

ABSTRACT
Large bone defects are ideally treated with autografts, which have many limitations. Therefore, osteoconductive scaffolds loaded with autologous bone marrow (BM) aspirate are increasingly used as alternatives. The purpose of this study was to compare the growth of multipotential stromal cells (MSCs) from unprocessed BM on a collagen‐containing bovine bone scaffold (Orthoss® Collagen) with a non‐collagen‐containing bovine bone scaffold, Orthoss®. Another collagen‐containing synthetic scaffold, Vitoss® was included in the comparison. Colonization of scaffolds by BM MSCs (n = 23 donors) was evaluated using microscopy, colony forming unit‐fibroblast assay and flow‐cytometry. The number of BM MSCs initially attached to Orthoss® Collagen and Vitoss® was similar but greater than Orthoss® (p = 0.001 and p = 0.041, respectively). Furthermore, the number of MSCs released from Orthoss® Collagen and Vitoss® after 2‐week culture was also higher compared to Orthoss® (p = 0.010 and p = 0.023, respectively). Interestingly, collagen‐containing scaffolds accommodated larger numbers of lymphocytic and myelomonocytic cells. Additionally, the proliferation of culture‐expanded MSCs on Orthoss® collagen and Vitoss® was greater compared to Orthoss® (p = 0.047 and p = 0.004, respectively). Collectively, collagen‐containing scaffolds were superior in supporting the attachment and proliferation of MSCs when they were loaded with unprocessed BM aspirates. This highlights the benefit of collagen incorporation into bone scaffolds for use with autologous bone marrow aspirates as autograft substitutes. © 2015 The Authors. Journal of Orthopaedic Research Published by Wiley Periodicals, Inc. on behalf of Orthopaedic Research Society. J Orthop Res 34:597–606, 2016.
Keywords: multipotential stromal cells (MSCs), bone marrow (BM), collagen, scaffolds, bone graft substitutes
Non‐union or delayed bone healing is still a problem in a considerable number of injuries associated with fractures and/or bone loss.1 The gold standard therapy for fracture non‐union remains the implantation of bone autograft.2 However, its use is associated with problems including limited accessibility, formation of haematoma, bleeding, infection, and harvest‐related pain.3, 4 The use of autograft substitutes, such as allograft scaffolds provides a valid alternative, particularly when these scaffolds are loaded with autologous bone marrow (BM) aspirates, growth factors or their combination.5, 6 Autologous BM aspirates contain osteogenic progenitors, multipotential stromal cells (MSCs), which are crucial players in the process of bone repair.7, 8 MSC‐loaded scaffolds have shown effectiveness in the experimental animal models of bone defects 9, 10, 11 and consequently have become a promising therapeutic method in the discipline of orthopaedic surgery.11, 12
To enhance the repair of complicated bone fractures, there are many types of osteoconductive scaffolds composed of either natural hydroxyapatite or synthetic materials, available for clinical use. The synthetic scaffolds are formed of inorganic calcium phosphate, polymers or composites of these materials.5, 13 Extracellular matrix (ECM) is formed of various proteins including collagen types I and II, fibronectin, biglycan, decorin, perlecan, and laminin. These components form the niche for BM MSCs in vivo and provide the mechanical and biological support for MSCs in order to respond to the surrounding signals favoring bone formation.14 As a major component of ECM, collagen is used as a natural biocompatible polymer for bone substitution but it has poor mechanical properties. Thus, collagen is usually combined with other bone substitute materials.5, 15
Studies analysing the biology of MSCs loaded on scaffolds particularly collagen‐containing ones have been most commonly performed using culture‐expanded MSCs.16, 17 Bone scaffolds are commonly soaked in unprocessed BM aspirates intra‐operatively prior to use for treatment of non‐union. However, little is known about how efficient is BM MSC attachment to scaffolds and how the modification of scaffold composition, such as collagen incorporation could affect colonization of these scaffolds by MSCs.
The aim of this study was to investigate the impact of collagen addition to a bovine bone allograft scaffold, on the attachment and proliferation of non‐cultured MSCs, i.e., derived from unprocessed BM aspirates. A bovine bone scaffold, Orthoss® (non‐collagen containing) was used as a control for the collagen‐containing one, Orthoss® collagen. Another collagen‐containing synthetic bone scaffold, Vitoss®, was also included in the study. Using unprocessed BM aspirates, we aimed to quantify rare MSCs, which were able to attach, survive and proliferate on these scaffolds.
MATERIALS AND METHODS
Scaffolds
The scaffolds used in this study were: Orthoss®, Orthoss® Collagen (Geistlich Surgery, Wolhusen, Switzerland) and Vitoss® (Stryker, Malvern, PA). Orthoss® is a bovine bone mineral, i.e., hydroxyapatite with nano pores (10–20 nm) and macro pores (100–300 μm). Orthoss® Collagen is a similar material as Orthoss® but is complemented with 10% porcine collagen. Vitoss® is a synthetic beta‐tricalcium phosphate with pores of variable size (1–1,000 μm) and is supplemented with type I bovine collagen. Orthoss® was provided as 2–4 mm granules of total 7 g and with an average volume of 8 mm3 per granule. The whole Orthoss® Collagen scaffold block (500 mg, 10 × 10 × 8 mm, 0.8 cc), was divided into eight equal pieces each of ∼100 mm3 volume. Vitoss® strip (25 × 100 × 4 mm, 10 cc) was punched using 4 mm sterile disposable biopsy punch with plunger (Miltex, NJ) into 50 mm3 particles. To standardize the volume of all scaffolds (100 mm3); 12 granules of Orthoss, one piece of Orthoss® Collagen and two particles of Vitoss® were used for each experiment.
Patients and Bone Marrow Samples
Following ethic committee approval (06/Q1206/127, National Research Ethics Committee Yorkshire & Humber–Leeds East), BM aspirates were obtained from the iliac crest of twenty three consented patients; 13 males and 10 females with median age of 47 years, range 14–82. All patients were undertaking elective orthopaedic surgery for various reasons but reported as having no underlying diseases. Ten millilitre of BM was aspirated using bevel tipped trocar needle (Stryker 306–111, 11‐gauge, MI) and a single draw method and then placed in two EDTA containing VACUETTE® blood collection tubes. In some experiments, culture‐expanded MSCs from seven donors were used. Culture‐expanded MSCs were generated as previously described18 and grown in Stem MACS MSC Expansion media (Miltenyi Biotec, Surrey, UK) supplemented with penicillin/streptomycin (Sigma–Aldrich, St. Louis) at 37°C/5% CO2 culture condition.
