

Abstract
The design of multitarget‐directed ligands is a promising strategy for discovering innovative drugs. Here, we report a mechanistic study that clarifies key aspects of the dual inhibition of the fatty acid amide hydrolase (FAAH) and the cyclooxygenase (COX) enzymes by a new multitarget‐directed ligand named ARN2508 (2‐[3‐fluoro‐4‐[3‐(hexylcarbamoyloxy)phenyl]phenyl]propanoic acid). This potent dual inhibitor combines, in a single scaffold, the pharmacophoric elements often needed to block FAAH and COX, that is, a carbamate moiety and the 2‐arylpropionic acid functionality, respectively. Molecular modeling and molecular dynamics simulations suggest that ARN2508 uses a noncovalent mechanism of inhibition to block COXs, while inhibiting FAAH via the acetylation of the catalytic Ser241, in line with previous experimental evidence for covalent FAAH inhibition. This study proposes the molecular basis for the dual FAAH/COX inhibition by this novel hybrid scaffold, stimulating further experimental studies and offering new insights for the rational design of novel anti‐inflammatory agents that simultaneously act on FAAH and COX.
Keywords: cyclooxygenase, drug design, fatty acid amide hydrolase, molecular dynamics, molecular modeling, multitarget ligands
Nonsteroidal anti‐inflammatory drugs (NSAIDs) are widely used to treat acute and chronic pain.1, 2 NSAIDs exert their action by inhibiting COX, which converts arachidonic acid (AA) into prostanoids that act as physio‐pathological effectors.3 COX exists in two isoforms, COX‐1 and COX‐2, and NSAIDs are classified into several classes, being either nonselective for COX‐1 and COX‐2 or selective for COX‐2.4 Unfortunately, NSAID action is accompanied by a number of side effects, especially at the gastrointestinal level, where peptic ulceration and dyspepsia can limit their clinical use.5 However, recent studies have indicated that the analgesic effect of NSAIDs is enhanced when administered in combination with drugs that inhibit FAAH.6, 7 FAAH is a serine hydrolase responsible for deactivating the bioactive lipid anandamide, which is the main endogenous neurotransmitter involved in the endocannabinoid‐mediated control of pain.8, 9, 10 FAAH inhibition greatly decreases the frequency and severity of gastric side effects caused by COX inhibition. A multitarget‐directed drug discovery strategy11 to simultaneously block FAAH and COX could thus generate new anti‐inflammatory therapeutics for the treatment of pain.12, 13, 14, 15
Recently, some members of our group first disclosed, in a patent application,15 a new class of systemically active agents that simultaneously inhibit FAAH, COX‐1, and COX‐2 with high potency and selectivity; ARN2508 was identified as the lead inhibitor (Figure 1, compound 12 in Ref. 15). ARN2508 shows high potency with an inhibitory concentration (IC50) of 0.031±0.002 μm against rat FAAH, 0.012±0.002 μm against COX‐1, and 0.43±0.025 μm against COX‐2. ARN2508 has been proven to exert profound therapeutic effects in in vivo models of intestinal inflammation, without exhibiting the typical side effects of classical NSAIDs.15
Figure 1.

Design of multitarget inhibitors of FAAH and COX‐1/2. By merging the key pharmacophoric elements of carbamate‐based FAAH inhibitors (URB524, top left) and 2‐arylpropionic acid COX‐1/2 inhibitors (flurbiprofen, top right), we generated a hybrid scaffold (ARN2508) active on both FAAH and COX‐1/2.
ARN2508 combines, in a single scaffold, the pharmacophoric elements that characterize two well‐known classes of inhibitors of FAAH and COX. It bears the pharmacophoric element needed for FAAH inhibition, i.e. a carbamate group also found in the potent FAAH inhibitor URB524.16 It also bears a pharmacophoric group needed for COX inhibition, i.e. the 2‐arylpropionic acid also found in the COX inhibitor flurbiprofen (FLP; Figure 1).17 Carbamate‐based inhibitors covalently inhibit FAAH by binding at the catalytic serine (Ser241).16 FLP tightly binds COX‐1/2 via its free carboxylate moiety, which establishes a network of polar interactions within the enzyme active site.18, 19 Accordingly, we hypothesize that ARN2508 covalently inhibits FAAH using the carbamate group, while blocking COX thanks to the carboxylate moiety. Notably, removing the carboxylate on ARN2508 results in the complete loss of activity toward both COX isoforms.15
FAAH catalyzes the hydrolysis of anandamide, generating AA, which is the substrate of COX. Both active sites are characterized by a long hydrophobic channel, which accommodates the long arachidonoyl chain of the substrates, and by a hydrophilic tip, which allows the polar head group of the substrate lipid to bind (Figure 2). The binding pockets of the COX and FAAH active sites share structural similarities, as previously demonstrated with a comparative study.14 This further rationalizes the activity of dual inhibitors such as ARN2508 (Figure 2).12, 14, 15
Figure 2.
