

Abstract
Acute or chronic viral infections can lead to generalized immunosuppression. Several mechanisms, such as immunopathology of CD8+ T cells, inhibitory receptors, or regulatory T (Treg) cells, contribute to immune dysfunction. Moreover, patients with chronic viral infections usually do not respond to vaccination, a finding that has not been previously explained. Recently, we reported that CD169+ macrophages enforce viral replication, which is essential for guaranteeing antigen synthesis and efficient adaptive immune responses. In the present study, we used a chronic lymphocytic choriomeningitis virus infection mouse model to determine whether this mechanism is affected by chronic viral infection, which may impair the activation of adaptive immunity. We found that enforced viral replication of a superinfecting virus is completely blunted in chronically infected mice. This absence of enforced viral replication in CD169+ macrophages is not explained by CD8+ T‐cell‐mediated immunopathology but rather by prolonged IFN‐I responses. Consequently, the absence of viral replication impairs both antigen production and the adaptive immune response against the superinfecting virus. These findings indicate that chronic infection leads to sustained IFN‐I action, which is responsible for the absence of an antiviral immune response against a secondary viral infection.
Keywords: CD169+ macrophages, Chronic infection, Enforced virus replication, Lymphocytic choriomeningitis virus, Vesicular stomatitis virus
Introduction
Many acute and chronic viral infections induce a generalized immunosuppression 1, 2 in a broad range of host species 3, 4. During acute infection, immunosuppression is usually transient but can last for several months after the initial replication has been controlled. In contrast, persistent infection with certain viruses can lead to lifelong immunosuppression 5.
In humans, acute infection with the measles virus can cause a generalized immunosuppression lasting for several months and can facilitate bacterial superinfection 6, 7, 8. Persistent infections with a low viral load, such as CMV and EBV infections, are known to negatively influence the immune response against other viruses (i.e. influenza virus) 9, 10. An increase in the number of regulatory T cells in mice chronically infected with Friend retrovirus suppresses immunity to a superinfection with murine CMV 11.
Infection with HIV or with human T‐cell lymphotropic virus leads to severe immunosuppression and a poor response against infection with several opportunistic pathogens 1, 12. This finding is explained primarily by the infection of CD4+ T cells. Additionally, several studies using the lymphocytic choriomeningitis virus (LCMV) model have shown that NK cells, the development or reduction in the number of dendritic cells (DCs), and CD8+ T‐cell‐mediated immunopathology contribute to immunosuppression 2, 13, 14, 15. This finding is in line with observations from studies of infections with other viruses showing that constant activation of CD8+ T cells in lymph nodes and spleen alters immune function 2, 11. It has been reported that LCMV infects B cells that produce neutralizing antibodies, which will be eliminated by immunopathological LCMV‐specific CD8+ T cells 2. This LCMV infection also induces a highly specific immunosuppression that delays the responses of neutralizing antibody and thereby enhances viral persistence 5, 13, 16. A comparison of various strains of LCMV found that a higher rate of viral replication is associated with the severity and duration of immunosuppression. For example, infection with the LCMV strain Docile, which causes chronic infection, leads to a strong suppression of the immune system, whereas infection with the LCMV strain Armstrong causes only slight immunosuppression 5. Although the immunosuppression is explained by the immunopathology related to CD8+ T cells, it still remains unclear why mice infected with LCMV‐Docile exhibit immunosuppression despite the exhaustion of CD8+ T cells.
Recent studies have shown that chronic infection with LCMV Clone 13 leads to an increase in the production of type I IFN, which in turn can increase the production of the immunosuppressive cytokine IL‐10 17, 18. Studies using monkeys such as sooty mangabeys and rhesus macaques found that the onset of acquired immunodeficiency syndrome (AIDS) is strongly correlated with levels of type I IFN 19. The mechanism by which type I IFN induces immunosuppression remains unknown.
Recently we reported that special APCs, such as metallophilic marginal zone macrophages (CD169+), in the spleen and DCs express Usp18, which regulates host cell susceptibility to type I IFN and supports viral replication 20, 21. This enforced viral replication is essential for initiating innate and adaptive immune responses. Whether the mechanism of enforced viral replication is influenced by chronic viral infection remains unknown.
Results
Chronic infection limits a secondary antiviral immune response
To determine the effect of a chronic infection on the suppression of the adaptive immune system, we infected C57BL/6 mice with LCMV strain Docile 30 days before superinfection with the vesicular stomatitis virus (VSV). We infected another group of C57BL/6 mice with VSV alone to serve as controls (Fig. 1A). We measured the production of VSV‐neutralizing antibodies in the serum at various time points. We found that chronic infection with LCMV Docile blunted the induction of total neutralizing antibodies (IgM and IgG) and of neutralizing IgG against VSV (Fig. 1B). This shows that chronic infection can lead to suppression of the activity of the adaptive immune system against a secondary viral infection.
Figure 1.