Loading Scaffolds With Bone Marrow Aspirates or Culture‐Expanded MSCs
BM aspirates were thoroughly mixed then 500 μl of the aspirate was incubated with 100 mm3 of each of three scaffolds. The same BM sample was used to load the three scaffolds in each experiment. Several replicates were performed depending on the type of experiment. The scaffolds were incubated with BM aspirate for one and half or three hours at 37°C with gentle rotational movement using a rotator with reciprocal turning angle, 45° and vibration angle, 5° (Biosan, Riga, Latvia), as described previously.19 In some experiments, scaffolds were similarly incubated with passage three culture‐expanded MSCs (average of 2 × 105 MSCs in 500 μl of media/100 mm3 of each scaffold). Following incubation, scaffolds were gently rinsed in phosphate‐buffered saline (PBS, Invitrogen, Paisley, UK) and then cultured in Stem MACS media. The culture media was half changed every 2–3 days to take the advantage of MSC autocrine activation of their proliferation.20
Colony Forming Unit‐Fibroblast Assay
The number of MSCs initially attached to scaffolds was detected using Colony Forming Unit‐Fibroblast (CFU‐F) assay to enumerate of the number of MSCs in BM aspirates before and after loading on scaffolds. To assess MSC numbers in BM aspirate before loading on scaffolds (pre‐loading control), 200 μl of BM aspirate was added to 15 ml of Stem MACS media and then seeded in duplicate in 10 cm diameter culture dishes (Corning Life Sciences, Amsterdam, Holland). The media were replaced 48 h later to remove non‐adherent cells and the culture was continued for 14 days.18 The resulting colonies were counted after staining with methylene blue. The assay was repeated on the BM aspirates left at the end of incubation with the scaffolds (post‐loading control). The numbers of MSCs attached to scaffolds were calculated using pre and post loading controls. As scaffolds are known to be different in their porosity and hence the amount of liquid that can be absorbed inside,21 the volume of non‐bound BM sample was recorded and used in the subsequent calculations of attached MSCs.
Environmental Scanning Electron Microscopy
Scaffolds loaded with BM aspirates were processed for Environmental Scanning Electron Microscopy (ESEM) to examine cell attachment on the surface of scaffolds after 2‐week cultivation, as previously described.19 Briefly, scaffolds were removed from the culture, washed twice with PBS and then fixed in 10% formalin. Empty scaffolds were similarly cultured and processed as negative controls. ESEM was performed using S‐3700N scanning electron microscope (Hitachi, Berkshire, UK). Images were captured of hydrated samples under low vacuum and variable pressure at −20°C.
Histology
Histology was used to assess the presence of attached MSCs on the surface and inside scaffolds after 2‐weeks of culture. After harvesting from culture, scaffolds were gently washed with PBS and subsequently, kept in 10% formalin for cell fixation for at least 24 h before being processed for histology. Empty scaffolds were included as negative controls. Specimens were embedded in paraffin and subsequently, cut into 5 μm sections using a Leica RM2255 microtome. Orthoss required decalcification to facilitate the sectioning of the scaffold, using EDTA solution (250 g disodium EDTA and 1750 ml deionised water, pH 7.0) for 2–3 days. The slides were processed for a routine staining with haematoxylin and eosin (H&E), and scanned under bright‐field mode using Nikon microscope E1000. Nuance V.3.0.1.2 software (Perkin Elmer) was used to acquire the images.
Digestion of Scaffolds and Release of Cells
To release the cells from the scaffolds following the culture, the scaffolds were gently washed with PBS and subsequently, treated with 1% trypsin (Sigma–Aldrich) for 10 min at 37°C. More tightly attached cells were subsequently released using 0.25% collagenase (Stem Cell Technologies, Grenoble, France). For each scaffold, 0.5 ml collagenase was added for 30 min and incubated at 37°C with slight agitation using VIBRAX‐VXR shaker (IKA, Staufen, Germany). The digested fraction was next passed through 70 μm cell strainer (BD falcon) to separate cells from the scaffold debris. Cells were then washed in PBS and re‐suspended in 50 μl of blocking buffer (10% of mouse serum and human IgG1, Sigma–Aldrich) to prevent nonspecific binding before being processed for flow‐cytometry staining.
Surface Phenotype and Quantification of MSCs Released From Scaffolds
Flow‐cytometry was performed to assess the standard phenotype of culture‐expanded MSCs.22, 23 The antibodies against CD90 (Bio‐Rad, Hertfordshire, UK), CD45, CD73, CD105, CD13, CD106, CD146, and CD166 (all from BD Biosciences) were used. To identify MSCs released after digestion of scaffolds, the antibodies against CD90, CD73, and CD45 were used. All antibodies were applied at the manufacturer's recommended concentrations and the staining was performed for 30 min at room temperature in the dark. Live/Dead® Violet Viability Kit (Molecular Probes, Life Technologies, Paisley, UK) was used to exclude debris from data analysis and to evaluate the proportions of live and dead cells. The total number of MSCs released from each scaffold was calculated using CountBright™ absolute counting beads (Molecular probes, Invitrogen, UK) using the manufacture's recommended formula: Cell count per μl = (number of live cell events/number of beads events) × (assigned bead count of the bead lot/Volume of sample). The data were acquired on LSRII (BD Biosciences) and analysed using BD FACS Diva™ software (BD Biosciences).
