

Abstract
Interleukin-8 (IL-8) is a potent neutrophil-activating chemokine which triggers the infiltration and migration of neutrophils into areas of bacterial infection. Helicobacter pylori-infected patient studies as well as animal models have revealed that H. pylori type I strains carrying an intact cytotoxin-associated gene pathogenicity island (cag-PAI) with a functional type IV secretion system (T4SS) induce IL-8 expression and secretion in gastric mucosa. This gastric mucosal IL-8 expression correlates with severe histological changes due to H. pylori infection.
In the present study, we explored a new recognition pattern on the bacterial adhesion protein CagL inducing IL-8 expression in H. pylori-infected host cells. To analyze the secreted IL-8 concentration, we performed IL-8 enzyme-linked immunosorbent assay (ELISA). To investigate the H. pylori-induced IL-8 expression on the transcriptional level, we transiently transfected gastric epithelial cells (AGS) with a human IL-8 luciferase reporter construct.
The results of this study demonstrate that specifically the C-terminal coiled-coil region of the H. pylori CagL protein, a protein described to be located on the tip of the T4SS-pilus, is responsible for several in vitro observations: 1) H. pylori-induced IL-8 secretion via the transforming growth factor (TGF)-α activated epidermal growth factor-receptor (EGF-R) signaling pathway; 2) H. pylori-induced elongation of the cells, a typical CagA-induced phenotype; and 3) the bridging of the T4SS to its human target cells. This novel bacterial-host recognition sequence allows a new insight into how H. pylori induces the inflammatory response in gastric epithelial cells and facilitates the development of precancerous conditions.
Keywords: Helicobacter pylori, IL-8, CagL, coiled-coil
Introduction
Helicobacter pylori is a gram-negative, spiral-shaped, microaerophilic, and highly motile bacterial pathogen which colonizes the human stomach in more than 50% of the world population. In about 10–20% of the infected individuals, H. pylori causes chronic gastric inflammation and severe sequelae such as gastroduodenal ulcers, mucosa-associated lymphoid tissue (MALT) lymphoma, and gastric adenocarcinoma [1, 2]. The outcome of severe forms of disease is dependent on bacterial factors produced by H. pylori type I strains as well as specific host susceptibility [3, 4]. In the H. pylori infection model of Mongolian gerbils, only animals treated with H. pylori type I strains developed severe gastrointestinal inflammation. It has been demonstrated that only H. pylori type I strains characterized by harboring an intact cytotoxin-associated gene pathogenicity island (cag-PAI) with a functional T4SS induce a severe outcome of gastric diseases. In parallel, this results in a strong induction of proinflammatory cytokines (e.g., IL-1β, IL-6, TNF-α, IFN-γ, and IL-8). Animals infected with an isogenic mutant strain carrying a nonfunctional T4SS did not display severe inflammatory responses in the corpus mucosa [5-7].
IL-8 belongs to a superfamily of secreted proteins, the chemokines (chemoattractant cytokines), which specialize in mobilizing leukocytes to areas of immune challenge [8]. The function of this protein family member is to potently stimulate leukocyte migration along the chemotactic gradient, subsequently leading to the adhesion and infiltration of inflammatory cells into the affected tissues.
During H. pylori infection, the increased IL-8 concentration causes a significant infiltration of neutrophils and lymphocytes into the gastric mucosa, resulting in chronic gastritis [9]. In vivo studies demonstrated a correlation between the gastric mucosal IL-8 levels and the histological severity of H. pylori-infected gastritis patients [10-13].
Furthermore, in vitro data showed that H. pylori-induced IL-8 secretion under serum-free conditions is dependent on a functional T4SS [14, 15] but not on the translocated CagA protein because isogenic cagA-mutant strains were not able to eliminate the ability of the bacterium to stimulate IL-8 expression [16-18]. Also, adhesion studies have revealed that direct contact of the bacterium with the epithelial cell is required for IL-8 induction [19]. Viala et al. elucidated that not only CagA is translocated via the T4SS but also peptidoglycan. This latter H. pylori-induced activation of a proinflammatory signaling cascade involves the host defense molecule Nod1 and the transcription factor NFκB [20, 21]. To date, the exact mechanism how H. pylori induces IL-8 expression is only partially understood. So far, it has been shown that the IL-8 promoter binding sites for both transcription factors, NFκB- and activating protein (AP)-1, are required for an optimal transcription induced by H. pylori infection [22, 23]. In response to H. pylori infection, the activated transcription factors NFκB and AP-1 attach to their DNA binding sites within the IL-8 promoter and induce its expression [24-27].
In this study, we identified a H. pylori factor domain that is essential for inducing IL-8 expression. Since direct contact to the host cell and a functional T4SS is required to induce IL-8 expression, we focused on the H. pylori surface proteins that could be potential binding partners to the host cell, especially the T4SS surface protein CagL, a possible bridging adhesin to the host cells [28, 29]. To investigate the H. pylori-induced IL-8 promoter activity, we used gastric epithelial cells (AGS) transiently transfected with a human IL-8 promoter luciferase reporter construct. In parallel, IL-8 secretion of stimulated gastric epithelial cells was quantified by applying a specific IL-8 ELISA. Our data reveal for the first time that a specific C-terminal coiled-coil region of CagL plays a crucial role in H. pylori adherence to the epithelial cells and the hummingbird phenotype as well as IL-8 expression. Furthermore, we could demonstrate a signaling cascade of IL-8 induction by a CagL-dependent but focal adhesion kinase (FAK)-β1-integrin-independent mechanism that involves the activation of a transforming growth factor (TGF)-α and epidermal growth factor (EGF)-receptor (EGF-R) complex. These findings were determined by applying isogenic H. pylori CagL-mutant strains lacking the specific C-terminal coiled-coil region. These mutants were completely unable to induce IL-8 expression and secretion.
Materials and methods
Bacteria and cell lines
H. pylori strains were grown on gas chromatography (GC) agar plates (Oxoid, Wesel, Germany) supplemented with horse serum (5%), vancomycin (10 μg/ml), trimethoprim (5 μg/ml), and nystatin (1 μg/ml) (serum plates) and incubated for 2–3 days under microaerobic conditions (85% N2, 10% CO2, 5% O2) at 37 °C. Campylobacter jejuni C64 [30] was grown on Columbia blood agar plates (Oxoid) under microaerobic conditions.