Chronic infection limits antiviral immune response. (A) Experimental protocol shows infection time points. (B) C57BL/6 WT mice were chronically infected with 2 × 104 PFU LCMV strain Docile (LCMV‐Docile) for 30 days or left uninfected. VSV‐neutralizing antibodies were measured in chronically LCMV‐infected or uninfected WT mice intravenously infected with 1 × 104 PFU VSV at the indicated time points by neutralizing assay. Data are expressed as mean ± SEM (n = 4–6 mice/group) and are pooled from two independent experiments. ***p < 0.001. Statistical significance was detected with ANOVA.
Enforced viral replication is blunted in chronically infected mice
In a previous study we found that enforced viral replication is essential for activating adaptive immunity and producing neutralizing antibodies against VSV 20. We questioned whether chronic infection may influence the mechanism of enforced viral replication. To study this hypothesis, we infected mice with LCMV‐Docile for 30 days. Mice were then superinfected with VSV for 7 h, and the viral titer in spleen and inguinal lymph nodes was determined. Control mice were infected with VSV alone. VSV replication was lower in mice infected with LCMV‐Docile than in control mice (Fig. 2A).
Figure 2.
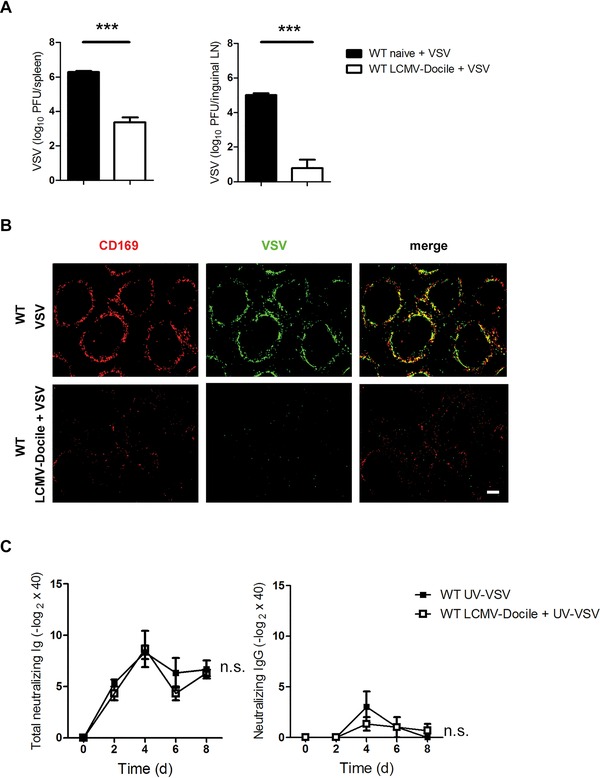
Enforced viral replication is blunted in chronically infected mice. (A) C57BL/6 WT mice were chronically infected with 2 × 104 PFU LCMV strain Docile (LCMV‐Docile) for 30 days or left uninfected. In addition, mice were infected with 2 × 108 PFU VSV for 7 h. VSV titers in spleen and inguinal lymph nodes were measured by plaque assay (n = 3–6). (B) C57BL/6 WT mice were intravenously infected with 2 × 104 PFU LCMV‐Docile for 30 days or left uninfected. Mice were then infected with 2 × 108 PFU VSV for 7 h. Immunofluorescence of spleen sections stained for VSV glycoprotein (green) and CD169 (red). Scale bar = 100 μm (n = 3). Fluorescent images were captured at 10x magnification using Keyence BZ‐9000E microscope. (C) C57BL/6 WT mice were chronically infected with 2 × 104 PFU LCMV‐Docile for 30 days or left uninfected. VSV‐neutralizing antibodies were measured in chronically LCMV‐infected or uninfected WT mice intravenously immunized with 2 × 108 PFU UV‐light inactivated VSV (n = 3). (A and C) Data are expressed as means ± SEM and are representative of two independent experiments. .n.s.: not significant; ***p < 0.001. Statistically significant differences were detected with Student's t‐test (A) or ANOVA.
The reduction of VSV replication by chronic LCMV infection was additionally assessed by immunohistological staining of spleen sections (Fig. 2B). To determine whether this immunosuppression was due to the inhibition of enforced viral replication, we used VSV that had been inactivated with UV light to immunized naive mice and mice with chronic LCMV infection. Both groups of mice exhibited similar titers of total neutralizing and IgG‐neutralizing antibodies (Fig. 2C), a finding suggesting that B cells can still produce antibodies during chronic infection. These findings suggest that the persistence of LCMV infection could inhibit enforced viral replication of the superinfected virus.