Proliferation of Culture‐Expanded MSCs
To assess the proliferation of culture‐expanded MSCs loaded on the bone scaffolds, these MSCs were first serum starved for 12 h in 1 g/l glucose containing DMEM medium to synchronize their cell cycle status before incubation with scaffolds.24 On the day of experiment, MSCs were trypsinised and then labeled with 1 μM of Carboxyfluorescein succinimidyl ester (CFSE) (Cell Trace™ CFSE Cell Proliferation Kit, Life technologies, Paisley, UK). Cells were then washed, re‐suspended in Stem MACS media and incubated with each of three scaffolds for 3 h before being transferred to the culture. A part of MSCs not incubated with scaffolds was processed for flow‐cytometry to determine “day 0” expression level of CFSE. After 4 days, the scaffolds were digested and MSCs were released and then stained for flow‐cytometry to determine their CFSE levels. Overlapping histogram panels for CFSE staining were performed using Kalusa 1.3 analysis software (Beckman coulter).
Statistical Analysis
The graphs were prepared using the GraphPad Prism 6. All statistics were performed using IBM SPSS® 21 statistics software. The comparison of the numbers and percentages of attached or proliferated MSCs, and other cells between the three scaffolds were analyzed using Friedman's Two‐way Analysis of Variance (ANOVA) test. Then, the pairwise comparisons were performed using Dunn–Bonferroni tests. To compare the number of aspirated BM MSCs between male/female or between younger/older donors, unpaired t‐test was applied. The correlation was performed using Spearman's rho test.
RESULTS
Attachment of Non‐Cultured BM MSCs to Scaffolds
The total number of non‐cultured MSCs attached to scaffolds was calculated by counting colony‐forming MSCs before and after 3 h incubation between unprocessed BM and scaffolds (Fig. 1).
Figure 1.
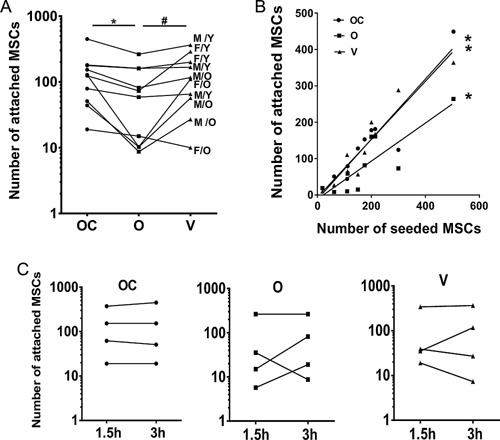
Attachment of unprocessed BM MSCs to Orthoss® collagen, Orthoss®, and Vitoss®; (A) The number of MSCs attached to the three scaffolds (3 h incubation, n = 10 donors, *p = 0.001, # p = 0.041). The numbers were calculated from MSC counts before and after loading of scaffolds with BM aspirate using CFU‐F assay. OC: Orthoss® collagen, O: Orthoss®, and V: Vitoss®. M: male, F: female, O: old, Y: young. (B) The correlation between the numbers of seeded MSCs and the number of MSCs attached to the three scaffolds. *Orthoss® collagen: r = 0.87, p = 0.001, Orthoss®: r = 0.83, p = 0.003, Vitoss®: r = 0.93, p < 0.001. (C) The number of MSCs attached to three scaffolds after 1.5 h compared to 3 h incubation (n = 4 donors).
Experiments performed on 10 independent BM aspirates revealed that the total number of BM MSCs attached to Orthoss® collagen and Vitoss®, was not significantly different (median values of 126 and 117 MSCs/100 mm3 of scaffold, respectively, p = 0.791) (Fig. 1A). However, the number of BM MSCs attached to Orthoss® collagen and Vitoss® was significantly higher (∼two fold, p = 0.001 and p = 0.041, respectively) compared to Orthoss® (median 60 MSCs/100 mm3 of scaffold, Fig. 1A). This indicated that the collagen‐containing scaffolds were superior compared to Orthoss® in supporting the attachment of rare MSCs from unprocessed BM aspirates.
The number of attached MSCs was widely variable between donors (Fig. 1A) and significantly correlated with the initial counts of MSCs in BM aspirates independently of the scaffold type (Orthoss® collagen: p = 0.001, Orthoss®: p = 0.003 and Vitoss®: p < 0.001) (Fig. 1B). There was no statistical significant difference in the frequencies of aspirated MSCs between male and female donors (mean values 405 and 315 MSCs/ml of BM), whereas BM aspirates from older donors (>45 years old) had fewer MSCs compared to younger donors (≤45 years old) (mean values 163 and 532 MSCs/ml of BM, respectively, p = 0.0145).
When MSC attachment data were reanalyzed according to the age and gender of donors, no preferential attachment of a certain group (male/female or young/old) was found. The numbers of attached BM MSCs from both male and female donors were similarly distributed on the scaffolds (Fig. 1A). In summary, scaffolds with high numbers of attached MSCs corresponded to BM aspirates from a “younger” group of donors, reflecting their higher initial frequencies in these donors.
The study of incubation times shorter than 3 h was clearly merited, considering scaffolds are loaded with BM aspirates in operating theatres. Using a subset of samples (n = 4), we next investigated if shortening the incubation time by half could impair MSC attachment. Interestingly, no decrease was found in the number of MSCs attached to the three scaffolds following 1.5 h long incubation, compared to 3 h (Fig. 1C).
Overall, these data revealed that, despite the infrequency and variable numbers of MSCs in BM aspirates, these rare MSCs were able to attach to the tested scaffolds. Interestingly, the collagen‐containing scaffolds (Orthoss® collagen and Vitoss®) have provided better support for the initial MSC attachment compared to the non‐collagen‐containing scaffold, Orthoss®.
Microscopic Visualization of MSC Attachment to Scaffolds
We next aimed to examine the surface of these scaffolds and to visualize cells, which attached and survived on the three scaffolds after 2‐weeks of culture using ESEM and histology (Figs. 2 and 3). Using ESEM on empty scaffolds revealed their basic surface architecture. The surface of both of Vitoss® and Orthoss® collagen was mostly covered by collagen fibers with visible parts of bone mineral, in contrast to Orthoss®, which had a smooth surface of pure bone mineral (Fig. 2, top panels). For this reason, cells attached to the surface of Orthoss® were clearly visible as flat and branched cells bridging scaffold pores (Fig. 2, bottom middle panel). It was hard to detect similar cells on the surface of Vitoss® or Orthoss® collagen due to their surface coverage with collagen (Fig. 2, bottom left and right panels). However, the smoothness of collagen fibres and increased coverage of mineral parts on the surface of these scaffolds when compared to controls suggested a presence of attached cells.
