

Abstract
Apolipoprotein E (ApoE) belongs to a large class of proteins that solubilize lipids for physiological transport. Humans have three different APOE alleles—APOE ε2, APOE ε3 and APOE ε4—and genetic studies identified ApoE4 as the strongest genetic risk factor for Alzheimer’s disease (AD). People who are homozygous for ApoE4 (i.e. ApoE4/E4) are an order of magnitude more likely to develop late-onset AD (LOAD) than ApoE3/E3 carriers. Several differences between ApoE3 and ApoE4 may contribute to AD including the observation that ApoE4 is degraded to a greater extent than ApoE3 in the human brain. Experiments with high-temperature requirement serine peptidase A1 (HtrA1), which is found in the nervous system, demonstrate that HtrA1 is an allele-selective ApoE-degrading enzyme that degrades ApoE4 more quickly than ApoE3. This activity is specific to HtrA1, as similar assays with HtrA2 showed minimal ApoE4 proteolysis and trypsin had no preference between ApoE4 and ApoE3. HtrA1 has also been reported to cleave the tau protein (Tau) and the amyloid protein precursor (APP) to hinder the formation of toxic amyloid deposits associated with AD. Competition assays with ApoE4, ApoE3, and Tau revealed that ApoE4 inhibits Tau degradation. Thus, the identification of ApoE4 as an in vitro HtrA1 substrate suggests a potential biochemical mechanism that links ApoE4 regulation of AD proteins such as Tau.
Graphical Abstract
INTRODUCTION
An aging populous will lead to a larger percentage of individuals with Alzheimer’s disease (AD). Research into AD hopes to fight this outcome by identifying targets that can treat or prevent AD. Genetic studies of families with early-onset (i.e. familial) AD led to the identification of mutations of amyloid precursor protein (APP) and the presenilins (PSEN1 and PSEN2)1,2. Mechanistic studies revealed that the presenilin processing of APP led to the formation of the amyloid beta peptide (Aβ)3–5, which is the major constituent of plaques in the post-mortem brains of AD patients6–8. Increased production of Aβ is associated with AD, and, therefore, AD drugs under development seek to target this pathway9,10. Unfortunately, early results have been disappointing11,12.
In addition to research into early onset AD, which comprises up to 5% of total AD cases13, genetics has also looked for clues for late-onset AD (LOAD), and these experiments identified ApoE4 as the strongest risk factor for LOAD14–16. People that are homozygous for APOE ε4 allele are 12 times more likely to suffer from AD at the age of 65 than corresponding non-APOE ε4 carriers17. ApoE is predominantly produced and secreted from astrocytes18,19 and this protein is responsible for trafficking cholesterol to neurons in the form of lipoprotein particles20,21. Furthermore, research into the role of APOE ε4 and AD has revealed several potential mechanisms that might explain the role of this gene in AD.
ApoE is a 299 amino acid protein with three different isoforms in humans, ApoE2, ApoE3, and ApoE422. The three isoforms differ at two amino acid positions, 112 and 158. ApoE2 has two cysteines at these positions, ApoE4 has two arginines at these positions, and ApoE3 has a cysteine at 112 and an arginine at 15823,24. ApoE4 is the least stable of all three isoforms25,26, and one hypothesis is that this instability leads to accelerated ApoE4 degradation. Indeed, analysis of human tissue from homozygous ApoE3 and ApoE4 genotypes revealed increased ApoE proteolytic fragments in ApoE4 background27,28. Whether the loss of ApoE4 or the generation of ApoE4 fragments are related to AD is not known28–30.
We became interested in trying to identify a candidate protease that might be responsible for ApoE4 breakdown. One study using cultures of primary rat neurons demonstrated that an extracellular serine protease is responsible for ApoE4 processing, though the enzyme was not identified31. High-temperature requirement serine peptidase A1 (HtrA1) belongs to the serine peptidase family. Biochemical experiments have revealed that amyloid precursor protein (APP) and Tau are both HtrA1 substrates32,33. Oligomerization of hyperphosphorylated Tau to form neurofibrillary tangles is another hallmark of AD, and in vitro experiments with HtrA1 showed that this protease can disentangle Tau fibrils and cleave Tau34. Here, we examined the possibility that ApoE4 may also be an HtrA1 substrate.