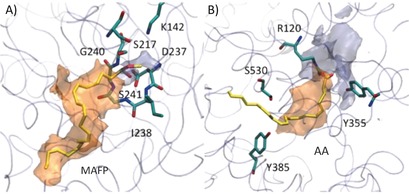
Active sites of A) FAAH (PDB code: 1MT5)6 and B) COX‐2 (PDB code: 3PGH)20 in complex with the substrate analogue methyl arachidonyl fluorophosphonate (MAFP) and with arachidonic acid (AA), respectively. The hydrophilic (light blue) and hydrophobic (orange) isocontour surfaces of the proteins are shown. The protein is shown as a transparent cyan tube. The ligand (yellow) and key protein residues (cyan) are shown as sticks. Adapted with permission from Ref. 14. Copyright the American Chemical Society, 2012.14
Here, we used molecular modeling and simulations to decipher the mechanism of binding of ARN2508 at the target level. This robust and informative approach has been applied to several other pharmaceutically relevant targets.21 Importantly, the mechanism of FAAH covalent inhibition of carbamate‐based agents, such as ARN2508, has been widely studied during the last years by several independent groups,8, 9, 10, 22, 23 while the mechanism of binding at the COX level of this compound class is less clear. Thus, in the present study, we first use molecular dynamics (MD) simulations and free energy calculations, to propose and analyze a plausible model for the binding of ARN2508 to COX, which represents the bottleneck for dual activity given the smaller size of the binding pocket of this enzyme, compared with FAAH.1, 2, 3, 6, 7 We compared our MD‐based results to existing structural data, including the X‐ray crystal structures of COX in complex with the inhibitor FLP19 and with AA,4 which is the main substrate of COX. Our results suggested a possible noncovalent inhibition of COX, providing a rationale for the potency of ARN2508 with respect to FLP.15 Then, we used molecular docking calculations to rationalize the binding of ARN2508 within the FAAH active site, confirming a possible covalent inhibition of the enzyme. This agrees well with the experimentally characterized mechanism of covalent FAAH inhibition by carbamate‐based inhibitors.24, 25, 26
In detail, we built three MD simulation systems of the COX‐1 protein in complex with either ARN2508 (i.e., COX‐1/ARN2508), the AA substrate (i.e. COX‐1/AA), or FLP (i.e. COX‐1/FLP). This was based on the X‐ray structure of sheep COX‐1 in complex with AA, solved at 3.0 Å resolution (PDB code: 1DIY).4 Importantly, we considered the COX‐1 isoform, which is a bottleneck for COX inhibition because its active site architecture is narrower than that of COX‐2.1 The initial binding poses of the ligands AA and FLP were derived from the available crystallographic data,4, 19 while ARN2508 was docked within the COX‐1 active site using Glide software27 from the Schrödinger suite.28 The initial binding pose of ARN2508 is based on the underlying assumption of a FLP‐like binding mode, which, again, is based on structural data. Full details on our model systems and simulation set‐ups are reported in the Supporting Information.
Simulations of ∼100 ns per system, with statistics collected over the two monomers, showed a stable protein framework, as determined by calculating the root mean square deviation (RMSD, Figure S1 A in the Supporting Information) for the protein heavy atoms with respect to the minimized X‐ray structure,21 which lie below the crystal structure resolution. During the dynamics of the three investigated systems, the ligands adopted a stable bound configuration (Figure 3 B; see also, Figure S1 B in the Supporting Information), which confirmed a tight binding at the active site of COX‐1. In particular, in the COX‐1/AA system, the ligand stably maintained the conformation and orientation of the original crystallographic structure, assuming an extended L‐shape conformation with two kinks in the center.4
Figure 3.