Figure 2.
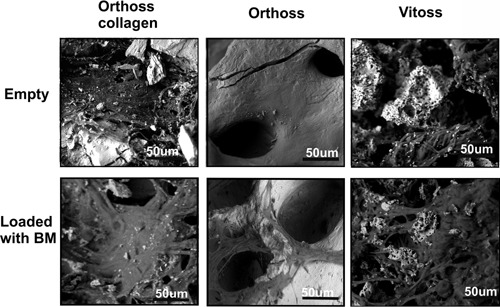
The surface topography and MSC attachment on scaffolds using ESEM; Empty (top panels) or BM aspirate‐loaded scaffolds (bottom panel) were examined after 2‐week culture. Scale bar = 50 μm.
Figure 3.
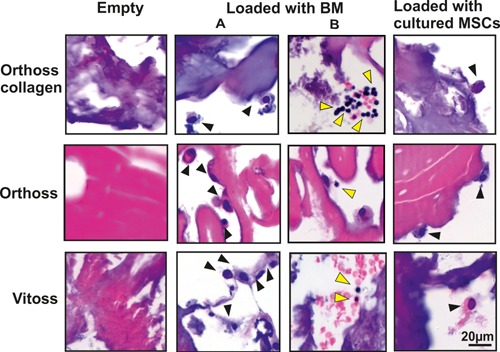
Histological assessment of MSC attachment to scaffolds using H&E staining; Empty (left panels), BM aspirate‐loaded (middle panels, A and B) and culture‐expanded MSC‐loaded (right panels) scaffolds after 2‐week culture. Black arrows indicate putative MSCs and yellow arrows indicate other BM cells. Scale bar = 20 μm.
The inner structure of scaffolds was examined using H&E‐stained sections of the empty scaffolds (Fig. 3, left panel). The scaffolds seeded with culture‐expanded MSCs and used as positive controls, showed a presence of elongated or rounded cells mostly attached to or near the surface of the three scaffolds (Fig. 3, right panel). Cells resembling culture‐expanded MSCs were also detected on the surface and inside the scaffolds loaded with BM aspirate (Fig. 3, middle panel, A). Additionally, smaller round cells were also found inside scaffolds loaded with BM aspirates (Fig. 3, middle panel, B). This indicated that a variety of BM‐derived cells could attach and survive inside these scaffolds.
Quantification of BM MSCs Released From Scaffolds Following In Vitro Culture
Cells released from scaffolds were next analyzed by flow‐cytometry in order to confirm the presence of MSCs, and detect which of the three examined scaffolds best supported the survival of MSCs (Fig. 4). Live cells were identified based on positive staining with live cell marker, Calcein violet and negative staining with Aqua dye, which only stains dead cells.25 The percentages of dead cells (Aqua positive cells after exclusion of debris) in relation to total released cells were similar in all three scaffolds (mean: 29%, 33%, and 30% for Orthoss® collagen, Orthoss® and Vitoss®, respectively). Based on cell scattering characteristics and CD45 expression, the lymphocytic lineage cells were detected as CD45brightSSClow and myelomonocytic lineage cells as CD45medSSCmed (Fig. 4A).26 Within the CD45‐negative fraction, MSCs were identified as CD73+CD90+ cells (Fig. 4A).
Figure 4.
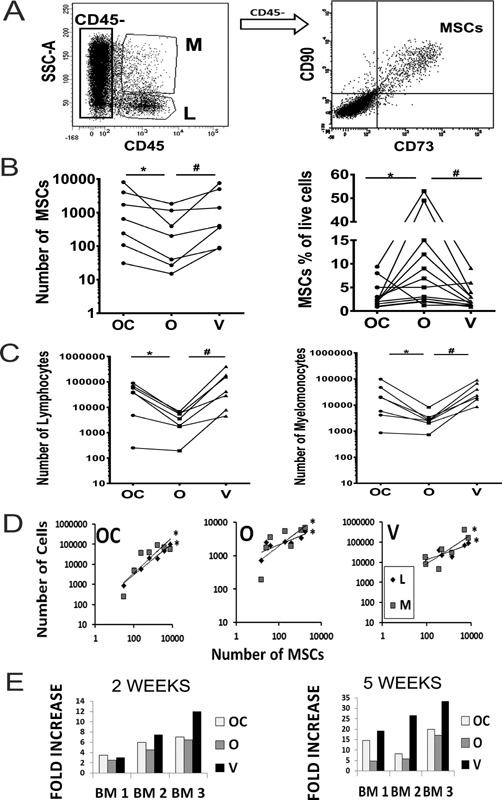
Quantification of MSCs and other BM cells released from BM‐loaded scaffolds; (A) Gating strategy using flow‐cytometry: Lymphocytic cells (L) as CD45brightSSClow, Myelomonocytic cells (M) as CD45medSSCmed and MSCs as CD45‐CD73+CD90+ cells. (B) Total numbers of MSCs released from the scaffolds following 2‐week culture (left panel, n = 7 donors) *p = 0.010, # p = 0.023. Counting beads were used for absolute quantification. The percentage of MSCs out of total live cells released from the scaffolds following 2‐week culture (right panel, n = 12 donors). *p = 0.032, # p = 0.024. OC: Orthoss® collagen, O: Orthoss®, and V: Vitoss®. (C) Total numbers of lymphocytic cells (left panel) and myelomonocytic cells (right panel) released from the scaffolds following 2‐week culture (n = 7 donors). For both; panels *p = 0.045, # p = 0.004. (D) The correlation between the number of MSCs and the number of lymphocytic (L) and myelomonocytic cells (M) released from the three scaffolds following 2‐week culture. Orthoss® collagen: R = 0.99, *p < 0.001 and r = 0.85, *p = 0.014. Orthoss: R = 0.82, *p = 0.023 and r = 0.89, *p = 0.007. Vitoss: R=0.85, *p = 0.014 and r = 0.8, *p = 0.036 for MSCs versus myelomonocytic and lymphocytic cells, respectively. (E) Fold increase in MSC numbers released from scaffolds cultured for 2 and 5 weeks relative to MSC number counted after 1‐week culture. Samples from three different BM donors are shown.
