167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.de Souza-Pinto N.C., Mason P.A., Hashiguchi K., Weissman L., Tian J., Guay D., Lebel M., Stevnsner T.V., Rasmussen L.J., Bohr V.A. Novel DNA mismatch-repair activity involving YB-1 in human mitochondria. DNA Repair. 2009;8:704–719. doi: 10.1016/j.dnarep.2009.01.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Stuart J.A., Hashiguchi K., Wilson D.M., 3rd, Copeland W.C., Souza-Pinto N.C., Bohr V.A. DNA base excision repair activities and pathway function in mitochondrial and cellular lysates from cells lacking mitochondrial DNA. Nucleic Acids Res. 2004;32:2181–2192. doi: 10.1093/nar/gkh533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ruchko M.V., Gorodnya O.M., Zuleta A., Pastukh V.M., Gillespie M.N. The DNA glycosylase Ogg1 defends against oxidant-induced mtDNA damage and apoptosis in pulmonary artery endothelial cells. Free Radic. Biol. Med. 2011;50:1107–1113. doi: 10.1016/j.freeradbiomed.2010.10.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.van Loon B., Samson L.D. Alkyladenine DNA glycosylase (AAG) localizes to mitochondria and interacts with mitochondrial single-stranded binding protein (mtSSB) DNA Repair (Amst) 2013;12:177–187. doi: 10.1016/j.dnarep.2012.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Bess A.S., Crocker T.L., Ryde I.T., Meyer J.N. Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res. 2012;40:7916–7931. doi: 10.1093/nar/gks532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shokolenko I., Venediktova N., Bochkareva A., Wilson G.L., Alexeyev M.F. Oxidative stress induces degradation of mitochondrial DNA. Nucleic Acids Res. 2009;37:2539–2548. doi: 10.1093/nar/gkp100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Harman D. The biologic clock: The mitochondria. J. Am. Geriatr. Soc. 1972;20:145–147. doi: 10.1111/j.1532-5415.1972.tb00787.x. [DOI] [PubMed] [Google Scholar]
- 78.Zheng W., Khrapko K., Coller H.A., Thilly W.G., Copeland W.C. Origins of human mitochondrial point mutations as DNA polymerase γ-mediated errors. Mutat. Res. 2006;599:11–20. doi: 10.1016/j.mrfmmm.2005.12.012. [DOI] [PubMed] [Google Scholar]
- 79.Bailey L.J., Cluett T.J., Reyes A., Prolla T.A., Poulton J., Leeuwenburgh C., Holt I.J. Mice expressing an error-prone DNA polymerase in mitochondria display elevated replication pausing and chromosomal breakage at fragile sites of mitochondrial DNA. Nucleic Acids Res. 2009;37:2327–2335. doi: 10.1093/nar/gkp091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Vermulst M., Wanagat J., Kujoth G.C., Bielas J.H., Rabinovitch P.S., Prolla T.A., Loeb L.A. DNA deletions and clonal mutations drive premature aging in mitochondrial mutator mice. Nat. Genet. 2008;40:392–394. doi: 10.1038/ng.95. [DOI] [PubMed] [Google Scholar]
- 81.Lee H.R., Johnson K.A. Fidelity of the human mitochondrial DNA polymerase. J. Biol. Chem. 2006;281:36236–36240. doi: 10.1074/jbc.M607964200. [DOI] [PubMed] [Google Scholar]
- 82.Johnson A.A., Johnson K.A. Exonuclease proofreading by human mitochondrial DNA polymerase. J. Biol. Chem. 2001;276:38097–38107. doi: 10.1074/jbc.M106046200. [DOI] [PubMed] [Google Scholar]
- 83.Stevnsner T., Thorslund T., de Souza-Pinto N.C., Bohr V.A. Mitochondrial repair of 8-oxoguanine and changes with aging. Exp. Gerontol. 2002;37:1189–1196. doi: 10.1016/s0531-5565(02)00142-0. [DOI] [PubMed] [Google Scholar]
