

Abstract
Background
Head and neck squamous cell carcinoma (HNSCC) exhibits high rates of recurrence, and with few approved targeted agents, novel treatments are needed. We analyzed a molecular profiling database for the distribution of biomarkers predictive of chemotherapies and targeted agents.
Methods
Seven hundred thirty‐five patients with advanced HNSCC (88 with known human papillomavirus [HPV] status), were profiled using multiple platforms (gene sequencing, gene copy number, and protein expression).
Results
Among the entire patient population studied, epidermal growth factor receptor (EGFR) was the protein most often overexpressed (90%), TP53 gene most often mutated (41%), and phosphatidylinositol 3‐kinase (PIK3CA) most often amplified (40%; n = 5). With the exception of TP53 mutation, other biomarker frequencies were not significantly different among HPV‐positive or HPV‐negative patients. PIK3CA mutations and phosphatase and tensin homolog (PTEN) loss are frequent events, independent of HPV status. The immune response‐modulating programmed cell death 1 (PD1) and programmed cell death ligand 1 (PDL1) axis was active across sites, stages, and HPV status.
Conclusion
Molecular profiling utilizing multiple platforms provides a range of therapy options beyond standard of care. © 2015 Wiley Periodicals, Inc. Head Neck 38: E1625–E1638, 2016
Keywords: head and neck squamous cell carcinoma, molecular profiling, DNA sequencing, protein expression, biomarkers
INTRODUCTION
Head and neck squamous cell carcinoma (HNSCC) accounts for more than 550,000 cases annually, worldwide,1 with incidence rates of certain subtypes (oropharyngeal) on the rise.2 Carcinogen (tobacco, alcohol) exposure and infection with the human papillomavirus (HPV) are described as the 2 major etiological causes of HNSCC. Differences in prognoses have been reported for HPV‐negative and HPV‐positive HNSCC, with HPV positivity being associated with improved clinical outcome and better response to therapy.1
TP53, which is inactivated through mutation or viral oncoprotein interactions in a large proportion of HNSCC, is not directly targetable.3 Standard therapy includes multimodal approaches consisting of radiation, chemotherapy (fluoropyrimidines, platinum analogs, taxanes, etc.),and surgery.4 The only Food and Drug Administration‐approved targeted agent for HNSCC is an epidermal growth factor receptor (EGFR) monoclonal antibody, cetuximab, with single agent overall response rates of 10% to 13%.5 EGFR overexpression in HNSCC ranges from 40% to 60%.6 Several biomarkers, including KRAS and NRAS status, are predictive of response to cetuximab in patients with colorectal cancer, however, there is no strong evidence supporting the predictive utility of any biomarkers (EGFR protein or gene copy number, and HPV status) for cetuximab use in HNSCC.7, 8
Active areas of research in HNSCC include the identification of novel targets, exploration of resistance mechanisms to current therapies, and identification of combination strategies. Recent studies report the high incidence (up to 30%) of phosphatidylinositol 3‐kinase (PIK3CA) pathway mutations in HNSCC.9 PIK3CA inhibitors, therefore, are a promising drug class that may provide treatment success, however, these agents have failed as monotherapy, in other tumor types, and resistance mechanisms have emerged.10, 11 Immunomodulatory agents also share promise as a therapeutic strategy for HNSCC due to the role of adaptive immune resistance to allow for tumor development in HPV‐associated HNSCC.12
Tumor molecular profile‐guided treatment has been successfully utilized to identify molecular targets in patients with metastatic solid tumors. A pilot study in patients with refractory metastatic solid tumors demonstrated improved progression‐free survival (PFS) on a molecular profiling‐guided regimen compared to the regimen that the patient had just previously failed.13 This concept has been confirmed by other groups, suggesting molecular analysis of cancer to guide treatment improves clinical outcomes.14
In the current study, we review a database of biomarker frequency data collected from a commercial molecular profiling service (Caris Life Sciences, Phoenix, AZ). The cases included were advanced, refractory, and/or metastatic HNSCC. Our purposes were to identify traditional and novel treatment options for patients with head and neck cancer that are advanced, refractory, and difficult to treat. To date, a large survey of protein‐based biomarkers that are predictive of traditional chemotherapies (cytotoxics), alongside an assessment of gene alterations (copy number and mutations) has not been performed for HNSCC.
This analysis identified numerous alterations that have potential to impact drug selection through a multiplatform approach. A profiling service that utilizes protein and molecular testing assays can provide options for combination strategies, which may include chemotherapy backbones to targeted agents, which is supported by an illustrative case report. In addition, the data supports the use of agents in clinical trials (eg, PIK3CA inhibitors, immunomodulatory therapies), combination strategies (eg, PIK3CA inhibitors with cetuximab), or agents approved for other solid tumors (eg, gemcitabine, irinotecan). Additionally, this detailed cataloging of protein and genomic alterations in HNSCC may shed light on opportunities for future clinical trial design.
MATERIALS AND METHODS
Patients and multiplatform molecular profiling
Molecular markers that have been associated with sensitivity or resistance to cytotoxic and targeted agents (theranostic biomarkers) were assayed by immunohistochemistry, in situ hybridization, and gene sequencing. The biomarker to drug associations are derived from prospective or retrospective clinical research studies in various solid tumors, including HNSCC, or are part of the National Comprehensive Cancer Network (NCCN) Biomarkers Compendium.15 For some biomarkers, therapeutic associations are suggested based on emerging data (eg, investigational agents in clinical trials).
This study includes data from patients with refractory, aggressive, and/or metastatic head and neck cancer, prospectively assayed by at least 1 platform (immunohistochemical [IHC], in situ hybridization [ISH], and Sanger/next‐generation sequencing [NGS]) by Caris Life Sciences (July 30, 2009 to February 24, 2015), and includes a subgroup of patients with known HPV status (n = 88). Formalin‐fixed paraffin‐embedded (FFPE) HNSCC samples were sent by treating physicians for analysis of theranostic biomarkers. All tumor samples were verified by board‐certified pathologists for sufficient tumor content, specimen quality, and confirmation of diagnosis. The testing performed for each patient may vary based on the physician's request, tissue availability, technology advancements (eg, Sanger vs. NGS), and emerging clinical evidentiary support for theranostic biomarkers.
Immunohistochemistry
IHC analysis of 24 proteins was performed on FFPE tumor samples using commercially available detection kits and automated staining techniques (Benchmark XT; Ventana Medical Systems, Tucson, AZ; and AutostainerLInk 48; Dako, Carpinteria, CA). Antibody clones and thresholds used are provided in Supplementary Table S1, online only. Appropriate positive and negative controls were used for all proteins tested. IHCs were scored manually by board‐certified pathologists using predefined thresholds consisting of intensity of staining (0, 1+, 2+, and 3+) and percentage of tumor cells that stained positive. Thresholds are derived from peer‐reviewed clinical literature, which associates response to treatment to biomarker status. Tests are interpreted as positive or negative, and the expression data are represented as a distribution (percentage) of positive or negative results observed in the cohort tested.
In situ hybridization
Gene copy number alterations of cMET, EGFR, HER2, PIK3CA, and TOP2A were analyzed by DNA ISH using fluorescence in situ hybridization and/or chromogenic in situ hybridization probes as part of the automated staining techniques (Benchmark XT; Ventana Medical Systems) and automated imaging systems (BioView, Billerica, MA). Cutoffs are provided in the Supplementary Table S1, online only. The ratio of gene to pericentromeric regions of chromosome 7 (EGFR, cMET), 17 (HER2, TOP2A), and 3 (PIK3CA) were used to determine increases in gene copy number. Ratios higher than defined cutoff were considered positive and ratios less than defined cutoff were considered negative. HPV DNA status was determined by ISH using the INFORM HPV III Family 16 probe (Ventana Medical Systems), which detects high‐risk HPV subtypes (16, 18, 31, 33, 39, 45, 51, 52, 56, 58, and 66). Observation of single or multiple punctate hybridization signals localized to tumor cell nuclei was scored as positive, and the lack of any hybridization signals was considered negative.
Sanger sequencing
Sanger sequencing included selected regions of BRAF, cKIT, EGFR, KRAS, NRAS, and PIK3CA and was performed using M13‐linked polymerase chain reaction primers designed to flank and amplify targeted sequences. Polymerase chain reaction products were bidirectionally sequenced using the BigDye Terminator version 1.1 chemistry, and analyzed using the 3730 DNA Analyzer (Applied Biosystems, Grand Island, NY). Sequence traces were analyzed using Mutation Surveyor software version 3.25 (Soft Genetics, State College, PA).
Next‐generation sequencing
NGS was performed on genomic DNA isolated from FFPE tumor tissue using the Illumina MiSeq platform. Specific regions of 47 genes were amplified using the Illumina TruSeq Amplicon Cancer Hotspot panel.16 All variants reported are detected with >99% confidence (based on mutation frequency and amplicon coverage) with an average sequencing depth of >1000 X.
Statistical methods
Retrospective analysis of biomarker frequency distributions was attained using standard descriptive statistics. The 2‐tailed Fisher's exact test was performed using JMP version 10.0 (SAS Institute, Cary, NC) to test where frequencies differed by subgroup. A 2‐tailed p value ≤ .05 was considered statistically significant and Bonferroni correction was used to correct for multiple comparisons.
Validation and institutional review board
All methods utilized in this study were clinically validated to at least Clinical Laboratory Improvement Amendments, College of American Pathologists, and International Organization for Standardization (ISO) 15189. This retrospective analysis utilized previously collected deidentified data created under the Caris honest‐broker policy and followed consultation with the Western Institutional Review Board (IRB), which is the IRB of record for Caris Life Sciences. The project was deemed exempt from IRB oversight and consent requirements were waived.
RESULTS
Patient and tumor characteristics
Patient and tumor characteristics are described in Table 1. The cohort is subdivided into 3 groups: (1) All HNSCC (n = 735; includes all prospectively assayed patients with HNSCC); (2) HPV‐positive (n = 39); and (3) HPV‐negative (n = 49). The population consisted of a median age of 60, 59, and 58 years, for all, HPV‐positive, and HPV‐negative, respectively. The male sex accounted for 78% of the patients assessed. Prevalence for HPV‐positivity in the male sex (85%) was observed compared to the HPV‐negative (69%) subgroup. Primary tumor site distribution favored the oropharynx in all subgroups, however, it was the highest in HPV‐positive group (79%). Followed by the oropharynx (51%), head and neck, not otherwise specified (15%), larynx (11%), oral cavity (9%), nasopharynx (9%), and pharynx (5%), comprised the remainder of the entire HNSCC population studied. More than half (56%) of the tumor samples utilized for profiling were attained from metastatic sites (stage IV disease).
Table 1.
Clinicopathological characteristics of 735 patients with head and neck squamous cell carcinoma molecularly profiled.
| Patient characteristic | All HNSCC (n = 735) | HPV‐positive (n = 39) | HPV‐negative (n = 49) |
|---|---|---|---|
| Age, y | |||
| Average (range) | 59.5 (19–90) | 59.2 (40–80) | 57.5 (27–87) |
| Sex (%) | |||
| Male | 570 (78) | 33 (85) | 34 (69) |
| Female | 165 (22) | 6 (15) | 15 (31) |
| Tumor sites in head and neck, no. (%) | |||
| Oropharynx | 372 (51) | 31 (79) | 25 (51) |
| Nasopharynx | 63 (9) | – | 4 (8.2) |
| Pharynx | 40 (5) | 1 (3) | 3 (6.1) |
| Larynx | 80 (11) | 2 (5) | 5 (10.2) |
| Oral cavity | 67 (9) | 1 (3) | 6 (12.25) |
| Head and neck, NOS | 113(15) | 4 (10) | 6 (12.25) |
| Site of tumor profiled, no. (%) | |||
| Primary sites | 321(44) | 19 (49) | 18 (37) |
| Metastatic sites* | 414 (56) | 20 (51) | 31 (63) |
Abbreviations: HNSCC, head and neck squamous cell carcinoma; HPV, human papillomavirus; NOS, not otherwise specified.
*Including: connective and soft tissue of head, face, and neck (n = 138); lymph nodes (n = 93); lung and bronchus (n = 75); liver (n = 34); bones and joints (n = 26); pleura (n = 7); chest and NOS (n = 6); skin (n = 3); colon (n = 5); mediastinum (n = 4); pelvis and NOS (n = 3); retroperitoneum and peritoneum (n = 5); stomach (n = 3); vertebra (n = 5); adrenal gland (n = 3); brain (n = 2); orbit (n = 1); and small bowel (n = 1).
Multiplatform testing
A total of 76 theranostic biomarkers were examined in this study. Testing varied from patient‐to‐patient, therefore, the total number of patients assayed by each platform and each specific biomarker are provided in Tables 2 and 3.
Table 2.
Frequencies of alterations of predictive biomarkers as measured by immunohistochemistry and in situ hybridization.
| All HNSCC | ||||
|---|---|---|---|---|
| Technology | Biomarker | No. tested | No. altered | % |
| IHC (expression is high or overexpressed, unless indicated by “*,” which indicates low or no expression) | AR | 664 | 27 | 4.066 |
| BCRP | 111 | 39 | 35.14 | |
| cKIT | 300 | 15 | 5.000 | |
| cMET | 391 | 63 | 16.11 | |
| COX2 | 18 | 5 | 27.778 | |
| EGFR | 123 | 111 | 90.24 | |
| ER | 668 | 42 | 6.29 | |
| ERCC1* | 381 | 190 | 49.87 | |
| HER2 | 690 | 12 | 1.74 | |
| MGMT* | 703 | 274 | 38.98 | |
| MRP1 | 293 | 260 | 88.74 | |
| PD1 | 182 | 125 | 68.68 | |
| PDL1 | 183 | 32 | 17.49 | |
| PDGFRA | 116 | 17 | 14.655 | |
| PGP | 591 | 19 | 3.21 | |
| PR | 666 | 33 | 4.955 | |
| PTEN* | 697 | 357 | 51.220 | |
| RRM1* | 653 | 356 | 54.518 | |
| SPARC | 683 | 196 | 28.697 | |
| TLE3 | 401 | 166 | 41.397 | |
| TOP2A | 596 | 485 | 81.376 | |
| TOPO1 | 645 | 387 | 60.000 | |
| TS* | 646 | 369 | 57.121 | |
| TUBB3* | 318 | 208 | 65.409 | |
| ISH (amplification rates) | cMET ISH | 312 | 2 | 0.64 |
| EGFR ISH | 236 | 50 | 21.186 | |
| HER2 ISH | 419 | 9 | 2.15 | |
| PIK3CA ISH | 5 | 2 | 40.000 | |
| TOP2A ISH | 68 | 3 | 4.41 | |
Abbreviations: HNSCC, head and neck squamous cell carcinoma; EGFR, epidermal growth factor receptor; PD1, programmed death‐1 PDL1, programmed death ligand‐1; PGP, permeability glycoprotein; PTEN, phosphatase and tensin homolog; SPARC, secreted protein acidic and rich in cysteine; IHC, immunohistochemical; ISH, in situ hybridization; PIK3CA, phosphatidylinositol 3‐kinase.
Table 3.
Mutation frequencies as measured by next‐generation sequencing and Sanger sequencing technologies (where indicated*).
| Technology | Gene tested | No. tested | No. mutated | % |
|---|---|---|---|---|
| Mutation (NGS or # (NGS + Sanger) | ABL | 321 | 2 | 0.62 |
| AKT | 335 | 5 | 1.49 | |
| ALK | 337 | 0 | 0.00 | |
| APC | 335 | 12 | 3.58 | |
| ATM | 333 | 5 | 1.50 | |
| BRAF# (336 tested by NGS) | 442 | 1 | 0.23 | |
| BRCA1 | 87 | 5 | 5.75 | |
| BRCA2 | 87 | 8 | 9.20 | |
| CDH1 | 337 | 1 | 0.30 | |
| cKIT# (335 tested by NGS) | 388 | 4 | 1.03 | |
| cMET | 336 | 11 | 3.27 | |
| CSF1R | 336 | 1 | 0.30 | |
| CTNNB1 | 336 | 2 | 0.60 | |
| EGFR# (337 tested by NGS) | 360 | 3 | 0.83 | |
| ERBB4 | 334 | 3 | 0.90 | |
| FBXW7 | 335 | 14 | 4.18 | |
| FGFR1 | 337 | 0 | 0.00 | |
| FGFR2 | 337 | 0 | 0.00 | |
| FLT3 | 337 | 1 | 0.30 | |
| GNA11 | 294 | 0 | 0.00 | |
| GNAQ | 229 | 0 | 0.00 | |
| GNAS | 337 | 0 | 0.00 | |
| HER2 | 334 | 0 | 0.00 | |
| HNF1A | 307 | 3 | 0.98 | |
| HRAS | 299 | 8 | 2.68 | |
| IDH1 | 337 | 2 | 0.59 | |
| JAK2 | 337 | 0 | 0.00 | |
| JAK3 | 337 | 2 | 0.59 | |
| KDR | 335 | 3 | 0.90 | |
| KRAS# (333 tested by NGS) | 448 | 11 | 2.46 | |
| MLH1 | 334 | 0 | 0.00 | |
| MPL | 335 | 0 | 0.00 | |
| NOTCH1 | 333 | 0 | 0.00 | |
| NPM1 | 334 | 0 | 0.00 | |
| NRAS# (335 tested by NGS) | 381 | 4 | 1.05 | |
| PDGFRA | 333 | 4 | 1.20 | |
| PIK3CA# (328 tested by NGS) | 421 | 53 | 12.59 | |
| PTEN | 324 | 13 | 4.01 | |
| PTPN11 | 337 | 0 | 0.00 | |
| RB | 333 | 5 | 1.50 | |
| RET | 330 | 1 | 0.30 | |
| SMAD4 | 336 | 4 | 1.19 | |
| SMARCB1 | 336 | 0 | 0.00 | |
| SMO | 280 | 0 | 0.00 | |
| STK11 | 326 | 6 | 1.84 | |
| TP53 | 335 | 137 | 40.90 | |
| VHL | 303 | 0 | 0.00 |
Abbreviations: NGS, next‐generation sequencing; EGFR, epidermal growth factor receptor; PIK3CA, phosphatidylinositol 3‐kinase; PTEN, phosphatase and tensin homolog.
Protein and gene copy number alterations
The distribution of protein expression measured by IHC and gene copy number by ISH is detailed in Table 2. Overall, the most frequently altered proteins as measured by IHC include: EGFR (90%), MRP1 (89%), TOP2A (81%), programmed death‐1 (PD‐1; 69%), and TUBB3 (65%). Additional therapeutically relevant biomarkers in HNSCC occurring at high frequency in our study included loss of phosphatase and tensin homolog (PTEN; 51%). For gene copy number changes, PIK3CA was most frequently amplified (40%; n = 5), followed by EGFR (21%; amplified + polysomy).
Mutational analyses
The detection of variants of up to 47 genes is described in Table 3 and specific allele changes are cataloged in Supplementary Table S2, online only. Our data confirm the common, key molecular changes (TP53, PIK3CA, PTEN, FBXW7, HRAS, etc.) reported by other comprehensive genomic sequencing studies for HNSCC.17, 18, 19, 20 TP53 was the most frequently mutated gene (41%), and was not detected in the HPV‐positive subgroup (see Table 4). After TP53, the PIK3CA pathway (PIK3CA, PTEN, AKT, and STK11) was the most frequently mutated oncogenic pathway (19.6%). Mutations in this pathway were found at slightly higher rates in HPV‐positive HNSCC (Table 4). Detailed analysis on the PIK3CA pathway, concurrent mutations in the mitogen‐activated protein kinase (MAPK) pathway, and incorporation of PTEN expression (IHC) data will be discussed later.
Table 4.
Comparison of biomarker frequencies (immunohistochemical, in situ hybridization, and next‐generation sequencing) in human papillomavirus–positive and negative head and neck squamous cell carcinoma subgroups.
| HPV+ | HPV‐ | Statistical analysis | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Biomarker | HPV+ no. tested | HPV+ no. altered | HPV+, % | HPV− no. tested | HPV− no. altered | HPV−, % | p values | Bonferroni corrected significance | Bonferroni corrected p value |
| AR | 38 | 1 | 2.632 | 45 | 2 | 4.444 | 1 | Not significant | 1 |
| BCRP | 2 | 1 | 50.000 | 4 | 0 | 0.000 | .3333 | Not significant | 1 |
| cKIT | 24 | 0 | 0.000 | 42 | 1 | 2.381 | 1 | Not significant | 1 |
| cMET | 16 | 3 | 18.750 | 10 | 2 | 20.000 | 1 | Not significant | 1 |
| COX2 | 1 | 1 | 100.000 | 4 | 0 | 0.000 | .2 | Not significant | 1 |
| EGFR | 7 | 7 | 100.000 | 6 | 6 | 100.000 | 1 | Not significant | 1 |
| ER | 38 | 5 | 13.158 | 46 | 1 | 2.174 | .0866 | Not significant | 1 |
| ERCC1* | 26 | 14 | 53.846 | 41 | 17 | 41.463 | .4511 | Not significant | 1 |
| HER2 | 39 | 2 | 5.128 | 47 | 0 | 0.000 | .2027 | Not significant | 1 |
| MGMT* | 39 | 13 | 33.333 | 49 | 27 | 55.102 | .0534 | Not significant | 1 |
| MRP1 | 24 | 20 | 83.333 | 42 | 39 | 92.857 | .246 | Not significant | 1 |
| PD1 | 21 | 14 | 66.667 | 20 | 5 | 25.000 | .0122 | Not significant | 1 |
| PDL1 | 21 | 4 | 19.048 | 21 | 8 | 38.000 | .3058 | Not significant | 1 |
| PDGFRA | 2 | 0 | 0.000 | 4 | 0 | 0.000 | 1 | Not significant | 1 |
| PGP | 39 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| PR | 38 | 1 | 2.632 | 46 | 6 | 13.043 | .1212 | Not significant | 1 |
| PTEN* | 39 | 23 | 58.974 | 49 | 34 | 69.388 | .2661 | Not significant | 1 |
| RRM1* | 39 | 19 | 48.718 | 48 | 29 | 60.417 | .2887 | Not significant | 1 |
| SPARC(m) | 38 | 13 | 34.211 | 48 | 10 | 20.833 | .2208 | Not significant | 1 |
| TLE3 | 17 | 10 | 58.824 | 11 | 7 | 63.636 | 1 | Not significant | 1 |
| TOP2A | 38 | 35 | 92.105 | 49 | 39 | 79.592 | .1352 | Not significant | 1 |
| TOPO1 | 39 | 19 | 48.718 | 47 | 17 | 36.170 | .277 | Not significant | 1 |
| TS* | 39 | 23 | 58.974 | 47 | 31 | 65.957 | .6544 | Not significant | 1 |
| TUBB3* | 15 | 11 | 73.333 | 6 | 4 | 66.667 | 1 | Not significant | 1 |
| cMET ISH | 15 | 0 | 0.000 | 11 | 0 | 0.000 | 1 | Not significant | 1 |
| EGFR ISH | 16 | 0 | 0.000 | 33 | 9 | 27.273 | .0208 | Not significant | 1 |
| HER2 ISH | 19 | 0 | 0.000 | 14 | 1 | 7.143 | .4242 | Not significant | 1 |
| PIK3CA ISH | 1 | 0 | 0.000 | 2 | 1 | 50.000 | 1 | Not significant | 1 |
| TOP2A ISH | 2 | 0 | 0.000 | 3 | 1 | 33.333 | 1 | Not significant | 1 |
| ABL | 36 | 0 | 0.000 | 47 | 0 | 0.000 | .41860465 | Not significant | 1 |
| AKT | 36 | 0 | 0.000 | 47 | 0 | 0.000 | .42261427 | Not significant | 1 |
| ALK | 37 | 0 | 0.000 | 47 | 0 | 0.000 | .42680615 | Not significant | 1 |
| APC | 36 | 3 | 8.333 | 47 | 4 | 8.511 | .5556178 | Not significant | 1 |
| ATM | 36 | 0 | 0.000 | 46 | 1 | 2.174 | .4254446 | Not significant | 1 |
| BRAF | 36 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| BRCA1 | 3 | 0 | 0.000 | 2 | 0 | 0.000 | 1 | Not significant | 1 |
| BRCA2 | 3 | 1 | 33.333 | 2 | 1 | 50.000 | 1 | Not significant | 1 |
| CDH1 | 37 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| cKIT | 36 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| cMET | 36 | 3 | 8.333 | 47 | 0 | 0.000 | .0777 | Not significant | 1 |
| CSF1R | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| CTNNB1 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| EGFR | 37 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| ERBB4 | 36 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| FBXW7 | 36 | 3 | 8.333 | 47 | 1 | 2.128 | .3117 | Not significant | 1 |
| FGFR1 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| FGFR2 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| FLT3 | 37 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| GNA11 | 36 | 0 | 0.000 | 45 | 0 | 0.000 | 1 | Not significant | 1 |
| GNAQ | 29 | 0 | 0.000 | 45 | 0 | 0.000 | 1 | Not significant | 1 |
| GNAS | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| HER2 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| HNF1A | 33 | 1 | 3.030 | 47 | 0 | 0.000 | .4125 | Not significant | 1 |
| HRAS | 33 | 0 | 0.000 | 45 | 1 | 2.222 | 1 | Not significant | 1 |
| IDH1 | 37 | 0 | 0.000 | 47 | 1 | 2.128 | 1 | Not significant | 1 |
| JAK2 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| JAK3 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| KDR | 36 | 1 | 2.778 | 47 | 2 | 4.255 | 1 | Not significant | 1 |
| KRAS | 36 | 2 | 5.556 | 47 | 0 | 0.000 | .1851 | Not significant | 1 |
| MLH1 | 36 | 0 | 0.000 | 45 | 0 | 0.000 | 1 | Not significant | 1 |
| MPL | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| NOTCH1 | 36 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| NPM1 | 36 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| NRAS | 37 | 1 | 2.703 | 47 | 0 | 0.000 | .4405 | Not significant | 1 |
| PDGFRA | 36 | 1 | 2.778 | 47 | 3 | 6.383 | .6294 | Not significant | 1 |
| PIK3CA | 36 | 3 | 8.333 | 46 | 1 | 2.174 | .3148 | Not significant | 1 |
| PTEN | 36 | 3 | 8.333 | 46 | 1 | 2.174 | .3148 | Not significant | 1 |
| PTPN11 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| RB | 36 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| RET | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| SMAD4 | 36 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| SMARCB1 | 37 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| SMO | 32 | 0 | 0.000 | 46 | 0 | 0.000 | 1 | Not significant | 1 |
| STK11 | 35 | 0 | 0.000 | 47 | 0 | 0.000 | 1 | Not significant | 1 |
| TP53 | 37 | 0 | 0.000 | 47 | 27 | 57.447 | 1.16E‐06 | Significant | 8.83E‐05 |
| VHL | 35 | 0 | 0.000 | 45 | 0 | 0.000 | 1 | Not significant | 1 |
Abbreviations: HPV+, human papillomavirus–positive; HPV‐, human papillomavirus–negative.
The p values in bold indicate statistical significance before correction for multiple comparisons.
BRCA1 and BRCA2 mutations were detected in 5.75% and 9.2%, respectively, however, the clinical significance of all but one of the variants detected in these genes is currently unknown (see Supplementary Table S2, online only). Also of note, BRCA1/2 mutations almost exclusively occurred in the context of co‐mutations with TP53 and/or PIK3CA pathway mutations (PIK3CA, PTEN; see Supplementary Table S3, online only).
Our analysis exhibited a low frequency of targetable gene alterations outside of cell cycle checkpoints (TP53, RB1), DNA repair pathways (ATM, BRCA1, BRCA2) and PIK3CA (PIK3CA, AKT, PTEN) pathways. Despite being detected at lower frequencies, several druggable mutations are found in the “Long Tail” of alterations, such as EGFR, which occurred at a frequency of 1% (Supplementary Figure S1, online only). Our analysis did not detect variants in the following genes: MPL, ALK, VHL, MLH1, NPM1, SMO, FGFR1, FGFR2, JAK2, NOTCH1, SMARCB1, GNAS, GNAQ, GNA11, HER2, and PTPN11.
Multiple mutations in single mitogenic pathways may indicate that these genes are “driving” cancer growth.9 Genes that fall into this category include TP53, PIK3CA, and PTEN. Another measure of whether these simultaneous events are important to HNSCC pathogenesis, and may consequently have therapeutic implications, is whether multiple mutations occurred in more than 1 patient, which is documented by the frequency for co‐occurring mutations (Supplementary Table S3, online only). Co‐mutations in PIK3CA and TP53 were the most frequent, observed in 10 patients. Simultaneous mutations in several signaling nodes indicate pathway cross‐talk, or feedback loops, and identify ideal candidates for dual‐targeting strategies.
Biomarker alterations and clinical relevance
A comprehensive listing of biomarker alterations and their potential clinical and therapeutic significance is found in Figure 1. All alterations, in a platform agnostic fashion, are sorted by frequency. Notably, biomarkers associated with response to “On NCCN compendium” therapies for HNSCC, filter toward the top of the list (TUBB321/TLE322 for taxanes, TS23 for 5‐fluorouracil, RRM124 for gemcitabine, and ERCC125 for platinum agents).
Figure 1.
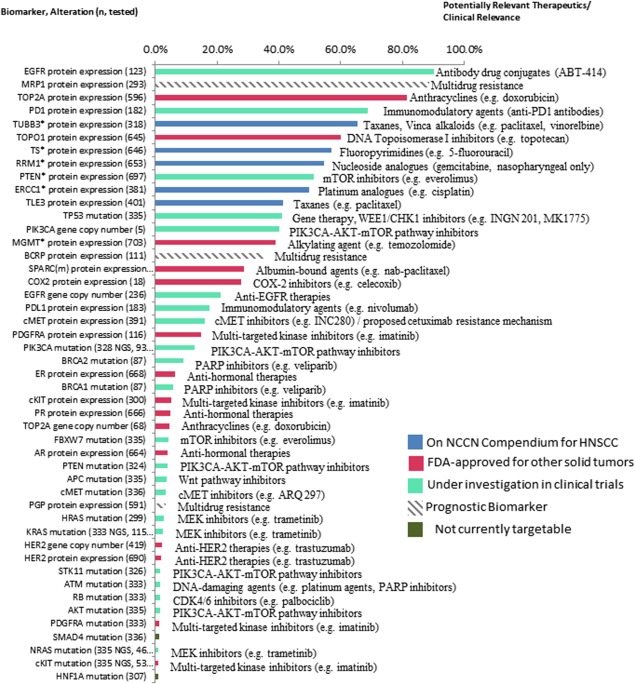
Biomarker alterations and associated clinical relevance, listed in order of frequency observed. Biomarkers are followed by alteration observed (eg, protein expression, mutation, amplification) and n, number of patients assayed. All frequencies are provided in terms of protein overexpression (immunohistochemical [IHC]), increased gene copy number/amplification (in situ hybridization [ISH]), or mutated (next‐generation sequencing [NGS]/Sanger). Biomarkers with * indicate frequency of low or lack of protein expression (IHC), which associates with benefit to associated therapy. Blue bars indicate therapy is On‐NCCN (National Comprehensive Cancer Network) Compendium for head and neck squamous cell carcinoma (HNSCC), red bars indicate therapy is Food and Drug Administration‐approved for other solid tumors, green bars indicate therapy is under investigation in clinical trials, gray hashed bar indicates a prognostic marker, and dark green bars indicate therapy is not currently targetable.
EGFR plays an important role in epithelial malignancies, including HNSCC.26 The high expression and gene copy number increases in EGFR (90% and 21% in our study, respectively) have been documented previously, with similarly observed overexpression (protein)27 and amplification (gene copy number)8 rates. EGFR mutations were detected in 1% of the total HNSCC cohort.
The second most frequently altered biomarker in this study was the overexpression of MRP1 in 89% of the population studied. MRP1, or multidrug resistance protein, is an ATP‐binding cassette (ABC) transporter that functions as “drug pump.”28 With exposure to many therapies, high expression of this family of transporters in a cohort of heavily treated patients with HNSCC is not entirely unexpected.29, 30 Unfortunately, the significance of ABC transporters in multidrug resistance in a clinical setting is largely understudied. It is noteworthy that permeability glycoprotein (PGP), a different ABC transporter, is observed in a significantly smaller portion of patients with HNSCC (3%).
Tailoring therapy based on differences in substrate specificities for these transporters is worthy of further research. For example, TOP2A was overexpressed in 81% of HNSCC, and although anthracyclines are used to target high TOP2A enzyme levels, high expression of MRP1 in a majority of HNSCC serves as a potential resistance mechanism for epirubicin and related therapeutics, which are substrates of MRP1.30 Alternatively, patients with HNSCC exhibiting TOPO1 overexpression (60%) may benefit from epipodophyllotoxins, such as irinotecan or topotecan, which are substrates of PGP but not MRP1.30 A previous prospective trial of 49 patients showed up to 20% response rate to single agent irinotecan in unselected refractory and/or metastatic HNSCC.31
Low frequency of overexpression of PGP (3%), a known drug efflux mechanism for taxanes, supports the utility of these agents in HNSCC. Previous trials demonstrate response rates of 20% to 42% for single agent paclitaxel or docetaxel in unselected refractory and/or metastatic HNSCC.5 An alternative solvent‐free formulation of taxanes is offered by albumin‐bound paclitaxel. Studies have shown that high tumor expression of secreted protein acidic cysteine‐rich (SPARC), an albumin‐binding matrix‐associated protein, may facilitate the accumulation of albumin‐bound paclitaxel. Our analysis demonstrates SPARC overexpression in 29% of patients. Desai et al32 describe preliminary evidence for the utility of SPARC staining as a predictive marker for response to nab‐paclitaxel in a retrospective analysis of 60 patients with HNSCC. The study demonstrated that the response to nab‐paclitaxel was higher for SPARC‐positive than SPARC‐negative patients (83% vs 25%). New albumin‐targeting agents are under development, and also may be beneficial in up to a third of patients with HNSCC based on SPARC overexpression in our cohort.
Harnessing the immune system has long represented an attractive therapeutic target in cancer. Major breakthroughs surrounding the PD1 and programmed cell death ligand 1 (PDL1) pathway as a key suppressor of immune response and recent application of anti‐PD1 and anti‐PDL1 drugs prove to be very promising options in patients with solid tumors. Importantly, PD1 staining on tumor infiltrating lymphocytes (TILs) was detected in 69%, whereas PDL1 staining in tumor cells was found in 18% of patients. A more thorough analysis on the patterns of expression of these proteins follows.
Forty‐one percent of patients in this study exhibited mutations in TP53. Despite its high penetrance in HNSCC, this gene has historically been challenging to target. Whether this drug is “druggable,” is an area of active research. Agents under clinical investigation include the WEE1 kinase inhibitor, MK1775, and adenoviral gene transfer (INGN 201). Another cell cycle checkpoint found to be altered in the lower spectrum of frequency (2%) is RB1, and cell cycle checkpoint inhibitors are under investigation for these aberrations. Of note, like TP53, RB1 mutations often co‐occur with mutations in additional signaling nodes (PIK3CA and RAS; see Supplementary Table S3, online only), which may have dual‐targeting implications.
The hepatocyte growth factor receptor, also known as cMET, is an additional oncogene with targeted agents under clinical investigation and found to be overexpressed in 16% of our cohort. However, in accordance with previous reports, cMET amplification and mutations occurred at much lower frequencies,33 1% (2 of 312) and 3% (11 of 336), respectively. Clinical data are lacking in determining the efficacy of cMET inhibitors for treatment of HNSCC; however, these agents are in all phases of clinical development for all solid tumors, including HNSCC.
Low levels of MGMT and RRM1, which are associated with improved responses to temozolomide34 and gemcitabine,35 were found in 39% and 55% of patients, respectively. Clinical data are limited for application of these agents in refractory and/or metastatic HNSCC. A phase II study of temozolomide in patients with aerodigestive tract cancers showed 1 patient with HNSCC displaying MGMT promoter methylation (promoter methylation leads to loss of MGMT expression) had a partial response.36 Gemcitabine has been shown to yield a 13% response rate in a cohort of 61 unselected refractory and/or metastatic HNSCC37 and more recently, in a small trial of heavily pretreated, unselected refractory and/or metastatic HNSCC demonstrated 1 complete response, 2 partial responses, and 3 stable diseases, yielding a response rate of 37.5%.38
Overexpression of HER2 was found in 1.7% and amplification events were found in 2.1% in our cohort. In the 9 patients with HER2 amplification by ISH, 6 patients had concurrent protein overexpression. Incidence rates for HER2‐positivity in HNSCC are conflicting, ranging from 0% to 47%, however, the scoring criteria applied in these studies is variable.39 Thresholds for IHC (≥3+ and 10%) and ISH (≥2 HER2:CEP17 signal ratio) in this analysis were derived from the guidelines for breast cancer, therefore, these data highlight true‐positive/amplified rates, which are amenable to HER2 directed therapies. The relatively low rates of protein overexpression and gene amplification suggest these therapies may have high therapeutic impact for a small subgroup of HNSCC. A recent case study of salivary duct cancer,40 as well as a small trial of 107 therapy‐naive locally advanced HNSCC,41 provide preliminary evidence to support HER2‐directed therapy (trastuzumab, lapatinib), in HER2 amplified or overexpressed HNSCC. Of note, the HER2‐positive/amplified patients in our study included patients with oropharyngeal, laryngeal, and nasopharyngeal subtypes of HNSCC.
Comparison of biomarker frequencies in human papillomavirus–positive/–negative head and neck squamous cell carcinoma subgroups
A comparison of the frequency of biomarker expression (IHC), gene copy number changes (ISH), and mutations (NGS/Sanger sequencing) according to HPV status are listed in Table 4. Low expression of MGMT by IHC, PD1‐positive TILs by IHC, EGFR amplification by ISH, and TP53 mutations by NGS were significantly different by the Fisher exact test, however, with the exception of TP53 mutation, all lost significance when adjusted for multiple testing effects. Importantly, mutations in PIK3CA and PTEN occurred in both HPV‐positive and HPV‐negative patients, with slightly higher rates in HPV‐positive patients (Table 4).
Comprehensive Analysis of phosphatidylinositol 3‐kinase pathway‐driven head and neck squamous cell carcinoma
The PIK3CA pathway has emerged as the most common targetable alteration in HNSCC; however, based on most recent data, the efficacy of these agents for this population, and other solid tumors, is currently unclear.42, 43 Our analysis into the specific aberrations in this pathway is with anticipation of providing more clues into how to incorporate PIK3CA‐targeted strategies for HNSCC.
Exceeding mutation rates in the PIK3CA pathway (13% PIK3CA, 4% PTEN, 2% STK11, and 1% AKT1; Table 3) are loss of PTEN expression as determined by IHC (51%; Table 2). The distribution of loss of PTEN expression by IHC according to PIK3CA pathway genotype is detailed in Figure 2. The important therapeutic implications of PTEN loss are being explored. In the context of wildtype genotypes (44% of PIK3CA wildtype patients exhibit PTEN loss; Figure 2), Janku et al44 report impressive PFS for 2 patients with PIK3CA wildtype HNSCC exhibiting loss of PTEN expression by IHC: 1 patient, treated with a PI3K inhibitor in combination with chemotherapy, experienced an 18.4‐month PFS, and another treated with an mTOR inhibitor in combination with a targeted agent, experienced an 11.4‐month PFS. The high rates of PTEN loss in HNSCC, therefore, indicate the potential widespread application of mTOR inhibitors and the importance of PTEN loss of expression as a predictive tool in selection of PI3K or mTOR inhibition.
Figure 2.
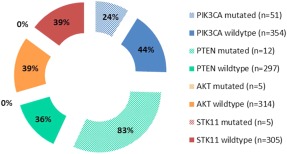
Percent of patients exhibiting phosphatase and tensin homolog (PTEN) loss (immunohistochemical [IHC]) according to phosphatidylinositol 3‐kinase (PIK3CA) pathway genotype. Solid pieces represent percentage of patients with head and neck squamous cell carcinoma (HNSCC) with wildtype PIK3CA, PTEN, AKT, and STK11 exhibiting concurrent loss of PTEN protein expression by IHC. Hashed pieces represent percentage of patients with HNSCC with mutated PIK3CA and PTEN genotypes exhibiting concurrent loss of PTEN protein expression by IHC. Patients with mutated AKT (n = 5) and STK11 (n = 5) retained PTEN expression, therefore are represented by “0%.”
In contrast, PTEN loss was identified as a resistance mechanism to PIK3CA inhibition in a recent study of patients with PIK3CA‐mutated breast cancer.45 Our data demonstrated 24% of patients with PIK3CA mutations exhibit PTEN loss. The significance of PTEN loss in PIK3CA‐mutated patients, before initiating PIK3CA‐directed therapy, is unknown at this time, however, it may have important therapeutic relevance.
Our data exhibited a higher proportion of exon 9 mutations, or helical domain mutations. The distribution of mutations within exons 9, 20, and other exons (see Figure 3) confirms previous reports, with highest mutation frequency in exon 9 (E545K>E542), followed by exon 20 (H1074R).9, 44 Recent studies demonstrate that responses to PIK3CA inhibitors may differ based on the specific PIK3CA mutations.44
Figure 3.
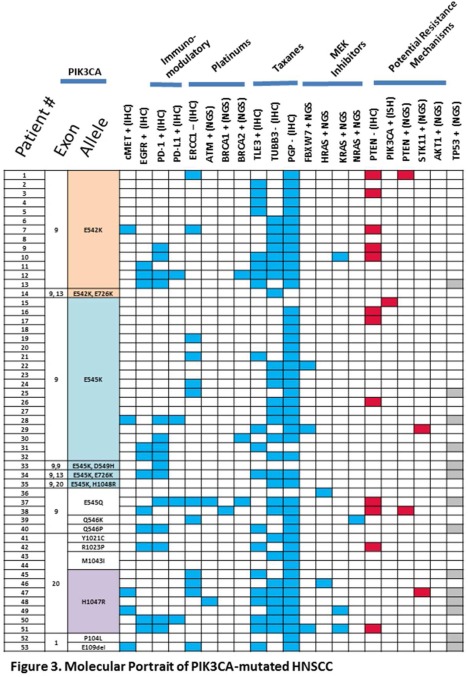
Molecular portrait of phosphatidylinositol 3‐kinase (PIK3CA)‐mutated head and neck squamous cell carcinoma (HNSCC). Mosaic plot of concurrent, targetable alterations in PIK3CA‐mutated HNSCC (each row represents 1 patient; n = 53). Plot is organized according to observed PIK3CA mutations (eg, exon 9, exon 20). Blue squares indicate the presence of a molecular alteration that may be used to incorporate additional therapies to a PIK3CA‐targeted combination regimen, based on current data. Red squares indicate a molecular alteration that may lead to a resistance mechanism, based on current data. The contribution of a TP53 mutation (gray squares) in PIK3CA‐targeted therapy is unknown at this time.
PIK3CA mutations rarely occur alone; therefore, the influence of aberrations in additional signaling molecules and their impact on PIK3CA‐directed therapy is under investigation. Figure 3 outlines the presence of concurrent molecular alterations that may be used to help strategize combination strategies for HNSCC. The data show the molecular heterogeneity of PIK3CA‐mutated HNSCC and highlights various combination approaches. For example, in patients who harbor mutations in RAS (6 of 53 or 11%), which are signaling feedback loops, resistance to cetuximab46, 47 have been described, therefore, dual‐targeting of cetuximab with PIK3CA/mTOR inhibitors may be a suitable precision therapy option. Another promising approach is the combination of chemotherapy plus PIK3CA inhibitor.48 Utilization of the predictive markers for taxanes, including TUBB3, TLE3, and PGP, may be helpful to enrich the patients most likely to benefit from a PIK3CA with chemotherapy backbone combination.
Programmed death‐1/programmed death ligand‐1
The role of the PD1 and PDL1 immunomodulatory axis in HNSCC, a cancer with viral and nonviral etiologies, was investigated. Previous reports indicate predilection for tonsillar cancers to harbor activation of the pathway12 because of viral immune surveillance with HPV‐positive HNSCC. We explored the patterns of PD1‐positivity on tumor infiltrating lymphocytes and expression of PDL1 in tumor cells in Figure 4, according to disease state (Figure 4A), primary site location (Figure 4B), and HPV status (Figure 4C).
Figure 4.
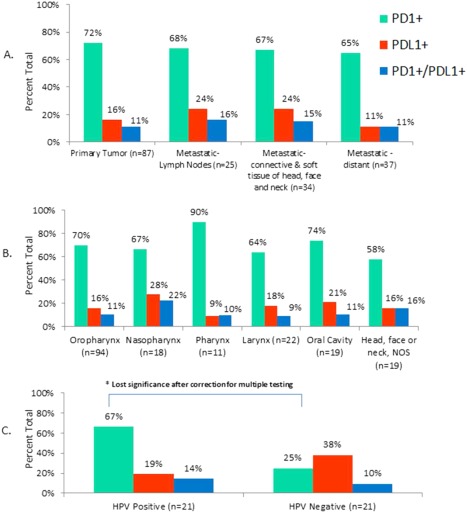
Percent positive expression of programmed death‐1 (PD‐1) and programmed death ligand‐1 (PDL1), according to (A) disease state, (B) primary site location, and (C) human papillomavirus (HPV) status. Green bars represent PD1‐positive expression on tumor infiltrating lymphocytes; red bars represent PDL1‐positive expression in tumor cells and blue bars represent concurrent PD1/PDL1‐positivity. (A) Percent expression according to disease stage of specimen used for profiling. (B) Percent expression according to primary tumor site location. (C). Percent expression according to HPV status.
PD1‐positive TILs were detected in a range of 65% to 72%, across disease stages, and PDL1‐positivity in tumor cells was detected at slightly higher levels (24%) in lymph node metastases and regional metastases (connective and soft tissues of the head, face, and neck) compared with distant metastases (11%; not statistically significant). Both PD1‐positive TILs and PDL1‐positivity in tumor cells was found across primary disease sites, with highest frequency of 90% PD1‐positivity occurring in pharyngeal cancers, and highest PDL1 levels (28%) detected in nasopharyngeal cancers. The Fisher exact test found significant differences in PD1‐positive TILs in HPV‐positive versus HPV‐negative HNSCC (Figure 4C), however, it lost significance after corrections for multiple testing.
Case illustration
A 68‐year‐old man, former smoker, diagnosed with stage IV hypopharyngeal and proximal esophageal SCC, locally advanced with regional lymph node involvement, and extension to the nasopharynx was referred for molecular profiling after progressing on 3 prior chemotherapies and localized radiation (cisplatin/5‐fluorouracil/radiation; paclitaxel/carboplatin; and gemcitabine). Testing using a multiplatform approach revealed several protein aberrations indicative of potential for responses to various chemotherapies (see Table 5). In addition, NGS revealed variants in APC (L1129S) and TP53 (R213X). The treating physician designed a combination treatment strategy based on these results, including cetuximab, pemetrexed, and nab‐paclitaxel.
Table 5.
Select molecular profiling results for head and neck squamous cell carcinoma case illustration.
| Test | Modality (IHC/ISH/NGS) | Alteration | Interpretation | Drug associations |
|---|---|---|---|---|
| PGP | IHC | 0+ 100% (intensity and cell staining) | Negative | Benefit from taxanes (docetaxel, paclitaxel, and nab‐paclitaxel) |
| SPARC | IHC | 2+ 35% (intensity and cell staining) | Positive | |
| TLE3 | IHC | 2+ 35% (intensity and cell staining) | Positive | |
| TS | IHC | 1+ 1% (intensity and cell staining) | Negative | Benefit from fluoropyrimidines (fluorouracil, pemetrexed) |
| TOPO1 | IHC | 2+ 40% (intensity and cell staining) | Positive | Benefit from camptothecin Derivatives (topotecan, irinotecan) |
| TOP2A | IHC | 1+ 2% (intensity and cell staining) | Negative | Lack of benefit from TOP2A‐targeted agents (doxorubicin) |
| APC | NGS | L1129S | Variant of unknown significance | Clinical trials |
| TP53 | NGS | R213X | Pathogenic mutation | Clinical trials |
Abbreviations: IHC, immunohistochemical; ISH, in situ hybridization; NGS, next‐generation sequencing; PGP, permeability glycoprotein; SPARC, secreted protein acidic and rich in cysteine.
After 2 months of treatment, a positron emission tomography (PET)/CT revealed a near‐total resolution of hypermetabolic soft tissue thickening involving the pharyngeal mucosa and complete resolution of metabolic activity previously seen within the right prevertebral space and deep cervical fascia consistent with a 90% to 95% response to therapy (see Figure 5). The patient was in near complete remission by PET/CT criteria when he developed bleeding from a cavity left from the previous tumor site. Direct visualization by endoscopy showed no residual tumor; however, mucosal bleeding and inflammation was observed. Bleeding was stopped by local therapy. Unfortunately, the patient presented with recurrent bleeding from the same site causing aspiration and asphyxia resulting in his death.
Figure 5.
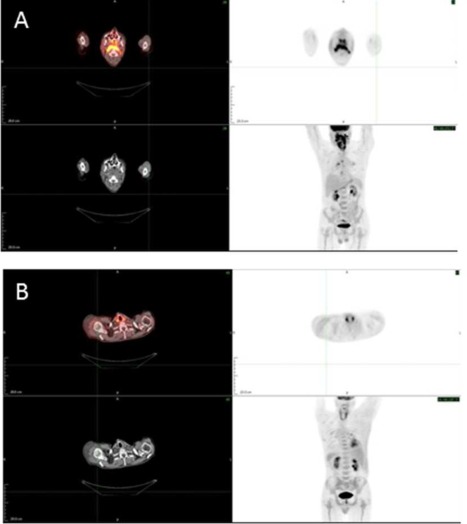
Response to combination treatment strategy (cetuximab, pemetrexed, and nab‐paclitaxel) prospectively designed by comprehensive molecular profiling. (A) Pretreatment fluorodeoxyglucose‐positron emission tomography (FDG‐PET)/CT displaying pharyngeal metabolic activity. (B) Posttreatment (2 months) FDG‐PET/CT demonstrating near complete resolution of metabolic activity.
DISCUSSION
Standard therapy for patients with HNSCC includes a variety of cytotoxic agents (cisplatin, fluorouracil, and paclitaxel) and biologic agents (cetuximab). Over the course of several lines of therapy, exposure to chemotherapeutics may lead to an upregulation of ABC transporters. Many advanced cancers, therefore, exhibit a multidrug resistant phenotype, in which tumor cells are more resistant to cancer agents that are substrates for the cellular transporters that are abundantly expressed and can be easily exuded before the cellular cytotoxic effects are observed. In HNSCC, low expression of PGP and high expression of MRP1 favors selection of chemotherapies that are substrates for PGP (taxanes, epipodophyllotoxins), and not MRP1 (anthracyclines).29, 30 Layering the multidrug resistant status with additional predictive biomarkers may further streamline selection of chemotherapies.
We showed that TOPO2A is overexpressed in a large majority (81%) of HNSCC, however, anthracyclines are considered substrates of MRP1, therefore, targeting a different topoisomerase, such as TOPO1 (60%) with epipodophyllotoxins, may be a more suitable approach because these agents are substrates of PGP. Selecting patients for topoisomerase inhibitor‐based treatment may improve response rates for these agents in HNSCC. In the UK MRC FOCUS trial, TOPO1 expression levels by IHC identified subgroups of patients with metastatic colorectal cancer who benefited from irinotecan, citing high expressers as having a major overall survival benefit from irinotecan.49
The taxanes are also important standard therapies for which predictive biomarkers, including PGP, TUBB3, and TLE3, may identify a potential range of responders. For example, some patients may exhibit low PGP, low TUBB3, and high TLE3, which, in theory, would identify the “best” responders. Additionally, SPARC was also found to be overexpressed in 29%. Studies in patients with HNSCC support the predictive role of SPARC, for selecting nab‐paclitaxel, a taxane with clinical advantages, including shorter infusion times, less hypersensitivity reactions, and fewer toxicities,50 however, larger studies are needed to confirm this observation. Our clinical illustration provides support for switching taxanes to a nanoparticle‐bound formulation of the drug, if SPARC is overexpressed, as this patient demonstrated response to regimen containing nab‐paclitaxel, after progressing on paclitaxel.
Currently, gemcitabine is considered an NCCN guideline‐endorsed chemotherapy for nasopharyngeal cancer,4 however, based on our cohort, low expression occurs in 55% of HNSCC. The high frequency of low RRM1 expression points to the expansion of gemcitabine to be “On‐NCCN compendium” across HNSCC subtypes, beyond nasopharyngeal cancers.
Temozolomide has shown little utility in refractory and/or metastatic HNSCC; however, the clinical data are from unselected cohorts. MGMT is frequently transcriptionally silenced in glioblastoma multiforme and a predictive marker for alkylating agents. Outside of glioblastoma multiforme, a case series of patients with colorectal cancer treated with temozolomide based on low rates of MGMT expression,51 supports the selection of patients based on a predictive biomarker. Our data demonstrated 39% of HNSCC exhibit low expression of MGMT, therefore, future clinical trial design for this agent, and, in particular, utilizing a predictive biomarker, may be worthy of investigation.
Our data demonstrated EGFR amplification in the HPV‐negative cohort only which is supported by recent genomic analyses of HNSCC.52, 53 The role of EGFR (protein or gene copy number) in predicting response to cetuximab, however, is conflicting.7, 8 HPV status was also investigated for its potential role as a predictive marker,54 however, subsequent analyses failed to confirm such observation.55 Recent data suggest the potential role of downstream effectors of the MAPK pathway.9, 56 An improved understanding of HNSCC disease biology is needed to define a predictive marker for cetuximab therapy.
The multiplatform approach provides a comprehensive approach to identifying molecular alterations that may occur at very low frequencies, but are therapeutically relevant. Further, utilization of multiple technologies significantly improves the chance of detecting druggable targets than mono‐platform (aka sequencing alone). Examples include alterations in cMET (overexpression or mutation), HER2 (overexpression or amplification), and mutations in EGFR, FBXW7, Ras family, and Wnt pathway. There may be open clinical trials with biomarker inclusion criteria (cMET), they may be relevant as feedback mechanisms in drug resistance (Ras), or they may be targetable with agents approved for other cancer types (HER2). Case reports in HNSCC demonstrating the clinical impact of these infrequent events are also available.40, 57
Molecular characterization of HNSCC through comprehensive genome sequencing has been reported by several groups.17, 53
We confirm the importance of TP53 and the PIK3CA pathway as major mutational events that are important for HNSCC tumorigenesis. This is captured by our mutation frequency data, as well as the observation of multiple “hits” within these pathways. With the future wider application of whole genome sequencing assays, an important research endeavor will be to distinguish driver mutations from neutral “passenger” mutations. That differentiation will be important for designing dual‐targeted treatment strategies, as well as defining key targets for drug discovery. Our analysis included an inventory of mutations that co‐occurred in our cohort of refractory and/or metastatic HNSCC. Some examples of co‐occurrence have been previously reported (FBXW7 + RAS), however, some have not (ERBB4 + IDH1).
Targeting the PIK3CA pathway has emerged as one of the most promising therapeutic targets in HNSCC. Inhibitors for PIK3CA are in clinical development for other cancer types and PIK3CA has been shown to be one of the most frequently mutated genes in HNSCC, regardless of HPV status (21% to 30%,9, 53 this study was 13%). The observed difference in mutation rates (also for TP53, observed 41%) may be explained by the utilization of hot spot sequencing (our analysis) compared to whole genome sequencing (TCGA). We also found a higher frequency of PIK3CA exon 9 mutations, which may require need for different PIK3CA‐targeted strategies. Exon 9 mutations predict inferior responses compared to exon 20 (H1047R) mutations, as described by Janku et al.44 Also important is the observation of co‐occurring PIK3CA + RAS mutations, which may attenuate response to PIK3CA inhibition.44
Strategizing treatment approaches in the setting of multiple PIK3CA pathway aberrations are currently being investigated. The role of PTEN loss as a resistance mechanism to the PI3K‐alpha inhibitor, BYL719, which is currently in clinical trials for HNSCC, was recently reported in breast cancer. Juric et al11, 45 demonstrated that loss PTEN expression was identified in progressing lesions of a patient with PIK3CA‐mutated metastatic breast cancer who initially responded to BYL719, however, the disease progressed rapidly, followed by death. Our data, indicating almost 24% of PIK3CA‐mutated patients also exhibit loss of PTEN expression, together with these clinical findings, suggest the importance of a multiplatform approach of molecular profiling, which includes assessment of PTEN protein levels by IHC.
Immune evasion is accomplished through deregulated expression of PD1 and PDL1. Recent data suggests this pathway plays an important role in virally driven cancers, including HPV‐positive HNSCC.12 The PD1/PDL1 immune checkpoint, however, is also an intense area of research for cancers with nonviral etiologies.58 Our data demonstrated that immune evasion through deregulation (overexpression) of the PD1/PDL1 axis is relevant to both viral (HPV) and nonviral (TP53) etiologies of HNSCC. Expression of the axis components is also prevalent across HNSCC tumor sites, therefore, is not specific to oropharyngeal subtypes. Elevated expression of PDL1 (24%) was seen at a higher frequency in metastatic HNSCC, which suggests a potential role in facilitating tumor progression.
The heterogeneity of tumor profiles included in this analysis was a major limitation to this study. Furthermore, treatment history and stage of disease at which tumors are sent for profiling (eg, at diagnosis or recurrence) would also greatly enhance this dataset and interpretation of the findings presented. Last, and most importantly, many of the biomarkers found to be altered in HNSCC lack the clinical data, specifically within head and neck cancer clinical studies. However, despite these limitations, we believe the data provided are of great importance in terms of clinical trial development of investigational agents and potential repurposing of traditional chemotherapies. In addition, we do provide a case illustration for which profiling of a patients with recurrent HNSCC allowed for the design of a combination treatment strategy that incorporated a chemotherapy backbone, based on protein biomarker expression, with the only approved targeted agent for HNSCC.
In summary, these data provide a comprehensive cataloging of targetable molecular alterations, which include protein expression, gene copy number, and genetic mutations. The data supports the use of agents in clinical trials (PIK3CA, PD1/PDL1), combination strategies (PIK3CA + EGFR), or agents approved for other solid tumors (MGMT, HER2). We propose a comprehensive molecular profiling approach to enhance personalized therapy options for HNSCC.
Supporting information
Supplementary Information Figure 1.
Supplementary Information Table 1.
Supplementary Information Table 2.
Supplementary Information Table 3.
Conflict of interest: R. Feldman, Z. Gatalica, S. Reddy are employees of Caris Life Sciences. There are no potential conflicts of interest for the other authors.
REFERENCES
- 1. Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer 2011;11:9–22. [DOI] [PubMed] [Google Scholar]
- 2. Chaturvedi AK, Engels EA, Pfeiffer RM, et al. Human papillomavirus and rising oropharyngeal cancer incidence in the United States. J Clin Oncol 2011;29:4294–4301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Stegh AH. Targeting the p53 signaling pathway in cancer therapy – the promises, challenges and perils. Expert Opin Ther Targets 2012;16:67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.National Comprehensive Cancer Network. Head and Neck Cancers, version 2.2014. Available at: http://www.nccn.org/professionals/physician_gls/pdf/head‐and‐neck.pdf. Accessed August 27, 2014.
- 5. Colevas AD. Chemotherapy options for patients with metastatic or recurrent squamous cell carcinoma of the head and neck. J Clin Oncol 2006;24:2644–2652. [DOI] [PubMed] [Google Scholar]
- 6. Szabó B, Nelhubel GA, Kárpáti A, Kenessey I, Jóri B, Szekely C. Clinical significance of genetic alterations and expression of epidermal growth factor receptor (EGFR) in head and neck squamous cell carcinomas. Oral Oncol 2011;47:487–496. [DOI] [PubMed] [Google Scholar]
- 7. Vermorken JB, Peyrade F, Krauss J, et al. Cisplatin, 5‐fluorouracil, and cetuximab (PFE) with or without cilengitide in recurrent/metastatic squamous cell carcinoma of the head and neck: results of the randomized phase I/II ADVANTAGE trial (phase II part). Ann Oncol 2014;25:682–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Licitra L, Mesia R, Rivera F, et al. Evaluation of EGFR gene copy number as a predictive biomarker for the efficacy of cetuximab in combination with chemotherapy in the first‐line treatment of recurrent and/or metastatic squamous cell carcinoma of the head and neck: EXTREME study. Ann Oncol 2011;22:1078–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Lui VW, Hedberg ML, Li H, et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov 2013;3:761–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Janku F, Tsimberidou AM, Garrido–Laguna I, et al. PIK3CA mutations in patients with advanced cancers treated with PI3K/AKT/mTOR axis inhibitors. Mol Cancer Ther 2011;10:558–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Castel P, Juric D, Won H, et al. Loss of PTEN leads to clinical resistance to the PI3Kα inhibitor BYL719 and provides evidence of convergent evolution under selective therapeutic pressure. Presented at the American Association for Cancer Research Annual Meeting 2014; April 5–9, 2014; San Diego, CA. Abstract LB‐327.
- 12. Lyford–Pike S, Peng S, Young GD, et al. Evidence for a role of the PD‐1:PD‐L1 pathway in immune resistance of HPV‐associated head and neck squamous cell carcinoma. Cancer Res 2013;73:1733–1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Von Hoff DD, Stephenson JJ Jr, Rosen P, et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–4883. [DOI] [PubMed] [Google Scholar]
- 14. Tsimberidou AM, Wen S, Hong DS, et al. Personalized medicine for patients with advanced cancer in the phase I program at MD Anderson: validation and landmark analyses. Clin Cancer Res 2014;20:4827–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.National Comprehensive Cancer Network. The NCCN Biomarkers Compendium (NCCN Compendium). Available at: http://www.nccn.org/professionals/biomarkers/content/. Accessed June 1, 2015.
- 16. Millis SZ, Bryant D, Basu G, et al. Molecular profiling of infiltrating urothelial carcinoma of both bladder and non‐bladder origin. Clin Genitourin Cancer 2015;13:e37–e49. [DOI] [PubMed] [Google Scholar]
- 17. Mountzios G, Rampias T, Psyrri A. The mutational spectrum of squamous‐cell carcinoma of the head and neck: targetable genetic events and clinical impact. Ann Oncol 2014;25:1889–1900. [DOI] [PubMed] [Google Scholar]
- 18. Agrawal N, Frederick MJ, Pickering CR, et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in NOTCH1. Science 2011;333:1154–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Stransky N, Egloff AM, Tward AD, et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011;333:1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cancer Genome Atlas Network . Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature 2015;517:576–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Sève P, Mackey J, Isaac S, et al. Class III beta‐tubulin expression in tumor cells predicts response and outcome in patients with non‐small cell lung cancer receiving paclitaxel. Mol Cancer Ther 2005;4:2001–2007. [DOI] [PubMed] [Google Scholar]
- 22. Kulkarni SA, Hicks DG, Watroba NL, et al. TLE3 as a candidate biomarker of response to taxane therapy. Breast Cancer Res 2009;11:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qiu LX, Tang QY, Bai JL, et al. Predictive value of thymidylate synthase expression in advanced colorectal cancer patients receiving fluoropyrimidine‐based chemotherapy: evidence from 24 studies. Int J Cancer 2008;123:2384–2389. [DOI] [PubMed] [Google Scholar]
- 24. Zhao LP, Xue C, Zhang JW, et al. Expression of RRM1 and its association with resistancy to gemcitabine‐based chemotherapy in advanced nasopharyngeal carcinoma. Chin J Cancer 2012;31:476–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Vilmar AC, Santoni–Rugiu E, Sørensen JB. ERCC1 and histopathology in advanced NSCLC patients randomized in a large multicenter phase III trial. Ann Oncol 2010;21:1817–1824. [DOI] [PubMed] [Google Scholar]
- 26. Cohen EE. Role of epidermal growth factor receptor pathway‐targeted therapy in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck. J Clin Oncol 2006;24:2659–2665. [DOI] [PubMed] [Google Scholar]
- 27. Licitra L, Störkel S, Kerr KM, et al. Predictive value of epidermal growth factor receptor expression for first‐line chemotherapy plus cetuximab in patients with head and neck and colorectal cancer: analysis of data from the EXTREME and CRYSTAL studies. Eur J Cancer 2013;49:1161–1168. [DOI] [PubMed] [Google Scholar]
- 28. Munoz M, Henderson M, Haber M, Norris M. Role of the MRP1/ABCC1 multidrug transporter protein in cancer. IUBMB Life 2007;59:752–757. [DOI] [PubMed] [Google Scholar]
- 29. Gottesman MM, Pastan I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu Rev Biochem 1993;62:385–427. [DOI] [PubMed] [Google Scholar]
- 30. Thomas H, Coley HM. Overcoming multidrug resistance in cancer: an update on the clinical strategy of inhibiting p‐glycoprotein. Cancer Control 2003;10:159–165. [DOI] [PubMed] [Google Scholar]
- 31. Gilbert J, Dang T, Cmelak A, et al. Single agent irinotecan for the treatment of metastatic or recurrent squamous carcinoma of the head and neck (SCCHN). Clin Med Oncol 2007;1:59–63. [Google Scholar]
- 32. Desai N, Trieu V, Damascelli B, Soon–Shiong P. SPARC expression correlates with tumor response to albumin‐bound paclitaxel in head and neck cancer patients. Transl Oncol 2009;2:59–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lacroix L, Post SF, Valent A, et al. MET genetic abnormalities unreliable for patient selection for therapeutic intervention in oropharyngeal squamous cell carcinoma. PLoS One 2014;9:e84319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Spiegl–Kreinecker S, Pirker C, Filipits M, et al. O6‐Methylguanine DNA methyltransferase protein expression in tumor cells predicts outcome of temozolomide therapy in glioblastoma patients. Neuro Oncol 2010;12:28–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gong W, Zhang X, Wu J, et al. RRM1 expression and clinical outcome of gemcitabine‐containing chemotherapy for advanced non‐small‐cell lung cancer: a meta‐analysis. Lung Cancer 2012;75:374–380. [DOI] [PubMed] [Google Scholar]
- 36. Hochhauser D, Glynne–Jones R, Potter V, et al. A phase II study of temozolomide in patients with advanced aerodigestive tract and colorectal cancers and methylation of the O6‐methylguanine‐DNA methyltransferase promoter. Mol Cancer Ther 2013;12:809–818. [DOI] [PubMed] [Google Scholar]
- 37. Catimel G, Vermorken JB, Clavel M, et al. A phase II study of gemcitabine (LY 188011) in patients with advanced squamous cell carcinoma of the head and neck. EORTC Early Clinical Trials Group. Ann Oncol 1994;5:543–547. [DOI] [PubMed] [Google Scholar]
- 38. Raguse JD, Gath HJ, Bier J, Riess H, Oettle H. Gemcitabine in the treatment of advanced head and neck cancer. Clin Oncol (R Coll Radiol) 2005;17:425–429. [DOI] [PubMed] [Google Scholar]
- 39. Khan AJ, King BL, Smith BD, et al. Characterization of the HER‐2/neu oncogene by immunohistochemical and fluorescence in situ hybridization analysis in oral and oropharyngeal squamous cell carcinoma. Clin Cancer Res 2002;8:540–548. [PubMed] [Google Scholar]
- 40. Falchook GS, Lippman SM, Bastida CC, Kurzrock R. Human epidermal receptor 2‐amplified salivary duct carcinoma: regression with dual human epidermal receptor 2 inhibition and anti‐vascular endothelial growth factor combination treatment. Head Neck 2014;36:E25–E27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Del Campo JM, Hitt R, Sebastian P, et al. Effects of lapatinib monotherapy: results of a randomised phase II study in therapy‐naive patients with locally advanced squamous cell carcinoma of the head and neck. Br J Cancer 2011;105:618–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chung CH. PIK3CA: the most common “targetable” genetic alteration in head and neck cancer – who might benefit? American Society of Clinical Oncology 2015 meeting. Chicago, IL.
- 43. Fruman DA, Rommel C. PI3K and cancer: lessons, challenges and opportunities. Nat Rev Drug Discov 2014;13:140–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Janku F, Hong DS, Fu S, et al. Assessing PIK3CA and PTEN in early‐phase trials with PI3K/AKT/mTOR inhibitors. Cell Rep 2014;6:377–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Juric D, Castel P, Griffith M, et al. Convergent loss of PTEN leads to clinical resistance to a PI(3)Kα inhibitor. Nature 2015;518:240–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Rampias T, Giagini A, Siolos S, et al. RAS/PI3K crosstalk and cetuximab resistance in head and neck squamous cell carcinoma. Clin Cancer Res 2014;20:2933–2946. [DOI] [PubMed] [Google Scholar]
- 47. Wang Z, Martin D, Molinolo AA, et al. mTOR co‐targeting in cetuximab resistance in head and neck cancers harboring PIK3CA and RAS mutations. J Natl Cancer Inst 2014;106:215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Hyman DM, Snyder AE, Carvajal RD, et al. Parallel phase Ib studies of two schedules of buparlisib (BKM120) plus carboplatin and paclitaxel (q21 days or q28 days) for patients with advanced solid tumors. Cancer Chemother Pharmacol 2015;75:747–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Braun MS, Richman SD, Quirke P, et al. Predictive biomarkers of chemotherapy efficacy in colorectal cancer: results from the UK MRC FOCUS trial. J Clin Oncol 2008;26:2690–2698. [DOI] [PubMed] [Google Scholar]
- 50. Desai N, Trieu V, Yao Z, et al. Increased antitumor activity, intratumor paclitaxel concentrations, and endothelial cell transport of cremophor‐free, albumin‐bound paclitaxel, ABI‐007, compared with cremophor‐based paclitaxel. Clin Cancer Res 2006;12:1317–1324. [DOI] [PubMed] [Google Scholar]
- 51. Shacham–Shmueli E, Beny A, Geva R, Blachar A, Figer A, Aderka D. Response to temozolomide in patients with metastatic colorectal cancer with loss of MGMT expression: a new approach in the era of personalized medicine? J Clin Oncol 2011;29:e262–e265. [DOI] [PubMed] [Google Scholar]
- 52. Seiwert TY, Zuo Z, Keck MK, et al. Integrative and comparative genomic analysis of HPV‐positive and HPV‐negative head and neck squamous cell carcinomas. Clin Cancer Res 2015;21:632–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Walter V, Yin X, Wilkerson MD, et al. Molecular subtypes in head and neck cancer exhibit distinct patterns of chromosomal gain and loss of canonical cancer genes. PLoS One 2013;8:e56823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Vermorken JB, Stöhlmacher–Williams J, Davidenko I, et al. Cisplatin and fluorouracil with or without panitumumab in patients with recurrent or metastatic squamous‐cell carcinoma of the head and neck (SPECTRUM): an open‐label phase 3 randomised trial. Lancet Oncol 2013;14:697–710. [DOI] [PubMed] [Google Scholar]
- 55. Vermorken JB, Psyrri A, Mesía R, et al. Impact of tumor HPV status on outcome in patients with recurrent and/or metastatic squamous cell carcinoma of the head and neck receiving chemotherapy with or without cetuximab: retrospective analysis of the phase III EXTREME trial. Ann Oncol 2014;25:801–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Burtness B. Activity of cetuximab (C) in head and neck squamous cell carcinoma (HNSCC) patients (pts) with PTEN loss or PIK3CA mutation treated on E5397, a phase III trial of cisplatin (CDDP) with placebo (P) or C. American Society of Clinical Oncology 2013 meeting. Chicago, IL.
- 57. Ganesan P, Ali SM, Wang K, et al. Epidermal growth factor receptor P753S mutation in cutaneous squamous cell carcinoma responsive to cetuximab‐based therapy. J Clin Oncol 2014. [Epub ahead of print]. [DOI] [PubMed] [Google Scholar]
- 58. Topalian SL, Hodi FS, Brahmer JR, et al. Safety, activity, and immune correlates of anti‐PD‐1 antibody in cancer. N Engl J Med 2012;366:2443–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information Figure 1.
Supplementary Information Table 1.
Supplementary Information Table 2.
Supplementary Information Table 3.