A) X‐ray structure of COX‐1 in complex with arachidonic acid (AA) (PDB code: 1DIY).4 COX‐1 comprises two monomers, represented by green (Mnr‐A) and cyan (Mnr‐B) ribbons. AA is shown with yellow sticks. B) Binding mode of AA, flurbiprofen (FLP), and putative binding pose of ARN2508 from our MD simulations of the COX‐1/AA, COX‐1/FLP, and COX‐1/ARN2508 systems. Key protein residues (cyan) and the ligands, AA (yellow), FLP (orange), and ARN2508 (magenta), are shown as sticks. The protein framework is shown as cyan ribbons.
We further characterized the binding of the compounds over the production runs, looking at the statistical distribution of the direct interactions (H‐bonds and hydrophobic contacts) between each ligand (AA, FLP, and ARN2508) and the COX‐1 residues that are in close contact with the natural AA substrate in the 1DIY X‐ray crystal structure (Figure 4).4 The interaction network formed by the substrate AA in the X‐ray structure was used as a reference for analyzing the ligand binding during our MD simulations. In particular, we focused on those interactions that mainly characterize the AA bound pose into COX‐1, namely the carboxyl, C2–C11, C12–C13, and C14–C20 interactions (Figure 4). Full details on the statistical analyses are reported in the Supporting Information.
Figure 4.
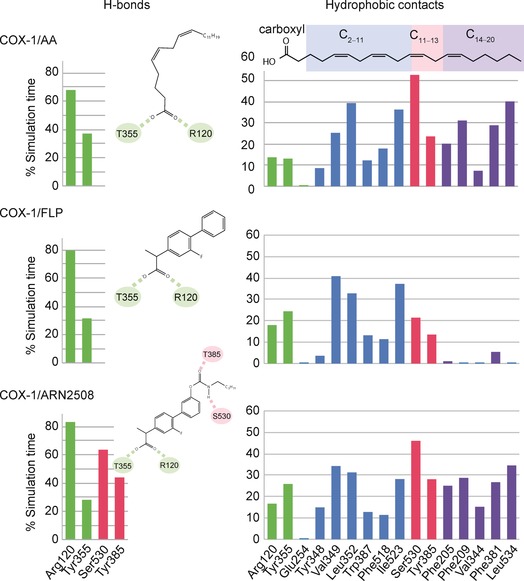
Statistical distribution (% of the total simulation time) over the whole production run of the H‐bonds (left) and the hydrophobic contacts (right) established by AA (top), FLP (middle), and ARN2508 (bottom) with the carboxyl (green bars), C2–C11 (blue bars), C12–C13 (red bars), and C14–C20 (violet bars) interaction regions of the COX‐1 active site. Interaction regions are defined based on the interaction network of the moieties of AA in the X‐ray structure (PDB code: 1DIY).4 Importantly, statistics were collected as a sum of the data arising from both COX‐1 subunits of each equilibrated system. Full details on statistical analysis are reported in the Supporting Information.
In the COX‐1/AA system, the AA carboxyl group forms a stable H‐bond network with Arg120 and Tyr355, which remains stable for 67.9 % and 37.0 % of the simulation time, respectively (first row/left column in Figure 4). Key hydrophobic contacts that involve the AA arachidonoyl chain and the COX‐1 residues are also well maintained (first row/right column in Figure 4). In detail, the C2–C11 chain of the AA substrate is in contact with Ile523, Leu352, Phe518, Trp387, and Val349. The C12–C13 moiety is in close proximity to Tyr385, which is likely the radical donor during COX catalysis, and the key Ser530, which is acetylated by aspirin for COX inhibition.1, 4, 29 The C14–C20 chain establishes multiple hydrophobic interactions with Leu354, Phe205, Phe209, Phe381, and Val344. Overall, key interactions between AA and the COX‐1 active site, as found in the crystal structure of the COX‐1/AA complex,4 are preserved throughout our simulations.