RESULTS AND DISCUSSION
ApoEs are HtrA1 substrates
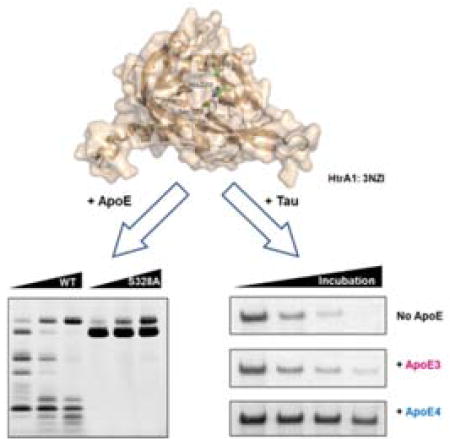
We tested whether HtrA1 is able to mediate the proteolysis of ApoE4. These initial experiments were carried out with purified recombinant ApoE proteins and HtrA1. We incubated recombinant ApoE3 and ApoE4 with various doses of human HtrA1 (10–50 μg/mL). Degradation of ApoE3 and ApoE4 was measured by SDS-PAGE followed by Coomassie staining to visualize remaining full-length ApoE protein and ApoE proteolytic fragments. Upon proteolysis of ApoE, we observed decreased levels of full-length ApoE with corresponding appearance of lower molecular weight proteolytic fragments (Figure 1). Higher concentrations of HtrA1 led to increased proteolysis of ApoEs and their proteolytic fragments (i.e. 20 kDa and 25 kDa in ApoE4 degradation). Mutation of the HtrA1 catalytic serine residue (Ser328) to alanine (HtrA1S328A) is unable to cleave ApoE proteins (Figure 1), which rules out any contaminating proteases as the source of the degradation. Thus, HtrA1 can cleave ApoEs in vitro.
Figure 1. In vitro degradation of ApoE proteins by human HtrA1.

Recombinant ApoE3 or ApoE4 (2 μg) was incubated with purified recombinant human ΔN-HtrA1 (aa 156–480, wild type or S328A mutant) for 2 hours at 37°C. Full-length ApoE proteins and their proteolytic fragments were visualized by SDS-PAGE followed by Coomassie staining.
HtrA1 cleavage of ApoE is allele selective
Comparison of the processing of ApoE3 and ApoE4 indicated that ApoE4 is a better substrate for HtrA1 than ApoE3. At every dose of HtrA1 there is less ApoE4 than ApoE3 and cleavage of ApoE4 generates more proteolytic fragments than ApoE3 (Figure 1).
We quantified any preference for ApoE4 by measuring the percent degradation of ApoE3 and ApoE4 in the presence of HtrA1 over a 22-hour period. Over this time period, we observed more ApoE4 than ApoE3 degradation (Figure 2A and 2B). ApoE3 and ApoE4 share three major proteolysis fragments at ~25, ~20, and ~12 kDa. Proteomics experiments identified these fragments as amino acids 1–195 (~25 kDa), 139–299 (~20 kDa), and 196–299 (~12 kDa) (Figure S1). With ApoE3 these fragments appear stable and continue to accumulate throughout the experiment (Figure 2A). By contrast, the similar ApoE4 fragments are less stable and the levels of most ApoE4 fragments decrease between 10 and 22 hours indicating further proteolysis of these fragments (Figure 2B). Furthermore, ApoE4 proteolysis results in an entire set of proteolytic fragments between 10–20 kDa and less than 10 kDa that are not observed for ApoE3 (Figure 2B). For example, we identified fragments between amino acids 1–125, and 222–299 that are unique to ApoE4 and not observed in the ApoE3 proteolysis (Figure S1).
Figure 2. Kinetic study of in vitro degradation of ApoE proteins by recombinant HtrA1 enzyme.

Recombinant A) ApoE3 and B) ApoE4 protein (5 μg) were incubated with 10 μg/mL human ΔN-HtrA1 (aa 156–480, 15 μL total volume) at 37°C for indicated times. ApoE full-length protein and its proteolytic fragments were Coomassie stained. C) Full-length proteins were quantified and used to calculate the degree of degradation. The percent degradation was determined by dividing band intensity of the full-length ApoE at each time point by the band intensity at the start of the experiment (i.e., t=0). The kinetic curves were fitted by nonlinear regression and the dash lines indicated the time where 50% of the ApoE was degraded.