CD8+ T cells do not prevent immunosuppression during chronic infection
Next, we questioned whether this suppression of the B‐cell response was due to CD8+ T‐cell‐mediated cytotoxicity during chronic LCMV infection. To answer this question, we infected perforin‐deficient mice (Prf−/−) with LCMV‐Docile, and 30 days later we superinfected them with VSV. A control group of Prf−/− mice was infected with VSV alone. Perforin is the primary cytotoxic effector protein in CD8+ T cells 22. We found that Prf−/− mice infected with chronic LCMV and then superinfected with VSV exhibit a much more impaired B‐cell response than do Prf−/− mice infected with VSV alone (Fig. 3A). Superinfected Prf− / − mice also exhibited an inhibition in VSV replication similar to that of coinfected WT mice (Figs. 2B and 3B). These findings exclude a cytotoxic role of CD8+ T cells in immunosuppression and suggest that another perforin‐independent mechanism leads to immunosuppression during chronic LCMV infection. In order to completely exclude the role of CD8+ T cells and not only perforin‐mediated cytotoxic effect, we infected MHC‐I‐deficient mice with LCMV‐Docile, and after 30 days we superinfected them with VSV. As control, we infected MHC‐I‐deficient mice only with VSV. Similar to Prf−/− mice, deletion of MHC‐I did not prevent the immunosuppression induced by chronic LCMV‐Docile infection (Fig. 3C). Additionally, superinfected MHC‐I−/− mice showed an inhibition in VSV replication similar to superinfected WT mice (Figs. 2B and 3D). We conclude that CD8+ T cells are not involved in the immunosuppression.
Figure 3.
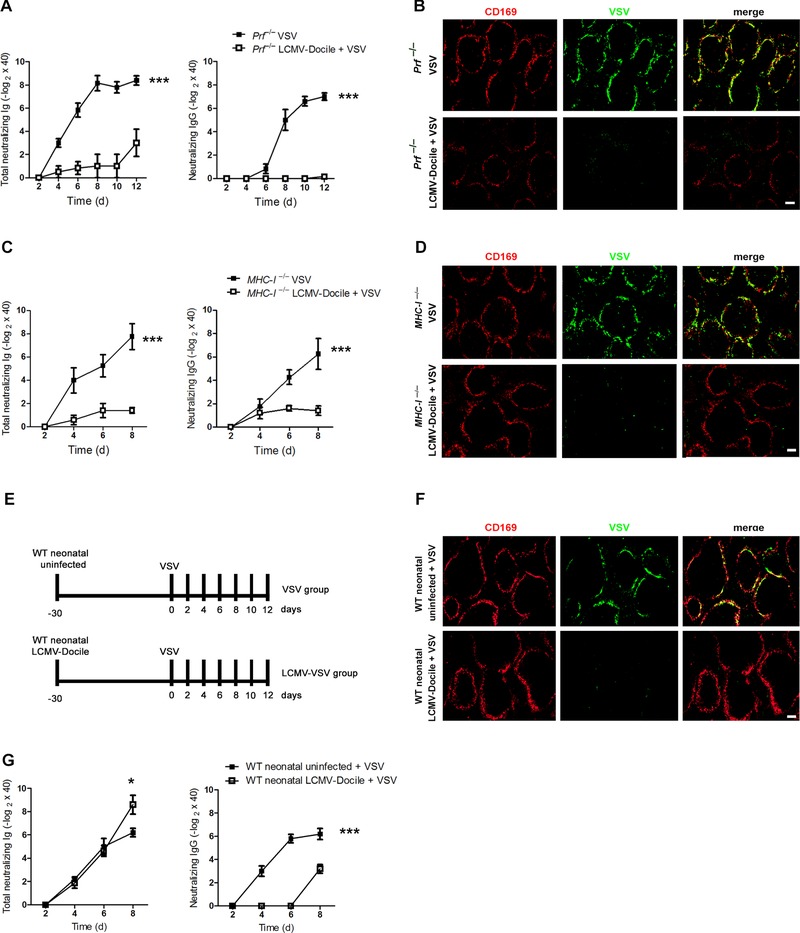
CD8+ T cells do not prevent immunosuppression during chronic infection. (A, C, and G): (A) Perforin‐deficient (Prf−/−), (C) MHC‐I‐deficient (MHC‐I−/−), or (G) C57BL/6 neonatal mice were chronically infected with 2 × 104 PFU LCMV strain Docile (LCMV‐Docile) or were left uninfected. After 30 days, mice were intravenously infected with 1 × 104 PFU VSV. VSV‐neutralizing antibodies were measured by neutralizing assay at the indicated time points ((A and C) n = 4–6; (G) n = 5–8). Data are expressed as mean ± SEM and are pooled from two independent experiments. *p < 0.5; ***p < 0.001. Statistical significance was detected with ANOVA; A, C, and G; IgG) or Student's t‐test (G; total Ig). (B, D, and F): (B) Prf−/−, (D) MHC‐I−/−, and (F) neonatal C57BL/6 WT mice were intravenously infected with 2 × 104 PFU LCMV‐Docile for 30 days or left uninfected. Mice were then infected with 2 × 108 PFU VSV for 7 h. Immunofluorescence of spleen sections stained for VSV glycoprotein (green) and CD169 (red). Fluorescent images were captured at 10x magnification using Keyence BZ‐9000E microscope. Scale bar = 100 μm. Images shown are representative of three independent experiments. (E) Experimental protocol shows infection time points.