In the COX‐1/FLP system, FLP is stable, as in its crystallographic pose, throughout the entire trajectory (Figure 3 B).19 The FLP carboxyl group stably H‐bonds Arg120 and Tyr355, with a high statistical abundance (79.6 % and 31.6 % of the simulation time, respectively; second row/left column in Figure 4). This confirms a tight binding at the COX‐1 active site.18, 19 Hydrophobic interactions with the residues in the C2–C11 interaction region were also statistically relevant. Notably, thanks to the biphenyl moiety, FLP shows improved hydrophobic contacts with Val394, with respect to the AA substrate (40.8 % vs 25.4 %, respectively). At the level of the catalytic residue Tyr385 and the key residue Ser530, the interactions are statistically less frequent. As expected, hydrophobic contacts are unlikely within the C14–C20 interaction region.4
In the COX‐1/ARN2508 system, the carboxyl group of ARN2508 H‐bonds Tyr355 and Arg120, suggesting a binding mode similar to that of the 2‐arylpropionic acid class of COX inhibitors (Figure 3 B).18, 19 This H‐bond network remains intact throughout the dynamics, as also observed for the COX‐1/AA and COX‐1/FLP systems. A thorough comparison between the binding mode of FLP and ARN2508 (Figure 3 B) shows that their common biphenyl moiety occupies the same region within the COX‐1 active site, involving the C2–C11 interaction region. This binding mode is well maintained throughout the dynamics. The statistical distribution of the hydrophobic contacts shows a similar interaction pattern for FLP and ARN2508 in the C2–C11 interaction region (second and third rows/right column in Figure 4). Moreover, the ARN2508 acyl chain establishes hydrophobic interactions with Phe205, Phe209, Val344, Phe381, and Leu354, occupying the C14–C20 interaction region. Here, the statistical distribution of the hydrophobic contacts shows a similar pattern to that of the AA substrate (right column in Figure 4). These data explain the potency of ARN2508 at the target level. ARN2508 benefits from the presence of the key structural components of FLP (a biphenyl moiety enhancing van der Waals contacts within the C2–C11 interaction region) as well as an arachidonoyl tail, which improves the hydrophobic interactions within the enzymatic cavity (within the C12–C13 and the C14–C20 interaction regions). Notably, structure–activity relationship (SAR) studies—recently reported in a patent application15—confirm the essential role of the terminal arachidonoyl moiety in ARN2508. Indeed, among 20 newly synthetized compounds sharing a hybrid scaffold active on FAAH and COX (Figure 1), the activity against the two enzymes decreases by substituting the terminal arachidonoyl chain with aliphatic/aromatic rings or longer acyl chains, likely because of steric hindrance, as suggested by our model. Thus, these extensive SAR studies agree with the mechanistic hypotheses and binding models proposed here.
During the dynamics, the ARN2508 carbamate group remains in close contact with Ser530, which is the key residue acetylated by aspirin for COX inhibition (Figure 3 B). To gain further insights into the possible mechanism of COX inhibition and to clarify whether the key Ser530 could act as a nucleophilic agent on the ARN2508 carbonyl, we monitored geometrical descriptors to analyze the formation of pre‐catalytic states to favor such a nucleophilic attack (Figure 5). In detail, we considered the so‐called “Bürgi–Dunitz trajectory” for the nucleophilic attack. According to this, the nucleophilic attack is likely to happen only when the distance (d nu) between the ARN2508 carbonyl carbon and the oxygen of Ser530 (C@ARN2508–O@Ser530) is lower than 3.4 Å and, concomitantly, when the attacking angle formed by the nucleophilic species (O@Ser530) and the ARN2508 carbonyl plane (θ nu) is within 110° ±20°.30
Figure 5.
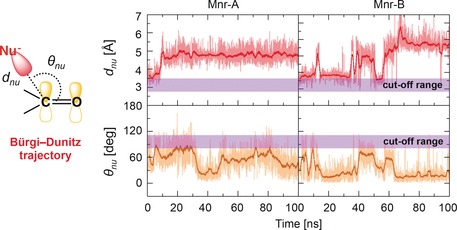
Time evolution, over the molecular dynamics (MD) production run of the COX‐1/ARN2508 system, of the distance (d nu, top graphs) and angle (θ nu, bottom graphs) for the nucleophilic attack between the Ser 530 nucleophile (O@Ser530) and the ARN2508 electrophile (C@ARN2508). Data are reported for both COX‐1 monomers Mnr‐A (left graphs) and Mnr‐B (right graphs). d nu and θ nu define the so‐called “Bürgi–Dunitz trajectory” for the nucleophilic attack, which is schematically represented on the left. Cut‐off ranges for d nu and θ nu favoring the nucleophilic attack are highlighted with a violet bar.
