

Summary
The acquisition of regulatory proteins is a means of blood‐borne pathogens to avoid destruction by the human complement. We recently showed that the gametes of the human malaria parasite Plasmodium falciparum bind factor H (FH) from the blood meal of the mosquito vector to assure successful sexual reproduction, which takes places in the mosquito midgut. While these findings provided a first glimpse of a complex mechanism used by Plasmodium to control the host immune attack, it is hitherto not known, how the pathogenic blood stages of the malaria parasite evade destruction by the human complement. We now show that the human complement system represents a severe threat for the replicating blood stages, particularly for the reinvading merozoites, with complement factor C3b accumulating on the surfaces of the intraerythrocytic schizonts as well as of free merozoites. C3b accumulation initiates terminal complement complex formation, in consequence resulting in blood stage lysis. To inactivate C3b, the parasites bind FH as well as related proteins FHL‐1 and CFHR‐1 to their surface, and FH binding is trypsin‐resistant. Schizonts acquire FH via two contact sites, which involve CCP modules 5 and 20. Blockage of FH‐mediated protection via anti‐FH antibodies results in significantly impaired blood stage replication, pointing to the plasmodial complement evasion machinery as a promising malaria vaccine target.
Introduction
The human complement is a first line of defence against microbial infections, triggering targeted attack and ultimately killing the microbial intruder. Major features of complement include C3b‐mediated cell opsonization for phagocytosis, immune cell recruitment mediated by the release of C3a and C5a and the formation of a terminal complement complex (TCC) to induce targeted lysis of the microbe.
While the classical and lectin pathways are initiated in response to bacterial molecular patterns or antigen–antibody immune complexes, the alternative complement pathway (ACP) is activated continuously at a low rate by spontaneous hydrolysis of complement factor C3, which can form a C3 convertase and then cleaves C3 to C3a and C3b. Factor C3b exposes a reactive thioester and attaches covalently to the surface of microbes, encouraged by its hydroxyl‐rich composition (Fig. 1A). C3b binding to cell surfaces in consequence triggers the ACP amplification loop via the assembly of C3b with activated factor B to form the C3 and C5 convertases, eventually resulting in TCC formation (Rother et al., 1998).
Figure 1.
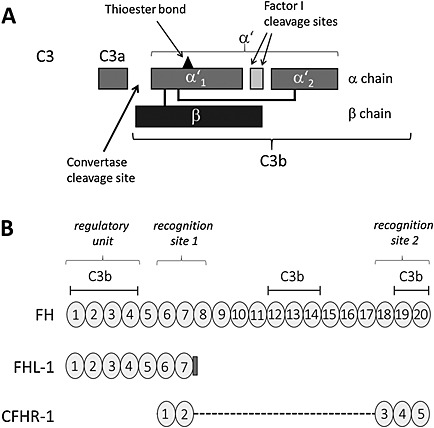
Schematic depicting the domain structure of C3 and the CCP domains of FH, FHL‐1 and CFHR‐1.
A. Domain structure of C3 and cleavage sites [modified from Simon et al. (2013)]. The peptide sizes are the following: C3 (180 kDa), C3a (9 kDa), C3b α′ (109 kDa), β (75 kDa), α′ (101 kDa), α′1 (67 kDa) and α′2 (40 kDa).
B. Domain structures of FH, FHL‐1 and CFHR‐1. FH is composed of 20 CCP domains (CCP 1‐20); FHL‐1 is identical in sequence with the seven N‐terminal CCP domains of FH (CCP 1‐7) and includes at its C‐terminal end an extension of four amino acids. CFHR‐1 has five CCP domains homologous with CCP6‐7 and CCP18‐20 of FH. The regulatory unit, the recognition sites and the binding sites for C3b in FH, FHL‐1 and CFHR‐1 are indicated.
In order to prevent any damage of self‐cells by complement, human cells use a variety of complement inhibitors, which either exhibit decay‐acceleration and/or co‐factor activity; thus, they either help disassembling the C3 and C5 convertase complexes or cleavage of C3b by factor I, or which prevent TCC assembly. Inhibitors include membrane‐bound regulators like complement receptor 1 (CR‐1), CD55 or CD59 and central fluid‐phase regulators like C4‐binding protein (C4BP) and factor H (FH). As the main regulator of the ACP, FH has an important role in discriminating between self and non‐self surfaces. The protein comprises 20 complement control protein (CCP) modules, beta‐sandwich domains containing about 60 amino acid residues. Besides FH, the FH family consists of six more proteins, i.e. FH‐like protein 1 (FHL‐1), an alternative splicing product of FH comprising CCP modules 1–7, as well as the FH‐related proteins CFHR‐1 to CFHR‐5, the genes of which originated from the FH gene via tandem duplication events (Fig. 1B) [reviewed in Józsi and Zipfel (2008), Skerka et al. (2013) and Józsi et al. (2015)].
Numerous microbial pathogens have evolved strategies to inhibit, control or prevent complement recognition, thus ensuring their survival in the human host. Such strategies include besides mimicking of host regulators and secretion of complement effector‐targeting proteases, the recruitment of host regulators to the pathogens' surfaces [reviewed in Blom et al. (2009) and Zipfel et al. (2013)]. In the past complement evasion mechanisms have been described for microbial pathogens like Streptococcus pneumoniae, Staphylococcus aureus, Neisseria meningitides, Borrelia burgdorferi and Candida albicans. While the immune evasion mechanisms of these pathogens have been studied extensively [reviewed in Zipfel et al. (2007, 2008, 2013), Józsi and Zipfel (2008), Blom et al. (2009), Luo et al. (2013) and Zipfel and Skerka (2014)], they have only recently been described by us for the sexual stages of Plasmodium parasites, the causative agents of the tropical disease malaria (Simon et al., 2013).
Malaria is a major global health burden, causing 198 million new cases and approximately 584 000 casualties annually worldwide (World Malaria Report, W.H.O, 2014). Malaria infections can persist for weeks or months in infected individuals, allowing the parasites to replicate in red blood cells (RBCs). Such compartmentalization within erythrocytes helps the parasites to evade recognition by the host immune system. Nonetheless, the parasites have to modify the RBC surface during growth in order to acquire nutrition from the blood plasma and to enable RBC sequestration on the blood vessel endothelium, making the infected erythrocyte assailable for the immune system. Further, the parasites have to leave the shelter of the RBC when the merozoites (MZs) explosively exit the host cell to initiate another replication cycle. During the few minutes that the free MZ circulate until they infect another RBC, they can easily be recognized and destroyed by the immune components, including complement [reviewed in Wirth and Pradel (2012), Chan et al. (2014) and Wright and Rayner (2014)].
The complement system can be activated during malaria infection through different pathways [reviewed in Greenwood and Brueton (1974), Phanuphak et al. (1985), Nyakoe et al. (2009) and Biryukov and Stoute (2014)]. For example, it was shown that the parasite digestive vacuole, which during schizont rupture is released together with the MZs into the blood stream, activates the ACP (Dasari et al., 2012, 2014). Malaria infection‐induced complement activation of the classical pathway via the formation of antigen–antibody immune complexes as well as activation of the mannose‐binding lectin pathway have also been reported (Jhaveri et al., 1997; Adam et al., 1981; Klabunde et al., 2002; Garred et al., 2003; Stoute et al., 2003).
Another point of attack for the human immune system to kill malaria parasites is the mosquito midgut stages. The malaria parasites are taken up by the mosquito during the blood meal in form of sexual precursor cells, the intraerythrocytic gametocytes. Within minutes following their uptake, these emerge from the enveloping RBCs in response to stimuli present in the mosquito midgut and transform into fertile gametes. The emerging gametes are vulnerable to the factors of the blood meal, including antibodies and complement proteins [reviewed in Pradel (2007), Kuehn and Pradel (2010) and Wirth and Pradel (2012)].
In a recent study, we identified FH as the factor responsible for protecting the extracellular gametes of Plasmodium falciparum from ACP‐mediated lysis (Simon et al., 2013). We showed that human complement is active in the mosquito midgut for 1 h after the blood meal and during this time represents a severe threat to the emerging gametes. In order to escape the ACP, gametes rapidly bind FH and FHL‐1 from the blood meal, once they have egressed from the enveloping erythrocyte. Binding of FH to the parasite surface then promotes inactivation of surface‐bound C3b via factor I.
We further identified the plasmodial transmembrane protein GAP50 (gliding‐associated protein 50) as the FH‐binding receptor of P. falciparum gametocytes. GAP50 was originally assigned to the parasite inner membrane complex (IMC), an alveolar double‐membrane structure underneath the parasite plasmalemma, which mediates both cell motility and stability. Whereas the C‐terminal transmembrane helix of GAP50 is anchored within the outer IMC membrane, the N‐terminal portion protrudes into the IMC lumen (Baum et al., 2006; Sanders et al., 2007; Frénal et al., 2010; Bosch et al., 2012). Although not motile, the gametocytes of P. falciparum possess an IMC, which here functions to stabilize their crescent shapes (Dearnley et al., 2012; Kono et al., 2012; Simon et al., 2013). The IMC disintegrates during gametogenesis, and then GAP50 relocates from the IMC to the plasmalemma, where it binds FH via CCP5‐7, thus protecting the extracellular gametes from attack by the human complement (Simon et al., 2013). In consequence, antibodies directed against GAP50 reduce FH binding to the gamete surface, leading to impaired gametogenesis and complement‐mediated destruction of the parasite. In accordance with these findings, standard membrane feeding assays demonstrated that the functional blockade of either GAP50 or FH by antibodies inhibits transmission of the parasite from the human to the mosquito (Simon et al., 2013).
While these data give a first glimpse of the complex mechanisms the malaria parasites use to evade the human complement system, there is hitherto no published evidence of ACP evasion by the asexual blood stages (ABSs). We now demonstrate that the intraerythrocytic schizonts as well as the free MZ bind FH, FHL1 and CFHR‐1 to their surfaces, thereby inactivating C3b to ensure their survival during the erythrocytic replication phase.
Results
Human complement affects the erythrocytic replication cycle of P. falciparum
The aim of this study was to investigate the protective effect of FH and FH family proteins on blood stage progeny of P. falciparum. We initially evaluated if the ABSs of P. falciparum are vulnerable to the human ACP during erythrocytic replication. ABSs with a starting parasitemia of 0.5% were cultivated in cell culture medium supplemented with 10 vol% of normal human serum (NHS) or heat‐inactivated serum (HIS) at 37°C over a period of three generation cycles, and the parasitemia was determined every 12 h. Medium was exchanged every 12 h to ensure the presence of active complement throughout the experiments. During this period the viability of the malaria parasites was impaired when these were cultivated in NHS. The differences in parasitemia between NHS‐treated and HIS‐treated cultures increased from generation 1 to 3 and at the beginning of generation 4 (120 h following seeding), the parasitemia of NHS‐treated parasite cultures was significantly reduced by 6% compared with the HIS‐treated control (Fig. 2A). The major reductions in parasitemia occurred during the transitions between the sequential cycles, indicating that active complement either blocks or kills the MZs before invasion. The percentages of ring stages, trophozoites, immature and mature schizonts at each time point, however, did not differ between NHS‐treated and HIS‐treated ABS cultures during the 120‐h cultivation period (Fig. S1).