Persistent viral infection can also occur if newborn mice are infected with virus. In this case, central tolerance is induced against the virus, and virus‐specific T cells are deleted 23. Therefore, we questioned whether even with the deletion of LCMV‐specific T cells, the immunosuppression and blunting of viral replication during neonatal infection is taking place. We therefore infected neonatal mice with LCMV‐Docile and then superinfected them with VSV. As controls, another group of C57BL/6 mice was infected with VSV alone (Fig. 3E). After superinfection with VSV, VSV replication was blunted in mice infected with LCMV as neonates (Fig. 3F). In addition, mice infected as neonates exhibited a slight reduction in the titers of total neutralizing antibody (IgM and IgG) and a massive reduction in the titers of neutralizing IgG against VSV (Fig. 3G). These findings led us to conclude that enforced viral replication is also blunted in mice infected as neonates, a conclusion that can explain the reduction in the adaptive immune response after secondary viral infection.
Prolonged activity of type I IFN inhibits enforced viral replication during chronic infection
Chronic LCMV infection leads to prolonged type I IFN responses 18, 24. We hypothesized that enhanced type I IFN responses would then induce an antiviral status via Oas1 and Mx1, which render APCs resistant to viral replication. We measured gene expression in spleens stimulated with type I IFN during chronic LCMV infection and compared it with gene expression under naïve conditions. Indeed, we found that the expression of Stat1, Irf7, Oas1, and Mx1 was higher during chronic infection (Fig. 4A). Moreover, chronically infected mice exhibited stronger expression of Ifnα4 and Ifnβ1 than did uninfected mice (Fig. 4A). To determine whether mice infected with LCMV as neonates also exhibit stronger type I IFN signaling and higher type I IFN expression, we measured the expression of these genes. As expected, mice infected as neonates exhibit sustained type I IFN activity (Fig. 4B).
Figure 4.
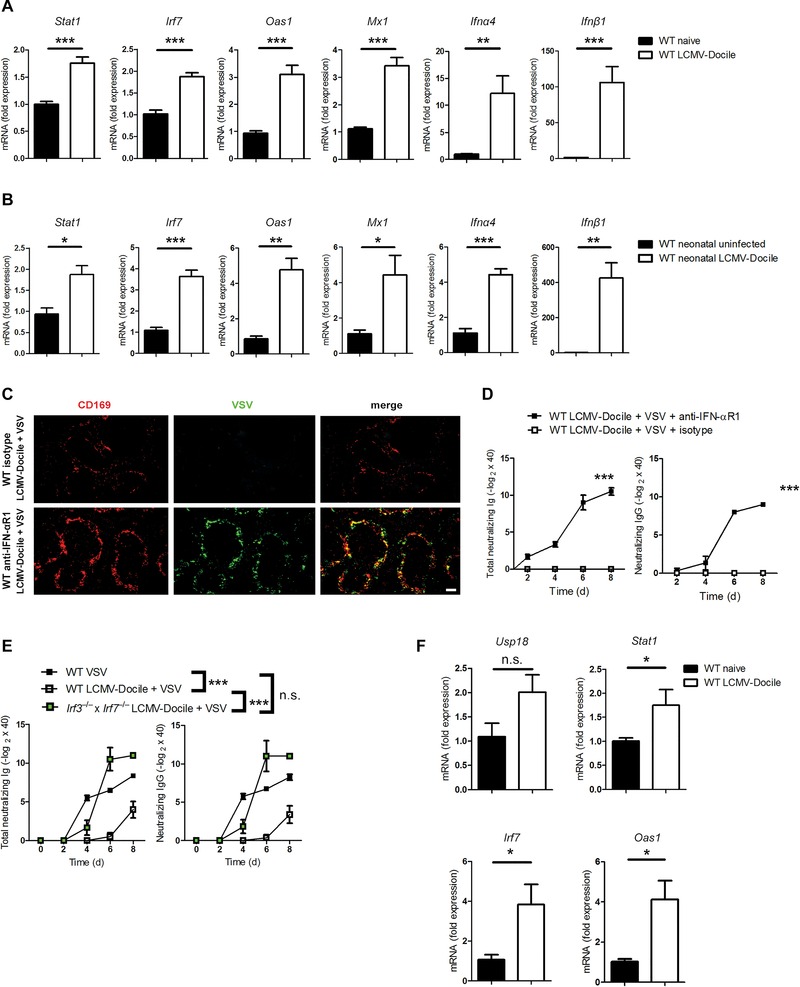
Prolonged action of type I IFN is responsible for the inhibition of enforced viral replication. (A) C57BL/6 WT mice were infected with LCMV strain Docile (LCMV‐Docile) or left uninfected. The expression of indicated genes was measured by quantitative real‐time PCR (qRT‐PCR) on day 30 after infection (n = 4–6). (B) The expression of indicated genes was measured by qRT‐PCR 30 days after neonatal C57BL/6 WT mice had been infected with LCMV‐Docile or left uninfected (n = 4). (C) C57BL/6 WT mice were intravenously infected with 2 × 104 PFU LCMV‐Docile for 30 days or left uninfected. Mice were then treated with anti‐IFN‐αR1 or isotype control antibody and infected with 2 × 108 PFU VSV for 7 h. Immunofluorescence of spleen sections stained for VSV glycoprotein (green) and CD169 (red). Fluorescent images were captured at 10x magnification using Keyence BZ‐9000E microscope. Scale bar = 100 μm (n = 3). (D) C57BL/6 WT mice were intravenously infected with 2 × 104 PFU LCMV‐Docile for 30 days or left uninfected. Mice were then treated with anti‐IFN‐αR1 or isotype control antibody and infected with 1 × 104 PFU VSV. VSV‐neutralizing antibodies were measured at the indicated time points (n = 3–6). (E) WT and Irf3−/− × Irf7−/− mice were chronically infected with LCMV‐Docile for 30 days. Control WT mice were left uninfected. Mice were then infected with 1 × 104 PFU VSV. VSV‐neutralizing antibodies were measured at the indicated time points (n = 6–8). (F) C57BL/6 WT mice were infected with LCMV‐Docile or left uninfected. The expression of indicated genes by FACS‐sorted CD169+ macrophages was measured by qRT‐PCR on day 30 after infection (n = 3). (A, B, and D–F) Data are expressed as means ± SEM and are pooled from two independent experiments. n.s.: not significant; *p < 0.5; **p < 0.01; ***p < 0.001. Statistical significance was detected by Student's t‐test (A, B, and F) or ANOVA (D and E).
To determine whether this prolonged type I IFN activity can inhibit the enforced viral replication of VSV, we infected WT mice with LCMV‐Docile and after 30 days we treated them with anti‐IFN‐αR1 or isotype control antibody. Afterward, we superinfected them with VSV. Immunohistological staining of spleen sections showed higher replication of VSV in superinfected WT mice that were treated with anti‐IFN‐αR1 antibody in comparison to control WT mice (Fig. 4C). This replication was comparable to WT mice that were infected only with VSV (Fig. 2B). To demonstrate that VSV replication in the absence of the type I IFN effect is sufficient to stimulate the adaptive immune system during chronic infection, we treated LCMV‐Docile‐infected mice with anti‐IFN‐αR1 antibody before superinfection with VSV and measured the production of VSV‐neutralizing antibodies. Indeed, blocking of IFN‐αR1 prevented immunosuppression and mice showed normal production of neutralizing antibodies comparing to control mice (Fig. 4D). Moreover, we used mice double‐deficient in Irf3 and Irf7 (Irf3−/− × Irf7−/−), which exhibit a defect in the production of type I IFN 25. Irf3−/− × Irf7−/− mice are less susceptible to death after infection with VSV than Ifnar−/− mice. We infected Irf3−/− × Irf7−/− mice with LCMV‐Docile and superinfected 30 days later with VSV. The production of neutralizing antibodies against VSV was comparable to that in WT mice that had been infected with VSV alone (Fig. 4E). Therefore, we concluded that chronic viral infection inhibits enforced viral replication by inducing antiviral effector genes. The absence of enforced viral replication limits the adaptive immune response.
In our previous work we found that naive CD169+ macrophages exhibit higher expression of Usp18 than do F4/80+ macrophages, and this increase in expression guarantees enforced viral replication 20. We then questioned why Usp18 expression in CD169+ macrophages could not guarantee VSV replication during chronic LCMV infection. We sorted CD169+ macrophages from naive mice and from mice chronically infected with LCMV (Supporting Information Fig. 1) and compared the expression of type I IFN‐induced genes by these macrophages with their expression by naive CD169+ macrophages. We found a slight increase in Usp18 expression after chronic LCMV infection (Fig. 4F) but a substantial increase in the expression type I IFN‐induced genes (Fig. 4F). This finding explains the blunting of enforced viral replication in these cells during chronic infection.
Discussion
In this study, we found that constant antiviral action of type I IFN during chronic infection inhibits the enforced viral replication of a secondary viral infection in specific APCs, such as CD169+ macrophages. The resulting reduction in the level of the antigen limits the activation of the adaptive immune responses against the secondary viral pathogen and consequently leads to immune failure independent of CD8+ T cells. Earlier studies showed that immunosuppression is due to cytotoxic CD8+ T cells 2, 26. The conclusion was that immunopathological LCMV‐specific CD8+ T cells mediate the elimination of LCMV‐specific and LCMV‐nonspecific infected B cells, and that this elimination prevents the reaction of the immune system against a secondary infection. In the present study we found that early enforced viral replication, which is essential for the activation of the adaptive immune system, is absent in chronically infected mice.
As we showed in a previous study, CD169+ macrophages are essential for enforced viral replication of VSV, and this replication guarantees the activation of the adaptive immune system and the control of the virus 20. Protecting CD169+ macrophages from the cytotoxicity of CD8+ T cells in Pfr −/− mice or MHC‐I − / − mice did not abrogate either immunosuppression or the reduction in viral replication. Moreover, central deletion of LCMV‐specific CD8+ T cells in mice infected as neonates did not prevent immunosuppression, and enforced viral replication was blunted in these mice. Taken together, these findings exclude the hypothesis that CD8+ T cells cause a general immunosuppression in chronically infected mice. However, the prolonged activity of type I IFN during chronic viral infection inhibits the enforced viral replication of a secondary virus and, consequently, inhibits an important mechanism of efficient antigen presentation and activation of the adaptive immune system. In the end, chronic infection leads not to immunosuppression but rather to chronic activation of the immune system.
In conclusion, the results presented here may change our understanding of the reaction of the immune system during chronic infection. Indeed, the failure to generate adaptive immune responses is not due to immunosuppression. Rather, the adaptive immune response is overwhelmed by the ongoing activation of the innate immune system. This condition prevents the secondary virus from replicating efficiently and, in the end, eliminates the virus before it can achieve specific activation of the adaptive immune system. This mechanism may explain why some chronically infected patients do not respond to vaccination. However, additional studies examining the importance of enforced viral replication in achieving successful vaccination are needed.
Materials and methods
Mice and viruses
All experiments were performed with animals housed in single ventilated cages, under the authorization of the Veterinäramt Nordrhein Westfalen (Düsseldorf, Germany) and in accordance with the German law for animal protection. Prf − / − and MHC‐I − / − mice were maintained on a C57BL/6 background. Irf3 − / − × Irf7 − / − mice were bred at Charité‐University Medicine Berlin, Germany, and were also maintained on a C57BL/6 background. VSV, Indiana strain (VSV‐IND, Mudd‐Summers isolate), was originally obtained from Prof. D. Kolakofsky (University of Geneva, Switzerland). Virus was propagated on baby hamster kidney 21 cells at an MOI of 0.01 and was then plaqued onto Vero cells 27. Mice were infected intravenously with VSV (2 × 108 PFU) for histological analysis and plaque assay or with (1 × 104 PFU) for neutralizing antibody measurement. Viral titers were measured with a plaque‐forming assay, as previously described. LCMV strain WE was originally obtained from Prof. Dr. F. Lehmann‐Grube (Heinrich Pette Institute, Hamburg, Germany), and LCMV strain Docile as gift from Prof. Dr. R. Zinkernagel (University of Zurich, Zurich, Switzerland) was propagated on L929 cells. Mice were infected intravenously with (2 × 104 PFU) LCMV. Neonatal C57BL/6 WT mice were infected intracerebrally 2 days after birth with LCMV‐Docile or were left uninfected. After 30 days, WT mice were additionally infected with VSV.
UV‐light inactivation
UV‐light inactivation of VSV was carried out in 35‐mm Petri dishes (Greiner Bio‐One, Frickenhausen, Germany) with a 1‐mL volume of virus solution (virus concentration, 1 × 109 PFU per mL of Dulbecco's Modified Eagle's Medium with 2% fetal calf serum (DMEM 2% FCS). The virus solution was exposed to a 12‐W germicidal lamp (Thermo Scientific, Wilmington, DE, USA) for 10 min at a distance of 10 cm. Mice were infected with 2 × 108 PFU UV‐light‐inactivated VSV.
Antibody treatment
LCMV‐Docile chronic infected mice were treated with anti‐IFN‐αR1 (clone MAR1‐5A3) or mouse IgG1 isotype control antibody (clone MOPC21; Bioxcell, NH, USA) on day 4 (1 mg) and day 2 (500 μg) before VSV infection for histological analysis and additionally on day 5 (250 μg) for neutralizing antibody measurement.
Neutralizing antibody assay
For analysis of IgG kinetics, samples were pretreated with β‐mercaptoethanol (0.1 M) for 1 h for removal of IgM and were afterward prediluted (1:40) in DMEM 2% FCS. For analysis of total Ig, serum was directly prediluted (1:40) in medium, after which the complement system was inactivated for 30 min at 56°C. Serum was titrated 1:2 over 12 steps and incubated with 1 × 103 PFU VSV. After 90 min of incubation at 37°C, the virus‐serum mixture was plaqued onto Vero cells. An overlay was added after 1 h, and the cells were again incubated at 37°C. Plaques were counted 24 h later by crystal violet staining.
Plaque assay
VSV titers were measured by smashing organs in DMEM 2% FCS. Smashed organs were titrated 1:3 over 12 steps and plaqued onto Vero cells. After 2 h of incubation at 37°C, an overlay was added, and the cells were incubated overnight at 37°C. Plaques were counted 24 h later by crystal violet staining. LCMV titers were measured with a plaque‐forming assay using MC57 cells, as previously described 28.
Sorting of CD169+ macrophages
Splenocytes were digested with Liberase DNAse (Roche, Basel, Switzerland) and stained with anti‐mSiglec‐1 (CD169; R&D Systems, Minneapolis, MN, USA), anti‐CD3 (eBioscience, San Diego, CA, USA), anti‐CD19 (eBioscience), anti F4/80 (eBioscience), anti‐CD11b (eBioscience), and DAPI (Life Technologies, CA, USA). After 30‐min incubation, CD169+ cells were sorted by FACS (BD FACS Aria III, BD Biosciences).
Histology
Histological analyses were performed on snap‐frozen tissues. Spleen sections were stained for the vesicular stomatitis glycoprotein with the anti‐VSV‐G antibody (Vi10; made in‐house). Metallophilic marginal zone macrophages were stained with mSiglec‐1 (CD169, R&D Systems).
Total RNA extraction, cDNA synthesis, and quantitative real‐time PCR
RNA was isolated from spleen tissue with the RNA Mini Kit (Qiagen, Hilden, Germany). Quantitation of RNA was performed with a NanoDrop ND‐1000 spectrophotometer (Thermo Scientific). The RNA was reverse‐transcribed to cDNA with the Quantitect Reverse Transcription Kit (Qiagen). Gene expression analysis was performed with GAPDH, Stat1, Irf7, Oas1, Mx1, Ifnα4, Ifnβ1, and Usp18 assays (Qiagen). For analysis, the expression levels of all target genes were normalized against GAPDH (dCt). Gene expression values were then calculated by the delta‐delta‐Ct (ddCt) method, with the mean of the control group as the calibrator to which all other samples were compared. Relative quantities (RQs) were determined with the equation RQ = 2− ddCt.
Statistical analysis
Unless stated otherwise, data are expressed as means ± SEM. Student's t‐test was used to detect statistically significant differences between groups. Significant differences between several groups were detected by two‐way analysis of variance (ANOVA). The level of statistical significance was set at p < 0.05.
Conflict of interest
The authors declare no financial or commercial conflict of interest.
Abbreviations
- LCMV
lymphocytic choriomeningitis virus
- VSV
vesicular stomatitis virus
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Peer Review Correspondence
Supporting Figure
Acknowledgments
We thank Konstanze Schättel, Patricia Spieker, and Sandra Francois for technical support. This study was supported by the Alexander von Humboldt Foundation (SKA‐2008 to K.S.L. and SKA 2010 to P.A.L.) and the Deutsche Forschungsgemeinschaft (DFG; CRC974, CRC/TRR60, LA2558/5‐1, and LA1419/5‐1). Mike Recher was supported by a professorship of the Swiss National Science Foundation (PP00P3_144863). FACS sorting was performed by Prof. M. Gunzer, West German Tumor Center (WTZ), at the Imaging Center Essen (IMCES) of University Hospital Essen.
See accompanying Commentary by John R. Teijaro
See accompanying Commentary: http://dx.doi.org/10.1002/eji.201546224
References
- 1. Klenerman, P. and Hill, A. , T cells and viral persistence: lessons from diverse infections. Nat. Immunol. 2005. 6: 873–879. [DOI] [PubMed] [Google Scholar]
- 2. Zinkernagel, R. M. , Planz, O. , Ehl, S. , Battegay, M. , Odermatt, B. , Klenerman, P. and Hengartner, H. , General and specific immunosuppression caused by antiviral T‐cell responses. Immunol. Rev. 1999. 168: 305–315. [DOI] [PubMed] [Google Scholar]
- 3. Heidari, M. , Sarson, A. J. , Huebner, M. , Sharif, S. , Kireev, D. and Zhou, H. , Marek's disease virus‐induced immunosuppression: array analysis of chicken immune response gene expression profiling. Viral Immunol. 2010. 23: 309–319. [DOI] [PubMed] [Google Scholar]
- 4. Jeklova, E. , Leva, L. , Matiasovic, J. , Kovarcik, K. , Kudlackova, H. , Nevorankova, Z. , Psikal, I. et al., Characterisation of immunosuppression in rabbits after infection with myxoma virus. Vet. Microbiol. 2008. 129: 117–130. [DOI] [PubMed] [Google Scholar]
- 5. Roost, H. , Charan, S. , Gobet, R. , Ruedi, E. , Hengartner, H. , Althage, A. and Zinkernagel, R. M. , An acquired immune suppression in mice caused by infection with lymphocytic choriomeningitis virus. Eur. J. Immunol. 1988. 18: 511–518. [DOI] [PubMed] [Google Scholar]
- 6. Schneider‐Schaulies, S. , Schneider‐Schaulies, J. , Niewiesk, S. and Ter Meulen, V. , Measles virus: immunomodulation and cell tropism as pathogenicity determinants. Med. Microbiol. Immunol. 2002. 191: 83–87. [DOI] [PubMed] [Google Scholar]
- 7. Griffin, D. E. , Measles virus‐induced suppression of immune responses. Immunol. Rev. 2010. 236: 176–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Braciale, T. J. , Sun, J. and Kim, T. S. , Regulating the adaptive immune response to respiratory virus infection. Nat. Rev. Immunol. 2012. 12: 295–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Khan, N. , Hislop, A. , Gudgeon, N. , Cobbold, M. , Khanna, R. , Nayak, L. , Rickinson, A. B. et al., Herpesvirus‐specific CD8 T cell immunity in old age: cytomegalovirus impairs the response to a coresident EBV infection. J. Immunol. 2004. 173: 7481–7489. [DOI] [PubMed] [Google Scholar]
- 10. Wang, X. , Yang, K. , Wei, C. , Huang, Y. and Zhao, D. , Coinfection with EBV/CMV and other respiratory agents in children with suspected infectious mononucleosis. Virol. J. 2010. 7: 247–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Duppach, J. , Francois, S. , Joedicke, J. J. , Dittmer, U. and Kraft, A. R. , Expanded regulatory T cells in chronically friend retrovirus‐infected mice suppress immunity to a murine cytomegalovirus superinfection. J. Virol. 2014. 88: 13892–13896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McGill, N. K. , Vyas, J. , Shimauchi, T. , Tokura, Y. and Piguet, V. , HTLV‐1‐associated infective dermatitis: updates on the pathogenesis. Exp. Dermatol. 2012. 21: 815–821. [DOI] [PubMed] [Google Scholar]
- 13. Waggoner, S. N. , Daniels, K. A. and Welsh, R. M. , Therapeutic depletion of natural killer cells controls persistent infection. J. Virol. 2014. 88: 1953–1960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Borrow, P. , Evans, C. F. and Oldstone, M. B. , Virus‐induced immunosuppression: immune system‐mediated destruction of virus‐infected dendritic cells results in generalized immune suppression. J. Virol. 1995. 69: 1059–1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Lee, L. N. , Burke, S. , Montoya, M. and Borrow, P. , Multiple mechanisms contribute to impairment of type 1 interferon production during chronic lymphocytic choriomeningitis virus infection of mice. J. Immunol. 2009. 182: 7178–7189. [DOI] [PubMed] [Google Scholar]
- 16. Mims, C. A. and Wainwright, S. , The immunodepressive action of lymphocytic choriomeningitis virus in mice. J. Immunol. 1968. 101: 717–724. [PubMed] [Google Scholar]
- 17. Teijaro, J. R. , Ng, C. , Lee, A. M. , Sullivan, B. M. , Sheehan, K. C. , Welch, M. , Schreiber, R. D. et al., Persistent LCMV infection is controlled by blockade of type I interferon signaling. Science 2013. 340: 207–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wilson, E. B. , Yamada, D. H. , Elsaesser, H. , Herskovitz, J. , Deng, J. , Cheng, G. , Aronow, B. J. et al., Blockade of chronic type I interferon signaling to control persistent LCMV infection. Science 2013. 340: 202–207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mandl, J. N. , Barry, A. P. , Vanderford, T. H. , Kozyr, N. , Chavan, R. , Klucking, S. , Barrat, F. J. et al., Divergent TLR7 and TLR9 signaling and type I interferon production distinguish pathogenic and nonpathogenic AIDS virus infections. Nat. Med. 2008. 14: 1077–1087. [DOI] [PubMed] [Google Scholar]
- 20. Honke, N. , Shaabani, N. , Cadeddu, G. , Sorg, U. R. , Zhang, D. E. , Trilling, M. , Klingel, K. et al., Enforced viral replication activates adaptive immunity and is essential for the control of a cytopathic virus. Nat. Immunol. 2012. 13: 51–57. [DOI] [PubMed] [Google Scholar]
- 21. Honke, N. , Shaabani, N. , Zhang, D. E. , Iliakis, G. , Xu, H. C. , Haussinger, D. , Recher, M. et al., Usp18 driven enforced viral replication in dendritic cells contributes to break of immunological tolerance in autoimmune diabetes. PLoS Pathog. 9: e1003650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Naneh, O. , Avcin, T. and Bedina Zavec, A. , Perforin and human diseases. Subcell. Biochem. 2014. 80: 221–239. [DOI] [PubMed] [Google Scholar]
- 23. Cihak, J. and Lehmann‐Grube, F. , Persistent infection of mice with the virus of lymphocytic choriomeningitis: virus‐specific immunological tolerance. Infect. Immun. 1974. 10: 1072–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Saron, M. F. , Riviere, Y. , Hovanessian, A. G. and Guillon, J. C. , Chronic production of interferon in carrier mice congenitally infected with lymphocytic choriomeningitis virus. Virology 1982. 117: 253–256. [DOI] [PubMed] [Google Scholar]
- 25. Lang, P. A. , Cervantes‐Barragan, L. , Verschoor, A. , Navarini, A. A. , Recher, M. , Pellegrini, M. , Flatz, L. et al., Hematopoietic cell‐derived interferon controls viral replication and virus‐induced disease. Blood 2009. 113: 1045–1052. [DOI] [PubMed] [Google Scholar]
- 26. Planz, O. , Seiler, P. , Hengartner, H. and Zinkernagel, R. M. , Specific cytotoxic T cells eliminate B cells producing virus‐neutralizing antibodies [corrected]. Nature 1996. 382: 726–729. [DOI] [PubMed] [Google Scholar]
- 27. Fink, K. , Lang, K. S. , Manjarrez‐Orduno, N. , Junt, T. , Senn, B. M. , Holdener, M. , Akira, S. et al., Early type I interferon‐mediated signals on B cells specifically enhance antiviral humoral responses. Eur. J. Immunol. 2006. 36: 2094–2105. [DOI] [PubMed] [Google Scholar]
- 28. Lang, P. A. , Contaldo, C. , Georgiev, P. , El‐Badry, A. M. , Recher, M. , Kurrer, M. , Cervantes‐Barragan, L. et al., Aggravation of viral hepatitis by platelet‐derived serotonin. Nat. Med. 2008. 14: 756–761. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Peer Review Correspondence
Supporting Figure