After 2‐weeks of culture, the numbers of live MSCs released from Orthoss® collagen and Vitoss® were significantly higher (nearly threefold and twofold respectively, p = 0.010 and p = 0.023, respectively) compared to Orthoss® (Fig. 4B, left panel). In contrast, the difference between the number of live MSCs released from Orthoss® collagen and Vitoss was minimal (Fig. 4B, left panel). This indicated that the collagen‐containing scaffolds provided a greater support for the survival of BM MSCs. However, the percentages of MSCs in relation to total live cells released from Orthoss® were significantly higher compared to Orthoss® collagen (threefold, p = 0.032) and Vitoss® (3.5‐fold, p = 0.024) (Fig. 4B, right panel).
The number of lymphocytic and myelomonocytic cells released from Orthoss® collagen and Vitoss® was higher than those released from Orthoss® (nearly 10‐fold & 11‐fold, p = 0.045 and p = 0.004 for both lymphocytic and myelomonocytic cells, respectively) (Fig. 4C). However, there was no significant difference between the number of these cells released from Orthoss® collagen and Vitoss®. Additionally, we observed significant positive correlations between the number of released MSCs and lymphocytic or myelomonocytic cells, respectively (Orthoss® collagen: p < 0.001 and p = 0.014, Orthoss®: p = 0.023 and p = 0.007 and Vitoss®: p = 0.014 and p = 0.036) (Fig. 4D). These data demonstrated that the collagen‐containing scaffolds played a positive role in supporting the survival of MSCs as well as different types of BM cells.
Comparing Figures 1 and 4, it was clear that the numbers of BM MSCs attached to the scaffolds were lower than the numbers of BM MSCs released from scaffolds after 2‐weeks of culture. This indicated that BM MSCs were proliferating inside these scaffolds. To compare how effectively the scaffolds supported the proliferation of MSCs, three different BM aspirates were used to load scaffolds and subsequently, cultured for 1, 2, and 5 weeks in a time‐course fashion (Fig. 4E). We then compared the numbers of MSCs released from the scaffolds after 2 and 5 weeks of culture relative to the numbers measured after 1‐week of culture. The data demonstrated that the increase of MSC counts either after 2 or 5 weeks culture on Vitoss® or Orthoss® collagen was greater compared to that on Orthoss® (Fig. 4E). These data indicated that all three scaffolds supported the proliferation of attached BM MSCs, but collagen‐containing scaffolds outperformed Orthoss® within that respect.
The Proliferation of Culture‐Expanded MSCs Inside Scaffolds
To confirm that collagen‐containing scaffolds support the proliferation of MSCs better than Orthoss®, we seeded the three scaffolds with a pure population of culture‐expanded MSCs. Before seeding on scaffolds, culture‐expanded MSCs were characterized by flow‐cytometry using the standard MSC markers.22, 23 All culture‐expanded MSCs used in these experiments were >90% positive for CD73, CD90, CD105, CD166, CD146, and CD13, 80% positive for CD106, and were negative for CD45 (Fig. 5A).
Figure 5.
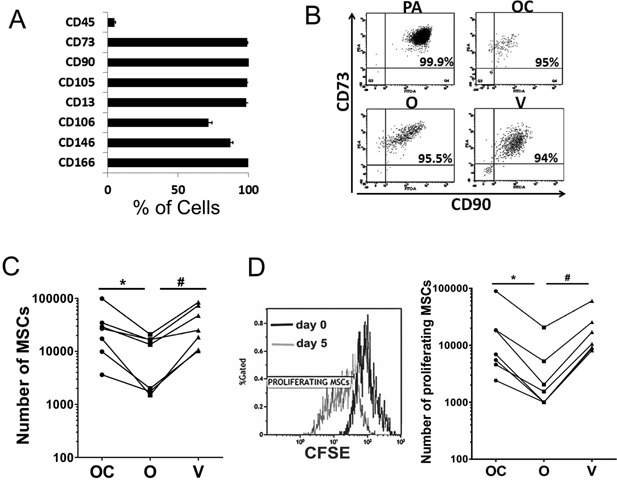
The proliferation of culture‐expanded MSCs inside scaffolds; (A) The surface phenotype of culture‐expanded MSCs before being seeded on scaffolds (n = 4 donors). Error bars indicate standard deviation. (B) The percentages of CD90+ CD73+ MSCs released from scaffolds following 2‐week culture compared to plastic adherent MSCs. OC: Orthoss® collagen, O: Orthoss®, V: Vitoss®, and PA: plastic adherent. (C) Total numbers of culture‐expanded MSCs after 4 days of culture on scaffolds (n = 7 donors). *p = 0.046, # p = 0.004. (D) Left panel: CFSE‐staining levels of culture‐expanded MSCs released from a scaffold on day 5 (grey histogram) compared to its levels before seeding (day 0, black histogram). Right panel: The total numbers of MSCs that proliferated during 4 days of in vitro culture on scaffolds (n = 7 donors) *p = 0. 047, # p = 0.004.
Culture‐expanded MSCs seeded on scaffolds were released and characterized using Live/Dead cell markers, CD45, CD73, and CD90. As expected, MSC population was predominant (Fig. 5B). Consistent with data obtained with BM aspirates, the total number of culture‐expanded MSCs released from Orthoss® collagen and Vitoss® was significantly higher compared to those extracted from Orthoss® (p = 0.046 and p = 0.004, respectively) (Fig. 5C) confirming that collagen‐containing scaffolds support better the survival of MSCs.
To quantify the proliferation of MSCs inside each of the three scaffolds, CFSE‐labeled MSCs were incubated with scaffolds then cultured for 4 days. The proportions of MSCs that have proliferated at day 5, i.e., have a reduced expression of CFSE following cell division compared to day 0 (Fig. 5D, left panel) were quantified. The data demonstrated that the numbers of proliferated MSCs inside Orthoss® collagen and Vitoss® were comparable, however, significantly greater compared to Orthoss® (4.3 and 6.8‐fold, respectively, p = 0.047 and p = 0.004, respectively) (Fig. 5D, right panel). Altogether, these data have confirmed that the presence of collagen as a component of bone scaffolds provided a better support for the proliferation of MSCs.