A plot of degradation percentage against incubation time showed that cleavage of ApoE4 was much faster than ApoE3. At 8 hours, for instance, over 80% of the starting ApoE4 was degraded while less than 40% ApoE3 had undergone proteolysis (Figure 2C). And after 22 hours, almost all ApoE4 was cleaved, while only slightly more than 60% of the ApoE3 was gone. To quantitate the differences between ApoE alleles, we defined the half-life as the time where 50% of the ApoE was degraded. With a half-life of about 2 hours for ApoE4 and a half-life of nearly 13 hours for ApoE3, HtrA1 has a clear allele selectivity for ApoE4 over ApoE3 (Figure 2C). We also calculated reaction rates by a linear fit of the first four time points, and found that the reaction rate for ApoE4 proteolysis is about 7.4 times faster than that for ApoE3 (5.72 and 0.77 × 10−8 M•s−1 respectively, Figure S2).
Specificity of HtrA1-ApoE4 enzyme-substrate interaction
In addition to the knowledge that ApoE4 is a better HtrA1 substrate than ApoE3, we wanted to know whether ApoE4 is a better substrate than other proteins too. We tested this by performing the cleavage reaction in the presence of bovine serum albumin (BSA, 0.1 mg/mL). If HtrA1 binds to and/or cleaves BSA, we would expect to observe a slowing in the ApoE4 processing. Comparison of the ApoE4 proteolysis by HtrA1 in the presence and absence of BSA showed little difference in ApoE4 proteolysis (Figure S3), indicating that HtrA1 recognizes specific features of ApoE4 protein.
Next, we wanted to know whether other peptidases are also able to cleave ApoE4. We tested HtrA2, the 26S proteasome, and trypsin. HtrA2 has an almost identical catalytic domain to HtrA1 (Figure 3A and S4), however, our biochemical studies indicate that HtrA2 cannot cleave either ApoE3 or ApoE4 (Figure 3B and 3C). We also found no proteolysis of ApoE4 in the presence of the 26S proteasome (Figure S5). The fact that HtrA2 and 26S proteasome are not ApoE degrading enzymes indicates that ApoE4 degradation not general.
Figure 3. Comparison of ApoE proteolysis by human HtrA1 and HtrA2.

A) Structural overlay of human HtrA1 (Wheat, PDB id: 3NZI) and HtrA2 (Cyan, PDB id: 1LCY) catalytic domains. The three catalytic triad residues are highlighted in stick representation. H220, D250 and S328 of HtrA1 are shown in green, and H198, D228 and S306 of HtrA2 are shown in magenta. B) Recombinant ApoE3 and C) ApoE4 were incubated with purified human ΔN-HtrA1 (aa 156–480) or human ΔN-HtrA2 (aa 134–458) for 2 hours at 37°C. D) Recombinant ApoE3 and ApoE4 (2 μg) were incubated with indicated amount of trypsin for 30 min at 37°C. ApoE full-length protein and its proteolytic fragments were visualized by Coomassie staining.
Furthermore, trypsin cleaves both ApoE3 and ApoE4, but we found no allele specificity with trypsin (Figure 3D). Comparison of HtrA1 and trypsin indicates unique features about HtrA1-ApoE4 interaction, and indicates that ApoE4 specific degradation is more complicated than ApoE4 being less stable than ApoE3. In addition, the proteolytic fragments of ApoE3 and ApoE4 generated by trypsin differ from HtrA1, indicating that these enzymes use different cleavage sites, which could be part of the explanation of the allele selectivity.
ApoE is degraded by HtrA1 in cell culture
Next, we tested whether HtrA1 can process ApoE4 in cell culture. We transfected full-length HtrA1 with a C-terminal myc tag into HEK293T cells. HtrA1 has a validated N-terminal signaling sequence that directs this enzyme through the secretory pathway, and western blot analysis revealed that most of the myc-tagged HtrA1 is secreted into the cell culture media (Figure 4A). Incubation of ApoE4 with the cell lysate or conditioned media showed that ApoE4 is only degraded in the conditioned media, which contains the highest levels of HtrA1 (Figure 4B and S6).
Figure 4. In vitro degradation of ApoE proteins by HtrA1 transfected HEK293T cells.