The configurations respecting the abovementioned requirements are classified as “catalytically significant conformations” and can be properly correlated to the formation of pre‐reactive states of the system with the propensity to react. This approach has been successfully employed for clarifying the covalent binding propensity of FAAH inhibitors.8, 10 We note that these structural parameters only relate to the propensity of ARN2508 to undergo nucleophilic attack given the proper relative orientation of the ligand with respect to the nucleophilic Ser530 in the binding pocket of COX‐1. We cannot correlate the propensity of ARN2508 to undergo nucleophilic attack with the enzymatic barrier for this reaction.8, 9 As shown in Figure 5, the d nu distance and the θ nu angle for the nucleophilic attack are out of range, suggesting that a covalent mechanism for COX inhibition is unlikely. Inversely, Ser530 stably H‐bonds to the ARN2508 carbonyl oxygen, further stabilizing the noncovalent COX‐1/ARN2508 complex (third row/left column in Figure 4).15
Using numerous structural snapshots from our MD simulations (see the Supporting Information), binding free energies (ΔG Bind) for AA, FLP, and ARN2508 in complex with the COX‐1 protein were estimated using the molecular mechanics/generalized Born–Poisson–Boltzmann surface area (MM/GB‐PBSA) method (Figure 6).31 As expected, the AA substrate shows a remarkable binding affinity for the COX‐1 protein, given a calculated free energy of binding (ΔG Bind‐PB) of −91.2±1.1 kcal mol−1. Interestingly, a comparable ΔG Bind‐PB value of −102.8±0.8 kcal mol−1 was found for ARN2508, whereas a lower ΔG Bind‐PB value was found for FLP (ΔG Bind‐PB=−72.6±0.7 kcal mol−1).
Figure 6.
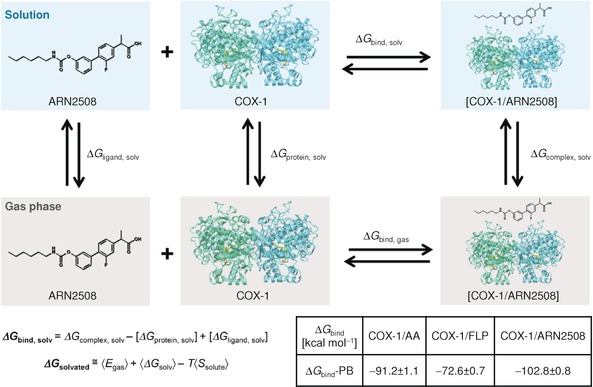
Schematic representation of a thermodynamic cycle for calculating the binding free energy (ΔG Bind) for a protein–ligand complex (shown for the COX‐1/ARN2508 system). Full details on deriving the ΔG Bind are in the Supporting Information. The solvated systems are shown in blue boxes, while systems in the gas phase are in gray boxes. The table reports the calculated ΔG Bind values following the molecular mechanics/Poisson Boltzmann (MM/PB) formalisms; the MM/GB‐PBSA method was used implemented in the Amber 12 package.31
Although only qualitative, these ΔG Bind‐PB values confirm the high affinity of the hybrid ARN2508 for the COX‐1 active site. Moreover, the ΔG Bind‐PB value for ARN2508 is higher than the ΔG Bind‐PB value for the endogenous AA substrate (by ∼12 kcal mol−1). This reflects how the biphenyl moiety in ARN2508 enhances the stability of the compound within the COX‐1 active site, by improving the van der Waals interactions (Figure 4). This energetic analysis further justifies the potency of ARN2508 as a COX‐1 inhibitor.15
As previously introduced, the binding of ARN2508 at the FAAH active site was here investigated via molecular docking calculations. Again, this is due to the fact that the covalent mechanism of carbamate‐based inhibitors, such as ARN2508, to block FAAH has been largely explained already.8, 9, 10, 22, 23 Toward this aim, as a target structure, we used the rat FAAH X‐ray crystal structure solved at 2.8 Å resolution (PDB code: 1MT5).6 This X‐ray structure was used in previous docking studies on dual FAAH/COX inhibitors as well.14 To date, the human FAAH enzyme has not been crystallized, and only a “humanized” structure of the rat FAAH, in which six amino acids were mutated into those found in the human FAAH (namely, L192F, F194Y, A377T, S435N, I491V, and V495M), is available (PDB code: 2VYA).25 In this respect, a recent comparative study, based on microsecond time scale MD simulations of the rat and “humanized” variants of FAAH, has shown that the mechanism of ligand binding is not likely affected by those six point mutations.10