DISCUSSION
The choice of scaffolds for the treatment of bone defects, as biomechanical fillers or as carriers for MSCs, is critical.27 As a prerequisite for bone tissue regeneration, MSCs need to attach and proliferate on scaffolds in order to subsequently differentiate and integrate into the surrounding tissues. Here, we used as a model, two scaffolds of the same basic composition (bovine hydroxyapatite) to explore the effect of collagen incorporation on scaffold colonization by BM MSCs. Using unprocessed BM aspirates, our data showed that the addition of collagen to Orthoss® collagen improved the attachment, survival and proliferation of BM MSCs compared to Orthoss®. A synthetic scaffold, Vitoss®, was included as an additional collagen‐containing scaffold and showed similar results to Orthoss® collagen. To best of our knowledge, this study is the first to quantitatively assess the attachment, survival and proliferation of rare human non‐cultured (unprocessed) BM MSCs on these scaffolds.
The numbers of MSCs in BM aspirates are well known to be low and variable because of sampling and donor related factors particularly aging, which negatively affects the number of BM MSCs.28, 29 Our results revealed a large variation in the numbers of MSCs attached to scaffolds, independent on the scaffold type, which in turn was related to the differences of the initial quantity of MSCs in BM aspirates. The increase in donor age was associated with the lower numbers of aspirated and hence attached MSCs.
We have confirmed the morphology of BM MSCs growing on Orthoss® as flat and interconnected cells bridging the pores of scaffold.19 However, cell attachment was not clear on Orthoss® collagen and Vitoss®. For quantification of MSCs following their enzymatic release from cultured scaffolds, we therefore developed a flow‐cytometry based method. In contrast to other studies, in which MSC growth inside scaffolds was tested using confocal/electron microscopy or via measurement of total cellular DNA,19, 30, 31 the use of flow‐cytometry in our study has enabled us to quantitatively assess scaffold colonization by MSCs as well as by other BM cells. In one previous study, a flow‐cytometry assay was used to assess the viability of culture‐expanded MSCs seeded on 3D construct made of a composite of hydroxyapatite and β‐TCP, but the assay was not quantitative.32
Our data shows that the number of BM MSCs released from Orthoss® collagen and Vitoss® after 2‐week in vitro culture was higher than Orthoss®, clearly indicating that collagen‐containing scaffolds provided a more favourable environment for MSC attachment and proliferation. Interestingly, the attachment and proliferation of MSCs from BM aspirates into Orthoss® collagen and Vitoss® were similar despite the difference in their mineral composition. Complementary to our results, Orthoss® collagen and Vitoss® have been shown to equally support the osteogenic differentiation of MSCs.33 The osteogenic potential of MSCs colonizing the scaffolds was not tested in this study but we have shown previously that Orthoss® supports osteogenic differentiation of MSCs after 3‐weeks in culture.19 Additionally, it has been shown that Vitoss® supports the in vitro and in vivo osteogenic differentiation of MSCs.34
It has been previously demonstrated that collagen‐mediated induction of MSC survival is a result of its interaction with membrane‐bound integrins on MSC surface.35 Our study demonstrates that the incorporation of collagen into a natural bovine bone scaffold supports the survival and proliferation of both culture‐expanded and non‐expanded MSCs. Consistently, the addition of collagen to nanocellulose‐ or chitosan‐made scaffolds has been shown to promote the attachment of culture‐expanded MSCs into these scaffolds.36, 37 Uniquely, we have evaluated the colonization of scaffolds by other BM cells, and not only MSCs. Our results demonstrated that relative percentages of BM MSCs inside Orthoss® were higher compared to collagen‐containing scaffolds because the latter scaffolds were more supportive for the attachment and survival of other BM cells (myelomonocytic or lymphocytic cells) in addition to MSCs. In agreement, a collagen‐containing scaffold has been shown to provide an attachment to MSC as well as neutrophils in an animal model of dental bone loss.38 Importantly, regardless of scaffold type, we detected positive correlations between the quantities of BM MSCs and myelomonocytic or lymphocytic cells released from scaffolds. These findings support the possibility that BM‐derived hematopoietic cells could have an additional positive effect on MSC growth on different bone scaffolds. Immune cells have been shown to enhance the proliferation of animal BM MSCs.39, 40 The inclusion of other BM cells such as endothelial cells could also support MSC proliferation.41 Therefore, our study provides the platform for future investigation of the effect of other BM cells on MSC survival, proliferation and functions when loaded on osteoconductive scaffolds.
CONCLUSION AND FUTURE PERSPECTIVE
The preparation of culture‐expanded MSCs for clinical use is expensive and requires a complicated process of regulation to ensure the clinical safety.42 Furthermore, culture‐expanded MSCs are seeded into scaffolds at very high densities, which do not represent their physiological densities in human bone23 or BM aspirates.28, 43, 44 Our investigation reflects real clinical procedure and shows that non‐cultured MSCs from unprocessed BM aspirates, despite their known low frequency as well as donor and aspiration related variation,28, 43, 45, 46 can attach and proliferate on bone scaffolds. Importantly, our data revealed that the attachment, survival and proliferation of MSCs were superior on collagen‐containing scaffolds, a finding which supports their broader use for orthopaedic clinical applications.
AUTHORS’ CONTRIBUTIONS
All authors have a substantial contribution to this work. PVG, EJ, and JE were responsible for the study conception and design. JE and CS have conducted all experimental work and data acquisition. Data analysis and statistics were performed by JE, CS, and EJ. Manuscript preparation and writing was completed by JE, EJ, and PVG. All authors have read and approved the final submitted version.