A) Expression of full-length HtrA1 (wildtype and S328A mutant) in HEK293T cells indicated that the majority of HtrA1 protein is secreted. B) Recombinant ApoE4 protein (5 μg) were incubated with conditioned media (C.M.) from HEK293T cells transfected with wildtype full-length HtrA1 or S328A mutant at 37 °C for 4 hours. C) Recombinant ApoE3 and ApoE4 (2 μg) were incubated with indicated amount of conditioned media (C.M.) from HEK293T cells transfected with wildtype full-length HtrA1 for 2 hours at 37 °C. ApoE full-length protein and its proteolytic fragments were visualized by western immunoblotting.
To control for the possibility that another extracellular protease is responsible for ApoE4 proteolysis, we repeated the experiment using a catalytically inactive HtrA1 mutant (HtrA1S328A). HtrA1S328A was highly expressed in HEK293T cells and, like wildtype HtrA1, was predominately secreted from the cell (Figure 4A). Conditioned media and cell lysate from HEK293T cells transfected with HtrA1S328A had no activity indicating that HtrA1 is responsible for the ApoE4 proteolysis (Figure 4B and S6). Exogenous HtrA1 expression and secretion is sufficient to mediate ApoE4 cleavage in cell culture, and given that ApoE4 is secreted from astrocytes supports a potential interaction between these two proteins in tissues.
In addition, we compared ApoE3 and ApoE4 cleavage using conditioned media from HEK293T cells transfected with wildtype full-length HtrA1. Both ApoE3 and ApoE4 showed a dose-dependent degradation, but ApoE4 has more proteolytic fragments (Figure 4C). The fragments are similar in size as degradation in vitro by recombinant ΔN-HtrA1. We also performed ApoE degradation over a 26-hour time course by full-length HtrA1 from HEK293T conditioned media (Figure S7). Similarly as ΔN-HtrA1 cleavage, several ApoE4, but not ApoE3, fragments underwent further proteolysis.
Next, we wanted to test whether endogenous HtrA1 is sufficient to induce allele selective degradation of ApoE proteins. U-87 MG human brain glioblastoma cells have the highest HtrA1 mRNA level according to Human Protein Atlas35. We observed robust HtrA1 secretion in conditioned media from U-87 cells (Figure S8). Incubation of ApoE proteins with U-87 conditioned media showed a dose-dependent proteolysis for ApoE4, while ApoE3 cleavage was limited, and the cleavage pattern was similar as purified ΔN-HtrA1 and full-length HtrA1 from HEK293T conditioned medium (Figure 5A).
Figure 5. In vitro degradation of ApoE proteins by U-87 conditioned media.

A) Recombinant ApoE3 and ApoE4 proteins (2 μg) were incubated with U-87 conditioned medium (C.M.) at 37 °C for 3 hours. B) Chemical structure of the HtrA1 inhibitor. C) Recombinant ApoE4 (2 μg) was incubated with U-87 conditioned medium (1 mg/mL) in presence of indicated amount of HtrA1 inhibitor for 3 hours at 37 °C. ApoE full-length protein and its proteolytic fragments were visualized by western blotting.
We then sought to block in vitro ApoE degradation by an HtrA1 inhibitor (HtrA1 Boronic Acid Inhibitor, HBAI) (Figure 5B), which has an IC50 about 0.21 μM36. We chose chemical inhibition over siRNA because this was certain to block the majority of HtrA1 activity. We validated the specificity of HBAI by activity-based proteomics37. We added HBAI at several doses (5–100 uM) to mouse brain lysate in the absence or presence of recombinant ΔN-HtrA1 (for easier analysis) followed by addition of the activity based probe FP-TAMRA. Analysis of the reactions by SDS-PAGE revealed HtrA1 was inhibited by HBAI (Figure S9), but no other proteins showed reduced FP-TAMRA labeling which indicates that HBAI specifically inhibits HtrA1 in the brain proteome. HBAI was also able to block in vitro ApoE degradation by recombinant ΔN-HtrA1 (Figure S10). Next, we used HBAI to test whether HtrA1 is responsible for ApoE4 in U-87 by adding the inhibitor at different doses (0–5 μM) and measuring ApoE4 degradation. HtrA1 inhibition blocked ApoE4 degradation to indicate that HtrA1 is the primary enzyme responsible for ApoE4 proteolysis in U-87 conditioned media (Figure 5C).