Human gastric adenocarcinoma AGS (ATCC CRL 1739) were obtained from the American Type Culture Collection (Rockville, MD). The cells were cultured in RPMI 1640 (Invitrogen, Germany) supplemented with horse serum (10%) under standard conditions. Cells at 70% confluence were starved for 12 h in Nutrient Mixture F12 (Invitrogen) and then infected with a multiplicity of infection (MOI) of 100 for 5 h. The cell culture supernatants were preserved at –70 °C for quantification of IL-8.
Construction of ΔcagL mutants
Standard techniques were used for routine DNA manipulation, subcloning, and plasmid construction as previously described in Sambrook’s molecular cloning. To produce a H. pylori B128 ΔcagL mutant, the plasmid pSH6 was constructed. pSH6 is carrying a kanamycin cassette in a XbaI site between two adjacent nucleotide regions of about 1000 bp up- and downstream of the cagL gene. The polymerase chain reaction (PCR) products were generated by using genomic DNA from H. pylori B128 strain applying the primers for 5′-cagL fragment: FP 5′-CGGAGCT-CAGGTTCAGACATCTTGCTTGG-′3 and RP 5′-GCTC-TAGAGCATCTTCTTCACCCATTTC-′3 and 3′-cagL frag ment: FP 5′-CCTCTA GAGCCAATTTTGAAGC-GAATGAG-′3 and RP 5′-CGGAATTCGACAACACTT-GAGT GGTTTAAAAC-′3. The amplified products were cloned into the restriction sites ScaI and EcoRI of pBluescript SK+ (Stratagene, La Jolla, CA).
To construct the H. pylori B128 ΔcagL(1-206) and ΔcagL(1-224) mutants C-terminal shortened by 31 and 13 amino acids, respectively, PCR products were amplified covering the remaining 206 and 224 amino acids followed by an additional stop codon. For amplifying the ΔcagL(1-206) and ΔcagL(1-224), the following primers were used: FP for both 5′-GATCGAGCTCGGGAT-CAATGGAGAAATCAAAACC-′3 and RP ΔcagL(1-206): 5′-GATCGCATGCCTAATTTAAAAAGACCTTGTT-GTGAGC-′3, and RP ΔcagL(1-224): 5′-GATCGCAT-GCCTATTGCCGCTTACTTTGTTCTAGG-′3. For both constructs, the PCR product downstream of the cagL gene was amplified with following primer set: FP 5′-GATC-GCATGCTATGATTTCTAACCTTTGGGA-′3 and RP 5′-GATCGAATTCGCCACTAACGCTTTGAGAG-′3. The additional SphI site between the two PCR fragments is located in the noncoding region between the cagL and cagN genes. This restriction site was used for inserting the kanamycin cassette. All PCR products were cloned into the pBluescript SK+ vector opened with SacI and EcoRI to generate the plasmids pGR-LD1 (1-224) and pGR-LD2 (1-206).
The resulting plasmids were transformed into Escherichia coli DH5α and the reisolated plasmids (QIAprep Spin Miniprep Kit, Qiagen, Hilden, Germany) subjected to DNA sequencing to verify sequence integrity.
The deletion mutants were obtained by transforming the constructed plasmids pSH6, pGR-LD1, and pGR-LD2 into the natural competent H. pylori B128 cells kept in liquid culture medium (brucella broth plus 10% FCS). The transformed bacteria were selected on serum plates supplemented with kanamycin.
Quantification of IL-8
The concentration of IL-8 in the cell culture supernatants was determined by sandwich ELISA as described previously [31].
IL-8 promoter assays
Cells, grown to a confluency of 90% in RPMI 1640 (Gibco, Karlsruhe, Germany) supplemented with horse serum (10%), were transfected with the human IL-8 promoter (gift of A. Sing) and pE1 (β-Galactosidase) at a rate of 10:1 using the Lipofectamine 2000 reagent (Invitrogen, Karlsruhe, Germany) according to manufacturer’s protocol. After incubation with the transfection solution, cells were washed and then starved for 12 h in Nutrient Mixture F12 (HAM; GIBCO). Following stimulation with H. pylori for 5 h, cells were harvested in lysis buffer (14 g/l K2HPO4, 2.67 g/l KH2PO4, 0.74 g/l EDTA, 1 g/l Triton X-100, 1 nM DTT) and luciferase activity was measured (MicroLumat Plus LB 96 V; Berthold Technologies, Bad Wildbad, Germany). The results were normalized to β-galactosidase levels measured using the β-Gal Assay Kit (Invitrogen) according to manufacturer’s instructions.
RNA interference
AGS were transfected with 50 nM siRNA using Lipofectamine RNAiMAX Reagent (Invitrogen) according to manufacturer’s instructions. siRNAs against FAK, β1-integrin, and control siRNA (with equal GC content) were purchased from Invitrogen. Cells were harvested 24 h post-transfection with FAK, β1-integrin, or control siRNA in RLT buffer (Qiagen RNeasy Mini Kit) + 1% β-mercaptoethanol. RNA isolation was performed as described in the Qiagen RNeasy Mini Kit protocol. Using the TaqMan Reverse Transcription Reagents (Roche, Germany) with random primers according to the kit protocol, we transcribed 1 µg mRNA into cDNA. Oligonucleotide primers specific for FAK, β1-integrin, and the housekeeping gene 18S rRNA were applied for real-time reverse transcription (RT)-PCR (ABI PRISM 7000, Applied Biosystems). For the amplification step, the FastStart Universal SYBR Green Master (ROX) kit (Roche) was used according to manufacturer’s instructions (data not shown). Immunoblotting was performed to verify the FAK- and β1-integrin-knockdown rate on protein level.