- 84.Dai D.F., Santana L.F., Vermulst M., Tomazela D.M., Emond M.J., MacCoss M.J., Gollahon K., Martin G.M., Loeb L.A., Ladiges W.C., et al. Overexpression of catalase targeted to mitochondria attenuates murine cardiac aging. Circulation. 2009;119:2789–2797. doi: 10.1161/CIRCULATIONAHA.108.822403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kennedy S.R., Salk J.J., Schmitt M.W., Loeb L.A. Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 2013;9:e1003794. doi: 10.1371/journal.pgen.1003794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Itsara L.S., Kennedy S.R., Fox E.J., Yu S., Hewitt J.J., Sanchez-Contreras M., Cardozo-Pelaez F., Pallanck L.J. Oxidative stress is not a major contributor to somatic mitochondrial DNA mutations. PLoS Genet. 2014;10:e1003974. doi: 10.1371/journal.pgen.1003974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Muftuoglu M., Mori M.P., Souza-Pinto N.C.d. Formation and repair of oxidative damage in the mitochondrial DNA. Mitochondrion. 2014;17:164–181. doi: 10.1016/j.mito.2014.03.007. [DOI] [PubMed] [Google Scholar]
- 88.Dizdaroglu M., Holwitt E., Hagan M.P., Blakely W.F. Formation of cytosine glycol and 5,6-dihydroxycytosine in deoxyribonucleic acid on treatment with osmium tetroxide. Biochem. J. 1986;235:531–536. doi: 10.1042/bj2350531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Wang D., Kreutzer D.A., Essigmann J.M. Mutagenicity and repair of oxidative DNA damage: Insights from studies using defined lesions. Mutat. Res. 1998;400:99–115. doi: 10.1016/s0027-5107(98)00066-9. [DOI] [PubMed] [Google Scholar]
- 90.Masaoka A., Terato H., Kobayashi M., Ohyama Y., Ide H. Oxidation of thymine to 5-formyluracil in DNA promotes misincorporation of dGMP and subsequent elongation of a mismatched primer terminus by DNA polymerase. J. Biol. Chem. 2001;276:16501–16510. doi: 10.1074/jbc.M008598200. [DOI] [PubMed] [Google Scholar]
- 91.Jaruga P., Dizdaroglu M. Repair of products of oxidative DNA base damage in human cells. Nucleic Acids Res. 1996;24:1389–1394. doi: 10.1093/nar/24.8.1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Feig D.I., Sowers L.C., Loeb L.A. Reverse chemical mutagenesis: identification of the mutagenic lesions resulting from reactive oxygen species-mediated damage to DNA. Proc. Natl. Acad. Sci. U.S.A. 1994;91:6609–6613. doi: 10.1073/pnas.91.14.6609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Graziewicz M.A., Bienstock R.J., Copeland W.C. The DNA polymerase γ Y955C disease variant associated with PEO and parkinsonism mediates the incorporation and translesion synthesis opposite 7,8-dihydro-8-oxo-2′-deoxyguanosine. Hum. Mol. Genet. 2007;16:2729–2739. doi: 10.1093/hmg/ddm227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Spelbrink J.N., Toivonen J.M., Hakkaart G.A.J., Kurkela J.M., Cooper H.M., Lehtinen S.K., Lecrenier N., Back J.W., Speijer D., Foury F., et al. In vivo functional analysis of the human mitochondrial DNA polymerase POLG expressed in cultured human cells. J. Biol. Chem. 2000;275:24818–24828. doi: 10.1074/jbc.M000559200. [DOI] [PubMed] [Google Scholar]
- 95.Stumpf J.D., Copeland W.C. MMS exposure promotes increased MtDNA mutagenesis in the presence of replication-defective disease-associated DNA polymerase γ variants. PLoS Genet. 2014;10:e1004748. doi: 10.1371/journal.pgen.1004748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kasiviswanathan R., Minko I.G., Lloyd R.S., Copeland W.C. Translesion synthesis past acrolein-derived DNA adducts by human mitochondrial DNA polymerase γ. J. Biol. Chem. 2013;288:14247–14255. doi: 10.1074/jbc.M113.458802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wallace D.C. Mitochondria and cancer. Nat. Rev. Cancer. 2012;12:685–698. doi: 10.1038/nrc3365. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.