Figure 2.
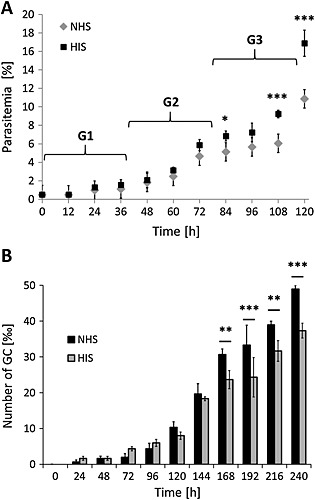
Effect of human complement on ABS replication and gametocytogenesis.
A. Effect of human complement on ABS replication. Ring stage cultures of strain NF54 with a starting parasitemia of 0.5% were maintained in cell culture medium supplemented with either 10 vol% NHS or HIS at 37°C over a period of 0–120 h to follow three generations (G1–3). The parasitemia was evaluated every 12 h via Giemsa smears. Significant differences in parasitemia are indicated (* p < 0.05, *** p < 0.001; Bonferroni post‐test, two‐way ANOVA). The experiment was performed in triplicate (mean ± SD); the data are representative for one of two independent experiments.
B. Effect of human complement on gametocytogenesis. Ring stage cultures of strain NF54 with a starting parasitemia of 5% were maintained in cell culture medium supplemented with either 10 vol% NHS or HIS at 37°C over a period of 0–240 h. The gametocytemia was evaluated every 24 h via Giemsa smears. Significant differences in gametocyte numbers are indicated (** p < 0.01, *** p < 0.001; Bonferroni post‐test, two‐way ANOVA). The experiment was performed in triplicate (mean ± SD); the data are representative for one of two independent experiments.
To confirm that NHS used in the previously mentioned experiment was active, frozen pooled NHS samples were thawed and added to cell culture medium at 20 vol%. The medium was then kept at 37°C for different time periods, ranging from 30 min to 24 h, and was subsequently added to a fixed number of pig RBCs in RPMI medium at 37°C. After 1 h the absorbance of the supernatant was measured at OD405 nm and compared with the supernatant of pig erythrocyte samples incubated in cell culture medium supplemented with HIS. Pig erythrocytes lysed by distilled water were used as a positive control. In NHS‐treated pig erythrocyte samples taken at 30 min to 12 h, the absorbance was significantly increased compared with the HIS‐treated erythrocyte samples (Fig. S2), demonstrating that the pig erythrocytes were partially lysed during this time period. The data indicate that NHS kept its complement‐lytic activity for more than 12 h after thawing.
Because gametocytogenesis is induced in ABS parasites by stress factors like drug pressure or immune response molecules [reviewed in Talman et al. (2004), Alano (2007) and Pradel (2007)], we further investigated if NHS represents a stress factor that induces gametocyte formation. ABSs at a starting parasitemia of 5% were cultivated in cell culture medium either supplemented with 10 vol% NHS or HIS over a period of 240 h. Medium was exchanged every 12 h to ensure the presence of active complement throughout the experiments. While in both cultures, small amounts of gametocytes were detectable as early as 24 h after seeding, the numbers of gametocytes were significantly increased between 168 and 240 h after seeding in cultures treated with NHS (Fig. 2B). In NHS‐treated cultures, furthermore, a higher percentage of stage V gametocytes were detectable compared with HIS‐treated cultures (Fig. S3).
We then investigated if human complement causes deposition of C3b on the surface of infected RBCs (iRBCs), which can progress to TCC formation. As the decisive factor of the ACP, C3b continously forms because of the spontaneous hydrolysis of C3 into C3a and C3b and triggers complement activation leading to TCC formation by complement components C5b‐9 at the surfaces of target cells. Indirect immunofluorescence assays (IFAs) using anti‐C3 antibody demonstrated deposition of C3b on the surfaces of schizont‐iRBCs when these were cultivated in the presence of NHS. The ABSs were counter‐labelled with antibodies against the parasite plasmalemma‐anchored MZ surface protein MSP1 (Holder and Freeman, 1984; Blackman et al., 1991; reviewed in Kadekoppala and Holder, 2010). In order to distinguish between intact schizont‐iRBCs and freed MZs, we either incubated the ABSs with 20 vol% NHS for 1 h and then fixed with paraformaldehyde before immunolabelling or the live schizont‐iRBCs were treated with saponin to permeabilize the RBC and parasitophorous vacuole membranes and to gain access to the MZs before incubation with NHS for 1 h, followed by paraformaldehyde‐fixation and immunolabelling. The IFAs revealed increased C3b labelling of intact schizont‐iRBCs as compared with the C3b deposition on the surface of non‐infected RBCs (niRBCs) (Fig. 3A). Noteworthy, a higher C3b labelling was also observed for freed MZs, demonstrating that C3b accumulates both on the surfaces of schizont‐iRBCs and of MZs. No labelling was observed, when sera of non‐immunized mice (NMS) was used as a negative control in the IFAs (Fig. S4A).
Figure 3.
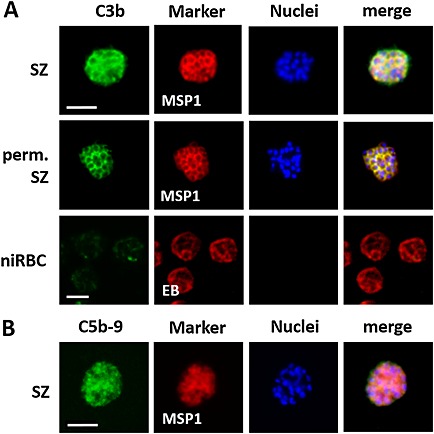
Complement factor deposition on the surfaces of ABS parasites.
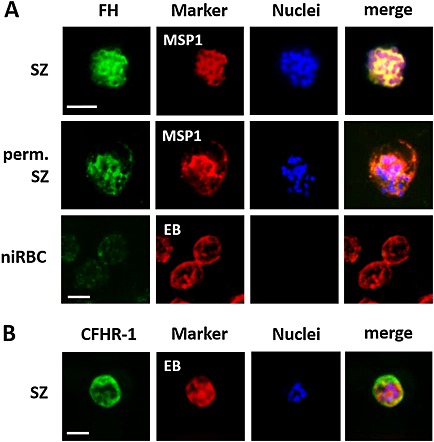
A. C3b deposition on the surfaces of ABS parasites. Intact schizonts (SZ), saponin‐permeabilized (perm.) schizonts and niRBC as control were incubated with 20 vol% NHS for 1 h and subjected to IFAs. C3b deposition was demonstrated by immunolabelling with anti‐C3 antibody (green). ABS parasites were detected by labelling with anti‐MSP1 antibody; the niRBCs were highlighted by Evans Blue (EB) staining (red).
B. TCC formation on the surfaces of iRBCs. Intact schizonts were incubated with 20 vol% NHS for 1 h and subjected to IFAs. TCC formation was detected by labelling with mAb anti‐C5b‐9 (green). ABS parasites were detected by labelling with anti‐MSP1 antibody (red). The nuclei were counterstained with Hoechst stain (blue). The data are representative for one of three independent experiments each. In the IFAs, strain F12 was used, similar results were observed for strain NF54. Bar, 5 µm.
To verify that saponin‐permeabilization of live schizont‐iRBCs renders the MZs accessible to molecules like the complement factors, we performed a control experiment, in which schizont‐iRBCs were treated with saponin before the cells were fixed with glutaraldehyde in suspension. Subsequently, an IFA was performed, using anti‐MSP1 antibody to highlight the MZ plasmalemma, without any further permeabilization step during this process. The IFA resulted in dominant MSP1 labelling of MZs, when the schizont‐iRBCs were permeabilized by saponin before fixation. When untreated schizont‐iRBCs were fixed and used in the IFA as a negative control, no MSP1‐labelling was observed (Fig. S4B).
Subsequently, lysates of schizont‐iRBCs or niRBCs incubated with 20 vol% NHS for 1 h were subjected to gel electrophoresis, followed by Western blot analysis, and probed with anti‐C3 antibodies. Purified C3b was loaded on the gel in order to demonstrate the unprocessed α′ and β chains of C3b having mobilities of 101 and 75 kDa respectively (Fig. 1A). The Western blots demonstrated an increased labelling for the β chain in the schizont‐iRBC lysate compared with niRBC lysate (Fig. 4A). Noteworthy, in the iRBC lysate, the band of the α′ chain was very weak, while the α′1 and α′2 peptides with molecular weights of 67 and 40 kDa were visible. The presence of α′1 and α′2 demonstrates processing of α′ in these samples, indicating that the iRBC‐bound C3b is cleaved by factor I (Fig. 4A).
Figure 4.
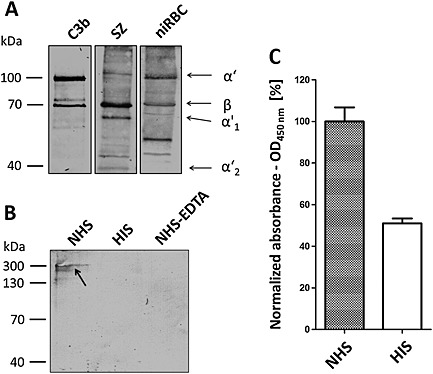
C3b deposition and TCC formation in ABS parasites.
A. C3b deposition and processing on the surfaces of ABS parasites. SZ of strain F12 or niRBC were incubated with 20 vol% NHS for 1 h; lysates were prepared and subjected to gel electrophoresis followed by Western blotting with anti‐C3 antibody. Purified C3b was loaded for positive control. Immunoblotting of the samples with anti‐C3 antibody highlighted the unprocessed α′ and β chains (101 and 75 kDa respectively), as well as processed α′1 and α′2 (67 and 40 kDa respectively).
B. TCC detection in ABS parasites. Schizonts of strain F12 were incubated with 20 vol% NHS, HIS or EDTA‐inactivated NHS; lysates were prepared and subjected to non‐reducing gel electrophoresis followed by Western blotting with mAb anti‐C5b‐9 to detect the TCCs (approximately 330 kDa; arrow).
C. NHS‐dependent TCC formation. Schizonts of strain F12 were incubated with 20 vol% NHS or HIS for 1 h and subjected to ELISA. Immunolabelling of the TCCs with mAb anti‐C5b‐9 was determined by measuring the relative absorbance at OD450 nm (NHS set to 100%). The experiment was performed in triplicate (mean ± SD); the data are representative for one of two independent experiments.
We further demonstrated the presence of TCCs on the surfaces of schizont‐iRBCs cultivated in 20 vol% NHS via IFA, using a monoclonal antibody (mAb) directed against C5b‐9 (Fig. 3B). The schizonts were counterlabelled with rabbit anti‐MSP1 antibody. In total, 90 ± 1.2% of schizont‐iRBCs (n = 100, in triplicate) stained positive for the C5b‐9 complex. The TCCs were also detectable in lysates of schizont‐iRBCs, when gel electrophoresis under non‐reducing conditions followed by Western blotting with anti‐C5b‐9 antibody was performed. In the blots, the TCC was running at a broad smear with an expected mobility of approximately 330 kDa (Fig. 4B). When the schizonts were cultured in HIS instead of NHS or when the NHS was inactivated by addition of 20 mM EDTA (Des Prez et al., 1975; Johnson et al., 2008), no TCC‐positive band was detected in the schizont‐iRBC lysates. TCC formation on the surfaces of schizont‐iRBCs, cultivated either in 20 vol% NHS or HIS for 1 h, was further quantified via ELISA. The absorbance was twofold increased in samples of NHS‐cultivated ABS cultures compared with the HIS control, when the mAb C5b‐9 was used for immunodetection (Fig. 4C), indicating increased TCC formation on the iRBC surface in the presence of NHS.