Immunoblotting
To prepare the cell lysates for immunoblotting, cells were harvested in Dulbecco’s phosphate-buffered saline (DPBS) buffer (Invitrogen) and boiled in Laemmli sample buffer for 10 min [32]. The lysates were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) using a minigel apparatus (Bio-Rad, Germany) and blotted onto a polyvinylidene difluoride (PVDF) membrane using a semi-dry blot system (Biotec Fischer, Reis kirchen, Germany). The membranes were blocked with 3% bovine serum albumin (BSA) in Trisbuffered saline (TBS) buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl) and incubated overnight with a polyclonal anti-CagL antibody (AK271) [14]. Alkaline phosphatasecoupled protein A was used to visualize bound anti-CagL antibody and a peroxidase (POX)-labeled secondary anti-mouse antibody for detecting bound anti-β1-integrin (Millipore, Germany), anti-FAK (Santa Cruz), and anti α-tubulin (Upstate).
Immunofluorescence
AGS were infected with H. pylori at optical density (OD) of 0.2 for 5 h. The cells were fixed by 1% paraformaldehyde (PFA). After washing the cells with PBS and blocking with goat-serum, we applied an anti-H. pylori antibody (AK 175) [33]. Alexa488-conjugated secondary antibody (Molecular Probes, Germany) was used to visualize rabbit antibodies. To depict the whole cells, a counter stain with phalloidin (Sigma-Aldrich, Germany) was applied. The stained specimens were examined with Olympus BX61 microscope. Quantification of adherent H. pylori strains to AGS was performed by applying the quantification software “cell P” of Olympus.
Statistics
Data are presented as mean ± SEM. The results were statistically analyzed using the Student’s t test with Sigma-Stat statistical software. (***: P value < 0.001 was considered as highly significant; #: P value >0.05 was considered not significant).
Results
H. pylori-specific CagL-dependent IL-8 induction
Previous studies have described that H. pylori type I strains carrying a cag-PAI with a functional T4SS are able to induce IL-8 secretion in gastric epithelial cells [34, 35]. In 2001, Fischer et al. analyzed the importance of each single protein of the cag-PAI on IL-8 induction using a systematic mutagenesis approach [14]. In contrast to the importance of the T4SS, it could be shown that several major H. pylori virulence factors, such as CagA and VacA, play no crucial role in IL-8 induction [14, 17, 18, 36].
Our study supports that H. pylori transcriptional up-regulation (Fig. 1A) and H. pylori-induced IL-8 secretion (Fig. 1B) are not dependent on CagA but dependent on an intact T4SS, since there was only a small basal IL-8 expression in wells stimulated with the isogenic B128ΔcagY-mutant strain. The mutant strain carries a deletion in the cagY locus which is essential for assembly of the T4SS. In parallel, we verified that CagL, a protein localized on the tip of the T4SS-pilus [28], seems to play an essential role in the induction of IL-8 expression, since AGS stimulated with a B128ΔcagL-mutant strain did not express and secret IL-8. We also transfected AGS with a CagA overexpression plasmid in serum-free media and could not measure any secreted IL-8 in the supernatant (data not shown).
Fig. 1.
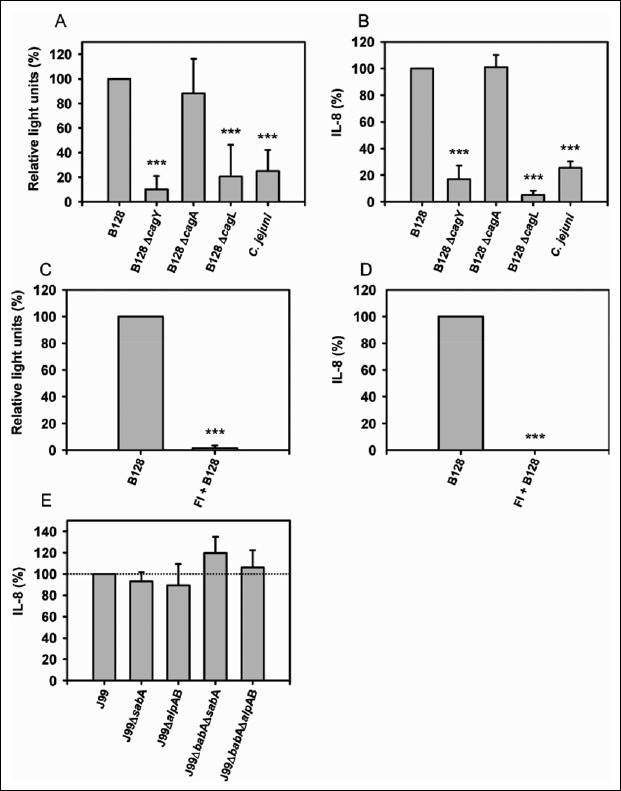
H. pylori type-I strain specific IL-8 expression. (A + B) AGS were stimulated with H. pylori B128 WT, isogenic mutant strains, and C. jejuni. (A, C) IL-8-promoter induction and (B, D) IL-8 secretion were determined in relation to H. pylori B128 WT (represents 100%). (C + D) Filter inserts (FI) were placed in each well, and AGS were stimulated with H. pylori B128 WT. (E) AGS were stimulated with H. pylori J99 WT and isogenic adhesin-mutant strains. IL-8 secretion was determined in relation to H. pylori J99 WT (represents 100%; interpolated dotted line). The mean values ± SEM of at least three independent experiments are shown. Statistical analysis (Student’s t test) was applied using SigmaStat software. ***P ≤ 0.001
To evaluate whether the increased IL-8 secretion is H. pylori-specific, we stimulated cells with another gastrointestinal pathogen C. jejuni. The results demonstrated a H. pylori-specific IL-8 induction because C. jejuni was not able to stimulate IL-8 expression (Fig. 1A, B).
Furthermore, we have elucidated whether the direct contact of the bacterium to the host cell is necessary to induce the IL-8 promoter activity as well as IL-8 secretion. The results demonstrated that the induction of the IL-8 promoter and the IL-8 secretion is dependent on a direct contact to the host cell. Parallel to that, by applying filter inserts, we were able to exclude that soluble H. pylori factors induce IL-8 expression without binding the bacterium to the epithelial cells (Fig. 1C, D). To establish persistent infection in the gastric mucosa, H. pylori mainly binds to the epithelial cell via the major bacterial adhesins BabA, SabA, and AlpAB [37–39]. We tested these adhesins on the ability to induce IL-8 secretion and revealed that H. pylori-induced IL-8 expression is independent of the major adhesins BabA, SabA, and AlpAB (Fig. 1E).