Thus, we report here the binding of ARN2508 to FAAH, as calculated from docking, which returned favorable interaction energy and docking score (−68 kcal mol−1). This plausible binding pose of ARN2508 within the FAAH binding site suggests a common covalent mechanism for inhibition as for other carbamate‐based FAAH inhibitors.16 In fact, in this configuration (Figure 7), the 2‐arylpropionic acid moiety of ARN2508 occupies the long hydrophobic channel of the FAAH catalytic site and establishes van der Waals interactions with Leu192, Leu380, Val270, and Ile238, while the terminal propionic acid H‐bonds Gln273. The carbamate functional group is the key pharmacophoric element needed for FAAH inhibition.24, 25, 26 Upon binding, it moves close to the catalytic Ser241, ready to undergo nucleophilic attack (Ser241, Ser217, and Lys142).9, 32 The ARN2508 carbonyl oxygen points toward the FAAH oxyanion hole (comprising Ile238, Gly239, and Gly240), which draws electron density away from the substrate′s carbonyl, favoring substrate hydrolysis.9, 22, 32 Remarkably, the obtained configuration of the carbamate functionality resembles the binding mode of the crystallized carbamate‐based FAAH inhibitors, which inhibit FAAH by covalently binding the catalytic Ser241.24, 26 Taken together, these data suggest that ARN2508 blocks FAAH through covalent inhibition.
Figure 7.
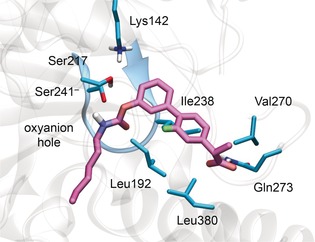
Putative binding mode of ARN2508 within the rat FAAH active site (PDB code: 1MT5),6 according to molecular docking calculations. The protein is shown as transparent ribbons, highlighting the oxyanion hole in blue. Key protein residues (cyan) and ARN2508 (magenta) are shown as sticks.
In summary, our study proposes an atomically detailed mechanism for dual FAAH/COX inhibition by the hybrid dual inhibitor ARN2508.15 We propose that ARN2508 noncovalently inhibits COX, while blocking FAAH via the acetylation of the catalytic Ser241, in agreement with the experimentally characterized mechanism of FAAH inhibition by several other carbamate‐based compounds.24, 25, 26 This mechanism of dual FAAH/COX inhibition merits further experimental validation, which could aid the development of novel anti‐inflammatory agents that simultaneously act on FAAH and COXs enzymes to treat pain and other inflammatory diseases.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
A.D.F. and M.D.V. are co‐authors of a patent application (WO2014023643A1),15 which covers the compound class presented in this article. M.D.V. thanks the Italian Association for Cancer Research (AIRC) for financial support through the “MFAG n. 14140” grant. G.P. thanks Prof. Ursula Roethlisberger (EPFL, Switzerland) for her support. The authors also thank the Partnership for Advanced Computing in Europe (PRACE) for HPC computing time, and Grace Fox for proofreading and copyediting the manuscript.
G. Palermo, A. D. Favia, M. Convertino, M. De Vivo, ChemMedChem 2016, 11, 1252.
References
- 1. Blobaum A. L., Marnett L. J., J. Med. Chem. 2007, 50, 1425–1441. [DOI] [PubMed] [Google Scholar]
- 2. Kurumbail R. G., Kiefer J. R., Marnett L. J., Curr. Opin. Struct. Biol. 2001, 11, 752–760. [DOI] [PubMed] [Google Scholar]
- 3. Malkowski M. G., Ginell S. L., Smith W. L., Garavito R. M., Science 2000, 289, 1933–1937. [DOI] [PubMed] [Google Scholar]
- 4. Rouzer C. A., Marnett L. J., J. Lipid Res. 2009, 50, S29–S34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Naesdal J., Brown K., Drug Safety 2006, 29, 119–132. [DOI] [PubMed] [Google Scholar]
- 6. Bracey M. H., Hanson M. A., Masuda K. R., Stevens R. C., Cravatt B. F., Science 2002, 298, 1793–1796. [DOI] [PubMed] [Google Scholar]
- 7. Cravatt B. F., Giang D. K., Mayfield S. P., Boger D. L., Lerner R. A., Gilula N. B., Nature 1996, 384, 83–87. [DOI] [PubMed] [Google Scholar]
- 8. Palermo G., Bauer I., Campomanes P., Cavalli A., Armirotti A., Girotto S., Rothlisberger U., De Vivo M., PLoS Comput. Biol. 2015, 11, e1004231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Palermo G., Campomanes P., Cavalli A., Rothlisberger U., De Vivo M., J. Phys. Chem. B 2015, 119, 789–801. [DOI] [PubMed] [Google Scholar]
- 10. Palermo G., Campomanes P., Neri M., Piomelli D., Cavalli A., Rothlisberger U., De Vivo M., J. Chem. Theory Comput. 2013, 9, 1202–1213. [DOI] [PubMed] [Google Scholar]
- 11.