ApoE4 inhibits Tau proteolysis by HtrA1
One AD associated pathology is the accumulation of neurofibrillary tangles generated by abnormal aggregation of Tau proteins. In vitro biochemical studies into the regulation of Tau led to the identification of HtrA1 as a Tau degrading enzyme. HtrA1 has recently been reported to have a non-proteolytic activity that enables it to disentangle Tau neurofibrillary tangles, suggesting that it can regulate the levels and aggregation of Tau34.
Since ApoE4 is a major genetic risk factor for Alzheimer’s disease and HtrA1 is able to cleave both ApoE4 and Tau, we wondered whether ApoE4 might compete with Tau as a substrate for HtrA1. To test this hypothesis, we incubated recombinant Tau protein with pure HtrA1 enzyme in the presence of ApoE proteins and analyzed the results at various times (Figure 6). Without ApoE present, HtrA1 degraded Tau leading to a steady decrease in full-length Tau levels over time. And after 90 minutes all of the Tau protein is gone. In the presence of ApoE proteins, we observed a slowdown of Tau degradation, but ApoE4 was a better inhibitor than ApoE3. After 90 minutes, about 40% of full-length Tau protein was still intact in the presence of ApoE4, while only about 20% remained in the ApoE3-containing reaction. A great deal more work needs to be performed in vivo to determine whether ApoE4 and Tau are endogenous substrates of HtrA1. Nevertheless, the concept that ApoE4 proteolysis, which is more prevalent than ApoE2 or ApoE3, is competing with the degradation of AD-related proteins like Tau is compelling whether or not HtrA1 is involved.
Figure 6. Inhibition of Tau degradation by ApoE.

A) Recombinant Tau (600 nM) was incubated with ApoE3 or ApoE4 (600 nM) and purified human ΔN-HtrA1 (156–480, 30 nM) for indicated times at 37°C. Full-length Tau protein was visualized by silver staining. B) The percent degradation was determined by dividing band intensity of the full-length Tau at each time point by the band intensity at the start of the experiment (i.e. t=0) and plotted against incubation time.
Taken together, ApoE and Tau are likely competitive substrates of HtrA1 enzyme, and the preference of ApoE4 over ApoE3 resulted in delayed degradation of Tau aggregates, which provides a new possible mechanism leading to Alzheimer’s disease.
CONCLUSION
As the most significant genetic risk factor for late-onset Alzheimer’s disease, understanding the biochemical, cellular, and physiological function and regulation of ApoE4 is of paramount importance. Here, we tested the possibility that the serine protease HtrA1, which had previously been linked to APP and Tau, is able to proteolytically degrade ApoE4. Our biochemical results indicate that HtrA1 is a candidate ApoE4 regulating enzyme, but additional work in vivo will be necessary to test whether this connection takes place in tissues. A proteomics study using the Tg-SwID mouse model revealed that HTRA1 is the most upregulated gene between 3 to 9 months of age38, and this presents a unique model where we can test the role of HtrA1 on ApoEs in vivo.
Furthermore, we observed that ApoE4 and Tau are competitive substrates for HtrA1. To our knowledge, the concept of ApoE4 inhibition of Tau or Aβ degradation has not been explored. Although Tau is predominantly an intracellular protein, emerging evidence suggests that Tau is secreted to extracellular space in vitro and in vivo39, where it could interact with HtrA1. Therefore, by elucidating this possible biochemical link between ApoE4 and Tau, this work has the potential of introducing a new mechanistic avenue to investigate in vivo. In pursuing these biochemical hypotheses, we will increase our understanding of the regulation and function of ApoE4 in late-onset AD, and this may eventually lead to new opportunities for treating the oncoming AD epidemic.
EXPERIMENTAL SECTION
Materials
Recombinant human HtrA2 (aa 134–458) and human 26S Proteasome Protein were purchased from R&D Systems (1458-HT-100 and E-365). Recombinant Tau protein (2N4R) was obtained from rPeptide (T-1001-2).
Plasmids
ApoE3 cDNA clone was kindly provided by Dr. Zhijiang Chen at the Salk Institute. The ApoE4 cDNA was obtained by site-directed mutagenesis of the ApoE3 sequence using QuikChange II kit (Agilent Technologies). ApoE3 and ApoE4 coding sequences were then subcloned into pMAL vector (New England Biolabs) with a TEV cleavage site between ApoE protein and MBP fusion for bacterial expression. Mammalian expression constructs of HtrA1 cDNA clones with a C-terminal myc tag were obtained from Origene. Human ΔN-HtrA1 (aa 156–480) was subcloned into pET21a vector with a C-terminal His6-tag for bacterial expression. HtrA1S328A mutant was generated by site-directed mutagenesis using QuikChange II kit (Agilent Technologies).
Does-dependent ApoE degradation by pure enzymes
Recombinant ApoE3 and ApoE4 were incubated for 2 hours at 37°C with purified human ΔN-HtrA1 (aa 156–480, wild type or S328A mutant) or human ΔN-HtrA2 (aa 134–458) in 50 mM Tris pH 8.0 with total volume of 15 μL. After reactions were completed, 5 μL of 4×SDS loading dye were added. Samples were boiled at 95°C for 10 min and subjected to SDS-PAGE using Bolt™ 4–12% Bis-Tris Plus polyacrylamide gels (Life Technologies). ApoE full-length protein and its proteolytic fragments were visualized by Coomassie staining.
Time-dependent ApoE degradation by human HtrA1
Recombinant ApoE3 and ApoE4 (5 μg) were incubated with purified human ΔN-HtrA1 (aa 156–480) at 37°C in 50 mM Tris pH 8.0 with total volume of 15 μL. 5 μL 4×SDS loading dye were added at the time points indicated in Figure 2A and 2B. Samples were boiled at 95°C for 10 min and subjected to SDS-PAGE using Bolt™ 4–12% Bis-Tris Plus polyacrylamide gels (Life Technologies). ApoE full-length protein and its proteolytic fragments were visualized by Coomassie staining.
ApoE degradation with HEK293T lysate and conditioned media
HEK293T cells (3×106) were seeded on a 10-cm plate. 24 hours after seeding, cells were transfected in Opti-MEM (Gibco) with 12 μg of plasmid DNA using Lipofectamine 2000 (Life Technologies) according to manufacturer’s protocol. 6 hours post-transfection, media were changed to 10 mL DMEM without phenol red. 72 hours post-transfection, cell culture media were collected and centrifuged at 1,000 rpm for 5 minutes to remove residual cells. The supernatant was then concentrated to 100 μL using a 10 kDa MWCO filter (Millipore). Meanwhile, cells were harvested and lysed in 50 mM Tris pH 8.0, 150 mM NaCl, 0.1% SDS, 0.5% NP-40, 0.5% sodium deoxycholate. Expression of corresponding proteins was tested by western blotting of 20 μg of cell lysate or conditioned medium using anti-myc antibody. Recombinant ApoE4 (5 μg) was incubated with either cell lysate or conditioned medium for 4 hours at 37°C in 50 mM Tris pH 8.0 with total volume of 15 μL. After reactions were completed, 5 μL of 4×SDS loading dye were added. Samples were boiled at 95°C for 10 min and subjected to SDS-PAGE using Bolt™ 4–12% Bis-Tris Plus polyacrylamide gels (Life Technologies). ApoE full-length protein and its proteolytic fragments were visualized by western blotting.
Tau degradation in the presence of ApoE
600 nM of Tau were incubated with 30 nM of purified human ΔN-HtrA1 (aa 156–480) and 600 nM of ApoE3 or ApoE4 in 50 mM Tris pH 8.0 at 37 °C. 10 μL of reaction was taken every 30 minutes, mixed with 5 μL of 4×SDS loading dye. Samples were boiled at 95°C for 10 min and subjected to SDS-PAGE using Bolt™ 4–12% Bis-Tris Plus polyacrylamide gels (Life Technologies). Full-length Tau protein was visualized by silver staining.
Supplementary Material
Acknowledgments
We thank Dr. Zhijiang Chen, Dr. Laura Tan and Professor Paul Sawchenko for fruitful discussions. U-87 MG cell line is a generous gift from Dr. Natalie Luhtala and Professor Tony Hunter at the Salk Institute. We also thank Matthew Kolar for running ABPP gels. Q.C. is a postdoctoral fellow funded by the George E. Hewitt Foundation for medical research. This study was supported by the National Institutes of Health AG042985, AG0476669, the Clayton Foundation, the Schlink Foundation, the Gemcon Family Foundation, the Joe W. and Dorothy Dorsett Brown Foundation and Helen McLoraine Chair in Molecular Neurobiology (K.-F.L.), the NCI Cancer Center Support Grant P30 (CA014195 MASS core, A.S.), the Leona M. and Harry B. Helmsley Charitable Trust grant (#2012-PG-MED002, A.S.), and Dr. Frederick Paulsen Chair/Ferring Pharmaceuticals (A.S.).
Footnotes
Notes
The authors declare no competing financial interest.
Supplementary methods and figures are described. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Kim DH, Yeo SH, Park JM, Choi JY, Lee TH, Park SY, Ock MS, Eo J, Kim HS, Cha HJ. Gene. 2014;545:185. doi: 10.1016/j.gene.2014.05.031. [DOI] [PubMed] [Google Scholar]
- 2.Chouraki V, Seshadri S. Adv Genet. 2014;87:245. doi: 10.1016/B978-0-12-800149-3.00005-6. [DOI] [PubMed] [Google Scholar]
- 3.O’Brien RJ, Wong PC. Annu Rev Neurosci. 2011;34:185. doi: 10.1146/annurev-neuro-061010-113613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Haass C, Kaether C, Thinakaran G, Sisodia S. Cold Spring Harb Perspect Med. 2012;2:a006270. doi: 10.1101/cshperspect.a006270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Newman M, Musgrave IF, Lardelli M. Biochim Biophys Acta. 2007;1772:285. doi: 10.1016/j.bbadis.2006.12.001. [DOI] [PubMed] [Google Scholar]
- 6.Cras P, Kawai M, Lowery D, Gonzalezdewhitt P, Greenberg B, Perry G. Proc Natl Acad Sci USA. 1991;88:7552. doi: 10.1073/pnas.88.17.7552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hardy J, Selkoe DJ. Science. 2002;297:353. doi: 10.1126/science.1072994. [DOI] [PubMed] [Google Scholar]
- 8.Thinakaran G, Koo EH. J Biol Chem. 2008;283:29615. doi: 10.1074/jbc.R800019200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mucke L. Nature. 2009;461:895. doi: 10.1038/461895a. [DOI] [PubMed] [Google Scholar]
- 10.Yamin G, Ono K, Inayathullah M, Teplow DB. Curr Pharm Des. 2008;14:3231. doi: 10.2174/138161208786404137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Teich AF, Arancio O. Biochem J. 2012;446:165. doi: 10.1042/BJ20120653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cummings JL, Morstorf T, Zhong K. Alzheimers Res Ther. 2014;6:37. doi: 10.1186/alzrt269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanzi RE. J Alzheimers Dis. 2013;33:S5. doi: 10.3233/JAD-2012-129044. [DOI] [PubMed] [Google Scholar]
- 14.Mahley RW, Weisgraber KH, Huang Y. Proc Natl Acad Sci USA. 2006;103:5644. doi: 10.1073/pnas.0600549103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Science. 1993;261:921. doi: 10.1126/science.8346443. [DOI] [PubMed] [Google Scholar]
- 16.Strittmatter WJ, Roses AD. Proc Natl Acad Sci USA. 1995;92:4725. doi: 10.1073/pnas.92.11.4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kim J, Basak JM, Holtzman DM. Neuron. 2009;63:287. doi: 10.1016/j.neuron.2009.06.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pitas RE, Boyles JK, Lee SH, Foss D, Mahley RW. Biochim Biophys Acta. 1987;917:148. doi: 10.1016/0005-2760(87)90295-5. [DOI] [PubMed] [Google Scholar]
- 19.DeMattos RB, Brendza RP, Heuser JE, Kierson M, Cirrito JR, Fryer J, Sullivan PM, Fagan AM, Han X, Holtzman DM. Neurochem Int. 2001;39:415. doi: 10.1016/s0197-0186(01)00049-3. [DOI] [PubMed] [Google Scholar]
- 20.Ikonen E. Nat Rev Mol Cell Biol. 2008;9:125. doi: 10.1038/nrm2336. [DOI] [PubMed] [Google Scholar]
- 21.Puglielli L, Tanzi RE, Kovacs DM. Nat Neurosci. 2003;6:345. doi: 10.1038/nn0403-345. [DOI] [PubMed] [Google Scholar]
- 22.Liu CC, Kanekiyo T, Xu H, Bu G. Nat Rev Neurol. 2013;9:106. doi: 10.1038/nrneurol.2012.263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mahley RW, Rall SC. Annu Rev Genomics Hum Genet. 2000;1:507. doi: 10.1146/annurev.genom.1.1.507. [DOI] [PubMed] [Google Scholar]
- 24.Roses AD. Annu Rev Med. 1996;47:387. doi: 10.1146/annurev.med.47.1.387. [DOI] [PubMed] [Google Scholar]
- 25.Clement-Collin V, Barbier A, Dergunov AD, Visvikis A, Siest G, Desmadril M, Takahashi M, Aggerbeck LP. Biophys Chem. 2006;119:170. doi: 10.1016/j.bpc.2005.07.009. [DOI] [PubMed] [Google Scholar]
- 26.Morrow JA, Segall ML, Lund-Katz S, Phillips MC, Knapp M, Rupp B, Weisgraber KH. Biochemistry. 2000;39:11657. doi: 10.1021/bi000099m. [DOI] [PubMed] [Google Scholar]
- 27.Jones PB, Adams KW, Rozkalne A, Spires-Jones TL, Hshieh TT, Hashimoto T, von Armin CAF, Mielke M, Bacskai BJ, Hyman BT. PLoS ONE. 2011;6:e14586. doi: 10.1371/journal.pone.0014586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mahley RW, Huang YD. Neuron. 2012;76:871. doi: 10.1016/j.neuron.2012.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rohn TT. Int J Mol Sci. 2013;14:14908. doi: 10.3390/ijms140714908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tolar M, Marques MA, Harmony JAK, Crutcher KA. J Neurosci. 1997;17:5678. doi: 10.1523/JNEUROSCI.17-15-05678.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tamboli IY, Heo D, Rebeck GW. PLoS One. 2014;9:e93120. doi: 10.1371/journal.pone.0093120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Grau S, Baldi A, Bussani R, Tian X, Stefanescu R, Przybylski M, Richards P, Jones SA, Shridhar V, Clausen T, Ehrmann M. Proc Natl Acad Sci USA. 2005;102:6021. doi: 10.1073/pnas.0501823102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tennstaedt A, Popsel S, Truebestein L, Hauske P, Brockmann A, Schmidt N, Irle I, Sacca B, Niemeyer CM, Brandt R, Ksiezak-Reding H, Tirniceriu AL, Egensperger R, Baldi A, Dehmelt L, Kaiser M, Huber R, Clausen T, Ehrmann M. J Biol Chem. 2012;287:20931. doi: 10.1074/jbc.M111.316232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Poepsel S, Sprengel A, Sacca B, Kaschani F, Kaiser M, Gatsogiannis C, Raunser S, Clausen T, Ehrmann M. Nat Chem Biol. 2015;11:862. doi: 10.1038/nchembio.1931. [DOI] [PubMed] [Google Scholar]
- 35.Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S, Alm T, Edqvist PH, Berling H, Tegel H, Mulder J, Rockberg J, Nilsson P, Schwenk JM, Hamsten M, von Feilitzen K, Forsberg M, Persson L, Johansson F, Zwahlen M, von Heijne G, Nielsen J, Ponten F. Science. 2015;347:1260419. doi: 10.1126/science.1260419. [DOI] [PubMed] [Google Scholar]
- 36.Grau S, Richards PJ, Kerr B, Hughes C, Caterson B, Williams AS, Junker U, Jones SA, Clausen T, Ehrmann M. J Biol Chem. 2006;281:6124. doi: 10.1074/jbc.M500361200. [DOI] [PubMed] [Google Scholar]
- 37.Saghatelian A, Jessani N, Joseph A, Humphrey M, Cravatt BF. Proc Natl Acad Sci USA. 2004;101:10000. doi: 10.1073/pnas.0402784101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Searcy JL, Le Bihan T, Salvadores N, McCulloch J, Horsburgh K. PLoS ONE. 2014;9:e89970. doi: 10.1371/journal.pone.0089970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang Y, Mandelkow E. Nat Rev Neurosci. 2016;17:5. doi: 10.1038/nrn.2015.1. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.