ACKNOWLEDGMENTS
We would like to thank the clinical orthopaedic team at Leeds general infirmary hospital particularly Mr. Boon Tan for providing samples. The authors gratefully acknowledge Dr. Thomas Baboolal, Mrs. Jackie Hudson, Mr. Mike Shires and Dr. Sally Boxall for the technical help and advice. EJ and PVG are partly supported by NIHR‐Leeds Musculoskeletal Biomedical Research Unit (LMBRU). JE was supported by a grant from Geistlich Pharma; EJ and PVG were grant holders but received no salary from this grant. JE is also affiliated with the department of clinical pathology, Faculty of Medicine, Mansoura University, Egypt. CS was a beneficiary of Fellowships by Fundación Profesor Novoa Santos (INIBIC‐CHUAC, Spain) and Erasmus from the University of A Coruna, Spain.
REFERENCES
- 1. Gomez‐Barrena E, Rosset P, Lozano D, et al. 2015. Bone fracture healing: cell therapy in delayed unions and nonunions. Bone 70:93–101. [DOI] [PubMed] [Google Scholar]
- 2. De Long WG Jr, Einhorn TA, Koval K, et al. 2007. Bone grafts and bone graft substitutes in orthopaedic trauma surgery. A critical analysis. J Bone Joint Surg Am 89:649–658. [DOI] [PubMed] [Google Scholar]
- 3. Qvick LM, Ritter CA, Mutty CE, et al. 2013. Donor site morbidity with reamer‐irrigator‐aspirator (RIA) use for autogenous bone graft harvesting in a single centre 204 case series. Injury 44:1263–1269. [DOI] [PubMed] [Google Scholar]
- 4. Calori GM, Colombo M, Mazza EL, et al. 2014. Incidence of donor site morbidity following harvesting from iliac crest or RIA graft. Injury 45:S116–S120. [DOI] [PubMed] [Google Scholar]
- 5. Giannoudis PV, Dinopoulos H, Tsiridis E. 2005. Bone substitutes: an update. Injury 36:S20–S27. [DOI] [PubMed] [Google Scholar]
- 6. Oryan A, Alidadi S, Moshiri A, et al. 2014. Bone regenerative medicine: classic options, novel strategies, and future directions. J Orthop Surg Res 9:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Jones E, Yang X. 2011. Mesenchymal stem cells and bone regeneration: current status. Injury 42:562–568. [DOI] [PubMed] [Google Scholar]
- 8. Pittenger MF. 2008. Mesenchymal stem cells from adult bone marrow. Methods Mol Biol 449:27–44. [DOI] [PubMed] [Google Scholar]
- 9. Bruder SP, Kraus KH, Goldberg VM, et al. 1998. The effect of implants loaded with autologous mesenchymal stem cells on the healing of canine segmental bone defects. J Bone Joint Surg Am 80:985–996. [DOI] [PubMed] [Google Scholar]
- 10. Liu G, Zhao L, Zhang W, et al. 2008. Repair of goat tibial defects with bone marrow stromal cells and beta‐tricalcium phosphate. J Mater Sci Mater Med 19:2367–2376. [DOI] [PubMed] [Google Scholar]
- 11. Crowley C, Wong JM, Fisher DM, et al. 2013. A systematic review on preclinical and clinical studies on the use of scaffolds for bone repair in skeletal defects. Curr Stem Cell Res Ther 8:243–252. [DOI] [PubMed] [Google Scholar]
- 12. Marcacci M, Kon E, Moukhachev V, et al. 2007. Stem cells associated with macroporous bioceramics for long bone repair: 6‐ to 7‐year outcome of a pilot clinical study. Tissue Eng 13:947–955. [DOI] [PubMed] [Google Scholar]
- 13. Campana V, Milano G, Pagano E, et al. 2014. Bone substitutes in orthopaedic surgery: from basic science to clinical practice. J Mater Sci Mater Med 25:2445–2461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen XD. 2010. Extracellular matrix provides an optimal niche for the maintenance and propagation of mesenchymal stem cells. Birth Defects Res C Embryo Today 90:45–54. [DOI] [PubMed] [Google Scholar]
- 15. Reilly GC, Engler AJ. 2010. Intrinsic extracellular matrix properties regulate stem cell differentiation. J Biomech 43:55–62. [DOI] [PubMed] [Google Scholar]
- 16. Yang XB, Bhatnagar RS, Li S, et al. 2004. Biomimetic collagen scaffolds for human bone cell growth and differentiation. Tissue Eng 10:1148–1159. [DOI] [PubMed] [Google Scholar]
- 17. Qu Z, Yan J, Li B, et al. 2010. Improving bone marrow stromal cell attachment on chitosan/hydroxyapatite scaffolds by an immobilized RGD peptide. Biomed Mater 5:065001. [DOI] [PubMed] [Google Scholar]
- 18. Cox G, Boxall SA, Giannoudis PV, et al. 2012. High abundance of CD271(+) multipotential stromal cells (MSCs) in intramedullary cavities of long bones. Bone 50:510–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kouroupis D, Baboolal TG, Jones E, et al. 2013. Native multipotential stromal cell colonization and graft expander potential of a bovine natural bone scaffold. J Orthop Res 31:1950–1958. [DOI] [PubMed] [Google Scholar]
- 20. Kim J, Ma T. 2012. Regulation of autocrine fibroblast growth factor‐2 signaling by perfusion flow in 3D human mesenchymal stem cell constructs. Biotechnol Prog 28:1384–1388. [DOI] [PubMed] [Google Scholar]
- 21. Ana Rita Duartea CJFM, Rui Reisa L. 2010. Enzymatic degradation of 3D scaffolds of starch‐poly‐(ɛ‐caprolactone) prepared by supercritical fluid technology. Polym Degrad Stabil 95:2110–2211. [Google Scholar]
- 22. Dominici M, Le Blanc K, Mueller I, et al. 2006. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 8:315–317. [DOI] [PubMed] [Google Scholar]
- 23. Jones E, English A, Churchman SM, et al. 2010. Large‐scale extraction and characterization of CD271+ multipotential stromal cells from trabecular bone in health and osteoarthritis: implications for bone regeneration strategies based on uncultured or minimally cultured multipotential stromal cells. Arthritis Rheum 62:1944–1954. [DOI] [PubMed] [Google Scholar]
- 24. Chen M, Huang J, Yang X, et al. 2012. Serum starvation induced cell cycle synchronization facilitates human somatic cells reprogramming. PLoS ONE 7:e28203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Baboolal TG, Boxall SA, El‐Sherbiny YM, et al. 2014. Multipotential stromal cell abundance in cellular bone allograft: comparison with fresh age‐matched iliac crest bone and bone marrow aspirate. Regen Med 9:593–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Pattanapanyasat K, Sukapirom K, Kowawisatsut L, et al. 2008. New BD FACSCount CD4 reagent system for simultaneous enumeration of percent and absolute CD4 T‐lymphocytes in HIV‐1‐infected pediatric patients. Cytometry Part B Clin Cytom 74:S98–106. [DOI] [PubMed] [Google Scholar]
- 27. Janicki P, Schmidmaier G. 2011. What should be the characteristics of the ideal bone graft substitute? Combining scaffolds with growth factors and/or stem cells. Injury 42:S77–S81. [DOI] [PubMed] [Google Scholar]
- 28. Cuthbert R, Boxall SA, Tan HB, et al. 2012. Single‐platform quality control assay to quantify multipotential stromal cells in bone marrow aspirates prior to bulk manufacture or direct therapeutic use. Cytotherapy 14:431–440. [DOI] [PubMed] [Google Scholar]
- 29. Stolzing A, Jones E, McGonagle D, et al. 2008. Age‐related changes in human bone marrow‐derived mesenchymal stem cells: consequences for cell therapies. Mech Ageing Dev 129:163–173. [DOI] [PubMed] [Google Scholar]
- 30. Polini A, Pisignano D, Parodi M, et al. 2011. Osteoinduction of human mesenchymal stem cells by bioactive composite scaffolds without supplemental osteogenic growth factors. PLoS ONE 6:e26211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tsiridis E, Ali Z, Bhalla A, et al. 2009. In vitro proliferation and differentiation of human mesenchymal stem cells on hydroxyapatite versus human demineralised bone matrix with and without osteogenic protein‐1. Expert Opin Biol Ther 9:9–19. [DOI] [PubMed] [Google Scholar]
- 32. Gamblin AL, Renaud A, Charrier C, et al. 2014. Osteoblastic and osteoclastic differentiation of human mesenchymal stem cells and monocytes in a miniaturized three‐dimensional culture with mineral granules. Acta Biomater 10:5139–5147. [DOI] [PubMed] [Google Scholar]
- 33. Payer M, Lohberger B, Stadelmeyer E, et al. 2010. Behaviour of multipotent maxillary bone‐derived cells on beta‐tricalcium phosphate and highly porous bovine bone mineral. Clin Oral Implants Res 21:699–708. [DOI] [PubMed] [Google Scholar]
- 34. Roberts SJ, Geris L, Kerckhofs G, et al. 2011. The combined bone forming capacity of human periosteal derived cells and calcium phosphates. Biomaterials 32:4393–4405. [DOI] [PubMed] [Google Scholar]
- 35. Popov C, Radic T, Haasters F, et al. 2011. Integrins alpha2beta1 and alpha11beta1 regulate the survival of mesenchymal stem cells on collagen I. Cell Death Dis 2:e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kuzmenko V, Samfors S, Hagg D, et al. 2013. Universal method for protein bioconjugation with nanocellulose scaffolds for increased cell adhesion. Mater Sci Eng C Mater Biol Appl 33:4599–4607. [DOI] [PubMed] [Google Scholar]
- 37. Ragetly GR, Griffon DJ, Lee HB, et al. 2010. Effect of collagen II coating on mesenchymal stem cell adhesion on chitosan and on reacetylated chitosan fibrous scaffolds. J Mater Sci Mater Med 21:2479–2490. [DOI] [PubMed] [Google Scholar]
- 38. Araujo MG, Liljenberg B, Lindhe J. 2010. Dynamics of Bio‐Oss Collagen incorporation in fresh extraction wounds: an experimental study in the dog. Clin Oral Implants Res 21:55–64. [DOI] [PubMed] [Google Scholar]
- 39. Seifert M, Lubitz A, Trommer J, et al. 2012. Crosstalk between immune cells and mesenchymal stromal cells in a 3D bioreactor system. Int J Artif Organs 35:986–995. [DOI] [PubMed] [Google Scholar]
- 40. Baddoo M, Hill K, Wilkinson R, et al. 2003. Characterization of mesenchymal stem cells isolated from murine bone marrow by negative selection. J Cell Biochem 89:1235–1249. [DOI] [PubMed] [Google Scholar]
- 41. Saleh FA, Whyte M, Genever PG. 2011. Effects of endothelial cells on human mesenchymal stem cell activity in a three‐dimensional in vitro model. Eur Cell Mater 22:242–257. [DOI] [PubMed] [Google Scholar]
- 42. Torre ML, Lucarelli E, Guidi S, et al. 2015. Ex vivo expanded mesenchymal stromal cell minimal quality requirements for clinical application. Stem Cells Dev 24:677–685. [DOI] [PubMed] [Google Scholar]
- 43. Veyrat‐Masson R, Boiret‐Dupre N, Rapatel C, et al. 2007. Mesenchymal content of fresh bone marrow: a proposed quality control method for cell therapy. Br J Haematol 139:312–320. [DOI] [PubMed] [Google Scholar]
- 44. Guimaraes JA, Duarte ME, Fernandes MB, et al. 2014. The effect of autologous concentrated bone‐marrow grafting on the healing of femoral shaft non‐unions after locked intramedullary nailing. Injury 45:S7–S13. [DOI] [PubMed] [Google Scholar]
- 45. Alvarez‐Viejo M, Menendez‐Menendez Y, Blanco‐Gelaz MA, et al. 2013. Quantifying mesenchymal stem cells in the mononuclear cell fraction of bone marrow samples obtained for cell therapy. Transplant Proc 45:434–439. [DOI] [PubMed] [Google Scholar]
- 46. Hernigou P, Homma Y, Flouzat Lachaniette CH, et al. 2013. Benefits of small volume and small syringe for bone marrow aspirations of mesenchymal stem cells. Int Orthop 37:2279–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]