CagL C-terminal coiled-coil region is essential for IL-8 induction
CagL is a protein located at the tip of the T4SS that is proposed to be necessary for CagA translocation [28, 40]. To predict the 2D and 3D structure of this important protein, we submitted the CagL amino acid sequence of H. pylori B128 to the Expasy program (www.expasy.org) for analysis by the protein homology/analogy recognition engine (Phyre) software. The selected “Predict Secondary Structure” (PSIPRED) method of the protein structure prediction server at the UCL Bioinformatics Group, Bloomsbury Centre for Bioinformatics, London, UK (www.sbg.bio.ic.ac.uk/phyre/) is a highly accurate method for protein secondary structure prediction because it uses a very stringent cross validation method to evaluate the method’s performance [41]. The PSIPRED analysis of the CagL protein sequence determined a basic α-helix structure separated by eight coiled-coil motifs and one β-sheet region as well as an N- and C-terminal coiled-coil motif (Fig. 2A). Considering the predicted 3D structure obtained from the same program with the highest estimated precision of 95% (Fig. 2B), we concentrated on the C-terminal end of the protein with its distant α-helix arm ending in a coiled-coil structure. To identify a putative IL-8-inducing domain of the CagL protein, we constructed two isogenic mutant strains: one deprived of the whole C-terminal protein arm, named H. pylori B128 ΔcagL(1-206) (Fig. 2A, D), and the other mutant strain shortened by 13 amino acids covering the C-terminal coiled-coil region, named H. pylori B128 ΔcagL(1-224) (Fig. 2A, C). Both mutant strains, still expressing 206 and 224 amino acids of the CagL protein (Fig. 2E), were used together with the wild-type (WT) strain and an isogenic ΔcagL knock-out mutant for stimulating AGS directly or transiently transfected with the IL-8 promoter luciferase reporter construct. All three ΔcagL-mutant strains revealed a highly significant reduction of the IL-8 promoter expression (Fig. 2F) as well as IL-8 secretion (Fig. 2G) compared to the WT strain. Our data suggest that the C-terminal coiled-coil structure of the CagL protein is essential for IL-8 induction.
Fig. 2.

H. pylori-induced IL-8 expression is dependent on a C-terminal coiled-coil region of CagL. (A) Schematic view of the H. pylori CagL protein and its isogenic mutants DCagL(1-224) and DCagL(1-206). The proteins are drawn to scale as bars, with the number of amino acid residues from the genome sequence of strain H. pylori B128 WT. Blue color represents predicted a-helices, red color for coiled-coils, and yellow color for b-sheets analyzed by PSIPRED (www.sbg.bio.ic.ac.uk/phyre/). The numbers above the bars indicate the respective amino acids. (B–D) Three-dimensional structure of the H. pylori (B) CagL protein and its isogenic mutants (C) DCagL(1-224) and (D) DCagL(1-206) predicted by Phyre software with 95% estimated precision. (E) Western blot was performed to demonstrate that CagL was still expressed in B128DcagL(1-206) and B128DcagL(1-224)-mutant strains. (F, G) AGS were stimulated with H. pylori B128 WT and isogenic H. pylori B128DcagL, B128DcagL(1-206), and B128DcagL(1-224)-mutant strains. (F) IL-8-promoter induction and (G) IL-8 secretion were determined in relation to H. pylori B128 WT (represents 100%). The mean values ± SEM of at least three independent experiments are shown. Statistical analysis (Student’s t test) was applied using SigmaStat software. ***P ≤ 0.001
CagL C-terminal coiled-coil region is crucial for H. pylori-adherence
To examine whether the CagL protein and especially the C-terminal coiled-coil region play a crucial role in H. pylori adhesion to the epithelial cells, we tested the previously described CagL-mutant strains on their ability to adhere to gastric epithelial cells (AGS). Therefore, AGS were stimulated with the H. pylori B128 WT strain and its isogenic mutant strains B128ΔcagL, B128ΔcagL(1-224), and B128ΔcagL(1-206). The adherence patterns of the bacteria to the cell surface were analyzed via immunofluorescence. To visualize the cell-bound bacteria and the epithelial cells, we applied a fluorescein isothiocyanate (FITC)-labeled anti-H. pylori antibody and the actin-staining phalloidin, respectively. By quantification of the fluorescent adherent H. pylori, it could be demonstrated that all three CagL-mutant strains showed similar adherence patterns with a significantly reduced binding capability of approximately 80%, compared to the H. pylori B128 WT strain (Fig. 3A, B). Additionally, the CagL-mutant strains lost their ability to induce cell elongations (“hummingbird” phenotype) in comparison to the WT strain (Fig. 3C). These data reveal that the loss of the C-terminal coiled-coil region of the CagL protein is sufficient to considerably reduce H. pylori-binding efficiency to the epithelial cells as well as diminish cell elongation.
Fig. 3.

CagL C-terminal coiled-coil region is involved in H. pylori-adhesion. (A + C) AGS grown on chamber slides were stimulated with H. pylori B128 WT and isogenic H. pylori B128 ΔcagL, B128ΔcagL(1-206) and B128ΔcagL(1-224) mutant strains. The AGS were stained with phalloidin (red), the bacteria detected with anti-H. pylori antibody stained with FITC-labeled secondary antibody (green), and merged at 1:100 and 1:400 magnification (yellow: overlap of bacteria and cells). (B) Quantification of adherent H. pylori strains (B128 WT and isogenic B128 ΔcagL, B128ΔcagL(1-206) and B128ΔcagL(1-224) mutants) to AGS. The mean values ± SEM of at least three independent experiments are shown. Statistical analysis (Student’s t test) was applied using SigmaStat software. ***P ≤ 0.001. (C) AGS stimulated with H. pylori strains were detected as described above for “hummingbird” phenotype appearance
β1-integrin-independent IL-8 induction via TGF-α and EGF-R
Upon binding of H. pylori to the host cell, clustered integrins assemble into actin-rich structures called focal adhesions (FAs), where integrin signaling is mediated predominantly by Src and the focal adhesion kinase (FAK) [42]. In 2007, Kwok et al. were able to demonstrate that the translocation of CagA is dependent on binding of the H. pylori CagL protein to the β1-integrin receptor of the host cell [28]. Hence, we examined whether the binding to β1-integrin and the signal transduction via FAK play a central role in H. pylori CagL-mediated IL-8 induction.
To elucidate the role of FAK and β1-integrin in H. pylori-activated IL-8 expression, we applied a siRNA approach. The obtained data revealed a FAK- and β1-integrin-independent IL-8 induction mechanism (Fig. 4A–C). In parallel, a specific Src-kinase inhibitor was not able to inhibit H. pylori-stimulated IL-8 secretion (data not shown). With the knowledge that neither the β1-integrin host receptor nor FAK is involved in H. pylori IL-8 induction, we focused on host-cell signaling, especially on epidermal growth factor (EGF)-receptor (EGF-R) and its ligands activated upon H. pylori-binding. Therefore, the EGF-R and its major ligands EGF and transforming growth factor (TGF)-α were tested as potential candidates involved in the H. pylori-induced IL-8 signal transduction pathway. Measurements with the EGF-R tyrosine kinase inhibitor Gefitinib and a specific TGF-α catching antibody showed a significant decrease in H. pylori-induced IL-8 secretion. These data established the involvement of TGF-α and its receptor EGF-R in H. pylori-induced IL-8 signaling (Fig. 4D). In contrast, the natural agonist EGF was not important for H. pylori-induced IL-8 secretion.
Fig. 4.
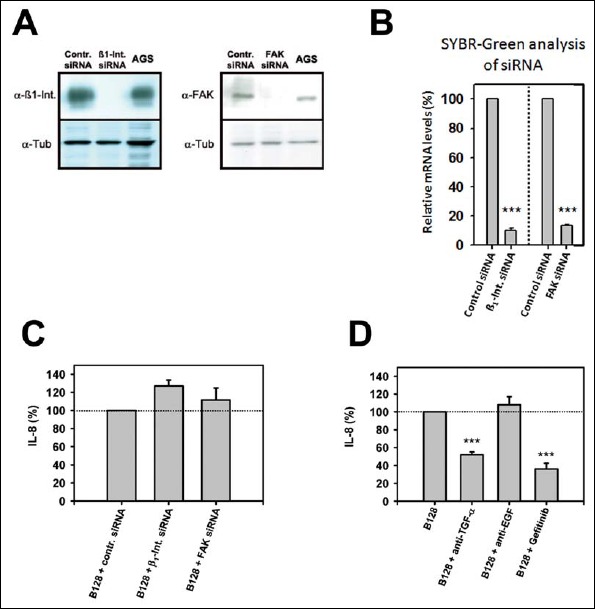
H. pylori-induced IL-8 secretion is EGF-R dependent. (A + B) AGS were transfected with β1-integrin siRNA (50 nM), FAK siRNA (50 nM), or control siRNA (50 nM) for 72 h and subsequently stimulated with H. pylori B128 WT. (A) Western blot was performed to demonstrate efficient β1-integrin and FAK silencing. (B) Relative mRNA levels of SYBR-Green analysis of β1-integrin and FAK siRNA in relation to control siRNA (100%). (C) IL-8 secretion was determined. (D) AGS were pre-incubated with anti-TGF-α (10 μg/ml) and anti-EGF (10 μg/ml) antibodies or EGF-R-inhibitor gefitinib (5 μM) and stimulated with H. pylori B128 WT. IL-8 secretion was determined in relation to H. pylori B128 WT (represents 100%; interpolated dotted line). The mean values ± SEM of at least three independent experiments are shown. Statistical analysis (Student’s t test) was applied using SigmaStat software. ***P ≤ 0.001
Discussion
In this study, we identified the domain of the H. pylori CagL protein in bacterial triggered IL-8 expression of gastric epithelial cells. Based on the following facts, we decided to investigate which domain of the bacterial ligand CagL is involved in H. pylori-induced IL-8 induction: 1) only cag-PAI-positive H. pylori strains with direct contact to gastric epithelial cells are capable of inducing IL-8 expression [14, 19, 34, 43], 2) the systematic mutagenesis screen of the H. pylori cag-PAI revealed an abolished IL-8 secretion in ΔcagL mutants [14, 44], 3) CagL is located on the tip of the T4SS [28], and 4) CagL is a bridging adhesion to host cell integrin [28, 45, 46].
In our study, we identified for the first time a C-terminal coiled-coil region in CagL which is responsible for H. pylori-induced IL-8 expression as well as host-cell adhesion. Our data prove that a deletion of the last 13 C-terminal amino acids of CagL is sufficient to block H. pylori-induced IL-8 expression and diminish bacterial adhesion and cell elongation. Our data support a protein interaction study by Kutter et al. who described predicted coiled-coil regions in nearly all T4SS proteins and speculated that their interactions are mediated by these domains [47].
The coiled-coil motif is a highly versatile assembly motif found in a wide range of structural proteins with functions ranging from the assembly of macromolecular complexes, signal transduction, and vesicular trafficking up to molecular recognition [48, 49]. In several gramnegative bacteria (e.g., Shigella flexneri, Salmonella spp., Yersinia spp., Pseudomonas aeroginosa), the presence and functions of coiled-coils in structural and regulatory proteins have been thoroughly explored [50, 51]. This is in sharp contrast to H. pylori where little is presently known about coiled-coil motifs and its functions.
To determine the host-cell receptor recognizing the CagL coiled-coil motif, we primarily focused on the described interaction of CagL with the β1-integin host receptor. This interaction based on the CagL arginine-glycine-aspartic acid (RGD) motif was demonstrated to be essential for CagA translocation into the gastric epithelial cell and activation of downstream cascades via FAK and Src [28, 40]. Recently, it could be elucidated that H. pylori T4SS exploits β1-integrin also in an RGD-independent manner [52]. It was also reported that other gram-negative pathogenic bacteria (e.g., Yersinia enterocolitica) use the β1-integin host receptor to induce IL-8 expression [53]. In contrast, our data revealed that neither β1-integin nor FAK is involved in H. pylori-associated IL-8 induction. Additionally, several p-integrin blocking antibodies were applied (β1, β2, β3, β4, β5, and β7) to screen for a putative CagL coiled-coil receptor on the host-cell surface which could be involved in IL-8 induction (data not shown). Consequently, it could be demonstrated that all tested integrins play no remarkable role in H. pylori-triggered IL-8 expression. Therefore, we assume that integrin is not the only interaction partner of CagL. This is in accordance with the findings of Backert et al. that it can also bind to human fibronectin in a RGD-independent manner [45].
Coiled-coil associations, with crucial roles in many physiological and pathological processes, structural simplicity, and a reversible nature, are promising targets for pharmacological interference, as already successfully established by botulinum toxins and viral fusion inhibitors [48].
In conclusion, our data lead us to propose a model of IL-8 induction whereby the C-terminal coiled-coil domain of the T4SS surface protein CagL is essential for bridging the T4SS to its host target cell using another receptor than integrins. Following the signal cascades, shedded TGF-α is binding to EGF-R [54, 56] and activates downstream regulatory elements such as NFκB, an important transcription factor for IL-8 induction. On the other side, we could demonstrate that this specific C-terminal coiled-coil domain is a bacterial adhesion ortholog to VirB5 of Agrobacterium spp. [45]. A deletion of this specific domain reduces the adhesion of the H. pylori strains to gastric epithelial cells and diminishes the cell elongation, since a ΔcagL mutant is deficient in CagA phosphorylation [57, 58].
Thus, infection with H. pylori induces the chemotactic cytokine IL-8 that is associated with severe gastric inflammation demonstrated by recruitment, migration, and activation of neutrophils into the sites of infection. Therefore, it is of great interest to clarify the molecular mechanisms involved in H. pylori CagL-triggered IL-8 induction. In the future, the major focus should concentrate on the elucidation of the host receptor for the CagL-mediated IL-8 induction via coiled-coil structures. The interference of this protein–protein interaction might offer an auspicious therapeutical approach to impair H. pylori binding to the gastric epithelial cells and to prevent H. pylori CagL-induced IL-8 secretion.
Funding Statement
Funding sources This work was supported by grants from the German Research Foundation (RI 972/3-1) and Förderprogramm für Forschung und Lehre (FöFoLe) (50/2007) to GR.
References
- 1.Houghton J, Wang TC: Helicobacter pylori and gastric cancer: a new paradigm for inflammation-associated epithelial cancers. Gastroenterology 128(6), 1567–1578 (2005) [DOI] [PubMed] [Google Scholar]
- 2.Lee A, Fox J, Hazell S: Pathogenicity of Helicobacter pylori: a perspective. Infect Immun 61(5), 1601–1610 (1993) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.El-Omar EM, Carrington M, Chow WH, McColl KE, Bream JH, Young HA, Herrera J, Lissowska J, Yuan CC, Rothman N, Lanyon G, Martin M, Fraumeni JF, Jr, Rabkin CS: Interleukin-1 polymorphisms associated with increased risk of gastric cancer. Nature 404(6776), 398–402 (2000) [DOI] [PubMed] [Google Scholar]
- 4.Rieder G, Fischer W, Haas R: Interaction of Helicobacter pylori with host cells: function of secreted and translocated molecules. Curr Opin Microbiol 8(1), 67–73 (2005) [DOI] [PubMed] [Google Scholar]
- 5.Ogura K Maeda S Nakao M Watanabe T Tada M Kyutoku T et al.: Virulence factors of Helicobacter pylori responsible for gastric diseases in Mongolian gerbil. J Exp Med 192(11), 1601–1610 (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rieder G, Merchant JL, Haas R: Helicobacter pylori cag-type IV secretion system facilitates corpus colonization to induce precancerous conditions in Mongolian gerbils. Gastroenterology 128(5), 1229–1242 (2005) [DOI] [PubMed] [Google Scholar]
- 7.Wiedemann T, Loell E, Mueller S, Stoeckelhuber M, Stolte M, Haas R, Rieder G: Helicobacter pylori cag-pathogenicity island-dependent early immunological response triggers later precancerous gastric changes in Mongolian gerbils. PLoS One 4(3), e4754 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baggiolini M, Loetscher P, Moser B: Interleukin-8 and the chemokine family. Int J Immunopharmacol 17(2), 103–108 (1995) [DOI] [PubMed] [Google Scholar]
- 9.Crabtree JE, Lindley IJ: Mucosal interleukin-8 and Helicobacter pylori-associated gastroduodenal disease. Eur J Gastroenterol Hepatol 6 Suppl 1, S33–38 (1994) [PubMed] [Google Scholar]
- 10.Ando T, Kusugami K, Ohsuga M, Shinoda M, Sakakibara M, Saito H, Fukatsu A, Ichiyama S, Ohta M: Interleukin-8 activity correlates with histological severity in Helicobacter pylori-associated antral gastritis. Am J Gastroenterol 91(6), 1150–1156 (1996) [PubMed] [Google Scholar]
- 11.Lee KE, Khoi PN, Xia Y, Park JS, Joo YE, Kim KK, Choi SY, Jung YD: Helicobacter pylori and interleukin-8 in gastric cancer. World J Gastroenterol 19(45), 8192–81202 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Siddique I, Al-Qabandi A, Al-Ali J, Alazmi W, Memon A, Mustafa AS, Junaid TA: Association between Helicobacter pylori genotypes and severity of chronic gastritis, peptic ulcer disease and gastric mucosal interleukin-8 levels: evidence from a study in the Middle East. Gut Pathog 6(1), 41 (2014) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chongruksut W, Limpakan Yamada S, Chakrabandhu B, Ruengorn C, Nanta S: Correlation of Helicobacter pylori and interleukin-8 mRNA expression in high risk gastric cancer population prediction. World J Gastrointest Oncol 8(2), 215–221 (2016) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fischer W, Puls J, Buhrdorf R, Gebert B, Odenbreit S, Haas R: Systematic mutagenesis of the Helicobacter pylori cag pathogenicity island: essential genes for CagA translocation in host cells and induction of interleukin-8. Mol Microbiol 42(5), 1337–1348 (2001) [DOI] [PubMed] [Google Scholar]
- 15.Barrozo RM, Cooke CL, Hansen LM, Lam AM, Gaddy JA, Johnson EM, Cariaga TA, Suarez G, Peek RM, Jr, Cover TL, Solnick J V: Functional plasticity in the type IV secretion system of Helicobacter pylori. PLoS Pathog 9(2), e1003189 (2013) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar Pachathundikandi S, Brandt S, Madassery J, Backert S: Induction of TLR-2 and TLR-5 expression by Helicobacter pylori switches cagPAI-dependent signalling leading to the secretion of IL-8 and TNF-alpha. PLoS One 6(5), e19614 (2011) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Nozawa Y, Nishihara K, Peek RM, Nakano M, Uji T, Ajioka H, Matsuura N, Miyake H: Identification of a signaling cascade for interleukin-8 production by Helicobacter pylori in human gastric epithelial cells. Biochem Pharmacol 64(1), 21–30 (2002) [DOI] [PubMed] [Google Scholar]
- 18.Owen RJ, Sharp S, Lawson AJ, Durrani Z, Rijpkema S, Kidd M: Investigation of the biological relevance of Helicobacter pylori cagE locus diversity, presence of CagA tyrosine phosphorylation motifs and vacuolating cytotoxin genotype on IL-8 induction in gastric epithelial cells. FEMS Immunol Med Microbiol 36(3), 135–140 (2003) [DOI] [PubMed] [Google Scholar]
- 19.Rieder G, Hatz RA, Moran AP, Walz A, Stolte M, Enders G: Role of adherence in interleukin-8 induction in Helicobacter pylori-associated gastritis. Infect Immun 65(9), 3622–3630 (1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viala J, Chaput C, Boneca IG, Cardona A, Girardin SE, Moran AP, Athman R, Mémet S, Huerre MR, Coyle AJ, DiStefano PS, Sansonetti PJ, Labigne A, Bertin J, Philpott DJ, Ferrero RL: Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat Immunol 5(11), 1166–1174 (2004) [DOI] [PubMed] [Google Scholar]
- 21.Irving AT, Mimuro H, Kufer TA, Lo C, Wheeler R, Turner LJ, Thomas BJ, Malosse C, Gantier MP, Casillas LN, Votta BJ, Bertin J, Boneca IG, Sasakawa C, Philpott DJ, Ferrero RL, Kaparakis-Liaskos M: The immune receptor NOD1 and kinase RIP2 interact with bacterial peptidoglycan on early endosomes to promote autophagy and inflammatory signaling. Cell Host Microbe 15(5), 623–635 (2014) [DOI] [PubMed] [Google Scholar]
- 22.Aihara M, Tsuchimoto D, Takizawa H, Azuma A, Wakebe H, Ohmoto Y, Imagawa K, Kikuchi M, Mukaida N, Matsushima K: Mechanisms involved in Helicobacter pylori-induced interleukin-8 production by a gastric cancer cell line, MKN45. Infect Immun 65(8), 3218–3224 (1997) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Brandt S, Kwok T, Hartig R, Konig W, Backert S: NF-kappaB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc Natl Acad Sci U S A 102(26), 9300–9305 (2005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chu SH, Kim H, Seo JY, Lim JW, Mukaida N, Kim KH: Role of NF-kappaB and AP-1 on Helicobater pylori-induced IL-8 expression in AGS cells. Dig Dis Sci 48(2), 257–265 (2003) [DOI] [PubMed] [Google Scholar]
- 25.Seo JH, Lim JW, Kim H, Kim KH: Helicobacter pylori in a Korean isolate activates mitogen-activated protein kinases, AP-1, and NF-kappaB and induces chemokine expression in gastric epithelial AGS cells. Lab Invest 84(1), 49–62 (2004) [DOI] [PubMed] [Google Scholar]
- 26.Naumann M, Wessler S, Bartsch C, Wieland B, Covacci A, Haas R, Meyer TF: Activation of activator protein 1 and stress response kinases in epithelial cells colonized by Helicobacter pylori encoding the cag pathogenicity island. J Biol Chem 274(44), 31655–31662 (1999) [DOI] [PubMed] [Google Scholar]
- 27.Sharma SA, Tummuru MK, Blaser MJ, Kerr LD: Activation of IL-8 gene expression by Helicobacter pylori is regulated by transcription factor nuclear factor-kappa B in gastric epithelial cells. J Immunol 160(5), 2401–2407 (1998) [PubMed] [Google Scholar]
- 28.Kwok T, Zabler D, Urman S, Rohde M, Hartig R, Wessler S, Misselwitz R, Berger J, Sewald N, König W, Backert S: Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 449(7164), 862–866 (2007) [DOI] [PubMed] [Google Scholar]
- 29.Pham KT, Weiss E, Jimenez Soto LF, Breithaupt U, Haas R, Fischer W: CagI is an essential component of the Helicobacter pylori Cag type IV secretion system and forms a complex with CagL. PLoS One 7(4), e35341 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gebert B, Fischer W, Weiss E, Hoffmann R, Haas R: Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 301(5636), 1099–1102 (2003) [DOI] [PubMed] [Google Scholar]
- 31.Yasumoto K, Okamoto S, Mukaida N, Murakami S, Mai M, Matsushima K: Tumor necrosis factor alpha and interferon gamma synergistically induce interleukin 8 production in a human gastric cancer cell line through acting concurrently on AP-1 and NF-kB-like binding sites of the interleukin 8 gene. J Biol Chem 267(31), 22506–22511 (1992) [PubMed] [Google Scholar]
- 32.Laemmli UK: Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227(5259), 680–685 (1970) [DOI] [PubMed] [Google Scholar]
- 33.Odenbreit S, Puls J, Sedlmaier B, Gerland E, Fischer W, Haas R: Translocation of Helicobacter pylori CagA into gastric epithelial cells by type IV secretion. Science 287(5457), 1497–1500 (2000) [DOI] [PubMed] [Google Scholar]
- 34.Selbach M, Moese S, Meyer TF, Backert S: Functional analysis of the Helicobacter pylori cag pathogenicity island reveals both VirD4-CagA-dependent and VirD4-CagA-independent mechanisms. Infect Immun 70(2), 665–671 (2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Naumann M: Pathogenicity island-dependent effects of Helicobacter pylori on intracellular signal transduction in epithelial cells. Int J Med Microbiol 295(5), 335–341 (1995) [DOI] [PubMed] [Google Scholar]
- 36.Huang J, O’Toole PW, Doig P, Trust TJ: Stimulation of interleukin-8 production in epithelial cell lines by Helicobacter pylori. Infect Immun 63(5), 1732–1738 (1995) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Linden S, Nordman H, Hedenbro J, Hurtig M, Boren T, Carlstedt I: Strain- and blood group-dependent binding of Helicobacter pylori to human gastric MUC5AC glycoforms. Gastroenterology 123(6), 1923–1930 (2002) [DOI] [PubMed] [Google Scholar]
- 38.Mahdavi J, Sonden B, Hurtig M, Olfat FO, Forsberg L, Roche N, Angstrom J, Larsson T, Teneberg S, Karlsson KA, Altraja S, Wadström T, Kersulyte D, Berg DE, Dubois A, Petersson C, Magnusson KE, Norberg T, Lindh F, Lundskog BB, Arnqvist A, Hammarström L, Borén T: Helicobacter pylori SabA adhesin in persistent infection and chronic inflammation. Science 297(5581), 573–578 (2002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Odenbreit S, Till M, Hofreuter D, Fal ler G, Haas R: Genet ic and functional characterization of the alpAB gene locus essential for the adhesion of Helicobacter pylori to human gastric tissue. Mol Microbiol 31(5), 1537–1548 (1999) [DOI] [PubMed] [Google Scholar]
- 40.Con radi J, Tegtmeyer N, Wozna M, Wissbrock M, Michalek C, Gagell C, Cover TL, Frank R, Sewald N, Backert S: An RGD helper sequence in CagL of Helicobacter pylori assists in interactions with integrins and injection of CagA. Front Cell Infect Microbiol 2, 70 (2012) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jones DT: Protein secondary structure prediction based on position-specific scoring matrices. J Mol Biol 292(2), 195–202 (1999) [DOI] [PubMed] [Google Scholar]
- 42.Mitra SK, Schlaepfer DD: Integrin-regulated FAK-Src signaling in normal and cancer cells. Curr Opin Cell Biol 18(5), 516–523 (2006) [DOI] [PubMed] [Google Scholar]
- 43.Nilsson C, Sillen A, Eriksson L, Strand ML, Enroth H, Normark S, Falk P, Engstrand L: Correlation between cag pathogenicity island composition and Helicobacter pylori-associated gastroduodenal disease. Infect Immun 71(11), 6573–6578 (2003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Covacci A, Rappuoli R: Tyrosine-phosphorylated bacterial proteins: Trojan horses for the host cell. J Exp Med 191(4), 587–592 (2000) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Backert S, Fronzes R, Waksman G: VirB2 and VirB5 proteins: specialized adhesins in bacterial type-IV secretion systems? Trends Microbiol 16(9), 409–413 (2008) [DOI] [PubMed] [Google Scholar]
- 46.Barden S, Lange S, Tegtmeyer N, Conradi J, Sewald N, Backert S, Niemann HH: A helical RGD motif promoting cell adhesion: crystal structures of the Helicobacter pylori type IV secretion system pilus protein CagL: structure 21(11), 1931–1941 (2013) [DOI] [PubMed] [Google Scholar]
- 47.Kutter S, Buhrdorf R, Haas J, Schneider-Brachert W, Haas R, Fischer W: Protein subassemblies of the Helicobacter pylori Cag type IV secretion system revealed by localization and interaction studies. J Bacteriol 190(6), 2161–2171 (2008) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strauss HM, Keller S: Pharmacological interference with protein–protein interactions mediated by coiled-coil motifs. Handb Exp Pharmacol (186), 461–482 (2008) [DOI] [PubMed] [Google Scholar]
- 49.Lupas A: Coiled coils: new structures and new functions. Trends Biochem Sci 21(10), 375–382 (1996) [PubMed] [Google Scholar]
- 50.Gazi AD, Charova SN, Panopoulos NJ, Kokkinidis M: Coiled-coils in type III secretion systems: structural flexibility, disorder and biological implications. Cell Microbiol 11(5), 719–729 (2009) [DOI] [PubMed] [Google Scholar]
- 51.Delahay RM, Frankel G: Coiled-coil proteins associated with type III secretion systems: a versatile domain revisited. Mol Microbiol 45(4), 905–916 (2002) [DOI] [PubMed] [Google Scholar]
- 52.Jimenez-Soto LF, Kutter S, Sewald X, Ertl C, Weiss E, Kapp U, et al. : Helicobacter pylori type IV secretion apparatus exploits beta1 integrin in a novel RGD-independent manner. PLoS Pathog 5(12), e1000684 (2009) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Grassl GA, Kracht M, Wiedemann A, Hoffmann E, Aepfelbacher M, von Eichel-Streiber C, Bohn E, Autenrieth IB: Activation of NF-kappaB and IL-8 by Yersinia enterocolitica invasin protein is conferred by engagement of Rac1 and MAP kinase cascades. Cell Microbiol 5(12), 957–971 (2003) [DOI] [PubMed] [Google Scholar]
- 54.Keates S, Keates AC, Katchar K, Peek RM, Jr, Kelly CP: Helicobacter pylori induces up-regulation of the epidermal growth factor receptor in AGS gastric epithelial cells. J Infect Dis 196(1), 95–103 (2007) [DOI] [PubMed] [Google Scholar]
- 55.Joh T Kataoka H Tanida S Watanabe K Ohshima T Sasaki M et al.: Helicobacter pylori-stimulated interleukin-8 (IL-8) promotes cell proliferation through transactivation of epidermal growth factor receptor (EGFR) by disintegrin and metalloproteinase (ADAM) activation. Dig Dis Sci 50(11), 2081–2089 (2005) [DOI] [PubMed] [Google Scholar]
- 56.Wallasch C, Crabtree JE, Bevec D, Robinson PA, Wagner H, Ullrich A: Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HBEGF. Biochem Biophys Res Commun 295(3), 695–701 (2002) [DOI] [PubMed] [Google Scholar]
- 57.Backert S, Selbach M: Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol 10(8), 1573–1581 (2008) [DOI] [PubMed] [Google Scholar]
- 58.Mimuro H, Berg DE, Sasakawa C: Control of epithelial cell structure and developmental fate: lessons from Helicobacter pylori. Bioessays 30(6), 515–520 (2008) [DOI] [PubMed] [Google Scholar]