- 11a. Cavalli A., Bolognesi M. L., Minarini A., Rosini M., Tumiatti V., Recanatini M., Melchiorre C., J. Med. Chem. 2008, 51, 347–372; [DOI] [PubMed] [Google Scholar]
- 11b. Zimmermann G. R., Lehar J., Keith C. T., Drug Discovery Today 2007, 12, 34–42. [DOI] [PubMed] [Google Scholar]
- 12. Bertolacci L., Romeo E., Veronesi M., Magotti P., Albani C., Dionisi M., Lambruschini C., Scarpelli R., Cavalli A., De Vivo M., Piomelli D., Garau G., J. Am. Chem. Soc. 2013, 135, 22–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cipriano M., Bjorklund E., Wilson A. A., Congiu C., Onnis V., Fowler C. J., Eur. J. Pharmacol. 2013, 720, 383–390. [DOI] [PubMed] [Google Scholar]
- 14. Favia A. D., Habrant D., Scarpelli R., Migliore M., Albani C., Bertozzi S. M., Dionisi M., Tarozzo G., Piomelli D., Cavalli A., De Vivo M., J. Med. Chem. 2012, 55, 8807–8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. De Vivo M., Scarpelli R., Cavalli A., Migliore M., Habrant D. Piomelli D., Favia A., (Fondazione Istituto Italiano Di Tecnologia, Italy; The Regents of the University of California, USA; Alma Mater Studiorum–Universita′ Di Bologna, Italy), PCT Int. Pat. Appl. WO 2014/023643 A1, 2014.
- 16. Seierstad M., Breitenbucher J. G., J. Med. Chem. 2008, 51, 7327–7343. [DOI] [PubMed] [Google Scholar]
- 17. Bayly C. I., Black W. C., Leger S., Ouimet N., Ouellet M., Percival M. D., Bioorg. Med. Chem. Lett. 1999, 9, 307–312. [DOI] [PubMed] [Google Scholar]
- 18. Kurumbail R. G., Stevens A. M., Gierse J. K., McDonald J. J., Stegeman R. A., Pak J. Y., Gildehaus D., Miyashiro J. M., Penning T. D., Seibert K., Isakson P. C., Stallings W. C., Nature 1996, 384, 644–648 [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Gupta K., Selinsky B. S., Loll P. J., Acta Crystallogr. Sect. D 2006, 62, 151–156; [DOI] [PubMed] [Google Scholar]
- 19b. Picot D., Loll P. J., Garavito R. M., Nature 1994, 367, 243–249; [DOI] [PubMed] [Google Scholar]
- 19c. Selinsky B. S., Gupta K., Sharkey C. T., Loll P. J., Biochemistry 2001, 40, 5172–5180; [DOI] [PubMed] [Google Scholar]
- 19d. Sidhu R. S., Lee J. Y., Yuan C., Smith W. L., Biochemistry 2010, 49, 7069–7079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duggan K. C., Hermanson D. J., Musee J., Prusakiewicz J. J., Scheib J. L., Carter B. D., Banerjee S., Oates J. A., Marnett L. J., Nat. Chem. Biol. 2011, 7, 803–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.