Asexual blood stage parasites bind FH and FH family proteins on their surfaces
The previously mentioned experiments pointed to an inactivation of C3b deposited on the surfaces of schizont‐iRBCs, which suggests an involvement of FH in complement evasion by the ABSs. To examine FH‐mediated complement evasion in more detail, we initially performed IFAs to demonstrate FH binding to the surface of schizont‐iRBCs. We either incubated the ABS parasites with 20 vol% NHS for 1 h before paraformaldehyde fixation, or we treated the live schizont‐iRBCs with saponin to permeabilize the enveloping membranes in order to gain access to the MZs before NHS incubation for 1 h, followed by paraformaldehyde fixation and immunolabelling. The IFAs using anti‐CCP1‐20 antibody revealed binding of FH (and FH family proteins) both to the surfaces of intact schizont‐iRBCs as well as to the freed MZs, which was increased compared with binding of FH to the surface of niRBCs (Fig. 5A). The binding of FH to schizont‐iRBCs was quantified, and a total of 92 ± 3.5% of schizonts (n = 100, in triplicate) were FH‐positive. For negative control, we labelled the paraformaldehyde‐fixed ABSs with antibodies directed against C4BP, a regulator of the classical and lectin complement pathways, and no labelling was observed (Fig. S5). For positive control in this experiment, Escherichia coli were used in the IFAs and a strong immunolabelling for C4BP was detectable.
Figure 5.
Binding of FH and CFHR‐1 to the surfaces of ABS parasites.
A. Binding of FH to the surfaces of ABS parasites. Intact SZ, saponin‐permeabilized (perm.) schizonts and niRBCs as control were incubated with 20 vol% NHS for 1 h and subjected to IFAs. FH binding was demonstrated by immunolabelling with anti‐CCP1‐20 antibody (green). ABS parasites were detected by labelling with anti‐MSP1 antibody; the niRBCs were highlighted by Evans Blue (EB) staining (red).
B. Binding of CHFR‐1 to the surfaces of ABS parasites. Intact schizonts were incubated with 20 vol% NHS for 1 h and subjected to IFAs. CHFR‐1 binding was detected by labelling with anti‐CHFR‐1 antibody (green). ABS parasites were detected by labelling with anti‐MSP1 antibody (red). The nuclei were counterstained with Hoechst stain (blue). The data are representative for one of three independent experiments each. In the IFAs, strain F12 was used; similar results were observed for strain NF54. Bar, 5 µm.
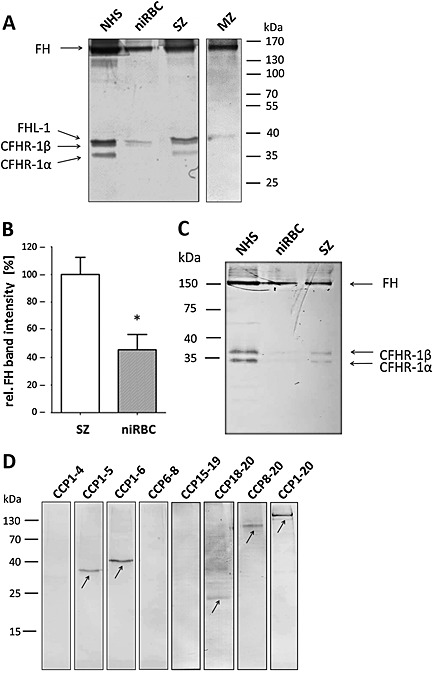
Subsequently, Western blot analyses were performed to investigate binding of FH and FH family proteins in more detail. Schizont‐iRBCs and niRBCs were incubated in 20 vol% NHS for 1 h, and lysates were prepared. Furthermore, MZs were isolated following a protocol by Boyle et al. (2010) before NHS incubation and lysate preparation. Lysates were subjected to non‐reducing gel electrophoresis followed by Western blotting using anti‐CCP1‐20 antibody. Western blotting revealed that both the intact schizont‐iRBCs as well as the isolated MZ bind FH and FHL‐1, which migrated with the expected molecular weights of 155 and 37 kDa (Fig. 6A). Noteworthy, the schizont‐iRBCs but not the niRBCs additionally bound CFHR‐1; the α and β variants of CFHR‐1 migrated with the expected molecular weights of 34 and 36 kDa. NHS was used as a positive control in the Western blots. Quantification of band intensities between FH bound to schizont‐iRBCs and niRBCs demonstrated a significantly increased binding of FH to the parasitized RBCs (Fig. 6B).
Figure 6.
Binding of FH and family proteins to ABS parasites.
A. Acquisition of FH and family proteins by ABS parasites. SZ and purified merozoites (MZ) of strain F12 or niRBCs for control were incubated with 20 vol% NHS for 1 h; lysates were prepared and subjected to non‐reducing gel electrophoresis followed by Western blotting with anti‐CCP1‐20 antibody. NHS was loaded for positive control. Immunoblotting demonstrated FH (155 kDa), FHL‐1 (37 kDa) and CFHR‐1β (36 kDa) and CFHR‐1α (34 kDa) in the samples. The data are representative for one of five independent experiments.
B. Quantification of FH‐binding to ABS parasites. Relative band intensities for FH from lysates of schizonts or niRBCs after incubation with NHS were measured from five independent Western blot experiments (mean ± SD) using the imageJ programme (NHS set to 100%). Significant differences in intensities are indicated (* p < 0.05; Student's t‐test).
C. Binding of CFHR‐1 to ABS parasites. SZ of strain F12 or niRBCs for control were incubated with 20 vol% NHS for 1 h; lysates were prepared and subjected to non‐reducing gel electrophoresis followed by Western blotting with anti‐CFHR antibody. NHS was loaded for positive control. Immunoblotting demonstrated CFHR‐1β (36 kDa) and CFHR‐1α (34 kDa) in the samples. The data are representative for one of two independent experiments.
D. Identification of CCP modules involved in FH‐binding to ABS parasites. Schizonts of strain F12 were incubated in cell culture medium in the presence of the indicated recombinant FH deletion mutants at a concentration of 10 ng µl‐1 before being subjected to non‐reducing gel electrophoresis and Western blotting, using polyclonal anti‐CCP1‐20 antibody. Incubation with FH (CCP1‐20) was used as a positive control. Arrows indicate the peptide bands. The data are representative for one of two independent experiments.
We investigated the CFHR‐1 binding in more detail, using a specific anti‐CFHR‐1 antibody, which recognizes CFHR‐1 and FH, but not FHL‐1. In IFAs, a total of 96 ± 0.6% of schizont‐iRBCs (n = 100, in triplicate) were positive for labelling with the anti‐CFHR‐1 antibody (Fig. 5B). Western blotting confirmed that schizont‐iRBCs but not niRBCs bind CFHR‐1, and the α and β variants of CFHR‐1 were detected in the schizont‐iRBC lysates (Fig. 6C).
We then aimed to determine which of the CCP modules of FH are involved in binding to the ABS parasites. Seven recombinant deletion mutants, comprising different CCP modules, were investigated for binding. Recombinant FH, comprising CCP1‐20, was used as a positive control in the assays. Initially, we separated the purified fragments by gel electrophoresis in order to determine purity and molecular mobilities of the respective proteins (Fig. S6). Subsequently, schizont‐iRBCs were incubated in cell culture medium supplemented with different recombinant FH deletion mutants (10 ng µl−1). The schizont‐iRBCs were thorougly washed, and the respective lysates were then subjected to non‐reducing gel electrophoresis followed by Western blot analysis, using anti‐CCP1‐20 antisera, in order to detect the FH fragments. Western blotting demonstrated binding of the peptides CCP1‐5, CCP1‐6 CCP18‐20, CCP8‐20 and of full‐length FH, CCP1‐20, used as a positve control, while the FH delection mutants CCP1‐4, CCP6‐8 and CCP15‐19 did not bind (Fig. 6D). Altogether, the data point to the involvement of two CCP modules, CCP5 and CCP20, in binding of FH and FH family proteins to the schizont‐iRBCs.
Because FH was particularly bound by the schizont‐iRBCs, we aimed to investigate the potential correlation between FH binding and the formation of knobs on the erythrocyte surface in these stages. During schizont maturation, the P. falciparum erythrocyte membrane protein 1 (PfEMP1) family proteins localize on knobs and bind endothelial cell surface molecules, like CD36, via their variable extracellular domains in order to adhere to the capillary walls and in consequence to escape clearance by the spleen [reviewed in e.g. Kirchgatter and Del Portillo (2005)]. Firstly, we investigated the percentile of knob‐infected RBCs in the P. falciparum F12 strain used in our studies. Noteworthy, this laboratory strain has never not been selected by us for knob‐formation. Knob‐infected RBCs were identified using a previously published gelatin flotation purification method (Goodyer et al., 1994) and resulted in roughly 42% of the schizont‐iRBCs showing a gelatin‐flotation behaviour known for knob‐infected erythrocytes (Fig. S7A). We further studied the efficiency of the schizont‐iRBCs to bind to recombinant CD36 via a microplate adhesion assay (Prudhomme and Sherman, 1999) using the Malstat reaction as a read‐out for parasite abundance. The absorbance was approximately fourfold increased in samples of schizont‐iRBCs bound to CD36‐coated wells as compared with niRBC samples (Fig. S7B). Non‐coated wells were used for negative control. Because the PfEMP1‐based knobs are sensitive to treatment with trypsin (Leech et al., 1984), we lastly investigated the FH‐binding behaviour of schizont‐iRBCs with and without prior trypsin treatment. Quantitative Western blot analysis demonstrated that treatment of the schizont‐iRBCs with trypsin prior to incubation with 20 vol% NHS has no effect on binding of FH by these cells (Figs 7 and S8A). For control, niRBCs with or without trypsin‐treatment were used, and here a significantly lower FH binding compared with the schizont‐iRBCs was observed, confirming our earlier observations. The activity of trypsin was tested by demonstrating the cleavage of glycophorin A from the schizont‐iRBC surface (Fig. S8B). In conclusion we postulate that binding of FH by the schizont‐iRBCs is not mediated by knobs.
Figure 7.
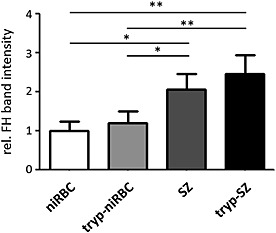
Binding of FH by ABS parasites following trypsin‐treatment. Schizonts of strain F12 were treated with 100 µg ml−1 trypsin for 30 min prior to incubation with 20 vol% NHS for 1 h. Lysates were prepared and subjected to non‐reducing gel electrophoresis followed by quantitative Western blotting using anti‐CCP1‐20 antibody. Relative band intensities for FH from lysates of schizonts or niRBCs with and without trypsin‐treatment were measured from five independent Western blot experiments (mean ± SD) using the imageJ programme (niRBC set to 1). Signficant differences in intensities are indicated (* p < 0.05, ** p < 0.01; Student's t‐test).
FH binding mediates C3b inactivation and protects the asexual blood stages from lysis
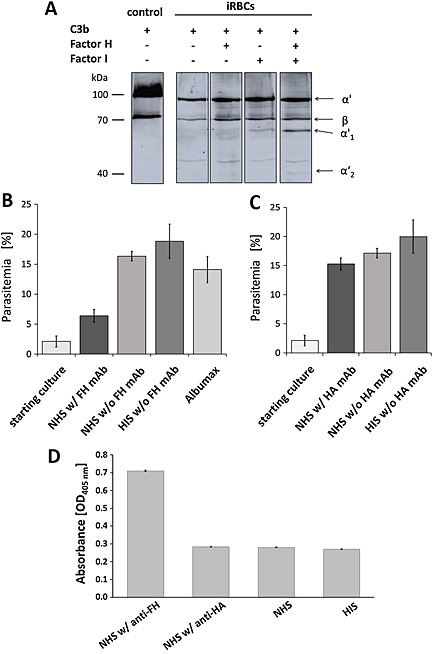
In a final set of experiments we aimed to investigate if FH binding protects the ABS parasites from complement‐mediated destruction. A cofactor assay was employed to investigate if the surface‐bound C3b is inactivated by FH and factor I. Schizont‐iRBCs were cultivated in Albumax‐complemented cell culture medium in the absence of human FH. Then purified FH was added to part of the cultures, and these were incubated with FH for 30 min. The parasite cultures were subsequently washed and incubated with purified C3b in the presence or absence of factor I for 30 min. Lysates were prepared and subjected to gel electrophoresis, followed by Western blotting, and probed with anti‐C3 antisera. C3b was loaded as a positive control. In schizont‐iRBCs, to which FH and factor I were added in order to inactivate C3b, the α′ chain was processed by factor I, and the peptides α′1 and α′2 (67 and 40 kDa) appeared in addition to the protein bands of α′ and β (101 and 75 kDa) (Fig. 8A). In samples without FH and/or factor I, however, the α′ chain was not cleaved to α′1 and α′2. A minor degradation of C3b was observed in schizont‐iRBC cultures supplemented with factor I, but lacking human FH, which might be due either to non‐specific proteolysis or caused by remnants of bovine FH of the Albumax medium, which might have bound to the iRBCs (Fig. 8A).
Figure 8.
Protection of ABS parasites from complement‐mediated lysis by FH.
A. FH‐mediated processing of C3b by factor I. Schizonts of strain F12 were incubated in cell culture medium in the presence or absence of purified FH for 30 min. The parasite cultures were subsequently washed and incubated with purified C3b in the presence or absence of factor I for 30 min. Lysates were prepared and subjected to gel electrophoresis followed by Western blotting and probed with anti‐C3 antibodies to visualize schizont‐bound C3b products α′ (101 kDa) and β (75 kDa), and the processing products α′1 (67 kDa) and α′2 (40 kDa). C3b was loaded as a positive control. The data are representative for one of three independent experiments.
B. Effect of functional inhibition of FH on ABS growth. Schizont cultures of strain F12 with a starting parasitemia of 2.5% were maintained in cell culture medium supplemented with either 20 vol% NHS or HIS or in 10 vol% Albumax‐supplemented medium in the presence or absence of mAb anti‐FH over a period of 0–24 h, and the parasitemia was subsequently evaluated via Giemsa smears. The experiment was performed in triplicate (mean ± SD); the data are representative for one of two independent experiments.
C. ABS growth in the presence of anti‐HA control antibody. Experiments were performed as described in B using a mAb against HA as negative control for B. The experiment was performed in triplicate (mean ± SD); the data are representative for one of two independent experiments.
D. Effect of functional inhibition of FH on iRBC lysis. Experiments were performed as described in B and C. The absorbance of the culture supernatants was measured at OD405 nm to determine lysis of iRBCs.
To evaluate a potential role of FH in protection against the ACP‐mediated growth impairment, schizont‐iRBC cultures at a parasitemia of 2.5% were incubated with 20 vol% NHS in the presence of a mAb directed against FH modules CCP8‐15 known to exhibit functional inhibition of the complement regulator (Oppermann et al., 2006; Simon et al., 2013) at a concentration of 100 µg ml−1. Controls were incubated with NHS or HIS without mAb anti‐FH or with Albumax‐supplemented cell culture medium. At 24 h post‐seeding, the parasitemia of the ABS cultures was decreased, when these were treated with the mAb anti‐FH compared with controls, and a 10% reduction in parasitemia was measured compared with NHS‐treated cultures lacking mAb anti‐FH (Fig. 8B). When a mAb directed against hemagglutinin (HA)‐tag was used as a negative control in the assays, no significant reduction of parasitemia was observed compared with the respective controls (Fig. 8C). Noteworthy, ABS cultures incubated with NHS in the presence of mAb anti‐FH further showed a more than two‐fold higher absorbance for haemoglobin at OD405 nm compared with the controls lacking mAb anti‐FH, indicating that here a higher number of iRBCs were lysed (Fig. 8D). In conclusion, the combined data show that FH protects the intraerythrocytic malaria parasites from lysis by the ACP by mediating inactivation of C3b on the surfaces of the iRBCs, an effect that can be derogated by antibody‐mediated blockage of FH function.
Discussion
While the mechanism of FH recruitment as a means to evade destruction by human ACP is well known for bacterial pathogens like Streptococcus pneumoniae, Staphylococcus aureus, Neisseria meningitides or Borrelia burgdorferi [reviewed in Zipfel et al. (2007, 2008, 2013), Józsi and Zipfel (2008), Blom et al. (2009), Luo et al. (2013) and Zipfel and Skerka (2014)), the role of human complement during the infection with malaria parasites has only been investigated in detail within the past few years.
In a recent study, we identified FH as the factor responsible for protecting the extracellular gametes of P. falciparum from the human ACP, which is active in the mosquito midgut for 1 h after the blood meal and which during this time represents a severe threat to the emerging gametes. In order to escape the ACP, the gametes bind FH and FHL‐1 from the blood meal, which then support inactivation of surface‐bound C3b via factor I (Simon et al., 2013).
In the same study, we identified the plasmodial transmembrane protein GAP50 as the FH‐binding receptor. While in MZs and in non‐activated intraerythrocytic gametocytes GAP50 localizes to the IMC, the protein relocates to the gamete plasmalemma upon activation (Simon et al., 2013). During this time, the IMC disintegrates to leave the newly formed gametes defined by a single membrane (Sologub et al., 2011). Consequently, antibodies directed against GAP50 are able to reduce FH binding to the gamete surface and inhibit parasite transmission from the human to the mosquito, making GAP50 a promising new candidate for transmission blocking vaccines (Simon et al., 2013).
In accordance with our observations that the ACP is as a major threat for the malaria parasites in the mosquito midgut, transmission blocking antibodies against prominent sexual stage surface proteins like Pfs230 or Pfs25 require active human complement to kill the mosquito midgut stages, as was previously shown in standard membrane feeding assays (e.g. Read et al., 1994; Williamson et al., 1995; Healer et al., 1997; Beiss et al., 2015; Boes et al., 2015; reviewed in Pradel, 2007). Furthermore, in the presence of anti‐Pfs230 antibodies complement factor C1q binds to the gamete surface and activates the classical complement pathway (Simon et al., 2013).
Until recently, antibodies directed against merozoite surface proteins, like MSP1, AMA‐1 or EBA175, were thought to functionally inhibit RBC invasion by interfering with attachment and binding of the merozoite to its host cell (e.g. Hodder et al., 2001; Miura et al., 2009; Reiling et al., 2012; Schwartz et al., 2012). In a new study, however, Boyle et al. (2015) demonstrated that acquired invasion‐inhibitory antibodies act through binding of C1q and activation of the classical complement pathway rather than by functionally inhibiting invasion. Without active complement, the majority of antibodies against merozoite surface proteins like MSP1 were not effective.
Interestingly, RBC invasion involves the human complement receptor CR‐1, which is recognized by the reticulocyte‐binding‐like protein Rh4 located on the apical pole of the MZ (Spadafora et al., 2010; Tham et al., 2010). Approximately 1000 CR‐1 receptors cover the RBC surface, recognize C3b‐opsonized immune complexes and transport these to the liver‐resident Kupffer cells for phagocytosis (Taylor et al., 1997; van Lookeren Campagne et al., 2007). The binding site of Rh4 was mapped to CCP1 of CR‐1, and this region is known to be involved in binding C3b and C4b to accelerate decay of the C3 and C5 convertases (Park et al., 2014). In consequence, Rh4 binding specifically inhibits the convertase decay‐accelerating activity of CR‐1 (Tham et al., 2011).
Besides CR‐1, RBCs also expose GPI‐anchored complement regulators CD55 and CD59 on their surfaces, which promote acceleration of C3 convertase decay and TCC dysfunction respectively [reviewed in Zipfel and Skerka (2009)]. Both GPI‐anchored proteins are abundantly present on the niRBC; however, they become partially internalized by malaria parasites following infection and can then be found bound to the parasitophorous vacuole membrane (Lauer et al., 2000). Noteworthy, in this context, it was recently shown that the digestive vacuoles, when released during schizont rupture, activate the ACP, resulting among others in erythrophagocytosis of bystanders, particularly of iRBCs with reduced CD55 and CD59 levels (Dasari et al., 2012, 2014).
It has previously been postulated that the expansion of MZ invasion ligands, resulting in multiple and apparently redundant invasion pathways, and the presence of highly polymorphic merozoite surface proteins, as it is known for MSP1, are a means of immune evasion by the MZ [reviewed in Wright and Rayner (2014)]. We now show that besides this molecular distraction of the immune system through antigen diversity, the MZs also evolved a complement evasion mechanism by binding FH to their surfaces, resulting in C3b inactivation and in consequence in inhibition of TCC formation and parasite killing. Because we observed FH binding not only on the surface of the extracellular MZs but also on the schizont‐iRBCs, our combined data provide evidence that both stages are highly vulnerable to human complement and thus need to acquire FH for protection.
Contrary to FH acquisition by the extracellular gametes, which is mediated via the binding of GAP50 to CCP5‐7, the ABSs bind FH via CCP modules 5 and 20. Modules CCP18–20 represent a common binding site used by self‐cells as well as by a variety of microbes like Haemophilus influenzae, Neisseria gonorrhoeae, Staphylococcus aureus, Candida albicans or Borrelia burgdoferi to recruit host FH (see Fig. 1B; e.g. Oppermann et al., 2006; Józsi et al., 2007; Meri et al., 2013; reviewed in Zipfel et al., 2007, 2008, 2013; Józsi and Zipfel, 2008; Skerka and Zipfel, 2008; Ferreira et al., 2010). Interestingly, FH binding via CCP20 enhances C3b binding to FH and initiates the formation of a tripartite complex between FH, C3b and the microbial receptor, which then enhances the C3b‐inactiving activity of FH (Meri et al., 2013). Binding of FH via the N‐terminal portion (CCP1‐7), on the other hand, yields the benefit for the pathogen to additionally recruit FHL‐1 for complement evasion. Modules CCP6‐7 can also be used by microbes like Streptococcus pyogenes, Neisseria gonorrhoeae or N. meningitides for complement evasion (e.g. Poltermann et al., 2007; Malito et al., 2013; reviewed in Zipfel et al., 2007).
Interestingly in this context, CCP7 plays an important role in autoimmune regulatory processes, and CCP7 mutations caused by single nucleotide polymorphisms can cause autoimmune defects such as age‐related macular degeneration (AMD), a leading form of blindness. AMD is a chronic inflammatory disease associated with the replacement of tyrosine (Y) by histidine (H) at CCP7 position 402 [Y(402)H]. This exchange results in inappropriate ACP regulation in the retina, ultimately leading to the formation of drusen and vision loss (Skerka et al., 2007; reviewed in Skerka and Zipfel, 2008; Zipfel et al., 2010). Another FH polymorphism associated with AMD is the substitution of isoleucine with valine at position 62 [I(62)V], which results in and increased binding affinity for C3b and enhanced cofactor activity (Tortajada et al., 2009).
AMD is more likely to be found in Caucasians than in persons of African descent (Clemons et al., 2005). However, this conclusion was reached after studying African Americans within the United States, and only a few studies have started to investigate the prevalence of AMD in Africa (e.g. Ziskind et al., 2008; Nwosu, 2011; Oluleye, 2012). Importantly, studies on AMD‐related FH polymorphisms in sub‐Saharan African revealed a marked difference in geographical distribution suggesting that pathogen infections might represent a driving force for these regional differences (Ermini et al., 2012). In view of these findings, the implication of FH polymorphisms on the complement evasion efficiency of malaria parasites and potential links between the occurrence of FH polymorphisms and the incidences of malaria infections in malaria‐endemic regions of Africa would be worth exploring.
We also showed that besides FH and FHL‐1, the ABS parasites are able to bind CFHR‐1. Such binding was not detected by the extracellular gametes of P. falciparum, probably because of the fact that CFHR‐1 is quickly degraded in the mosquito midgut following the blood meal (Simon et al., 2013). The CFHR‐group comprises five members, CFHR‐1 to CFHR‐5; each member of this group binds to C3b and can interact with the surfaces of a variety of pathogens [reviewed in Józsi et al. (2015)]. The precise role of each CFHR protein in complement activation and the exact contribution to disease pathology, however, is still unclear (Skerka et al., 2013). For example, CFHR‐1, CFHR‐2 and CFHR‐5, which circulate in the blood as dimers or tetramers, do not exhibit co‐factor activity and thus are unable to confer protection from complement activation. Further a recent study described that CFHR‐3 acts as an antagonist of FH during complement evasion of the meningococcal pathogen Neisseria meningitidis and supports TCC formation (Caesar et al., 2014). It was further observed that the CFHR proteins preferentially bind to patterns of microbe surfaces (Blaum et al., 2015; reviewed in Józsi et al., 2015), an observation also made in this study, when we showed a prominent CFHR‐1 binding to iRBCs, but not to RBCs. In a recent review, Józsi et al. (2015) thus postulated that the CFHR proteins might have evolved as decoys to reduce the amount of FH that can be bound by the pathogens, in consequence supporting their elimination by the human complement. Noteworthy, homozygous deficiency of CFHR‐1 and CFHR‐3 is very high (up to 16%) in African populations as compared with European populations (about 4%) (Holmes et al., 2013); again, the effect of CFHR‐deficient sera on complement evasion by malaria parasites and potential links between CFHR deficiencies and incidences of malaria infections in malaria‐endemic regions of Africa might be worth investigating.
It is now our aim to identify the receptor or the receptors involved in binding of FH and FH family proteins to the ABS surface. We currently expect that more than one receptor is involved in FH binding by the parasite, because both the free MZs as well as the schizont‐iRBCs are able to bind FH, and to date, no plasmodial protein is known that is present on both surfaces.
Surface‐bound microbial complement receptors represent excellent vaccine targets, because antibodies binding to these receptors on the one hand impair complement evasion by the microbe and on the other hand activate the classical complement pathway. The use of microbial FH‐receptors as vaccine candidates is currently been studied for meningococcal infections (Malito et al., 2013; Rippa et al., 2015), and crucial for such approach the vaccine candidate has to be mutated such that the protein loses its FH‐binding function in order not to interfere with the human complement system.
In‐depth knowledge on complement evasion by the ABSs and the identification of FH‐binding receptors thus represents a first step towards measures to interfere with the erythrocytic replication cycle by promoting the human complement to eliminate the parasites. These pieces of evidence would contribute towards the development of a vaccine capable of inhibiting FH‐mediated complement evasion of ABS and sexual stage parasites, in consequence preventing both progression and transmission of falciparum malaria.
Experimental procedures
Complement factors and antibodies
Purified human FH and factor I were purchased from Sigma‐Aldrich. Polyclonal goat anti‐human FH antibodies (directed against CCP1–20) and purified C3b were purchased from Calbiochem. Mouse mAb anti‐human FH (isotype IgG2bκ; clone 131X; in PBS without preservatives; directed against CCP8‐15), mouse mAb anti‐C5b‐C9 and polyclonal goat anti‐human C3 antibody were purchased from Quidel. Polyclonal rabbit anti‐human C4BP antibody was kindly provided by A. Blom (Lund University) (Happonen et al., 2009). Mouse mAb anti‐human HA (clone 12CA5; without preservatives or stabilizers) was purchased from Roche. Expression of recombinant peptides CCP1‐4, CCP1‐5, CCP1‐6, CCP6‐8, CCP15‐19, CCP8‐20 and CCP18‐20 in Pichia pastoris was described elsewhere (Kühn et al., 1995; Kühn and Zipfel, 1996). Generation of the polyclonal rabbit anti‐CFHR‐1 antisera was described elsewhere (Heinen et al., 2009).
Parasite culture
P. falciparum ABSs of strains NF54 (gametocyte‐producing) or F12 (gametocyte‐deficient) were cultured in RPMI‐HEPES culture medium supplemented with 50 µg ml−1 hypoxanthine (Sigma‐Aldrich) and 10 µg ml−1 gentamicin (Gibco) at 5% hematocrit. The medium contained 0.5% Albumax II (Gibco) or 10–20 vol% human A+ serum (HIS, inactivated for 1 h at 55°C, or NHS) (Trager and Jensen, 1977; Ifediba and Vanderberg, 1981). All cultures were maintained at 37°C in an atmosphere of 5% O2, 5% CO2 and 90% N2. Human erythrocyte concentrate and serum were purchased from the Department of Transfusion Medicine, University Hospital Aachen, Germany. The erythrocyte and sera samples were pooled, and the donors remained anonymous; the work on human blood was approved by the ethics commission of RWTH Aachen University. To synchronize the ABSs, parasite cultures with 3–4% ring stages were centrifuged; the pellet was resuspended in five times pellet's volume of 5 vol% sorbitol (AppliChem) in ddH2O and incubated for 10 min at room temperature (RT) (Lambros and Vanderberg, 1979). The cells were washed once with cell culture medium to remove the sorbitol, diluted to 5 vol% hematocrit with cell culture medium and further cultivated as described in the preceding texts. Schizont‐iRBCs were purified using Percoll gradient centrifugation as described (Radfar et al., 2009). To gain access to live MZ, schizont‐iRBCs were either treated with 0.05% saponin in incomplete cell culture medium (without serum) for 5 min at 37°C or the MZ were purified using a published protocol (Boyle et al., 2010). Here, schizont‐iRBCs were incubated with incomplete medium containing 10 μM E64 (Sigma‐Aldrich) for 8 h at 37°C. When the mature schizont stage was reached, the cells were centrifuged for 10 min at 1900× g and resuspended in 5 ml of incomplete medium via a syringe. The MZ were then filtrated through a 1.2‐µm filter (VWR) and counted using a Neubauer chamber.
Replication and gametocytogenesis assays
To determine ABS replication, a synchronized ring stage culture of strain NF54 with a parasitemia of 0.5% was cultivated with cell culture medium containing 10 vol% NHS or HIS during a period of 0–120 h at 37°C. Medium was exchanged every 12 h to ensure the presence of active complement throughout the experiments. Simultaneously, samples were taken every 12 h and Giemsa smears were performed. The parasitemia was determined in 200 RBCs. To identify the blood stages present in the culture at a given time point, 100 iRBCs were counted per setting using a Zeiss LSM 510 at 100× magnification. To determine gametocytogenesis, a synchronized ring stage culture of strain NF54 with a parasitemia of 5% was cultivated in cell culture medium containing 10 vol% NHS or HIS at 37°C during a period of 0–240 h. Simultaneously, medium was exchanged and samples were taken every 24 h for Giemsa smear preparation. Gametocytemia was determined per 1000 RBCs, and the gametocyte stages II–V at the different time points were recorded. The experiments were performed in triplicate; data analysis was performed using MS Excel 2010 and GraphPad Prism 5.
Pig erythrocyte lysis assay
Cell culture medium with 20 vol% of freshly thawed NHS or of HIS was incubated for different time periods (30 min, 1, 6, 12, 16, 20 and 24 h) at 37°C. A total number of 5 × 106 pig erythrocytes was added to 100 µl of cell culture medium containing NHS or HIS and incubated for 1 h at 37°C. Subsequently, samples were centrifuged at 3400× g for 1 min at RT. The supernatants were collected and transferred to a 96‐well plate. The amount of haemoglobin in the supernatant because of RBC lysis was determined by measuring the absorbance at OD405 nm. Erythrocyte lysis was expressed as a percentage of maximum haemoglobin release as compared with lysis of pig RBCs by suspension in ddH2O (set to 100%). The experiments were performed in triplicate; data analysis was performed using MS Excel 2010 and GraphPad Prism 5.
Indirect immunofluorescence assay
ABS cultures of strains NF54 and F12 containing intact or saponin‐permeabilized schizont‐iRBCs were incubated with cell culture medium containing 20 vol% NHS for 1 h at 37°C. After 3× washing with PBS, the cells were air‐dried on Teflon slides and fixed in 4% paraformaldehyde/PBS or the cells were fixed in 4% paraformaldehyde/0.0025% glutaraldehyde/PBS in suspension for 10 min at RT. The fixed cells were subsequently blocked with 3% BSA/PBS for 1 h at RT and then incubated for 2 h at 37°C with polyclonal goat anti‐CCP1‐20 antibody, polyclonal rabbit anti‐CFHR‐1 or anti‐C4BP antibody, mouse mAb anti‐C5b‐C9 or polyclonal goat anti‐human C3 antibody in 0.5% BSA/PBS. For negative control, the fixed cells were incubated with NMS. Binding of primary antibody was visualized using Alexa Fluor 488‐conjugated goat anti‐mouse, chicken anti‐goat or goat anti‐rabbit secondary antibody (Molecular Probes) depending on the primary antibody used. The ABSs were routinely double‐labelled with polyclonal rabbit anti‐MSP1 antibody in combination with Alexa Fluor 594‐conjugated goat anti‐rabbit secondary antibody for 2 h at 37°C. If no double‐labelling of the ABSs with anti‐MSP1 antibody occurred, the RBCs were counterstained with 0.05% Evans Blue/PBS (Sigma‐Aldrich) for 1 min at RT. Nuclei were stained with Hoechst 33342 according to the manufacturer's protocol (Invitrogen). Specimens were examined by Leica AF 6000 microscope. Digital images were processed using Adobe Photoshop CS software.
Western blot analysis
A total number of 2 × 107 per 100 µl schizont‐iRBCs or purified MZ of strain F12 were incubated in cell culture medium containing 20 vol% NHS, NHS inactivated by 20 mM EDTA (Des Prez et al., 1975; Johnson et al., 2008) or HIS for 1 h at 37°C. For trypsin treatment, the cells were incubated in cell culture medium containing 100 µg ml−1 trypsin for 30 min at 37°C, followed by 5× washing with incomplete medium, prior to incubation with 20 vol% NHS. Following 3× washing with PBS to eliminate unbound FH, the cell pellets were resuspended in 2× SDS sample buffer and heated for 10 min at 94°C. Under non‐reducing conditions, the sample buffer lacked DTT and β‐mercaptoethanol. Lysed niRBCs were used as a negative control. Following gel electrophoresis, separated parasite proteins were transferred to a nitrocellulose membrane according to the manufacturer's protocol (Carl Roth). Membranes were blocked to avoid unspecific binding by incubation with 5% skim milk powder/1% BSA/TBS, pH 7.5, followed by immunoblotting with the polyclonal goat anti‐CCP1‐20 antibody, polyclonal rabbit anti‐CFHR‐1 antibody, mouse mAb anti‐C5b‐C9, polyclonal goat anti‐glycophorin A antibody (Sigma‐Aldrich) or polyclonal goat anti‐human C3 antibody for 2 h at RT. After washing, membranes were incubated with the respective alkaline phosphatase‐conjugated secondary antibody (Sigma‐Aldrich) for 1 h at RT and developed in a solution of nitroblue tetrazolium chloride (NBT) and 5 brom‐4‐chlor‐3‐indoxylphosphate (BCIP; Sigma‐Aldrich). Scanned blots were processed using Adobe Photoshop CS software, and band intensities were measured using the ImageJ programme version 1.44 p (NIH); data analysis was performed using MS Excel 2010.
ELISA for TCC detection
A total number of 5 × 107 schizonts‐iRBCs of strain F12 was incubated with 100 µl of cell culture medium containing 20 vol% NHS or HIS for 1 h at 37°C and washed 3× with PBS. The cells were spun down; resuspended in sodium carbonate‐coating buffer, pH 9.0; and transferred to a 96‐well plate (ELISA high binding; Greiner). The wells were blocked by incubation with 3% skim milk powder/PBS over night at 4°C, followed by incubation with mAb anti‐C5b‐C9 for 2 h at 37°C. After 3× washing with PBS, the wells were incubated with an alkaline phosphatase‐conjugated secondary antibody (Sigma‐Aldrich) for 1 h at 37°C. The reaction was developed with p‐nitrophenylphosphate according to the manufacturer's protocol (Sigma‐Aldrich), and the absorbance was measured at OD450 nm. The experiments were performed in triplicate; data analysis was performed using MS Excel 2010.
FH deletion mutants binding assay
A total number of 2 × 107 schizont‐iRBCs of strain F12 was resuspended in 100 µl PBS containing 10 ng µl−1 of the FH deletion mutants (CCP1‐4, CCP1‐5, CCP1‐6, CCP6‐8, CCP15‐19, CCP18‐20 and CCP8‐20) or FH (CCP1‐20) and then incubated for 1 h at 37°C. The cells were washed 3× with PBS; the pellet was then subjected to non‐reducing gel electrophoresis followed by Western blot analysis using polyclonal goat anti‐CCP1‐20 antibody as described in the preceding texts.
Cofactor Assay
A total number of 2 × 107 schizont‐iRBCs of strain F12, cultivated in 50 µl of Albumax‐supplemented cell culture medium, were washed with incomplete cell culture medium and then incubated in the same volume of incomplete cell culture medium in the presence or absence of 125 ng purified FH/PBS for 30 min at 37°C. Schizonts were briefly washed with PBS, resuspended in 50 µl of incomplete cell culture medium and incubated with 250 ng purified C3b in the presence or absence of 30 ng purified factor I for another 30 min at 37°C. In the absence of complement factors, an equivalent volume of PBS was added. The cells were washed with PBS and subjected to gel electrophoresis followed by Western blot analysis using goat anti‐human C3 antisera.
Growth inhibition assay
A synchronized ABS culture of strain F12 containing schizont‐iRBCs with a parasitemia of 2.5% was cultivated in cell culture medium containing 20 vol% NHS or HIS or 10% Albumax as a control in the presence or absence of 100 µg ml−1 mAbs anti‐FH or anti‐HA for 24 h 37°C. In the absence of antibody, an equivalent volume of PBS was added to the cultures. Medium (containing the mAbs or PBS) was exchanged after 12 h to ensure the presence of active complement throughout the experiments. Giemsa smears were performed to determine the parasitemia at 24 h post‐seeding. The supernatant of the cultures was collected, and 100 µl were transferred to a 96‐well plate. The amount of haemoglobin in the supernatant because of RBC lysis was determined by measuring the absorbance at OD405 nm. The experiments were performed in in triplicate; data analysis was performed using MS Excel 2010.
Gelatin flotation purification
Knob‐infected RBCs were purified via gelatin purification as described (Goodyer et al., 1994). A total number of 5 × 107 schizont‐iRBCs of strain F12 were resuspended in cell culture medium containing 0.7% gelatin and incubated for 1 h at 37°C. Subsequently, the supernatant was removed carefully without touching the pellet. The cells of the supernatant were spun down for 1 min and washed 3× with incomplete medium. The gelatin‐purified schizont‐iRBCs were counted using a Neubauer chamber. Data analysis was performed using MS Excel 2010 and GraphPad Prism 5.
Microwell adhesion assay
The CD36‐binding behaviour of schizont‐iRBCs was investigated as described (Prudhomme and Sherman, 1999). A 96‐well plate (ELISA high binding; Greiner) was coated with 10 µg ml−1 of active recombinant CD36 (Thermo Fischer Scientific) overnight at 4°C. Following 3× washing with PBS, 1 × 107 schizont‐iRBCs per well, resuspended in 100 µl of binding buffer (50 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM CaCl2,50 µg ml−1 BSA), were added and incubated for 2 h at 37°C. After 3× washing with PBS, the relative numbers of bound parasites was determined using the Malstat assay, which assesses the plasmodial lactate dehydrogenase activity (Goodyer and Taraschi, 1997). A volume of 100 µl of the Malstat reagent (1% L‐lactate and 33% 3‐acetylpyridine adenine dinucleotide in Tris–HCl, pH 9; Sigma‐Aldrich) was added per well and mixed with 20 µl of a mixture of NBT (Nitro Blue Tetrazolium)/Diaphorase (1:1; 1 mg ml−1 stock each). The absorbance was measured at OD630 nm. The experiments were performed in in triplicate; data analysis was performed using MS Excel 2010 and GraphPad Prism 5.
Supporting information
Fig. S1: Effect of human complement on ABS differentiation.
Fig. S2: Pig RBC lytic activity of NHS.
Fig. S3: Effect of human complement on gametocyte differentiation.
Fig. S4: IFAs of NMS and saponin‐permeabilization controls.
Fig. S5: Binsding of C4BP to the surface of pathogens.
Fig. S6: Determination of purity of FH deletion mutant peptides.
Fig. S7: Evaluation of knob formation in P. falciparum strain F12.
Fig. S8: Binding of FH by ABS parasites following trypsintreatmen
Supporting info item
Acknowledgements
We thank Anna Blom and Vaibhav Agarwal (Lund University) for supplying all C4BP‐related reagents and for correction of the manuscript. We further thank Alexandra Golzmann and Martin Singheiser (RWTH Aachen University) for technical assistance and statistical support respectively. This research was partially funded by grants PR905/1‐3 and PR905/4‐1 from the Deutsche Forschungsgemeinschaft and by MALSIG of the EU 7th framework programme fund (to GP). TFAR received a fellowship from the Science without Borders Foundation Programme CAPES.
Rosa, T. F. A. , Flammersfeld, A. , Ngwa, C. J. , Kiesow, M. , Fischer, R. , Zipfel, P. F. , Skerka, C. , and Pradel, G. (2016) The Plasmodium falciparum blood stages acquire factor H family proteins to evade destruction by human complement. Cellular Microbiology, 18: 573–590. doi: 10.1111/cmi.12535.
References
- Adam, C. , Geniteau, M. , Gougerot‐Pocidalo, M. , Verroust, P. , Lebras, J. , Gibert, C. , and Morel Maroger, L. (1981) Cryoglobulins, circulating immune complexes, and complement activation in cerebral malaria. Infect Immun 31: 530–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alano, P. (2007) Plasmodium falciparum gametocytes: still many secrets of a hidden life. Mol Microbiol 66: 291–302. [DOI] [PubMed] [Google Scholar]
- Baum, J. , Richard, D. , Healer, J. , Rug, M. , Krnajski, Z. , Gilberger, T.W. , et al. (2006) A conserved molecular motor drives cell invasion and gliding motility across malaria life cycle stages and other apicomplexan parasites. J Biol Chem 281: 5197–5208. [DOI] [PubMed] [Google Scholar]
- Beiss, V. , Spiegel, H. , Boes, A. , Kapelski, S. , Scheuermayer, M. , Edgue, G. , et al. (2015) Heat‐precipitation allows the efficient purification of a functional plant‐derived malaria transmission‐blocking vaccine candidate fusion protein. Biotechnol Bioeng 112: 1297–1305. [DOI] [PubMed] [Google Scholar]
- Biryukov, S. , and Stoute, J.A. (2014) Complement activation in malaria: friend or foe? Trends Mol Med 20: 293–301. [DOI] [PubMed] [Google Scholar]
- Blackman, M.J. , Ling, I.T. , Nicholls, S.C. , and Holder, A.A. (1991) Proteolytic processing of the Plasmodium falciparum merozoite surface protein‐1 produces a membrane‐bound fragment containing two epidermal growth factor‐like domains. Mol Biochem Parasitol 49: 29–33. [DOI] [PubMed] [Google Scholar]
- Blaum, B.S. , Hannan, J.P. , Herbert, A.P. , Kavanagh, D. , Uhrín, D. , and Stehle, T. (2015) Structural basis for sialic acid‐mediated self‐recognition by complement factor H. Nat Chem Biol 11: 77–82. [DOI] [PubMed] [Google Scholar]
- Blom, A.M. , Hallström, T. , and Riesbeck, K. (2009) Complement evasion strategies of pathogens‐acquisition of inhibitors and beyond. Mol Immunol 46: 2808–2817. [DOI] [PubMed] [Google Scholar]
- Boes, A. , Spiegel, H. , Voepel, N. , Edgue, G. , Beiss, V. , Kapelski, S. , et al. (2015) Analysis of a multi‐component multi‐stage malaria vaccine candidate ‐ tackling the cocktail challenge. PLoS One 10: e0131456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosch, J. , Paige, M.H. , Vaidya, A.B. , Bergman, L.W. , and Hol, W.G. (2012) Crystal structure of GAP50, the anchor of the invasion machinery in the inner membrane complex of Plasmodium falciparum. J Struct Biol 178: 61–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle, M.J. , Wilson, D.W. , Richards, J.S. , David, T. , Riglar, D.T. , Tetteh, K.K.A. , et al. (2010) Isolation of viable Plasmodium falciparum merozoites to define erythrocyte invasion events and advance vaccine and drug development. Proc Natl Acad Sci U S A 107: 14378–14383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyle, M.J. , Reiling, L. , Feng, G. , Langer, C. , Osier, F.H. , Aspeling‐Jones, H. , et al. (2015) Human antibodies fix complement to inhibit Plasmodium falciparum invasion of erythrocytes and are associated with protection against malaria. Immunity 42: 580–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caesar, J.J. , Lavender, H. , Ward, P.N. , Exley, R.M. , Eaton, J. , Chittock, E. , et al. (2014) Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. Elife 3: 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, J.A. , Fowkes, F.J. , and Beeson, J.G. (2014) Surface antigens of Plasmodium falciparum‐infected erythrocytes as immune targets and malaria vaccine candidates. Cell Mol Life Sci 91: 3633–3657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clemons, T.E. , Milton, R.C. , Klein, R. , Seddon, J.M. , and Ferris, F.L. (2005) Risk factors for the incidence of advanced age‐related macular degeneration in the age‐related eye disease study. Ophthalmology 112: 533–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dasari, P. , Heber, S.D. , Beisele, M. , Torzewski, M. , Reifenberg, K. , Orning, C. , et al. (2012) Digestive vacuole of Plasmodium falciparum released during erythrocyte rupture dually activates complement and coagulation. Blood 119: 4301–4310. [DOI] [PubMed] [Google Scholar]
- Dasari, P. , Fries, A. , Heber, S.D. , Salama, A. , Blau, I.W. , Lingelbach, K. , et al. (2014) Malarial anemia: digestive vacuole of Plasmodium falciparum mediates complement deposition on bystander cells to provoke hemophagocytosis. Med Microbiol Immunol 203: 383–393. [DOI] [PubMed] [Google Scholar]
- Dearnley, M.K. , Yeoman, J.A. , Hanssen, E. , Kenny, S. , Turnbull, L. , Whitchurch, C.B. , et al. (2012) Origin, composition, organization and function of the inner membrane complex of Plasmodium falciparum gametocytes. J Cell Sci 125: 2053–2063. [DOI] [PubMed] [Google Scholar]
- Des Prez, R.M. , Bryan, C.S. , Hawiger, J. , and Colley, D.G. (1975) Function of the classical and alternate pathways of human complement in serum treated with ethylene glycol tetraacetic acid and MgCl2‐ethylene glycol tetraacetic acid. Infect Immun 11: 1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ermini, L. , Wilson, I.J. , Goodship, T.H. , and Sheerin, N.S. (2012) Complement polymorphisms: geographical distribution and relevance to disease. Immunobiol 217: 265–271. [DOI] [PubMed] [Google Scholar]
- Ferreira, V.P. , Pangburn, M.K. , and Cortésa, C. (2010) Complement control protein factor H: the good, the bad, and the inadequate. Mol Immunol 47: 2187–2197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frénal, K. , Polonais, V. , Marq, J.B. , Stratmann, R. , Limenitakis, J. , and Soldati‐Favre, D. (2010) Functional dissection of the apicomplexan glideosome molecular architecture. Cell Host Microbe 8: 343–357. [DOI] [PubMed] [Google Scholar]
- Garred, P. , Nielsen, M.A. , Kurtzhals, J.A. , Malhotra, R. , Madsen, H.O. , Goka, B.Q. , et al. (2003) Mannose‐binding lectin is a disease modifier in clinical malaria and may function as opsonin for Plasmodium falciparum‐infected erythrocytes. Infect Immun 71: 5245–5253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyer, I.D. , Johnson, J. , Eisenthal, R. , and Hayes, D.J. (1994) Purification of mature‐stage Plasmodium falciparum by gelatine flotation. Ann Trop Med Parasitol 88: 209–211. [DOI] [PubMed] [Google Scholar]
- Goodyer, I.D. , and Taraschi, T.F. (1997) Plasmodium falciparum: a simple, rapid method for detecting parasite clones in microtiter plates. Exp Parasitol 86: 158–160. [DOI] [PubMed] [Google Scholar]
- Greenwood, B.M. , and Brueton, M.J. (1974) Complement activation in children with acute malaria. Clin Exp Immunol 18: 267–272. [PMC free article] [PubMed] [Google Scholar]
- Happonen, K.E. , Sjoberg, A.P. , Morgelin, M. , Heinegard, D. , and Blom, A.M. (2009) Complement inhibitor C4b‐binding protein interacts directly with small glycoproteins of the extracellular matrix. J Immunol 182: 1518–1527. [DOI] [PubMed] [Google Scholar]
- Healer, J. , McGuinness, D. , Hopcroft, P. , Haley, S. , Carter, R. , and Riley, E. (1997) Complement‐mediated lysis of Plasmodium falciparum gametes by malaria‐immune human sera is associated with antibodies to the gamete surface antigen Pfs230. Infect Immun 65: 3017–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinen, S. , Hartmann, A. , Lauer, N. , Wiehl, U. , Dahse, H.M. , Schirmer, S. , et al. (2009) Factor H‐related protein 1 (CFHR‐1) inhibits complement C5 convertase activity and terminal complex formation. Blood 114: 2439–2447. [DOI] [PubMed] [Google Scholar]
- Hodder, A.N. , Crewther, P.E. , and Anders, R.F. (2001) Specificity of the protective antibody response to apical membrane antigen 1. Infect Immun 69: 3286–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holder, A.A. , and Freeman, R.R. (1984) The three major antigens on the surface of Plasmodium falciparum merozoites are derived from a single high molecular weight precursor. J Exp Med 160: 624–629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes, L.V. , Strain, L. , Staniforth, S.J. , Moore, I. , Marchbank, K. , Kavanagh, D. , et al. (2013) Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PLoS One 8: e60352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ifediba, T. , and Vanderberg, J. (1981) Complete in vitro maturation of Plasmodium falciparum gametocytes. Nature 294: 364–366. [DOI] [PubMed] [Google Scholar]
- Jhaveri, K.N. , Ghosh, K. , Mohanty, D. , Parmar, B.D. , Surati, R.R. , Camoens, H.M. , et al. (1997) Autoantibodies, immunoglobulins, complement and circulating immune complexes in acute malaria. Natl Med J India 10: 5–7. [PubMed] [Google Scholar]
- Johnson, J.B. , Capraro, G.A. , and Parks, G.D. (2008) Differential mechanisms of complement‐mediated neutralization of the closely related paramyxoviruses simian virus 5 and mumps virus. Virology 376: 112–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Józsi, M. , and Zipfel, P.F. (2008) Factor H family proteins and human diseases. Trends Immunol 29: 380–387. [DOI] [PubMed] [Google Scholar]
- Józsi, M. , Oppermann, M. , Lambris, J.D. , and Zipfel, P.F. (2007) The C‐terminus of complement factor H is essential for host cell protection. Mol Immunol 44: 2697–2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Józsi, M. , Tortajada, A. , Uzonyi, B. , Goicoechea de Jorge, E. , and Rodríguez de Córdoba, S. (2015) Factor H‐related proteins determine complement‐activating surfaces. Trends Immunol 36: 374–384. [DOI] [PubMed] [Google Scholar]
- Kadekoppala, M. , and Holder, A.A. (2010) Merozoite surface proteins of the malaria parasite: the MSP1 complex and the MSP7 family. Int J Parasitol 40: 1155–1161. [DOI] [PubMed] [Google Scholar]
- Kirchgatter, K. , and Del Portillo, H.A. (2005) Clinical and molecular aspects of severe malaria. An Acad Bras Cienc 77: 455–475. [DOI] [PubMed] [Google Scholar]
- Klabunde, J. , Uhlemann, A.C. , Tebo, A.E. , Kimmel, J. , Schwarz, R.T. , Kremsner, P.G. , and Kun, J.F. (2002) Recognition of Plasmodium falciparum proteins by mannan‐binding lectin, a component of the human innate immune system. Parasitol Res 88: 113–117. [DOI] [PubMed] [Google Scholar]
- Kono, M. , Herrmann, S. , Loughran, N.B. , Cabrera, A. , Engelberg, K. , Lehmann, C. , et al. (2012) Evolution and architecture of the inner membrane complex in asexual and sexual stages of the malaria parasite. Mol Biol Evol 29: 2113–2132. [DOI] [PubMed] [Google Scholar]
- Kuehn, A. , and Pradel, G. (2010) The coming‐out of the malaria gametocytes. J Biomed Biotechnol 2010: e976827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn, S. , and Zipfel, P.F. (1996) Mapping of the domains required for decay acceleration activity of the human factor H‐like protein 1 and factor H. Eur J Immunol 26: 2383–2387. [DOI] [PubMed] [Google Scholar]
- Kühn, S. , Skerka, C. , and Zipfel, P.F. (1995) Mapping of the complement regulatory domains in the human factor H‐like protein 1 and in factor H1. J Immunol 155: 5663–5670. [PubMed] [Google Scholar]
- Lambros, C. , and Vanderberg, J.P. (1979) Synchronization of Plasmodium falciparum erythrocytic stages in culture. J Parasitol 65: 418–420. [PubMed] [Google Scholar]
- Lauer, S. , VanWye, J. , Harrison, T. , McManus, H. , Samuel, B.U. , Hiller, N.L. , et al. (2000) Vacuolar uptake of host components, and a role for cholesterol and sphingomyelin in malarial infection. EMBO J 19: 3556–3564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leech, J.H. , Barnwell, J.W. , Miller, L.H. , and Howard, R.J. (1984) Identification of a strain‐specific malarial antigen exposed on the surface of Plasmodium falciparum‐infected erythrocytes. J Exp Med 159: 1567–1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, S. , Skerka, C. , Kurzai, O. , and Zipfel, P.E. (2013) Complement and innate immune evasion strategies of the human pathogenic fungus Candida albicans . Mol Immunol 56: 161–169. [DOI] [PubMed] [Google Scholar]
- Malito, E. , Faleri, A. , Lo Surdo, P. , Veggi, D. , Maruggi, G. , Grassi, E. , et al. (2013) Defining a protective epitope on factor H binding protein, a key meningococcal virulence factor and vaccine antigen. Proc Natl Acad Sci U S A 110: 3304–3309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meri, T. , Amdahl, H. , Lehtinen, M.J. , Hyvärinen, S. , McDowell, J.V. , Bhattacharjee, A. , et al. (2013) Microbes bind complement inhibitor factor H via a common site. PLoS Pathog 9: e1003308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura, K. , Zhou, H. , Diouf, A. , Moretz, S.E. , Fay, M.P. , Miller, L.H. , et al. (2009) Anti‐apical‐membrane‐antigen‐1 antibody is more effective than anti‐42‐kilodalton‐merozoite‐surface‐protein‐1 antibody in inhibiting Plasmodium falciparum growth, as determined by the in vitro growth inhibition assay. Clin Vaccine Immunol 16: 963–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nwosu, S.N. (2011) Age‐related macular degeneration in Onitsha, Nigeria. Niger J Clin Pract 14: 327–331. [DOI] [PubMed] [Google Scholar]
- Nyakoe, N.K. , Taylor, R.P. , Makumi, J.N. , and Waitumbi, J.N. (2009) Complement consumption in children with Plasmodium falciparum malaria. Malar J 8: 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oluleye, T.S. (2012) Is age‐related macular degeneration a problem in Ibadan, Sub‐Saharan Africa? Clin Ophthalmol 6: 561–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oppermann, M. , Manuelian, T. , Józsi, M. , Brandt, E. , Jokiranta, T.S. , Heinen, S. , et al. (2006) The C‐terminus of complement regulator Factor H mediates target recognition: evidence for a compact conformation of the native protein. Clin Exp Immunol 144: 342–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park, H.J. , Guariento, M. , Maciejewski, M. , Hauhart, R. , Tham, W.H. , Cowman, A.F. , et al. (2014) Using mutagenesis and structural biology to map the binding site for the Plasmodium falciparum merozoite protein PfRh4 on the human immune adherence receptor. J Biol Chem 289: 450–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phanuphak, P. , Hanvanich, M. , Sakulramrung, R. , Moollaor, P. , Sitprija, V. , and Phanthumkosol, D. (1985) Complement changes in falciparum malaria infection. Clin Exp Immunol 59: 571–576. [PMC free article] [PubMed] [Google Scholar]
- Poltermann, S. , Kunert, A. , von der Heide, M. , Eck, R. , Hartmann, A. , and Zipfel, P.F. (2007) Gpm1p is a factor H‐, FHL‐1‐, and plasminogen‐binding surface protein of Candida albicans. J Biol Chem 282: 37537–37544. [DOI] [PubMed] [Google Scholar]
- Pradel, G. (2007) Proteins of the malaria parasite sexual stages: expression, function and potential for transmission blocking strategies. Parasitology 134: 1911–1929. [DOI] [PubMed] [Google Scholar]
- Prudhomme, J.G. , and Sherman, I.W. (1999) A high capacity in vitro assay for measuring the cytoadherence of Plasmodium falciparum‐infected erythrocytes. J Immunol Methods 229: 169–176. [DOI] [PubMed] [Google Scholar]
- Radfar, A. , Méndez, D. , Moneriz, C. , Linares, M. , Marín‐García, P. , Puyet, A. , et al. (2009) Synchronous culture of Plasmodium falciparum at high parasitemia levels. Nat Protoc 4: 1899–1915. [DOI] [PubMed] [Google Scholar]
- Read, D. , Lensen, A.H. , Begarnie, S. , Haley, S. , Raza, A. , and Carter, R. (1994) Transmission‐blocking antibodies against multiple, non‐variant target epitopes of the Plasmodium falciparum gamete surface antigen Pfs230 are all complement‐fixing. Parasite Immunol 16: 511–519. [DOI] [PubMed] [Google Scholar]
- Reiling, L. , Richards, J.S. , Fowkes, F.J. , Wilson, D.W. , Chokejindachai, W. , Barry, A.E. , et al. (2012) The Plasmodium falciparum erythrocyte invasion ligand Pfrh4 as a target of functional and protective human antibodies against malaria. PLoS One 7: e45253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rippa, V. , Santini, L. , Lo Surdo, P. , Cantini, F. , Veggi, D. , Gentile, M.A. , et al. (2015) Molecular engineering of Ghfp, the gonococcal orthologue of Neisseria meningitidis factor H binding protein. Clin Vaccine Immunol 22: 769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother, K. , Till, G. , and Hänsch, G.M. (1998) The Complement System. Springer Science & Business Media Berlin. [Google Scholar]
- Sanders, P.R. , Cantin, G.T. , Greenbaum, D.C. , Gilson, P.R. , Nebl, T. , Moritz, R.L. , et al. (2007) Identification of protein complexes in detergent‐resistant membranes of Plasmodium falciparum schizonts. Mol Biochem Parasitol 154: 148–157. [DOI] [PubMed] [Google Scholar]
- Schwartz, L. , Brown, G.V. , Genton, B. , and Moorthy, V.S. (2012) A review of malaria vaccine clinical projects based on the WHO rainbow table. Malar J 11: 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon, N. , Lasonder, E. , Scheuermayer, M. , Kuehn, A. , Tews, S. , Fischer, R. , et al. (2013) Malaria parasites co‐opt human factor H to prevent complement‐mediated lysis in the mosquito midgut. Cell Host Microbe 13: 29–41. [DOI] [PubMed] [Google Scholar]
- Skerka, C. , and Zipfel, P.F. (2008) Complement factor H related proteins in immune diseases. Vaccine 26(Suppl 8): I9–14. [DOI] [PubMed] [Google Scholar]
- Skerka, C. , Lauer, N. , Weinberger, A.A. , Keilhauer, C.N. , Sühnel, J. , Smith, R. , et al. (2007) Defective complement control of factor H (Y402H) and FHL‐1 in age‐related macular degeneration. Mol Immunol 44: 3398–3406. [DOI] [PubMed] [Google Scholar]
- Skerka, C. , Chen, Q. , Fremeaux‐Bacchi, V. , and Roumenina, L.T. (2013) Complement factor H related proteins (CFHRs). Mol Immunol 56: 170–180. [DOI] [PubMed] [Google Scholar]
- Sologub, L. , Kuehn, A. , Kern, S. , Przyborski, J. , Schillig, R. , and Pradel, G. (2011) Malaria proteases mediate inside‐out egress of gametocytes from red blood cells following parasite transmission to the mosquito. Cell Microbiol 13: 897–912. [DOI] [PubMed] [Google Scholar]
- Spadafora, C. , Awandare, G.A. , Kopydlowski, K.M. , Czege, J. , Moch, J.K. , Finberg, R.W. , et al. (2010) Complement receptor 1 is a sialic acid‐independent erythrocyte receptor of Plasmodium falciparum. PLoS Pathog 6: e1000968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoute, J.A. , Odindo, A.O. , Owuor, B.O. , Mibei, E.K. , Opollo, M.O. , and Waitumbi, J.N. (2003) Loss of red blood cell‐complement regulatory proteins and increased levels of circulating immune complexes are associated with severe malarial anemia. J Infect Dis 187: 522–525. [DOI] [PubMed] [Google Scholar]
- Talman, A.M. , Paul, R.E. , Sokhna, C.S. , Domarle, O. , Ariey, F. , Trape, J.F. , and Robert, V. (2004) Influence of chemotherapy on the Plasmodium gametocyte sex ratio of mice and humans. Am J Trop Med Hyg 71: 739–744. [PubMed] [Google Scholar]
- Taylor, R.P. , Nardin, A. , and Sutherland, W.M. (1997) Clearance of blood‐borne pathogens mediated through bispecific monoclonal antibodies bound to the primate erythrocyte complement receptor. Cancer Immunol Immunother 45: 152–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham, W.H. , Wilson, D.W. , Lopaticki, S. , Schmidt, C.Q. , Tetteh‐Quarcoo, P.B. , Barlow, P.N. , et al. (2010) Complement receptor 1 is the host erythrocyte receptor for Plasmodium falciparum PfRh4 invasion ligand. Proc Natl Acad Sci U S A 107: 17327–17332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tham, W.H. , Schmidt, C.Q. , Hauhart, R.E. , Guariento, M. , Tetteh‐Quarcoo, P.B. , Lopaticki, S. , et al. (2011) Plasmodium falciparum uses a key functional site in complement receptor type‐1 for invasion of human erythrocytes. Blood 118: 1923–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tortajada, A. , Montes, T. , Martínez‐Barricarte, R. , Morgan, B.P. , Harris, C.L. , and de Córdoba, S.R. (2009) The disease‐protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum Mol Genet 18: 3452–3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trager, W. , and Jensen, J.B. (1977) Cultivation of erythrocytic stages. Bull World Health Organ 55: 361–365. [PMC free article] [PubMed] [Google Scholar]
- van Lookeren Campagne, M. , Wiesmann, C. , and Brown, E.J. (2007) Macrophage complement receptors and pathogen clearance. Cell Microbiol 9: 2095–2102. [DOI] [PubMed] [Google Scholar]
- Williamson, K.C. , Keister, D.B. , Muratova, O. , and Kaslow, D.C. (1995) Recombinant Pfs230, a Plasmodium falciparum gametocyte protein, induces antisera that reduce the infectivity of Plasmodium falciparum to mosquitoes. Mol Biochem Parasitol 75: 33–42. [DOI] [PubMed] [Google Scholar]
- Wirth, C.C. , and Pradel, G. (2012) Molecular mechanisms of host cell egress by malaria parasites. Int J Med Microbiol 302: 172–178. [DOI] [PubMed] [Google Scholar]
- World Malaria Report, W.H.O . (2014) http://www.who.int/malaria/publications/world_malaria_report_2014/en/
- Wright, G.J. , and Rayner, J.C. (2014) Plasmodium falciparum erythrocyte invasion: combining function with immune evasion. PLoS Pathog 10: e1003943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zipfel, P.F. , and Skerka, C. (2009) Complement regulators and inhibitory proteins. Nat Rev Immunol 9: 729–740. [DOI] [PubMed] [Google Scholar]
- Zipfel, P.F. , and Skerka, C. (2014) Staphylococcus aureus: the multi headed hydra resists and controls human complement response in multiple ways. Int J Med Microbiol 304: 188–194. [DOI] [PubMed] [Google Scholar]
- Zipfel, P.F. , Würzner, R. , and Skerka, C. (2007) Complement evasion of pathogens: common strategies are shared by diverse organisms. Mol Immunol 44: 3850–3857. [DOI] [PubMed] [Google Scholar]
- Zipfel, P.F. , Hallström, T. , Hammerschmidt, S. , and Skerka, C. (2008) The complement fitness factor H: role in human diseases and for immune escape of pathogens, like pneumococci. Vaccine 26(Suppl 8): I67–74. [DOI] [PubMed] [Google Scholar]
- Zipfel, P.F. , Lauer, N. , and Skerka, C. (2010) The role of complement in AMD. Adv Exp Med Biol 703: 9–24. [DOI] [PubMed] [Google Scholar]
- Zipfel, P.F. , Hallström, T. , and Riesbeck, K. (2013) Human complement control and complement evasion by pathogenic microbes‐tipping the balance. Mol Immunol 56: 152–160. [DOI] [PubMed] [Google Scholar]
- Ziskind, A. , Bardien, S. , van der Merwe, L. , and Webster, A.R. (2008) The frequency of the H402 allele of CFH and its involvement with age‐related maculopathy in an aged Black African Xhosa population. Ophthalmic Genet 29: 117–119. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1: Effect of human complement on ABS differentiation.
Fig. S2: Pig RBC lytic activity of NHS.
Fig. S3: Effect of human complement on gametocyte differentiation.
Fig. S4: IFAs of NMS and saponin‐permeabilization controls.
Fig. S5: Binsding of C4BP to the surface of pathogens.
Fig. S6: Determination of purity of FH deletion mutant peptides.
Fig. S7: Evaluation of knob formation in P. falciparum strain F12.
Fig. S8: Binding of FH by ABS parasites following trypsintreatmen
Supporting info item