- 21a. De Vivo M., Front. Biosci. 2011, 16, 1619–1633; [DOI] [PubMed] [Google Scholar]
- 21b. Palermo G., Cavalli A., Klein M. L., Alfonso-Prieto M., Dal Peraro M., De Vivo M., Acc. Chem. Res. 2015, 48, 220–228; [DOI] [PubMed] [Google Scholar]
- 21c. Palermo G., Stenta M., Cavalli A., Dal Peraro M., De Vivo M., J. Chem. Theory Comput. 2013, 9, 857–862; [DOI] [PubMed] [Google Scholar]
- 21d. Brunk E., Ashari N., Athri P., Campomanes P., de Carvalho F. F., Curchod B. F. E., Diamantis P., Doemer M., Garrec J., Laktionov A., Micciarelli M., Neri M., Palermo G., Penfold T. J., Vanni S., Tavernelli I., Rothlisberger U., Chimia 2011, 65, 667–671; [DOI] [PubMed] [Google Scholar]
- 21e. Palermo G., Riedel T., Davey C. A., Dyson P. J., Rothlisberger U., Biophys. J. 2015, 108, 59a; [Google Scholar]
- 21f. Palermo G., Minniti E., Greco M. R., Riccardi L., Simoni E., Convertino M., Marchetti C., Rosini M., Sissi C., Minarini A., De Vivo M., Chem. Commun. 2015, 51, 14310–14313. [DOI] [PubMed] [Google Scholar]
- 22. Tubert-Brohman I., Acevedo O., Jorgensen W. L., J. Am. Chem. Soc. 2006, 128, 16904–16913 [DOI] [PubMed] [Google Scholar]
- 23.
- 23a. Boger D. L., Miyauchi H., Du W., Hardouin C., Fecik R. A., Cheng H., Hwang I., Hedrick M. P., Leung D., Acevedo O., Guimaraes C. R., Jorgensen W. L., Cravatt B. F., J. Med. Chem. 2005, 48, 1849–1856; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Guimarães C. R., Boger D. L., Jorgensen W. L., J. Am. Chem. Soc. 2005, 127, 17377–17384; [DOI] [PubMed] [Google Scholar]
- 23c. Palermo G., Rothlisberger U., Cavalli A., Vivo M. De, Eur. J. Med. Chem. 2015, 91, 15–26; [DOI] [PubMed] [Google Scholar]
- 23e. Lodola A., Mulholland A. J., Methods Mol. Biol. 2013, 924, 67–89. [DOI] [PubMed] [Google Scholar]
- 24. Mileni M., Garfunkle J., Ezzili C., Kimball F. S., Cravatt B. F., Stevens R. C., Boger D. L., J. Med. Chem. 2010, 53, 230–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Mileni M., Johnson D. S., Wang Z., Everdeen D. S., Liimatta M., Pabst B., Bhattacharya K., Nugent R. A., Kamtekar S., Cravatt B. F., Ahn K., Stevens R. C., Proc. Natl. Acad. Sci. USA 2008, 105, 12820–12824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mileni M., Kamtekar S., Wood D. C., Benson T. E., Cravatt B. F., Stevens R. C., J. Mol. Biol. 2010, 400, 743–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Friesner R. A., Banks J. L., Murphy R. B., Halgren T. A., Klicic J. J., Mainz D. T., Repasky M. P., Knoll E. H., Shelley M., Perry J. K., Shaw D. E., Francis P., Shenkin P. S., J. Med. Chem. 2004, 47, 1739–1749. [DOI] [PubMed] [Google Scholar]
- 28. Madhavi Sastry G., Adzhigirey M., Day T., Annabhimoju R., Sherman W., J. Comput. Aid Mol. Des. 2013, 27, 221–234. [DOI] [PubMed] [Google Scholar]
- 29. Smith W. L., DeWitt D. L., Garavito R. M., Annu. Rev. Biochem. 2000, 69, 145–182. [DOI] [PubMed] [Google Scholar]
- 30. Burgi H. B., Dunitz J. D., Shefter E., J. Am. Chem. Soc. 1973, 95, 5065–5067. [Google Scholar]
- 31.
- 31a. Kuhn B., Gerber P., Schulz-Gasch T., Stahl M., J. Med. Chem. 2005, 48, 4040–4048; [DOI] [PubMed] [Google Scholar]
- 31b. Kuhn B., Kollman P. A., J. Med. Chem. 2000, 43, 3786–3791; [DOI] [PubMed] [Google Scholar]
- 31c. Miller B. R. I., McGee T. J. D., Swails J. M., Homeyer N., Gohlke H., Roitberg A. E., J. Chem. Theory Comput. 2012, 8, 3314–3321. [DOI] [PubMed] [Google Scholar]
- 32. Palermo G., Branduardi D., Masetti M., Lodola A., Mor M., Piomelli D., Cavalli A., De Vivo M., J. Med. Chem. 2011, 54, 6612–6623. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary